

Abstract
Zirconium-89 is an attractive metallo-radionuclide for use in immunoPET due to the favorable decay characteristics. Standardized methods for the routine production and isolation of high purity and high specific-activity 89Zr using a small cyclotron are reported. Optimized cyclotron conditions reveal high average yields of 1.52 ± 0.11 mCi/μA·h at a proton beam energy of 15 MeV and current of 15 μA using a solid, commercially available 89Y-foil target (0.1 mm, 100% natural abundance). 89Zr was isolated in high radionuclidic and radiochemical purity (>99.99%) as [89Zr]Zr-oxalate by using a solid-phase hydroxamate resin with >99.5% recovery of the radioactivity. The effective specific-activity of 89Zr was found to be in the range 5.28 – 13.43 mCi/μg (470 – 1195 Ci/mmol) of zirconium. New methods for the facile production of [89Zr]Zr-chloride are reported. Radiolabeling studies using the trihydroxamate ligand desferrioxamine B (DFO) gave 100% radiochemical yields in <15 min. at room temperature and in vitro stability measurements confirmed that [89Zr]Zr-DFO is stable with respect to ligand dissociation in human serum for >7 days. Small-animal PET imaging studies have demonstrated that free 89Zr(IV) ions administered as [89Zr]Zr-chloride accumulate in the liver whilst [89Zr]Zr-DFO is excreted rapidly via the kidneys within <20 min. These results have important implication for the analysis of immunoPET imaging of 89Zr-labeled monoclonal antibodies. The detailed methods described can be easily translated to other radiochemistry facilities and will facilitate the use of 89Zr in both basic science and clinical investigations.
Keywords: Zirconium-89, cyclotron targetry, hydroxamates, desferrioxamine, radiopharmaceuticals, positron emission tomography
1. Introduction
Zirconium-89 is perhaps one of the most promising metallo-radionuclides for developing new immuno-positron emission tomography (immunoPET) agents for in vivo imaging of cancer. [1–4] The physical decay properties of 89Zr (Figure 1: t1/2 = 78.41(12) h, EC = 76.6%, β+ = 22.3%, Emax(β+) = 897 keV, Eave.(β+) = 396.9 keV, Rave.(β+) = 1.18 mm, Eγ = 908.97(3) keV, Iγ = 100%) are ideally suited for use in the design of monoclonal antibody-based (mAb) radiotracers which require extended in vivo circulation times for optimal biodistribution and tumor targeting.[5, 6] The relatively low translational energy of the emitted positron also results in high resolution 89Zr images comparable to those observed with the 18F and 64Cu radionuclides (Rave.(β+) = 0.69 and 0.70 mm, respectively).[1, 7, 8]
Figure 1.

Nuclear decay scheme showing the main pathways for the decay of 89Zr and 89mZr to 89 Y.[5, 47]
Despite promising in vivo results, the nuclear medicine community has been slow to embrace the potential of 89Zr due to inefficient methods for its separation from the 89Y target material and challenging aqueous chelation chemistry. Separation methods involving solvent extraction,[9–12] cation and anion exchange chromatography,[10, 11, 13] and the use of solid-phase hydroxamate resins[6, 14–16] have been described. However, these lengthy procedures are technically challenging and often lead to low efficiencies for the recovery of 89Zr activity with highly variable radiochemical purity (21.7 – 97.5%).[11, 13, 16, 17] The low specific-activity of the recovered 89Zr results in further problems with the chelation chemistry and is likely to complicate quantitative analysis of in vivo PET imaging studies. Unlike the more commonly exploited metallo-radionuclides such as 68Ga, 86Y, and 60/61/62/64Cu, 89Zr has a preference to form 8-coordinate complexes and the group oxidation state of 4+ imparts a high propensity towards hydrolysis in aqueous solutions. The ionic nature of most Zr(IV) complexes means that the chemistry more closely resembles that of radiolanthanides such as 177Lu.
As a consequence of the challenges in targetry, separation and chelation chemistry, no standardized methods for the routine production of 89Zr have been reported. The work presented in this article describes validated benchmark procedures for the facile production of clinical-grade high specific-activity 89Zr by using a small cyclotron. In addition, in vitro and in vivo studies on the suitability of the hexadentate, trihydroxamate ligand, desferrioxamine B (Desferol, DFO) as a chelate for 89Zr(IV) are presented.
2. Materials and Methods
2.1 General experimental details
Yttrium-89 (100% naturally abundant) metal foil (0.1 mm thick, 4.47 g cm−3 at 20 °C, >99.9%) was purchased from American Elements (Los Angeles, CA). Trace Metal® Grade (<1 ppb metal impurity) 32 – 35% hydrochloric acid was purchased from Fisher Scientific (Pittsburgh, PA) and diluted to suitable concentrations (6, 3, 2, 1, 0.5 and 0.1 M) with >18.2 MΩ.cm−1 water (25 °C, Milli-Q, Millipore, Billerica, MA) which had been purified by passing through a 5 cm column of chelex resin (Bio-Rad Laboratories, Hercules, CA). Where appropriate, solvents that were pretreated with chelex resin to remove the metal ions are indicated throughout by the term “chelex” in parentheses. Dissolution of the 89Y irradiated target was performed in a 30 mL polytetrafluoroethylene (PTFE) tube which had pre-washed with Trace Metal c.HCl for >24 h, followed by H2O (chelex) and allowed to dry at room temperature. An aqueous solution of 1 M oxalic acid (4.50 g, 0.05 mol, Puriss grade >99.0%, SigmaAldrich, St. Louis, MO) was prepared by endothermic dissolution in H2O (50 mL, chelex). Desferrioxamine B mesylate was obtained from Calbiochem (Spring Valley, CA).89Zr-radiolabeling reactions were monitored by using silica-gel impregnated glass fibre instant thin-layer chromatography (ITLC-SG) paper (Pall Corp., East Hills, NY) and analyzed on a radio-TLC plate reader (Bioscan System 200 Imaging Scanner coupled to a Bioscan Autochanger 1000 (Bioscan Inc., Washington, DC) and Win-Scan Radio-TLC software version 2.2). Solvent systems included 0.05 M (DTPA) and phosphate buffered saline (1 × PBS). For more accurate quantification of activities experimental samples were counted for 1 min. on a calibrated Perkin Elmer (Waltham, MA) Automatic Wizard2 Gamma Counter using a dynamic energy windows of 800 – 1000 keV for 89Zr (909 keV emission) and 420 – 62 keV for 86Y (511 keV emission). All radioactive detection devices were calibrated, and maintained in accordance with previously reported routine quality-control procedures.[18]
Attenuated total internal reflectance (ATIR) spectroscopy was performed by using a Bruker Tensor 27 infra-red spectrometer (4000 – 650 cm−1) equipped with a Pike MIRacle single-bounce attenuated total reflectance (ATR) cell with a ZnSe single crystal (Bruker Daltonics Inc., Billerica, MA). The ATIR data were collected and processed by using OPUS version 5.5 software.
2.2 Cyclotron targetry and irradiation
Irradiations were conducted by using an EBCO TR19/9 variable beam energy (H−: 13 – 19 MeV, 1 – 150 μA, 5.0 × 10−7 Torr) cyclotron (Ebco Industries Inc., Richmond, British Columbia, Canada). Incident proton-beam energies between 15 – 16.5 MeV, and a beam current of 15 μA were used in 2 – 5 h target irradiations. The 89Zr was produced via the 89Y(p,n)89Zr transmutation reaction using a solid 89Y-foil (48 mm × 16 mm × 0.1 mm, approx. 0.33 g, 3.70 mmol) target mounted on a custom-made water-cooled target with a 10° angle of incidence. The radionuclidic purity of the product was determined by gamma-spectroscopy by using a high purity germanium (HPGe) detector (Canberra model GC2018) coupled to a calibrated multichannel analyzer (MCA, Canberra Inspector 2000, Canberra Industries, Oak Ridge, TN). The MCA was calibrated by using 133Ba (81.0, 302.8 and 356.0 keV), 109Cd (88.0 keV), 57Co (122.1 keV), 60Co (1173.2 and 1332.5 keV), 137Cs (661.6 keV) and 22Na (1274.5 keV) standard sources from Canberra Industries, Oak Ridge, TN and data were processed by using the Genie-2000 software. Yttrium-86 was produced and isolated as [86Y]-YCl3 in 0.1 M HCl by using the electrochemical methods described by Yoo et al.[19] Radioactivity measurements were made by using a Capintec CRC-15R Dose Calibrator (Capintec, Ramsey, NJ) with calibration factors of 465 and 711÷2 for 89Zr and 86Y, respectively.
2.3 Separation chemistry
89Zr was purified from the 89Y target material by use of a hydroxamate resin following a modified procedures described by Meijs et al.[16] and Verel et al.[6] The hydroxamate resin was prepared by functionalizing the carboxylate groups of the silica-based Waters Accell™ Plus CM weak cation exchange resin (an acrylic acid/acrylamide copolymer on Diol silica (Waters Corporation, Milford, MA). Surface functionality: –CO2−Na+, 300 Å pore size, 37 – 55 μm particle size, 0.35 mmol/g ligand density). Accell resin (1.0 g) was suspended in 8.0 mL of water (chelex) in Falcon tube (15 mL, Corning Life Sciences, Lowell, MA), to which was added 75 μL of 3 M HCl(aq.), 1.0 mL of 2,3,5,6-tetrafluorophenol (TFP, 200 mg/mL, 1.20 M, SigmaAldrich) in MeCN (chelex), and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDAC, 385 mg, 2.0 mmol, SigmaAldrich) coupling agent as a solid. CAUTION: TFP causes severe burns and is very hygroscopic. Fresh solutions of TFP are essential for successful reactions. The final pH was in the range 5.9 – 6.2 and was measured by using a calibrated pH meter (accumet AP84 meter with an accupHast pH electrode, Fisher Scientific). The reaction was mixed on an inversion mixer (ATR Inc.) at room temperature for 1 h after which time further 3 M HCl(aq.) (100 μL), TFP (1.0 mL, 1.20 M in MeCN) and EDAC (385 mg, 2.0 mmol) were added (pH5.8 – 6.1) and the reaction mixed by inversion at room temperature for 2 – 3 h. The resin was then collected by filtration and washed with water (chelex, 3 × 25 mL) and MeCN (3 × 25 mL) to remove any unreacted TFP and EDAC and the 1-(2-(dimethylamino)ethyl)-3-methylurea byproduct.
The resin which contained the activated TFP ester group was then converted into the hydroxamate resin by reaction with hydroxylamine. Hydroxylamine hydrochloride (0.695 g, 0.01 mol, SigmaAldrich) was dissolved in 1.0 mL of 1.0 M NaOH(aq.) and 2.0 mL of MeOH and after 15 min. the pH was adjusted to 5.3 – 5.4 by the addition of 25 – 50 μL of 1.0 M NaOH(aq.). This solution was added to the activated resin in a 15 mL Falcon tube, and the reaction (pH5.0 – 5.2) was mixed by inversion at room temperature for 18 h. The product was then collected by filtration and washed thoroughly with water (5 × 25 mL) and MeCN (5 × 25 mL) and dried in vacuo. Verel et al.[6] found that the hydroxamate resin was stable for up to 4 months and our investigations have confirmed that when stored at ambient temperature, the resin remains active for periods >12 months. The reaction can be scaled up to >5 g without compromising the efficiency of the coupling chemistry or the quality of the final hydroxamate resin obtained.
Approximately 1 h before the required separation of 89Zr, a custom made column consisting of a 4.0 mL Alltech SPE Extract-Clean reservoir (Alltech, Lancaster, PA) with hydroxamate resin (100 mg) packed between two frits was prepared. The column was activated by washing with MeCN (8 mL, chelex), water (15 mL, chelex and 2.0 M HCl(aq.) (2 mL). Our studies indicate that up to 50% less resin material can be used without compromising the separation efficiency and recovery of the 89Zr radioactivity.
The irradiated 89Y-foil target (approx. 0.33 g, 3.70 mmol) was transferred to a metal-ion free PTFE tube (50 mL) and was dissolved by the addition of 4 × 0.5 mL fractions of 6 M HCl(aq.). Attempts to dissolve this mass of target in 1 M HCl(aq.) as described by Verel et al.[6] were unsuccessful so higher concentrations of acid were used. To ensure complete oxidation H2O2(aq.) (25 μL, 30 – 32 wt. % in water, semiconductor grade, 99.999% trace metals basis, SigmaAldrich) can be added but was not found to affect the separation or subsequent chelation chemistry. The solution was then diluted with water (5 mL, chelex) to ensure that the final concentration of <2 M HCl(aq.). The solution was then transferred via pipette to the pre-washed hydroxamate column. The solution was eluted through the resin by placing the column under controlled positive pressure by using an improvised suba-seal pierced with a polyetheretherketone (PEEK) tubing connected to a Luer-lok syringe. This mechanism also facilitated minimal radiation exposure. After loading the 89Zr activity, the column was washed with 2 M HCl(aq.) (4 × 2.5 mL) and water (4 × 2.5 mL) to remove the soluble Y(III) ions and other impurities. Insoluble material remained trapped on the frit above the resin and did not break through. After blowing air through the column to remove the solvent, the 89Zr activity was slowly eluted with 5 successive portions of 1.0 M oxalic acid (4 × 0.5 mL, and 1 × 1.0 mL, elution time >2 min. for the first two fractions). The activity in each fraction and residual activity on the column were measured by using the dose calibrator (vide infra).
2.4 Resin binding assays
The binding affinity of 89Zr(IV) and 86Y(III) ions to the hydroxamate resin in HCl(aq.) solutions of varying concentration (6, 3, 2, 1, 0.5, 0.1 M) was assessed by conducting isotopic dilution assays. Briefly, 0.05 M stock solutions were prepared by dissolving anhydrous ZrCl4 (0.175 g, 0.75 mmol, >99.99% trace metal basis) and YCl3·6H2O (0.228 g, 0.75 mmol, >99.99% trace metal basis) in water (15 mL, chelex). Twelve reactions were prepared in 1.5 mL Eppendorf tubes via 1:2 serial dilution (500 μL samples) to give final non-radioactive Zr(IV) and Y(III) amounts in the range (25.0 – 0.01 μg). Stock solutions of [89Zr]Zr-chloride (vide infra) and [86Y]Y-chloride[19] (approx. 10 μCi/mL) were prepared by dissolution in various concentrations of HCl(aq.). Aliquots of the [89Zr]Zr-chloride or [86Y]Y-chloride stock solutions (50 μL, approx. 0.5 μCi) were added to each of the appropriate 500 μL solutions of non-radioactive ZrCl4(aq.) or YCl3(aq.). A 250 – 400 mg sample of hydroxamate resin was pre-washed in accordance with the protocol described above then resuspended in HCl(aq.) of varying concentration to give a final suspension of between 160 – 200 mg/mL. The binding assay was initiated by adding 50 μL aliquots of the hydroxamate resin suspension (8 – 10 mg, vortexed immediately prior to withdrawing the sample) to each of the reactions (total volume = 600 μL). The reactions were then incubated on a rotator at room temperature for 2 h. After incubation, the resin was allowed to settle to the bottom of the Eppendorf tubes before carefully withdrawing 550 μL of the supernatant. The radioactivity in the supernatant and resin pellet fractions was counted by using a calibrated Perkin Elmer Automatic Wizard2 Gamma Counter. The binding capacity corresponding to 100% loading of the hydroxamate resin with 89Zr(IV) ions was estimated by measuring the mass of amount of non-radioactive Zr at ca. 50% or 25% 89Zr binding, dividing the value by 2 or 4, respectively, then normalizing for the mass of resin in each reaction. Experiments were performed in triplicate on three separate occasions.
2.5 Specific-activity
The specific activity (mCi/μg) of 89Zr was determined experimentally via titration of the purified solution of [89Zr]Zr-oxalate in 1.0 M oxalic acid with the desferrioxamine B (DFO). The use of DFO as a ligand for 89Zr(IV) has been evaluated by Meijs et al.[20] Quantitative complexation of 89Zr with DFO was found to occur within 10 min. at room temperature (vide infra) which confirmed the suitability of DFO as a ligand for use in determination of specific-activity. Briefly, a stock solution of DFO (1.0 mg/mL, 1.77 mM) in water (chelex) and 500 μL was added to an Eppendorf tube (1.5 mL) containing 500 μL of water (chelex). Fifteen reactions were prepared via 1:2 serial dilution to give final DFO masses in the range (8.3 – 0.0005 μg). Solutions of [89Zr]Zr-oxalate (in 1.0 M oxalic acid) or [89Zr]Zr-chloride (in 0.1 M HCl(aq.)) were neutralized with the appropriate volume of 1.0 M Na2CO3 and stock solutions of activity were prepared by dilution with water (chelex). Aliquots of the [89Zr]Zr-oxalate or [89Zr]Zr-chloride stock solutions (50 μL, 25 – 30 μCi) were added and the reactions were vortexed for 15 s, then incubated on a rotator at room temperature for 1 h (final pH = 5 – 8). After incubation, 1 μL aliquots were withdrawn analyzed by ITLC using DTPA (50 mM, pH 7) as a mobile phase solvent. Free 89Zr forms an instantaneous complex with DTPA and eluted with the solvent from, whilst [89Zr]Zr-DFO remained at the origin. ITLC plates were measured by using radio-TLC plate reader. For more accurate quantification the strips were cut in half and the γ-rays emissions at 909 keV were counted on a calibrated Perkin Elmer Automatic Wizard2 Gamma Counter using a dynamic energy window of 800 – 1000 keV. The minimum ligand concentration for which 100% labeling occurred was estimated by multiplying the measured value at 50% by 2, and the specific-activity of 89Zr, in both mCi/μg and Ci/mmol of zirconium, was calculated by correcting for the total activity and molecular weight of DFO and 89Zr.
2.6 Preparation of [89Zr]Zr-chloride
[89Zr]Zr-oxalate solution in 1.0 M oxalic acid was loaded onto an activated Waters Sep-pak Light QMA strong anion exchange cartridge (an acrylic acid/acrylamide copolymer on Diol silica. Surface functionality: –C(O)NH(CH2)3N(CH3)3+ Cl−, 300 Å pore size, 37 – 55 μm particle size, 0.22 mmol/g ligand density), pre-washed with 6 mL MeCN, 10 mL 0.9% saline and 10 mL water (chelex). In all experiments >99.9% of the 89Zr activity remained trapped on the cartridge. The cartridge was then washed with water (>40 mL, chelex) and eluted with 1.0 M HCl(aq.) (300 – 500 μL). The amount of residual oxalic acid was estimated by collecting the eluate in a pre-weighed glass culture tube, and re-weighing after boiling the solution to dryness at 110 – 120 °C for 10 min. From the calculated mass of oxalic acid loaded onto the QMA cartridge, the estimated residual amount after elution was <0.2%. The [89Zr]Zr-chloride can be reconstituted in either water, 0.9% saline or 0.1 M HCl(aq.).
2.7 Chelation chemistry
[89Zr]Zr-DFO was prepared by the complexation of either [89Zr]Zr-oxalate (in 1.0 M oxalic acid) or [89Zr]Zr-chloride (in 0.1 M HCl(aq.)) with DFO. Aliquots of either [89Zr]Zr-oxalate or [89Zr]Zr-chloride (typically <20 μL, 300 – 500 μCi) were diluted in 100 μL of either water or 0.9% saline (NaCl, 4.5 g, 77 mmol, dissolved in 500 μL of water (chelex)) in 1.8 mL centrifuge tubes (Corning Life Sciences). The excess 1.0 M oxalic acid or 0.1 M HCl was neutralized by adding the appropriate volume of Na2CO3(aq.) (1.0 M or 0.1 M in water (chelex)) and the pH adjusted to 7 – 8 with <1 μL portions of 0.1 M HCl(aq.) and 0.1 M Na2CO3(aq.). An aliquot of DFO (10 μL, 1.0 mg/mL in water (chelex)) was then added, and the reaction mixture was vortexed for 30 s and left to react at room temperature for between 5 – 15 min. Samples were withdrawn at suitable time points and analyzed by ITLC using DTPA (50 mM, pH 7) as a mobile phase solvent. All reactions (in the pH range 6 – 9) were found to give 100% radiochemical yield within 15 min. as shown by the absence of any 89Zr activity at the solvent front in the ITLC. The lower bound of [89Zr]Zr-DFO specific-activity was in the range 30 – 50 μCi/μg of DFO ligand but typically specific activities between 2 and three orders of magnitude higher can be obtained.
In vitro stability of measurements of [89Zr]Zr-DFO (<15 μL, 30 – 50 μCi) in 500 μL of DTPA (50 mM pH7), PBS (pH7.4) and human serum (pH7) (filtered using a low protein-binding Acrodisc PF syringe filter, 0.8/0.2 μm Supor Membrane, Pall Corp.) were conducted. Solutions were incubated at 37.0 °C for 5 days and were analyzed at suitable time point by using ITLC and counting the plates using both a radio-TLC plate reader and gamma counter. Formulations of [89Zr]Zr-DFO or [89Zr]Zr-chloride (150 μL, 450 – 500 μCi) for in vivo studies were prepared in 0.9% saline at pH7.2 – 7.4.
2.8 PET imaging
All animal experiments were performed in accordance with the National Institutes of Health (NIH) and Institutional Animal Care and Use Committee (IACUC) guidelines. PET imaging was conducted on a microPET Focus 120 scanner (Concorde Microsystems).[21] Male athymic nu/nu mice (18 weeks old, Taconic) were first anesthetized by inhalation of 1% isoflurane (Baxter Healthcare)/oxygen gas mixture (flow rate of 2.0 L/min.) and placed on the scanner bed in the prone position. List-mode data were acquired for 60 minutes beginning immediately following tail-vein injection of either [89Zr]Zr-DFO or [89Zr]Zr-chloride (150 μL, 450 – 500 μCi, pH7.2 – 7.4) as a solution in 0.9% saline via intravenous (i.v.). An energy window of 350 – 750 keV and a coincidence timing window of 6 ns were used. The list-mode data were binned into 60 one-minute frames. For subsequently acquired static images a minimum of 20 million coincident events were recorded over 10 min. Data were sorted into 2-dimensional histograms by Fourier re-binning, and transverse images were reconstructed by filtered back-projection (FBP) into a 128 × 128 × 63 (0.72 × 0.72 × 1.3 mm) matrix. The reconstructed spatial resolution for 89Zr was 1.9 mm full-width half maximum (FWHM) at the center of the field-of-view (FOV). The image data were normalized to correct for non-uniformity of response of the microPET, dead-time count losses, positron branching ratio, and physical decay to the time of injection but no attenuation, scatter, or partial-volume averaging correction was applied. An empirically determined system calibration factor (in units of mCi/mL/cps/voxel) for mice was used to convert voxel count rates to activity concentrations. The resulting image data were then normalized to the administered activity to parameterize images in terms of percent of the injected doses per gram (%ID/gm). Manually drawn 2-dimensional regions-of-interest (ROIs) or 3-dimensional volumes-of-interest (VOIs) were used to determined the maximum %ID/g (decay corrected to the time of injection) in various organs.[22] Images were analyzed using ASIPro VM™ software (Concorde Microsystems).
3. Results and Discussion
Mealey reported the application of 89Zr (formulated in 1% sodium citrate) for the, “…external localization of human brain tumors…,” in 1957.[23, 24] Since these pioneering studies, several groups in the United States, Europe and Japan have explored the production, isolation and chelation chemistry of 89Zr(IV) ions for potential use in immunoPET.[8–13, 16, 17, 20] In particular, over the last two decades the research groups of Herscheid[16, 20, 25, 26] and van Dongen[6, 27–37] working at the Vrije University Medical Centre (Amsterdam, The Netherlands) have developed facile protocols for producing 89Zr-labeled mAbs for immunoPET.
In 2006, a pilot study involving 89Zr-labeled ibritumomab (89Zr-Zevalin) imaging of a male patient with B-cell non-Hodgkin’s Lymphoma (NHL) was reported.[32] In the same year Börjesson et al. described results from the first clinical trials of 89Zr-labeled chimeric mAb, U36 for the detection of lymph node metastases in 20 patients with squamous cell carcinoma of the head and neck (HNSCC).[33] These results showed that immunoPET imaging using 89Zr-DFO-U36 was able to detect all primary HNSCC tumors (n = 17) as well as lymph node metastases with high sensitivity (72%) and accuracy (93%). These data were comparable to the diagnostic results obtained by using [18F]-2-fluoro-2-deoxy-D-glucose ([18F]-FDG) PET, computed tomography (CT) and magnetic resonance imaging (MRI) in the same patients. In addition, 89Zr-labeled mAbs have been described as potential surrogate tracers for evaluating the in vivo dosimetry of 90Y and 177Lu-labeled mAbs for use in radioimmunotherapy (RIT).[1, 27, 28, 31, 32]
3.1 89Zr production and isolation
Zirconium-89 was produced from the 89Y(p,n)89Zr transmutation reaction by proton bombardment of commercially available, high-purity (99.99%, 0.1 mm thick) 89Y-foil on a small cyclotron. Proton and deuteron bombardment of 89Y-targets has been investigated by several research groups.[10, 11, 16, 17, 38–40] Recently, Mustafa et al. used the foil-activation method to obtain excitation functions for the 89Y(p,n)89Zr, 89Y(p,2n)88Zr and 89Y(p,pn)88Y transmutation reactions at proton beam energies up to 40 MeV.[40] Their results indicate that the p,n reaction for formation of 89Zr has an experimental threshold bombardment energy for formation around 4.59 MeV, which is close to the calculated Q-value of 2832.7(25) keV, and displays a maximum average cross-section (σave.) of 787.2 ± 2.6 mb at 13.80 MeV. In contrast, threshold energy values for the formation of 88Zr and 88Y occur around 13.30 MeV with σave. values of 786.9 ± 1.5 and 298.0 ± 5.7 mb, at 22.98 and 28.30 MeV, respectively. At incident proton beam energies <16 MeV, no contamination from 88Zr or 88Y was observed at the end-of bombardment (EOB) in our experiments.
A photograph of the custom-made water-cooled target assembly is shown in Figure 2 and details from 4 production representative runs are presented in Table 1. In total, over 15 irradiations have been conducted with typical total yields of 89Zr 40 – 60 mCi after 2 – 3 h irradiation. A proton beam energy of 15 MeV, with a 15 μA beam current was found to give optimum yields of 89Zr with an average production rate of 1.52 ± 0.11 mCi/μA·h. NB: The target must be allowed to cool for 1 h after the end-of-bombardment (EOB) to allow decay of the short-lived isomeric product, 89mZr (t1/2 = 4.161(17) min.).
Figure 2.

Photograph of the Memorial Sloan-Kettering Cancer Center (MSKCC) custom-made water-cooled solid-target assembly for the TR19/9 cyclotron used in the proton bombardment for the 89Y(p,n)89Zr transmutation reaction.
Table 1.
Experimental 89Zr production yields.a
| Run | Beam Energy (MeV) | Beam Current (μA) | Irradiation time (h) | Beam Charge μA·h | Decay Corrected EOB 89Zr Activity (mCi) | Yield (mCi/μA·h) |
|---|---|---|---|---|---|---|
| 1 | 16.5 | 15 | 3.42 | 51.25 | 59.18 | 1.15 |
| 2 | 15.0 | 15 | 2.08 | 31.25 | 44.99 | 1.44 |
| 3 | 15.0 | 15 | 2.50 | 37.50 | 54.82 | 1.46 |
| 4 | 15.0 | 15 | 2.67 | 40.00 | 65.92 | 1.65 |
The average yield for runs 2 – 4 is 1.52 ± 0.11 mCi/μA·h.
After cooling, the target was dissolved in 4 × 0.5 mL portions of 6 M HCl(aq.). Dissolution was complete within 10 min. However, a small trace of black insoluble material remained in suspension. This material is likely to be an insoluble form of yttrium chloride and was found not to contain any 89Zr activity. A small aliquot of H2O2 was added in accordance with the procedures described by Verel et al.[6] However, in aqueous solution Zr(IV) ions are the predominant species and the Zr(IV) + 4e− → Zr(0) reaction has a reduction potential of E° = −1.51 V versus the standard hydrogen electrode (SHE).[41] Experiments in which H2O2 was omitted showed no difference in the observed separation and chelation chemistry of the isolated 89 Zr samples.
The hydroxamate resin has been characterized by both spectroscopic and radiochemical binding studies. Functional group conversion of the carboxylate-to-hydroxamate ligand was characterized by using attenuated total internal-reflectance (ATIR) infra-red spectroscopy. Due to the nature of the solid polymer-coated resin and the low ligand density (<0.35 mmol/g) ATIR signal intensities were weak and as a consequence, only changes in very strongly absorbing carbonyl groups were observed. Difference spectra (hydroxamate – carboxylate resin spectra) revealed decreased absorption intensity at 1573 and 1407 cm−1, characteristic of the loss of the asymmetric ( νas) and symmetric (νs) C=O stretching modes of the carboxylate resin, respectively. An increase in absorption at 1737 and 1654 cm−1 was assigned to the νas and νs C=O stretching modes of –C(O)NHOH, respectively. As expected, the ATIR data indicate that the “hydroxamate resin” is actually present in the protonated hydroxamic acid form.
The separation efficiency and relative binding affinities of the hydroxamate resin towards Zr(IV) and Y(III) ions was measured by conducting isotopic dilution assays at different HCl(aq.) concentrations (Figure 3). For three separate resin preparations the estimated maximum loading capacity of Zr(IV) ions at 2 M HCl(aq.) was found to be in the range 0.13 – 0.31 (average: 0.25 ± 0.08) mmol/g. Assuming a 1:1 binding of hydroxamate-to-Zr(IV) ions, these data indicate that for the three preparations, the chemical reactions to convert the carboxylate resin into the hydroxamate form have overall estimated yields of 37%, 87% and 89% with an average of 71%. Lower yields are most likely due to inefficient formation of the activated tetrafluorophenol (TFP) ester and in these experiments it was found that use of freshly prepared TFP solutions in MeCN was essential for efficient TFP-ester formation. Similar isotopic dilution experiments performed with 89Y showed that in solutions of 1 – 6 M HCl(aq.), Y(III) ions do not bind to the resin.
Figure 3.

Measured loading capacity of the hydroxamate resin towards 89Zr(IV) ions in 2 M HCl(aq.) solution. Binding capacities measured in triplicate for 3 separate hydroxamate resin preparations were in the range 0.13 – 0.31 (average: 0.25 ± 0.08) mmol/g. Overall yields for the two-step carboxylate-to-hydroxamate functional group conversion reactions were found to be in the range 37 – 89%, with an average of 71%.
The loading of 89Zr(IV) ions was found to be sensitive to the concentration of HCl(aq.) used. At HCl(aq.) concentrations ≤2 M, the 89Zr(IV) loading efficiency was found to be >99.9%. However, at higher HCl(aq.) concentrations, (3 – 6 M) the binding affinity of 89Zr to the hydroxamate resin decreases and at 6 M HCl(aq.) <26.0% of the activity was bound to the column. Attempts to elute the 89Zr activity using higher concentrations of HCl(aq.) were unsuccessful which indicates that in contrast to resin loading, elution appears to be insensitive to acid concentration.
Elution of 89Zr was achieved with >99.5% recovery of the radioactivity by transchelation with 5 successive aliquots of 1.0 M oxalic acid (Table 2).[6, 16] Attempts to elute the activity with lower concentrations of oxalic acid revealed that concentrations <0.5 M were unable to effect elution. Only partial recovery of the 89Zr activity (13 – 34%) was achieved by using oxalic acid concentrations between 0.5 – 0.75 M. The nature of the aqueous phase Zr(IV)-oxalate species present at various pH values has recently been investigated by Kobayashi et al. and the [ZrIV(C2O4)4]4− was found to have a thermodynamic formation constant of log β = 29.7 ± 0.1.[42–44] Due to the low pKa of oxalic acid and the highly polarizing nature of the Zr(IV) ions, it likely that the tetra anionic species, [ZrIV(C2O4)4]4−, predominates in solution even at low pH values between 0 – 2. Reported labeling experiments are usually conducted in the pH range 5 – 9 and under these conditions [ZrIV(C2O4)4]4− will be the species present in solution.[6, 28, 30–37]
Table 2.
Typical data observed for the elution of 89Zr (19.82 mCi) from the hydroxamate resin.
| Fraction | Oxalic acid volume/mL | Activity/mCi | % |
|---|---|---|---|
| 1 | 0.5 | 13.08 | 66.00 |
| 2 | 0.5 | 5.35 | 27.00 |
| 3 | 0.5 | 0.850 | 4.30 |
| 4 | 0.5 | 0.352 | 1.77 |
| 5 | 1.0 | 0.080 | 0.40 |
| Column Residue | - | 0.106 | 0.53 |
In addition to inefficient separation chemistries, one of the main problems encountered in previous reports on the production of 89Zr has been contamination from the long-lived isotopes, 88Zr (t1/2 = 83.4(3) d) and 88Y (t1/2 = 106.65(4) d).[5, 8] In these studies, the radionuclidic purity of the isolated 89Zr fractions was found to be >99.99% which γ-ray spectroscopy confirming the absence of both 88Zr and 88Y (Figure 4). Also, unlike most other metallo-radionuclide preparations, which require the use of isotopically enriched targets (such as 64Ni(p,n)64Cu and [86Sr]SrCO3(p,n)86Y), 89Y occurs in 100% natural abundance which means complicated target-recycling steps are unnecessary.
Figure 4.

Spectrum of the γ ray emissions observed from a purified sample of 89Zr recorded 16 h after the end-of-bombardment (EOB). The spectrum is free of common and potential radioactive impurities as shown by the absence of peaks corresponding to 88Zr (392.87(9) keV, 100%)) and 88Y (898.042(3) and 1836.063(12) keV, with relative intensities of 93.7(3)% and 99.2(3)%, respectively) and the 89Zr was isolated in >99.99% radionuclidic purity.[5] Two small peaks are observed at 1461 and 1713 keV assigned to the decay of 40K (0.01% natural abundance) and 89Zr (Iγ = 0.76%, intensity relative to the emission at 909 keV), respectively. The isomeric excited state species 89mZr decays rapidly within 40 min. after the end of bombardment and was not observed.
The results presented here are consistent with other reported chemical separation studies which indicate that in contrast to separation using solvent exchange methods or cationic or anionic exchange resins, the hydroxamate column method (originally developed by Herscheid et al.[14, 16]), is the most efficient, providing 89Zr in >99.9% radiochemical yield.[6]
3.2 Chelation chemistry and specific-activity
For potential use in the labeling of mAbs, 89Zr solutions of high specific-activity and high radiochemical purity are essential. Specific-activities of the isolated 89Zr fractions have not been reported previously. The specific-activity of the isolated 89Zr was determined experimentally by isotopic dilution similar to methods described by McCarthy et al. and Yoo et al. for 64Cu and 86Y, respectively.[19, 45] Complexation of [89Zr]Zr-oxalate with DFO was found to occur in <15 min. at room temperature in with 100% radiochemical yield. The same reaction was used to measure specific-activity (Figure 5). In all isolation experiments, the effective specific-activity of 89Zr was found to be in the range 5.28 – 13.43 mCi/μg (470 – 1195 Ci/mmol) of zirconium. The maximum specific-activity (Smax in units of Ci/g) is given by Equation 1, where NA is Avogadro’s number (6.022 × 1023 particles/mol), λ is the decay constant (seconds) and Ar is the atomic mass (g/mol).
Figure 5.

DFO ligand titration experiment used in the determination of specific-activity of a purified 89Zr(IV) oxalate solution.
| (1) |
The calculated maximum specific-activity of 89Zr is 449035 Ci/g. Such high values are inaccessible and a more practical range suitable for labeling experiments for immunoPET can be estimated by comparing the measured specific-activity of 89Zr with reported range of “high” specific-activities for 64Cu.[45] By taking the ratio of S(89Zr)/S(64Cu) = 0.0732, the range of 888 – 1238 Ci/mmol was calculated for 89Zr. These data indicate that the isolated 89Zr is comparable to the high specific-activity samples of 64Cu produced at various institutions including Washington University School of Medicine (St. Louis, Missouri) and is suitable for use in immunoPET.[45]
Although the [89Zr]Zr-oxalate solution is suitable for complexation reactions involving the DFO ligand, oxalic acid is highly toxic (due to decalcification of blood and acute renal failure due to the obstruction of kidney tubules by calcium oxalate) and must be removed before performing any in vitro or in vivo studies. Therefore, we investigated methods for the efficient preparation of [89Zr]Zr-chloride. Meijs et al. have described lengthy procedures for the production of [89Zr]Zr-chloride from purified [89Zr]Zr-oxalate involving decarboxylation of the excess oxalic acid with H2O2 in 6 M HCl(aq) at 80 °C followed by drying of the reaction mixture at room temperature in vacuo or alternatively by the use of room temperature vacuum sublimation techniques at 2 × 10−4 Torr.[16] Our investigations found that [ZrIV(C2O4)4]4− can be removed from solution with 100% efficiency by trapping on an activated a Sep-pak QMA strong anion exchange cartridge. The majority (>99.8%) of the oxalic acid was then removed by washing the cartridge with a large volume of water, and the activity eluted with 100% recovery of 89Zr by chloride ion exchange with 300 – 500 μL of 1.0 M HCl(aq.). Standard 0.9% saline can also be used to elute the 89Zr activity but the recovery of activity was found less efficient (22 – 38% in 500 μL of saline). The acid can be removed rapidly by boiling the eluate at 110 °C under a continuous stream of argon, then reconstituted in 0.9% saline or 0.1 M HCl(aq.) for further labeling studies. The entire procedure for the formation of [89Zr]Zr-chloride requires <1 h.
In vitro stability investigations revealed that [89Zr]Zr-DFO is stable (>99%) with respect to ligand dissociation/demetalation for over 7 days at 37.0 °C in PBS and human serum. Stability measurements in the presence of DTPA (50 mM, pH7, 37.0 °C) indicated that transchelation of 25 – 30% of the 89Zr from DFO to DTPA occurred after 7 days. The formation constant of [ZrIV(DFO)]+ has not been reported but the complex is known to be more stable that that of [FeIII(DFO)]+ which has a log β around 32. DTPA represents a particularly strong ligand challenge and overall the stability results demonstrate that DFO is a suitable choice of ligand for complexation and conjugation of 89Zr(IV) ions to biologically active vectors such as mAbs.[6, 20] In contrast, to [89Zr]Zr-oxalate, the [89Zr]Zr-chloride in 0.1 M HCl(aq.) was found to react rapidly with phosphate and in PBS solution, insoluble species form which remain at the origin of the ITLC experiments. In this respect, [89Zr]Zr-chloride behaves in a similar way to [86/90Y]Y-chloride and it is anticipated that radiolabeling experiments with [89Zr]Zr-chloride may best be achieved in acetate buffers at pH4.5 – 6.0.
3.3 PET resolution measurements
The spatial resolution of 89Zr obtained by using the microPET Focus 120 small-animal imaging camera was assessed by using the Derenzo phantom and specific line-width measurements in a glass capillary (Figures 6a and b). The phantom contains 6 different sized pores with diameters increasing by 0.4 mm intervals between 1.2 – 3.2 mm, and separated by a distance equal to the diameter of the specified pore. From line-width measurements, the reconstructed spatial resolution for 89Zr was 1.94 mm full-width half maximum (FWHM) at the center of the field-of-view (FOV). The image demonstrates that excellent spatial resolution of objects separated by 2.0 mm can be achieved with PET imaging of 89Zr. Although the presence of the γ-ray at 909 keV (100% abundant) will affect dosimetry calculations for 89Zr-labeled radiotracers, it does not appear to inhibit the acquisition of high resolution PET images. These results are comparable to the resolution obtained with 18F and 64Cu radionuclides.[7, 28]
Figure 6.
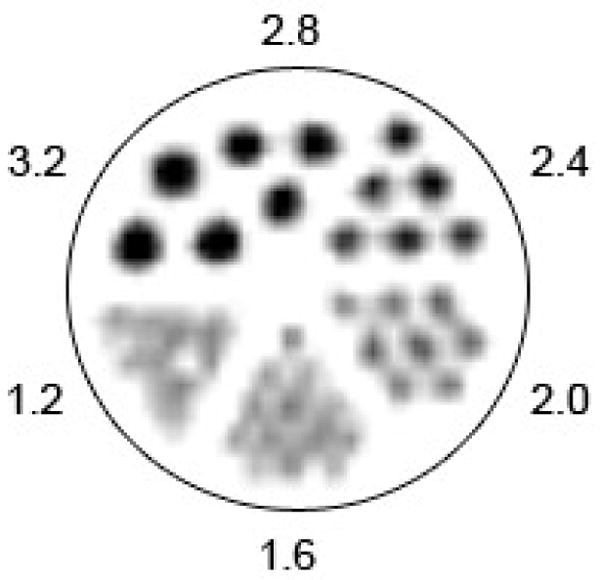
Spatial resolution measurements showing: a) a static 10 min. PET image recorded by using a “Derenzo phantom” filled with 6.0 mL of 89Zr (22.5 μCi/mL). Images were reconstructed by using filtered back-projection; b) the line-width profile and Gaussian-fit measured from a static 10 min. PET image of 89Zr (35 μCi) in a glass microcapillary pipette (10 μm diameter, Kimble Glass Inc.). The full-width at half-maximum (FWHM) is 1.94 mm which indicates that image resolution using 89Zr is approximately the same as the specified detector resolution of the Focus 120 microPET camera, and is comparable to the resolution obtained by using 18F or 64Cu radionuclides.
3.4 Small-animal imaging
Small-animal PET imaging experiments were conducted to examine the in vivo behaviour of [89Zr]Zr-DFO and [89Zr]Zr-chloride (Figures 7a and b). Dynamic PET imaging is consistent with the observations of Meijs et al.,[25] and showed that [89Zr]Zr-DFO is cleared very rapidly from the blood pool via glomerular filtration in the kidneys. Tissue-activity curves (TACs) generated from the 1 h dynamic PET experiment of [89Zr]Zr-DFO in a male, athymic nu/nu mouse demonstrate that virtually all of the injected activity is localized in the bladder within 20 min. post-administration. In contrast, [89Zr]Zr-chloride localizes rapidly in the liver and remains trapped for >8 h post-administration.
Figure 7.

Small-animal PET images of male athymic nu/nu mice injected with 89Zr-activity via the tail vein and showing the in vivo distribution of a) [89Zr]Zr-DFO recorded between 0 – 4 min., and b) [89Zr]Zr-chloride 8 h post-administration.
For immunoPET, two of the most common methods of excretion and/or degradation of the radiotracer are demetalation/ligand dissociation and hydrolysis of the linker group between, for example the mAb and the metallo-chelate. These PET imaging data demonstrate that if 89Zr ions are lost from potential [89Zr]Zr-DFO-mAb immunoPET conjugates via demetalation, high liver accumulation may be expected. In contrast, if the [89Zr]Zr-DFO complex remains intact, rapid renal excretion may be anticipated which would decrease background tissue accumulation and enhance image contrast. The exquisite images in both animal and human studies reported by van Dongen and co-workers suggest that [89Zr]Zr-DFO-mAb immunoPET agents have great potential as an alternative to other radionuclides including 124I, 64Cu, and 86Y for immunoPET.[6, 27, 28, 30–37, 46] Further investigations into the use of 89Zr-labeled mAbs, peptides and small molecules are underway.
4. Summary and Conclusions
Standardized methods for the production and isolation of high specific-activity 89Zr have been described. The targetry conditions and separation chemistry have been optimized to provide high yields of 89Zr with radionuclidic and radiochemical purities in excess of 99.99%. Specific-activities of the isolated 89Zr fractions have been measured and were found to be comparable to the reported specific-activities of other PET radionuclides including 64Cu and 86Y, which are sufficiently high for labeling of mAb-based radiotracers. In addition, new methods for the facile isolation of [89Zr]Zr-chloride are reported which will facilitate the development of new chelation chemistry. The methods described can be easily adapted to most nuclear medicine facilities and will facilitate the use of 89Zr in both basic science and clinical investigations of immunoPET. High specific-activity 89Zr is now produced routinely at the Memorial Sloan-Kettering Cancer Center (MSKCC) and further studies on the chelation chemistry are underway.
Figure 8.

Tissue-activity curves (TACs) of the heart, kidneys and bladder generated from volumes-of-interest (VOIs) drawn on the dynamic PET images of [89Zr]Zr-DFO recorded between 0 – 60 min. post-administration.
Acknowledgments
We thank Dr. NagaVaraKishore Pillarsetty and Dr. Peter M. Smith-Jones for assistance with the radiochemistry and also thank the staff of the Radiochemistry/Cyclotron Core at MSKCC. Technical services provided by the MSKCC Small-Animal Imaging Core Facility, supported in part by NIH Small-Animal Imaging Research Program (SAIRP) Grant No. R24 CA83084 and NIH Center Grant No. P30 CA08748, are gratefully acknowledged and we thank Dr. Pat Zanzonico, Valerie A. Longo and Dr. Gordon Roble. We also thank Bradley Beattie for assistance with the PET imaging. The generous support of Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research, and The Experimental Therapeutics Center of Memorial Sloan-Kettering Cancer Center are gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nayak TK, Brechbiel MW. Radioimmunoimaging with longer-lived positron-emitting radionuclides: potentials and challenges. Bioconjugate Chem. 2009 doi: 10.1021/bc800299f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boswell CA, Brechbiel MW. Development of radioimmunotherapeutic and diagnostic antibodies: an inside-out view. Nucl Med Biol. 2007;34:757–78. doi: 10.1016/j.nucmedbio.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Dongen GAMS, Visser GWM, Lub-de Hooge MN, de Vries EG, Perk LR. Immuno-PET: a navigator in monoclonal antibody development and applications. Oncologist. 2007;12:1379–89. doi: 10.1634/theoncologist.12-12-1379. [DOI] [PubMed] [Google Scholar]
- 4.Wu AM. Antibodies and antimatter: The resurgence of immuno-PET. J Nucl Med. 2009;50:2–5. doi: 10.2967/jnumed.108.056887. [DOI] [PubMed] [Google Scholar]
- 5.Mustafa MG, West HIJ, O’Brien H, Lanier RG, Benhamou M, Tamura T. Measurements and a direct-reaction plus Hauser-Feshbach analysis of 89Y(p,n)89Zr, 89Y(p,2n)88Zr, and 89Y(p,pn)88Y reactions up to 40 MeV. Phys Rev C. 1988;38:1624–37. doi: 10.1103/physrevc.38.1624. [DOI] [PubMed] [Google Scholar]
- 6.Verel I, Visser GWM, Boellaard R, Stigter-van Walsum M, Snow GB, van Dongen GAMS. 89Zr immuno-PET: Comprehensive procedures for the production of 89Zr-labeled monoclonal antibodies. J Nucl Med. 2003;44:1271–81. [PubMed] [Google Scholar]
- 7.Lewis JS, Singh RK, Welch Michael J. Long lived and unconventional PET radionuclides. In: Pomper MG, Gelovani JG, editors. Chapter 18 in Molecular Imaging in Oncology. Informa Healthcare; New York, NY: 2009. pp. 283–92. [Google Scholar]
- 8.Hohn A, Zimmermann K, Schaub E, Hirzel W, Schubiger PA, Schibli R. Production and separation of “non-standard” PET nuclides at a large cyclotron facility: the experiences at the Paul Scherrer Institute in Switzerland. Quarterly J Nucl Med Mol Imaging. 2008;52:145–50. [PubMed] [Google Scholar]
- 9.Inarida M, Shimamura A. Separation of carrier-free zirconium from yttrium target. Rikagaku Kenkyusho Hokoku. 1970;46:63–5. [Google Scholar]
- 10.Link JM, Krohn KA, Eary JF, Kishore R, Lewellen TK, Johnson MW, et al. 89Zr for antibody labelling and positron tomography. J Labeled Compd Radiopharm. 1986;23:1296–7. [Google Scholar]
- 11.DeJesus OT, Nickles RJ. Production and purification of 89Zr, a potential PET antibody label. Appl Radiat Isotopes. 1990;41:789–90. [Google Scholar]
- 12.Lahiri S, Mukhopadhyay B, Das NA. Simultaneous production of 89Zr and 90,91m,92mNb in alpha-particle activated yttrium and their separation by HDEHP. Appl Radiat Isotopes. 1997;48:883–6. [Google Scholar]
- 13.Kandil SA, Scholten B, Saleh ZA, Youssef AM, Qaim SM, Coenen HH. A comparative study on the separation of radiozirconium via ion-exchange and solvent extraction techniques, with particular reference to the production of 88Zr and 89Zr in proton induced reactions on yttrium. J Radioanal Nucl Chem. 2007;274:45–52. [Google Scholar]
- 14.Herscheid JDM, Vos CM, Hoekstra A. Manganese-52m for direct application: a new 52Fe/52mMn generator based on a hydroxamate resin. Int J Appl Radiat Isotopes. 1983;34:883–6. doi: 10.1016/0020-708x(83)90147-3. [DOI] [PubMed] [Google Scholar]
- 15.Fadeeva VI, Tikhomirova TI, Yuferova IB, Kudryavtsev GV. Preparation, properties and analytical applications of silica with chemically grafted hydroxamic acid groups. Anal Chim Acta. 1989;219:201. [Google Scholar]
- 16.Meijs WE, Herscheid JDM, Haisma HJ, Wijbrandts R, van Langevelde F, van Leuffen PJ, et al. Production of highly pure no-carrier added 89Zr for the labelling of antibodies with a positron emitter. Appl Radiat Isotopes. 1994;45:1143–7. [Google Scholar]
- 17.Zweit J, Downey S, Sharma HL. Production of no-carrier-added zirconium-89 for positron emission tomography. Appl Radiat Isotopes. 1991;42:199–201. [Google Scholar]
- 18.Zanzonico P. Routine quality controlof clinical nucliear medicine instrumentation: a brief review. J Nucl Med. 2009;49:1114–31. doi: 10.2967/jnumed.107.050203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoo J, Tang L, Perkins Todd A, Rowland Douglas J, Laforest R, Lewis Jason S, et al. Preparation of high specific activity (86)Y using a small biomedical cyclotron. Nucl Med Biol. 2005;32:891–7. doi: 10.1016/j.nucmedbio.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 20.Meijs WE, Herscheid JDM, Haisma HJ, Pinedo HM. Evaluation of desferal as a bifunctional chelating agent for labeling antibodies with Zr-89. Appl Radiat Isotopes. 1992;43:1443–7. doi: 10.1016/0883-2889(92)90170-j. [DOI] [PubMed] [Google Scholar]
- 21.Kim JS, Lee JS, Im KC, Kim SJ, Kim S-Y, Lee DS, et al. Performance measurement of the microPET Focus 120 scanner. J Nucl Med. 2007;48:1527–35. doi: 10.2967/jnumed.107.040550. [DOI] [PubMed] [Google Scholar]
- 22.Tseng J-C, Zanzonico PB, Levin B, Finn R, Larson SM, Meruelo D. Tumor-specific in vivo transfection with HSV-1 thymidine kinase gene using a sindbis viral vector as a basis for prodrug ganciclovir activation and PET. J Nucl Med. 2006;47:1136–43. [PubMed] [Google Scholar]
- 23.Mealey J. Turn-over of carrier-free zirconium-89 in man. Nature. 1957;179:673–4. doi: 10.1038/179673a0. [DOI] [PubMed] [Google Scholar]
- 24.Mealey J., Jr Application of positron-emitting zirconium-89 for potential use in brain tumor localization. Surg Forum. 1958;9:718–21. [PubMed] [Google Scholar]
- 25.Meijs WE, Haisma HJ, Van der Schors R, Wijbrandts R, Van den Oever K, Klok RP, et al. A facile method for the labeling of proteins with zirconium Isotopes. Nuc Med Biol. 1996;23:439–48. doi: 10.1016/0969-8051(96)00020-0. [DOI] [PubMed] [Google Scholar]
- 26.Meijs WE, Haisma HJ, Klok RP, van Gog FB, Kievit E, Pinedo HM, et al. Zirconium-labeled monoclonal antibodies and their distribution in tumor-bearing nude mice. J Nucl Med. 1997;38:112–8. [PubMed] [Google Scholar]
- 27.Verel I, Visser GWM, Boerman O, Van Eerd J, Finn R, Boellaard R, et al. Long-lived positron emitters Zirconium-89 and Iodine-124 for scouting of therapeutic radioimmunoconjugates with PET. Cancer Biother Radiopharmaceut. 2003;18:655–61. doi: 10.1089/108497803322287745. [DOI] [PubMed] [Google Scholar]
- 28.Verel I, Visser GWM, Boellaard R, Boerman O, Van Eerd J, Snow GB, et al. Quantitative 89Zr immuno-PET for in vivo scouting of 90Y-labeled monoclonal antibodies in Xenograft-bearing nude mice. J Nucl Med. 2003;44:1663–70. [PubMed] [Google Scholar]
- 29.Verel I, Visser GWM, Vosjan MJWD, Finn R, Boellaard R, Van Dongen GAMS. High-quality 124I-labelled monoclonal antibodies for use as PET scouting agents prior to 131I-radioimmunotherapy. Eur J Nucl Med Mol Imaging. 2004;31:1645–52. doi: 10.1007/s00259-004-1632-8. [DOI] [PubMed] [Google Scholar]
- 30.Brouwers A, Verel I, Van Eerd J, Visser G, Steffens M, Oosterwijk E, et al. PET Radioimmunoscintigraphy of renal cell cancer using 89Zr-labeled cG250 monoclonal antibody in nude rats. Cancer Biother Radiopharmaceut. 2004;19:155–63. doi: 10.1089/108497804323071922. [DOI] [PubMed] [Google Scholar]
- 31.Perk LR, Visser GWM, Vosjan MJWD, Stigter-van Walsum M, Tijink BM, Leemans CR, et al. 89Zr as a PET surrogate radioisotope for scouting biodistribution of the therapeutic radiometals 90Y and 177Lu in tumor-bearing nude mice after coupling to the internalizing antibody Cetuximab. J Nucl Med. 2005;46:1898–906. [PubMed] [Google Scholar]
- 32.Perk LR, Visser OJ, Stigter-van Walsum M, Vosjan MJWD, Visser GWM, Zijlstra JM, et al. Preparation and evaluation of 89Zr-Zevalin for monitoring of 90Y-Zevalin biodistribution with positron emission tomography. Eur J Nucl Med Mol Imaging. 2006;33:1337–45. doi: 10.1007/s00259-006-0160-0. [DOI] [PubMed] [Google Scholar]
- 33.Börjesson PKE, Jauw YWS, Boellaard R, de Bree R, Comans EFI, Roos JC, et al. Performance of immuno-Positron Emission Tomography with Zirconium-89-labeled chimeric monoclonal antibody U36 in the detection of lymph node metastases in head and neck cancer patients. Clin Cancer Res. 2006;12:2133–40. doi: 10.1158/1078-0432.CCR-05-2137. [DOI] [PubMed] [Google Scholar]
- 34.Nagengast WB, de Vries EG, Hospers GA, Mulder NH, de Jong JR, Hollema H, et al. In vivo VEGF imaging with radiolabeled bevacizumab in a human ovarian tumor xenograft. J Nucl Med. 2007;48:1313–9. doi: 10.2967/jnumed.107.041301. [DOI] [PubMed] [Google Scholar]
- 35.Perk LR, Stigter-van Walsum M, Visser GWM, Kloet RW, Vosjan MJWD, Leemans CR, et al. Quantitative PET imaging of Met-expressing human cancer xenografts with 89Zr-labelled monoclonal antibody DN30. Eur J Nucl Med Mol Imaging. 2008;35:1857–67. doi: 10.1007/s00259-008-0774-5. [DOI] [PubMed] [Google Scholar]
- 36.Perk LR, Visser GWM, Budde M, Vosjan MJWD, Jurek P, Kiefer GE, et al. Facile radiolabeling of monoclonal antibodies and other proteins with zirconium-89 or gallium-68 for PET imaging using p-isothiocyanatobenzyl-desferrioxamine. Nature Protocols. 2008 doi: 10.1038/nprot.2008.22. [DOI] [PubMed] [Google Scholar]
- 37.Aerts HJWL, Dubois L, Perk L, Vermaelen P, van Dongen GAMS, Wouters BG, et al. Disparity between in vivo EGFR expression and 89Zr-labeled cetuximab uptake assessed with PET. J Nucl Med. 2009;50:123–31. doi: 10.2967/jnumed.108.054312. [DOI] [PubMed] [Google Scholar]
- 38.Shure K, Deutsch M. Radiations from zirconium89. Physical Review. 1951;82:122. [Google Scholar]
- 39.Lightbody DB, Mitchell GE, Sayres A. 89Y(p,n)89Zr reaction. Phys Lett. 1965;15:155–7. [Google Scholar]
- 40.Mustafa MG, West HI, Jr, O’Brien H, Lanier RG, Benhamou M, Tamura T. Measurements and a direct-reaction-plus-Hauser-Feshbach analysis of 89Y(p,n)89Zr, 89Y(p,2n)88Zr, and 89Y(p,pn)88Y reactions up to 40 MeV. Phys Rev C. 1988;38:1624–37. doi: 10.1103/physrevc.38.1624. [DOI] [PubMed] [Google Scholar]
- 41.Bard AJ, Parsons R, Jordan J. Standard Potentials in Aqueous Solutions. IUPAC (Marcel Dekker); New York, USA: 1985. [Google Scholar]
- 42.Kobayashi T, Sasaki T, Takagi I, Moriyama H. Zirconium solubility in ternary aqueous system of Zr(IV)-OH-carboxylates\ J Nucl Science Tech (Tokyo, Japan) 2009;46:142–8. [Google Scholar]
- 43.Sasaki T, Kobayashi T, Takagi I, Moriyama H. Hydrolysis constant and coordination geometry of zirconium(IV) J Nucl Science Tech (Tokyo, Japan) 2008;45:735–9. [Google Scholar]
- 44.Kobayashi T, Sasaki T, Takagi I, Moriyama H. Solubility of zirconium(IV) hydrous oxides. J Nucl Science Tech (Tokyo, Japan) 2007;44:90–4. [Google Scholar]
- 45.McCarthy DW, Shefer RE, Klinkowstein RE, Bass LA, Margeneau WH, Cutler CS, et al. Efficient production of high specific activity 64Cu using a biomedical cyclotron. Nucl Med Biol. 1997;24:35–43. doi: 10.1016/s0969-8051(96)00157-6. [DOI] [PubMed] [Google Scholar]
- 46.Verel I, Visser Gerard WM, van Dongen Guus A. The promise of immuno-PET in radioimmunotherapy. J Nucl Med. 2005;46 (Suppl 1):164S–71S. [PubMed] [Google Scholar]
- 47.Hinrichsen PF. Decay of 78.4 hour zirconium-89 . Nucl Phys A. 1968;118:538–44. [Google Scholar]