

Abstract
The aim of this study was to improve the neuropathologic recognition and provide criteria for the pathological diagnosis in the neurodegenerative diseases grouped as frontotemporal lobar degeneration (FTLD); revised criteria are proposed. Recent advances in molecular genetics, biochemistry, and neuropathology of FTLD prompted the Midwest Consortium for Frontotemporal Lobar Degeneration and experts at other centers to review and revise the existing neuropathologic diagnostic criteria for FTLD. The proposed criteria for FTLD are based on existing criteria, which include the tauopathies [FTLD with Pick bodies, corticobasal degeneration, progressive supranuclear palsy, sporadic multiple system tauopathy with dementia, argyrophilic grain disease, neurofibrillary tangle dementia, and FTD with microtubule-associated tau (MAPT) gene mutation, also called FTD with parkinsonism linked to chromosome 17 (FTDP-17)]. The proposed criteria take into account new disease entities and include the novel molecular pathology, TDP-43 proteinopathy, now recognized to be the most frequent histological finding in FTLD. TDP-43 is a major component of the pathologic inclusions of most sporadic and familial cases of FTLD with ubiquitin-positive, tau-negative inclusions (FTLD-U) with or without motor neuron disease (MND). Molecular genetic studies of familial cases of FTLD-U have shown that mutations in the progranulin (PGRN) gene are a major genetic cause of FTLD-U. Mutations in valosin-containing protein (VCP) gene are present in rare familial forms of FTD, and some families with FTD and/or MND have been linked to chromosome 9p, and both are types of FTLD-U. Thus, familial TDP-43 proteinopathy is associated with defects in multiple genes, and molecular genetics is required in these cases to correctly identify the causative gene defect. In addition to genetic heterogeneity amongst the TDP-43 proteinopathies, there is also neuropathologic heterogeneity and there is a close relationship between genotype and FTLD-U sub-type. In addition to these recent significant advances in the neuropathology of FTLD-U, novel FTLD entities have been further characterized, including neuronal intermediate filament inclusion disease. The proposed criteria incorporate up-to-date neuropathology of FTLD in the light of recent immunohistochemical, biochemical, and genetic advances. These criteria will be of value to the practicing neuropathologist and provide a foundation for clinical, clinico-pathologic, mechanistic studies and in vivo models of pathogenesis of FTLD.
Keywords: Frontotemporal dementia, Semantic dementia, Progressive non-Xuent aphasia, Frontotemporal lobar degeneration, Motor neuron disease, Tauopathy, Ubiquitin, TDP-43 proteinopathy, Progranulin, Valosin-containing protein, Charged multivesicular body protein 2B, Neuronal intermediate filament inclusion disease, Neuropathologic diagnosis
Introduction
In this paper, we follow the convention that FTLD is an umbrella term that groups several different neurodegenerative diseases characterized by predominant destruction of the frontal and temporal lobes. After Alzheimer disease (AD) and dementia with Lewy bodies (DLB), frontotemporal lobar degeneration (FTLD) is the third most common neuro-degenerative cause of dementia in industrialized countries [59, 60, 69]. Most commonly, patients with FTLD present with frontotemporal dementia (FTD), a change in personal and social conduct, often associated with disinhibition, with gradual and progressive changes in language [53]. Other patients falling under the diagnostic umbrella of FTLD may present with early and progressive changes in language function, and two syndromes have been recognized: semantic dementia (SD) and primary progressive non-Xuent aphasia (PNFA) [40, 43, 53, 65, 67, 78]. In later stages of these particular syndromes, both behavioral and language dysfunction may be present. A proportion of patients with FTLD present with or develop parkinsonism as part of their disease process. Clinical amyotrophic lateral sclerosis/motor neuron disease (ALS/MND) may also be found in a proportion of patients with FTLD, especially those with FTD, indicating a spectrum of clinical phenotypes that relate to common neuropathologic lesions [3, 61, 69].
FTD, SD, or PNFA refer to the main clinical syndromes linked to the FTLD group. Typically, at least in the early course of the disease, patients with FTD do not have an amnestic syndrome, which distinguishes them clinically from AD [46, 53], but there are exceptions [29]. Although no pre-symptomatic biomarkers have been identified, at least in sporadic cases, clinical assessment, neuropsychology, and neuroimaging may help to distinguish FTD and the related disorders of SD and PNFA from other neurodegenerative causes of dementia [12, 43, 46]. The diagnosis of FTD, SD, or PNFA may only be considered when other potential causes of dementia including other nervous system diseases (e.g., small and large vessel disease), systemic conditions (e.g., hypothyroidism), tumors, and substance abuse have been excluded.
The apolipoprotein E (APOE) gene ε4 allele is a major risk factor for AD, though this is not the case in most association studies of FTLD (but see ref. [71]), and none of the autosomal dominant mutations in genes associated with some familial cases of AD [amyloid precursor protein (APP), presenilin 1 (PS1) and presenilin 2 (PS2)] acts as a risk factor for FTLD.
Recent developments in the molecular pathology and genetics of FTLD now dictate that a minimal panel of pathological investigations is required for correct diagnosis in this group of diseases. Standardization of nomenclature and approach will facilitate better understanding of clinico-pathologic correlations, provide insights into pathogenesis, and guide the construction and validation of in vivo models.
Neuropathologic evaluation
With the exception of those cases in which a gene defect has been identified, examination of the brain and neuropathology are essential in order to determine the disease entity underlying FTLD. Even in those cases that have been genetically characterized, it is not uncommon to find coexisting neurodegenerative disease and other pathology, which may have contributed to the clinical picture to a varying degree. The neuropathology of the brain, either on autopsy or, rarely, on biopsy, remains the “gold standard” for determining the neuropathologic diagnosis. Although most cases seen by a neuropathologist are likely to be cases of advanced disease, there is an increasing awareness that the molecular pathology of all neurodegenerative disease is present often several years prior to the onset of clinical symptoms, and this knowledge will inform the neuropathologist of pre-clinical FTLD in an otherwise cognitively and behaviorally normal subject.
Macroscopy
Examination of the brain of a patient with FTLD typically shows symmetrical focal atrophy of the frontal or temporal lobes, or both. In some patients there is asymmetry of atrophy, typically reflected in perisylvian loss on one side of the brain. Macroscopic atrophy of the basal ganglia and loss of pigmentation from the substantia nigra are seen in a proportion of cases. This focal atrophy is, not infrequently, the most dramatic in all of neuropathology. Conversely, in some individuals, for example, those who die at an earlier stage, the brain is unremarkable. The pattern of atrophy may assist in staging disease severity [11, 39, 41].
Microscopy
In most forms of FTLD, examination of the cerebral cortex with H&E staining shows microvacuolation and neuronal loss. In many cases, this is most evident around layer II of the affected cortical regions. In advanced cases, there is transcortical microvacuolation and neuronal loss. Swollen cortical neurons may be seen and highlighted with immunostaining for alpha B-crystallin; however, they are not specific for any disease subtype. White matter myelin loss and astrocytic gliosis may be seen. There may be significant neuronal loss from the basal ganglia and substantia nigra in some cases.
Specific diagnosis of disease within the broad group of FTLD now requires immunohistochemistry (IHC) to determine the molecular pathology, morphology, and distribution of lesions in the neuraxis, and thereby identify the neurodegenerative disease. In the routine microscopic evaluation of the brain of a patient with FTLD, other neurodegenerative diseases may be identified, most commonly AD [18], DLB [52], and, rarely, prion disease [33] and hereditary diffuse leukoencephalopathy with axonal spheroids [76].
Although some neurodegenerative diseases can be readily identified using conventional staining techniques (e.g., modified Bielschowsky and Gallyas silver impregnations and thio-Xavine-S for AD pathology [8, 18, 54]), more sensitive and reliable IHC techniques are now preferred. IHC methods are more consistent and dependable than are silver impregnation techniques, they have greater inter-rater reliability, as shown by the BrainNet Europe Consortium study [1], and IHC results can suggest or identify underlying molecular pathology. For example, antibodies raised against epitopes of tau readily label the neurofibrillary tangles, neuritic plaques, and neuropil threads of AD; anti-β-amyloid antibodies detect diffuse and compact β-amyloid deposits and cerebral amyloid angiopathy [10]; while anti-α-synuclein antibodies label Lewy bodies and Lewy neurites, the signature lesions of DLB [10, 52]. Neuropathologic staging schemes have been developed using tau, β-amyloid and α-synuclein IHC, and IHC is now replacing conventional stains in the neuropathologic diagnostic criteria for AD and DLB [10, 18, 52]. Prion IHC may be used reliably to detect or exclude prion disease in most cases [33]. Proteins targeted for degradation are ubiquitinated and several hallmark inclusions in neurodegenerative disease either in neurons or glia or both are detected by ubiquitin IHC [17, 19, 47, 48]. There is also age-related accumulation of ubiquitinated material in the brain [23], which can make detection of certain ubiquitin-related pathologies difficult. Until recently, ubiquitin IHC was the only marker for certain neuronal inclusions seen in FTLD and ALS/MND that contained neither tau nor α-synuclein epitopes. P62 (sequesto-some-1) IHC has recently been highlighted as an alternative method to detect a range of ubiquitin-immunoreactive structures in neurodegenerative diseases including ALS/MND, and FTLD. Like ubiquitin IHC, a range of pathological and age-related abnormalities are detected, but an advantage over anti-ubiquitin IHC appears to be that there is better contrast in the detection of intracellular pathology. More recently, TDP-43 has been identified as a major component of the inclusions of FTLD with ubiquitin-positive, tau- and α-synuclein-negative inclusions (FTLD-U) [3, 61], formerly called FTLD with MND-type inclusions, but without MND [53]. This protein now defines a novel class of neurodegenerative diseases collectively called TDP-43 proteinopathies [16], and TDP-43 IHC may be used to characterize a majority of FTLD-Us, but not all [16].
Although IHC is essential for determining the underlying molecular pathology of the majority of neurodegenerative diseases, other techniques may be available in dementia research centers and complement the routine neuropathologic diagnosis. The reliable and robust detection of abnormally aggregated proteins either within neurons or glia or both, or in the neuropil, is necessary for neuropathologic diagnosis. However, the density and distribution of abnormal protein aggregates, as identified by IHC, do not always correlate well with clinical symptoms. Other markers, such as synaptic and neuronal loss in affected brain areas, may correlate better with cognitive impairment and motor dysfunction. Thus, stereologic methods that assess synaptic and neuronal loss in an unbiased manner may be useful in clinico-pathologic studies in the dementia research center, but are not necessary, or usually feasible, for routine neuropathologic diagnosis.
Biochemistry is also useful, but not essential for diagnosis. Methods of fractionating brain homogenates may be used to rationally classify the tauopathies in a research setting [13]. In the adult brain, there are normally six isoforms of the microtubule-associated protein tau (MAPT): three isoforms with 0, 1, or 2 inserts contain three microtubule-binding repeats (3R tau) and three isoforms, also with 0, 1, or 2 inserts, contain four microtubule-binding repeats (4R tau) [28]. The tauopathies have a biochemical signature: tau protein in these disorders is relatively insoluble and these insoluble species can be detected by biochemical fractionation methods. The insoluble fractions may be further characterized according to the pattern of tau isoforms. For example, in AD, all six isoforms are abnormally hyperphosphorylated and migrate as three major bands and one minor band when visualized by immunoblotting. Treatment with the enzyme alkaline phosphatase removes phosphate groups, and the tau isoforms appear as six bands (3R and 4R tau). This biochemical signature may be used to distinguish AD from the FTLD tauopathies [13]. Thus, brain tissue from patients with FTLD where Pick bodies are present is characterized biochemically by predominantly 3R tau, while CBD, PSP, argyrophilic grain disease (AGD), and sporadic multiple system tauopathy with dementia (MSTD) are predominantly 4R tauopathies [13, 74], and neurofibrillary tangle dementia (NTD), also called tangle predominant form of senile dementia, has inclusions containing a mixture of 3R and 4R tau [34, 35]. FTLD with MAPT mutation, of which more than 40 have been described, is biochemically heterogeneous with different mutations being associated with 3R, 4R, or 3R and 4R tauopathy [15]. Monoclonal antibodies, which discriminate between 3R and 4R tau [22] are now commercially available; so, the molecular classification of tauopathies by isoform type may be easily undertaken in the histology laboratory that does not have access to biochemistry.
FTLD with ubiquitin-positive, tau-negative inclusions (FTLD-U), also known as FTLD with MND-type inclusions or MND inclusion dementia, is the most common underlying pathology in FTLD with and without clinical MND [45, 73]. TAR DNA-binding protein 43 (TDP-43), a nuclear protein implicated in exon skipping and transcription regulation, was recently identified as a major protein component of the ubiquitin-immunoreactive inclusions characteristic of sporadic and familial FTLD-U, with and without clinical MND, as well as in sporadic ALS [3, 16, 21, 61]. Biochemistry in these disorders shows TDP-43 to be abnormally phosphorylated, ubiquitinated and cleaved to generate C-terminal fragments, and is recovered only from areas with ubiquitin-immunoreactive inclusions including hippocampus, neocortex, and spinal cord [61]. The neuropathology of these conditions is characterized by ubiquitinand TDP-43-positive neuronal cytoplasmic inclusions (NCIs), neuronal intranuclear inclusions (NIIs), dystrophic neurites (DNs), and glial cytoplasmic inclusions (GCIs) that are negative for tau, α-synuclein, β-amyloid, neuronal intermediate filaments, and expanded polyglutamines [21, 61]. The variability in the morphologic types of neuronal inclusions, their distribution, density, and immunohistochemical profile has led to the proposed classification of FTLD-U into four pathologic subtypes [16, 63, 66]. Recently, mutations in the progranulin (PGRN) gene [4, 20, 57], the molecular genetic basis of non-tau familial FTD linked to chromosome 17, were discovered. The neuropathology in these cases is FTLD-U with ubiquitin-positive neurites, NCIs and, most characteristically, NIIs [4, 50] (but see ref. [37]). However, NIIs can be seen in other FTLD-U cases where PGRN mutations are not found [16] and therefore such NIIs cannot be considered pathognomonic for PGRN or other (i.e., valosin-containing protein VCP) mutations associated with FTLD. As demonstrated by IHC and biochemistry, the ubiquitinated pathologic protein in these cases is not progranulin, but TDP-43 [4, 57]. Pathologic TDP-43 is detected biochemically in both affected gray and white matter, suggesting that both glial and neuronal pathology may contribute to the pathogenesis of FTLD-U caused by PGRN mutations [61].
Frontotemporal lobar degeneration with VCP gene mutation, also called inclusion body myopathy associated with Paget's disease of bone and frontotemporal dementia (IBMPFD), is a rare autosomal dominant disorder caused by mutations in the VCP gene [77]. VCP, a member of the AAA-ATPase gene super family (ATPase associated with diverse cellular activities), has multiple cellular functions including acting as a molecular chaperone in endoplasmic reticulum-associated protein degradation, stress response, programmed cell death, and interactions with the ubiquitin-proteasome system. The neuropathology in FTLD with VCP mutation is a unique subtype of FTLD-U and is characterized by numerous NIIs and relatively few NCIs and DNs [26]; the ubiquitinated inclusions are not primarily composed of the mutated protein (VCP), but rather TDP-43 [26, 63].
Frontotemporal lobar degeneration with charged multivesicular body protein 2B (CHMP2B) gene mutation is the cause of FTD linked to chromosome 3 in a large Danish pedigree [68]. Human CHMP2B is a component of the endosomal secretory complex, which becomes dysregulated by the gene defects. Recent studies have revealed ubiquitin-positive, but TDP-43 negative, NCIs in the frontal neocortex and hippocampus, so that this disease is an FTLD-U, but not a TDP-43, proteinopathy [16].
A genetic locus on chromosome 9p for familial FTD-MND has been described [56]. In one family, candidate gene sequencing revealed the presence of a putative disease segregating stop codon mutation (Q342X) in the intraflagellar transport protein 74 (IFT74) gene [55]. IFT74 is a protein that localizes to the intracellular vesicle compartment and is a component of the intraflagellar transport system responsible for vesicular transport of material synthesized within the cell body into and along dendrites and axons. Neuropathology in a single case with the IFT74 mutation was reported as showing all the stigmata of FTLD-U (ubiquitinated NCIs, DNs, and NIIs) and TDP-43 proteinopathy similar to that seen in other reported families with FTD, with or without MND linked to chromosome 9p [16]. Nonetheless, it remains to be established in other families and patients that IFT74 is indeed a true locus for FTLD.
Neuropathologic classification of FTLDs
Following the principles of the previous consensus criteria for the neuropathologic diagnosis of FTLD [53], and the consensus criteria for the postmortem diagnosis of AD [18] and DLB [52], we acknowledge that only probabilistic statements can be made as to the causal relationship between the neuropathology and the clinical phenotype. Just as the constellation of clinical symptoms associated with FTD, SD, or PNFA do not predict reliably the underlying causative neurodegenerative disease, the presence of the neuropathology of FTLD does not predict with certainty one or other of the clinical phenotypes associated with FTLD, or even if the subject was demented. Small series of cases are inadequate to reliably and robustly determine clinico-pathologic correlations with any one form of FTLD. Multi-center collaborations are beginning to address this challenge [25], and it is only by pooling relatively rare cases from several research centers that reliable clinicopathologic correlations are likely to emerge.
The neuropathologic criteria proposed here (Table 1) are an evolution of the 2001 criteria proposed by McKhann et al. [53], and take into account more recent descriptions of novel disease entities [6, 14], the discovery of causative gene defects (PGRN, VCP, CHMP2B) and linkage to chromosome 9p [4, 5, 20, 55, 56, 57, 68, 75, 77], and the novel (TDP-43) proteinopathy, which is present in most cases of FTLD-U with or without MND [3, 16, 21, 61]. The neuropathologic diagnosis of FTLD requires the exclusion of other neurodegenerative and systemic diseases, tumors, and drugs of abuse, which may cause a clinical FTLD phenotype. The proposed rational classification of neurodegenerative diseases associated with a clinical FTLD phenotype comprises seven distinct neurohistological types, and is based on the underlying molecular pathology as far as it is known.
Table 1.
Comparison between the present proposed criteria and McKhann et al. [3] neuropathologic diagnostic criteria for FTLD
| Present criteria | McKhann et al. criteria |
|---|---|
| 1. Tauopathy (with associated neuron loss and gliosis) and insoluble tau with a predominance of 3R tau, the most likely diagnoses are: | 1. When the predominant neuropathological abnormalities are tau-positive inclusions (with associated neuron loss and gliosis) and insoluble tau has a predominance of tau with three microtubule-binding repeats, the most likely diagnoses are: |
| FTLD with Pick bodies | (a) Pick disease |
| FTLD with MAPT mutation | (b) Frontotemporal dementia with parkinsonism linked to chromosome 17 |
| (c) Other as yet unidentified familial and sporadic frontotemporal disorders | |
| 2. Tauopathy (with associated neuron loss and gliosis) and insoluble tau with a predominance of 4R tau, the most likely diagnoses are: | 2. When the predominant neuropathological abnormalities are tau-positive inclusions (with associated neuron loss and gliosis) and insoluble tau has a predominance of four microtubule-binding repeats, the most likely diagnoses are: |
| Corticobasal degeneration | (a) Corticobasal degeneration |
| Progressive supranuclear palsy | (b) Progressive supranuclear palsy |
| Argyrophilic grain disease | (c) Frontotemporal dementia with parkinsonism linked to chromosome 17 |
| Sporadic multiple system tauopathy with dementia FTLD with MAPT mutation | (d) Other as yet unidentified familial and sporadic frontotemporal disorders |
| 3. Tauopathy (with associated neuron loss and gliosis) and insoluble tau, with a predominance of 3R and 4R tau, the most likely diagnoses are: | 3. When the predominant neuropathological abnormalities are tau-positive inclusions (with associated neuron loss and gliosis) and insoluble tau has a predominance of three and four microtubule-binding repeats, the most likely diagnoses are: |
| Neurofibrillary tangle dementia | (a) Neurofibrillary tangle dementia |
| FTLD with MAPT mutation | (b) Frontotemporal dementia with parkinsonism linked to chromosome 17 |
| (c) Other as yet unidentified familial and sporadic frontotemporal disorders | |
| 4. Frontotemporal neuronal loss and gliosis without tau- or ubiquitin/P62-positive inclusions, the most likely diagnosis is: | 4. When the predominant neuropathological abnormalities are frontotemporal neuronal loss and gliosis without tau- or ubiquitin-positive inclusions and without detectable amounts of insoluble tau, the most likely diagnoses are: |
| FTLD (also known as dementia lacking distinctive histologic features) | (a) Frontotemporal lobar degeneration (also known as dementia lacking distinct histopathological features) |
| (b) Other as yet unidentified familial and sporadic frontotemporal disorders | |
| 5. TDP-43 proteinopathy with associated neuronal loss and ubiquitin-positive/P62-positive, tau-negative inclusions, with MND or without MND but with MND-type inclusions, the most likely diagnoses are: | 5. When the predominant neuropathological abnormalities are frontotemporal neuronal loss and gliosis with ubiquitin-positive, tau-negative inclusions and without detectable amounts of insoluble tau, with MND or without MND but with MND-type inclusions, the most likely diagnoses are: |
| FTLD-U with MND (FTLD-U types 1–3) | (a) Frontotemporal lobar degeneration with MND |
| FTLD-U but without MND (FTLD-U types 1–3) | (b) Frontotemporal lobar degeneration with MND-type inclusions but without MND, or |
| FTLD-U with PGRN mutation (FTLD-U type 3) | (c) Other as yet unidentified familial and sporadic frontotemporal disorders. |
| FTLD-U with VCP mutation (FTLD-U type 4) | |
| FTLD-U linked to chromosome 9p (FTLD-U type 2) | |
| Other as yet unidentified TDP-43 proteinopathies | |
| 6. Frontotemporal neuronal loss and gliosis with ubiquitin-positive/P62-positive, TDP-43- and tau-negative inclusions, the most likely diagnoses are: | |
| FTLD-U with CHMP2B mutation | |
| Basophilic inclusion body disease (BIBD) | |
| Other as yet unidentified FTLD-U, non-TDP-43 proteinopathies | |
| 7. Frontotemporal neuronal loss and gliosis with ubiquitin/P62 and α-internexin-positive inclusions, the most likely diagnosis is: | |
| Neuronal intermediate filament inclusion disease (NIFID) |
CHMP2B charged multivesicular body protein 2B gene, FTLD frontotemporal lobar degeneration, FTLD-U FTLD with ubiquitin-positive, tau-, α-synuclein-, TDP-43-, and neuronal intermediate filament protein-negative inclusions, MAPT microtubule-associated protein tau gene, MND motor neuron disease, neurofibrillary tangle dementia, also called tangle predominant form of senile dementia, PGRN progranulin gene, TDP-43 TAR DNA-binding protein 43, VCP valosin-containing protein gene
Algorithm for the neuropathologic diagnosis of FTLD
The proposed criteria for the neuropathologic diagnosis and nosology of FTLD builds on, and extends, the existing criteria to include neuropathologic assessment using disease-specific antibodies, biochemistry, and molecular genetics to arrive at the neuropathologic diagnosis of one of the disease entities causing FTLD. It is appreciated that it may not be practical, possible, or even necessary, to undertake sophisticated neuroanatomical investigations of neuron and synapse density, biochemistry or molecular genetics in every case. For these reasons, the following neuropathologic algorithm has been developed, which should be feasible at most dementia research centers. It is envisaged that this algorithm, with its inbuilt criteria for diagnosis, will supersede existing neuropathologic criteria [3], and become the standard operational protocol for the working neuropathologic diagnosis of FTLD (Figs. 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14).
Fig. 1.

Frontotemporal lobar degeneration neuropathology algorithm flow chart. AD Alzheimer's disease, AGD argyrophilic grain disease, AMYG amygdala, BIBD basophilic inclusion body disease, CBD corticobasal degeneration, CBLM cerebellum including the dentate nucleus (DN), CHMP2B charged multivesicular body protein 2B gene, CG cingulate gyrus, DLB dementia with Lewy bodies, DLDH dementia lacking distinctive histologic features, also called FTLD according to McKhann et al. [4] criteria, FTLD frontotemporal lobar degeneration, FTLD-U FTLD with ubiquitin-positive, tau-negative inclusions, GP globus pallidus, H&E hematoxylin and eosin, HIP hippocampus, IHC immunohistochemistry, INAα-internexin, MAPT microtubule-associated protein tau gene, MED medulla oblongata, MFG middle frontal gyrus, MID midbrain including the substantia nigra, MND motor neuron disease, MSTD sporadic multiple system tauopathy with dementia, NIFID neuronal intermediate filament inclusion disease, NF neurofilament; neurofibrillary tangle dementia, also called tangle predominant form of senile dementia, NOS not otherwise specified, OL occipital lobe, PGRN progranulin gene, FL frontal lobe, PL parietal lobe, PSP progressive supranuclear palsy, SC spinal cord, STG superior temporal gyrus, STR striatum, TDP-43 TAR DNA-binding protein 43, THAL/SUBTN thalamus and subthalamic nucleus, Ub ubiquitin, VCP valosin-containing protein gene, 3R, 4R, or 3R and 4R tau isoforms containing 3, 4, or 3 and 4 microtubule-binding repeats
Fig. 2.

Frontotemporal lobar degeneration with Pick bodies. Pick bodies (arrowheads) and a neurofibrillary tangle (arrow) in the subiculum (a) are immunolabeled by anti-phosphorylated tau antibodies (PHF1 immunohistochemistry). Pick bodies are not immunolabeled with anti-4R tau antibodies (arrowheads), while neurofibrillary tangles are immunolabeled (arrows) (b). Anti-3R tau antibodies clearly label Pick bodies (arrowheads) (c). b 4R tau (ET3) and c 3R tau (RD3) immunohistochemistry. Bars 10 μm
Fig. 3.

Corticobasal degeneration. a A swollen achromatic neuron (arrow) in the middle frontal gyrus. Hematoxylin and eosin (HE). b Tau-positive neurofibrillary tangles in the pyramidal neurons of the CA1 hippocampal subfield. c A globose neurofibrillary tangle (arrow) in the locus coeruleus. d An astrocytic plaque (asterisk), coiled body (arrow), and threads (arrowhead) in the deep cortical laminae and white matter of the parietal lobe. b, c, d Anti-phosphorylated tau (PHF1) immunohistochemistry. Bars 10 μm
Fig. 4.

Progressive supranuclear palsy. Neurofibrillary tangles in the subthalamic nucleus (a), occulomotor nucleus (b), and locus coeruleus (e). Tufted astrocytes in the putamen (c and d). a, b, c, e Anti-phosphorylated tau (PHF1) immunohistochemistry. d Gallyas silver impregnation. Scale bars a 50 μm, b, c, d, e 10 μm
Fig. 5.
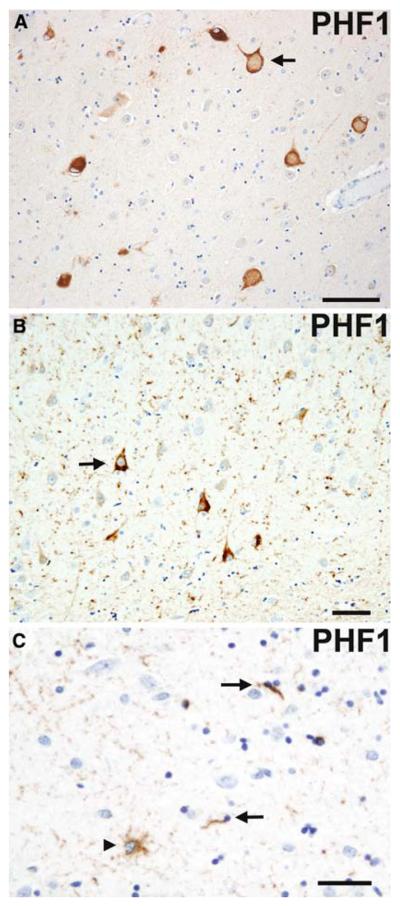
Argyrophilic grain disease. A swollen achromatic neuron (arrow) with pale center and more intense tau-immunoreactive periphery in the subiculum. Tau-immunoreactive grains in the neuropil and diffusely stained pyramidal neurons (arrow) indicating a preneurofibrillary tangle stage in the pyramidal layer of the hippocampus (b). A tau-immunoreactive astrocytic inclusion (arrowhead) and oligodendroglial cytoplasmic inclusions called coiled bodies (arrows) in the CA1 subfield of the hippocampus. (a, b, c) Anti-phosphorylated tau (PHF1) immunohistochemistry. Scale bars (a) 100 μm and (b and c) 50 μm
Fig. 6.

Sporadic multiple system tauopathy with dementia. Neuronal and glial globular inclusions at the gray/white junction. Anti-phosphorylated tau (PHF1) immunohistochemistry. Scale bar 100 μm
Fig. 7.

Neurofibrillary tangle dementia. a, b Numerous neurofibrillary tangles in the upper and lower pyramidal neurons of the occipitotemporal cortex; no neuritic plaques or amyloid deposits are present. a Gallyas silver impregnation. b Anti-phosphorylated tau (PHF1) immunohistochemistry. Scale bars 50 μm
Fig. 8.

Frontotemporal lobar degeneration (FTLD) with MAPT mutation. Inclusions in FTLD with MAPT G389R mutation (a) and FTLD with MAPT intron 10 + 16 mutation (b–g). a Numerous tau-immuno-reactive Pick body-like inclusions in the granule neurons of the dentate fascia. b A swollen achromatic neuron in the superior temporal gyrus. c A swollen neuron with a central area of pale anti-tau immunoreactivity surrounded by more intense staining. Fibrillary material surrounds the nucleus and extends into the apical dendrite. d An intraneuronal inclusion resembling a Pick body in the superior frontal gyrus. e A neurofibrillary tangle-like inclusion in layer V of the superior frontal gyrus. f A globose neurofibrillary tangle-like inclusion in the dorsal raphé nucleus. g An astrocytic fibrillary inclusion (a) and a coiled body (b) in an oligodendrocyte in the white matter of the frontal lobe. b Hematoxylin and eosin (HE); (a, c–g) anti-phosphorylated tau (AT8) immunohistochemistry. Scale bars 10 μm. (Adapted from Ref. [44]; reproduced with permission)
Fig. 9.

Frontotemporal lobar degeneration (FTLD)-U with or without MND: spectrum of TDP-43 pathology. Adjacent sections of superficial frontal neocortex showing neuronal cytoplasmic inclusions (NCIs), dystrophic neurites (DNs), and isolated neuronal intranuclear inclusions (NIIs) stain for both ubiquitin (a) and TDP-43 (b). NCI in the dentate granule cells stain for ubiquitin (c) and TDP-43 (d). Neuronal and glial inclusions include NCI (e), round and lentiform NIIs (f, g); skein-like (h), and compact round (i) NCI in the lower motor neurons; and glial cytoplasmic inclusion (GCI) (j). (a and c) ubiquitin immunohistochemisty (b, d, e–j TDP-43 immunohistochemistry). Scale bars 10 μm (a–d); 5 μm (e–j) (Adapted from Ref. [16]; reproduced with permission)
Fig. 10.

Frontotemporal lobar degeneration-U with MND. Diffuse perinuclear staining in a motor neuron (arrow). TDP-43 immunohistochemistry. Scale bar 10 μm
Fig. 11.

Frontotemporal lobar degeneration-U, subtypes 1–4. a–d Type 1 is characterized by long and tortuous dystrophic neurites (DNs) in lamiae II/III with relatively few neuronal cytoplasmic inclusions (NCIs) and no neuronal intranuclear inclusion (NII). b Type 2 has numerous NCIs, relatively few DNs, and no NII is present. c Type 3 has numerous NCIs and DNs and an occasional NII in lamina II. d Type 4 pathology in a case of FTD with VCP mutation is characterized by numerous NII and DN, but few NCI. TDP-43 immunohistochemistry. Scale bar 10 μm (a–d). (Adapted from Ref. [14]; reproduced with permission)
Fig. 12.
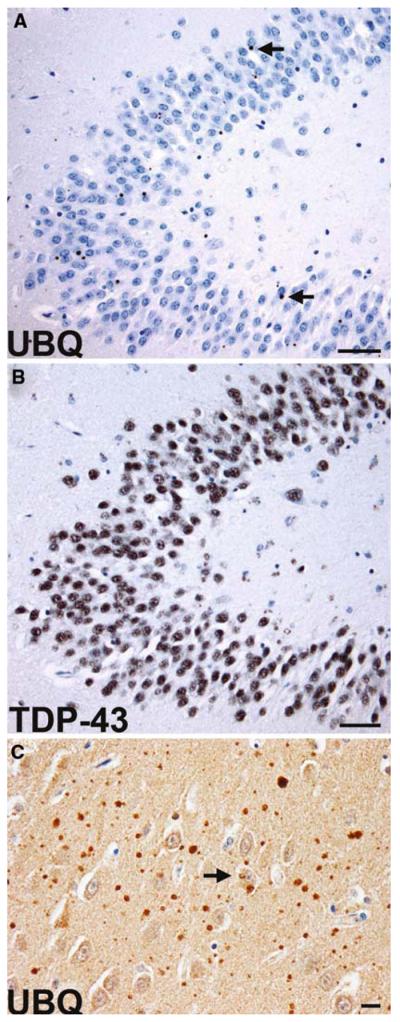
Frontotemporal lobar degeneration with CHMP2B mutation. a Sparse ubiquitin-immunoreactive NCIs (arrows) in the granule neurons of the dentate fascia. The NCIs are not labeled with anti-TDP-43 antibodies (b). Ubiquitin-positive neuropil aggregates and a sparse NCI (arrow) in the frontal lobe of an affected 61-year-old female (c). Scale bars a, b 50 μm, c 10 μm
Fig. 13.

Basophilic inclusion body disease. a A basophilic inclusion (BI) in the precentral gyrus (motor cortex), with a similar, weakly ubiquitin-immunoreactive inclusion in (b). c Neurons with basophilic inclusions showing fine granular perikaryal TDP-43 positivity in neurons with BIs on the left and negative in neurons with BIs on the right. a Hematoxylin and eosin; b ubiquitin, and c TDP-43 immunohisto-chemistry. Scale bars 20 μm
Fig. 14.

Neuronal intermediate filament inclusion disease. a Eosinophilic Lewy body-like NCI in a pyramidal neuron of the CA1 subfield of the hippocampus. b α-Internexin immunoreactive NCIs in layer III of the superior temporal gyrus. c Numerous α-internexin immunoreactive NCIs in the granule neurons of the dentate fascia. a Hematoxylin and eosin; b, c α-internexin immunohistochemistry. Scale bars 10 μm
Recently, staging schemes have been developed that no longer rely on capricious silver impregnation methods, but employ instead sensitive monoclonal and polyclonal antibodies that detect, by IHC, the pathologic proteins of the major neurodegenerative diseases [9, 10, 52]. Multicenter studies have confirmed the reproducibility and reliability of IHC over traditional staining methods, and IHC is recommended for the detection of the signature lesions of FTLD [1, 53], when appropriate. Thus, neurodegenerative diseases with α-synuclein pathology, with or without Aβ plaques and tau-positive neurofibrillary tangle (AD) pathology (i.e., DLB, Parkinson's disease, and multiple system atrophy), or those with plaque and tangle pathology (i.e. Alzheimer disease) are excluded using established sampling schemes and diagnostic criteria for these diseases [18, 52].
Tau-positive inclusions
Where neurofibrillary tangles alone are present, in the absence of Aβ plaques, in the context of neuronal loss and gliosis, NTD, also called tangle predominant form of senile dementia, which, like AD tangles, contain tau composed of all six isoforms, is a diagnostic possibility. Where the distribution of neuronal and glial tau pathology is more widespread and includes frontal, temporal, and parietal neocortex, basal ganglia, and brainstem nuclei, then sporadic MSTD may be indicated [6]. Neurofibrillary tangles in more subcortical regions including the basal ganglia, subthalamic nucleus, midbrain, and pontine nuclei indicate progressive supranuclear palsy [31, 72]. Distinguishing lesions in PSP are tau-positive tufted astrocytes and are found in affected neocortical and subcortical regions. Corticobasal degeneration is characterized by frontal and temporal atrophy that is not infrequently asymmetric, neuronal loss, gliosis, swollen achromatic neurons that are faintly tau-positive, and tau-positive neurofibrillary tangles in the neocortex, basal ganglia, and brainstem nuclei [24, 27]. Distinguishing lesions in CBD are tau-positive astrocytic plaques and threads found in the affected neocortex and subcortical white matter and in the basal ganglia. In both PSP and CBD, tau-positive oligodendroglial inclusions called coiled bodies are seen, but these are generally at a lower density than inclusions in astrocytes. Tau-positive ovoid structures (glial processes), astrocytes, and oligodendroglial inclusions (coiled bodies), when confined to the medial temporal lobe and limbic structures, indicate another tauopathy, AGD [7]. If globose tau-positive NCIs, called Pick bodies, are present in the non-pyramidal (dentate gyrus granule cells) and pyramidal neurons of area CA1 of the hippocampus, and pyramidal neurons of the temporal and frontal lobes, then FTLD with Pick bodies may be present [80]. Pick bodies are largely or wholly composed of 3R tau, which can be demonstrated by IHC or immunoblotting, while the tau-positive inclusions of PSP, CBD, MSTD, and AGD all contain 4R tau indicating that these latter disorders may represent a spectrum of 4R tauopathies [13, 74]. Finally, FTLD with MAPT mutation, also called FTD with parkinsonism linked to chromosome 17 (FTDP-17), is not only clinically and genetically heterogeneous (more than 40 mutations have been reported in the MAPT gene), but is also neuropathologically heterogeneous. The spectrum of neuronal and glial pathology seen in 3R, 4R, and combined 3R and 4R tauopathies is also found in such cases of familial tauopathy [15, 44, 70]. For the practicing neuropathologist, the presence of this spectrum of pathology in a case warrants further genetic investigation particularly if there is an autosomal dominant pattern of inheritance. Similar 3R and 4R tau heterogeneity is seen in some tauopathies that cannot be categorized as FTLD with Pick bodies, PSP, CBD, or AGD, and in individuals without MAPT mutations. Such a case may have tau pathology in the distribution described in sporadic MSTD, or may have to be categorized as “unclassifiable sporadic tauopathy.”
Tau-negative, ubiquitin-positive inclusions
TDP-43 proteinopathy
Immunohistochemistry for ubiquitin, P62, and TDP-43 in cases with FTLD generally reveals similar pathology that includes a spectrum of neuronal (NCIs, DNs, and NIIs) and glial, predominantly oligodendroglial, cytoplasmic inclusions (GCIs) [62, 61]. However, there are significant differences in the immunohistochemical findings with ubiquitin, P62, and TDP-43. Ubiquitin immunoreactivity is present in extensive age-related pathology in gray and white matter, e.g., dot-like bodies and granular degeneration of myelin, which can mask subtle neuronal and glial pathology and can be difficult to interpret. P62 immunostaining detects the same range of pathological structures as anti-ubiquitin, but highlights less age-related pathology, making interpretation somewhat less demanding. TDP-43 immunoreactivity is present in nuclei of most cells types and changes in distribution within affected neurons in disease. Screening cases using TDP-43 immunostaining as a primary diagnostic tool, as might be used for tau and α-synuclein accumulations, is more demanding. Four histologic subtypes of FTLD-U have been proposed, based on the predominant type of inclusion present as detected using anti-ubiquitin, its distribution in the cortex, and density [16, 63, 66]. Other types have been proposed as well and take into account involvement of other brain regions (e.g., hippocampus or corpus striatum) and the morphology of the lesions [2, 38]. Patterns of FTLD-U histology based solely on cortical pathology include a system proposed by Sampathu et al. [66], and Neumann et al. [63]; while Mackenzie et al. [49] have proposed a system that includes cortical and dentate fascia inclusions. In all these schemes the same descriptors were employed to differentiate subtypes, though the numbering of each subtype differed among schemes. In the present composite scheme, type 1 cases (as in [66], but known as type 2 in Mackenzie et al. [49]) are characterized by an abundance of long DNs, predominantly in the superficial cortical laminae, with few or no NCIs or NIIs. Type 2 cases (as in [66], but known as type 3 in Mackenzie et al. [49]) are characterized by numerous NCIs in both superficial and deep cortical laminae as well as infrequent DNs and sparse or no NIIs. Type 3 cases (as in [66], but known as type 1 in Mackenzie et al. [49]) are characterized by pathology predominantly in the superficial cortical layers with numerous NCIs, DNs, and variable numbers of NIIs. Type 4 cases [16, 63] are distinguished by numerous NIIs and infrequent NCIs and DNs in the neocortical areas with relative sparing of the hippocampus, consistent with the pathology described in cases of FTLD with VCP mutations (but see ref. [30]). Consensus on the validity, reliability, and reproducibility of the various proposed typing schemes, as well as their clinical significance, remain to be determined.
If TDP-43- and ubiquitin- or P62-positive, tau- and α-synuclein-negative inclusions are found in the superficial laminae of the frontal and temporal neocortex and neurons of the dentate gyrus, the most likely diagnosis is FTLD-U [21, 36, 45]. If in addition, there is TDP-43 proteinopathy and ubiquitin-positive inclusions in the lower motor neurons and evidence of motor neuron loss, gliosis, Bunina bodies, and corticospinal tract degeneration, FTLD with MND is the most likely diagnosis [53, 64, 79].
There is heuristic value in the sub-classification of FTLD-U beyond pathologic description alone, with clinical and genetic correlates of histologic patterns emerging. Cases with type 1 histology are associated with SD, whereas cases of FTLD with MND frequently show type 2 histology [49]. In genetic terms, cases with PGRN or VCP gene mutations, and in cases linked to chromosome 9, distinct patterns of ubiquitin- and TDP-43-positive inclusions are also seen. FTLD with PGRN mutation cases often display a PNFA clinical phenotype and exclusively show type 3 histology [16], whereas those with VCP mutations have type 4 histology. Cases linked to chromosome 9 have type 2 histology [16] and the majority of such cases also have ubiquitin- and TDP-positive inclusions in the upper and lower motor neurons, identical to those encountered in sporadic MND [16] or sporadic FTLD with MND where a similar type 2 histology is often present [49]. These latter data indicate that the pathology of FTLD linked to chromosome 9p is a specific subtype of FTLD-U (type 2) and that TDP-43 is the disease-associated protein. Biochemistry of sporadic cases of FTLD with and without MND, cases of sporadic ALS, and familial cases of FTLD-U with PGRN and VCP mutations, and those linked to chromosome 9p, all have a characteristic biochemical signature: TDP-43 is detected in the detergent-insoluble urea fractions from affected regions and is abnormally phosphorylated, with additional protein bands of ~25 and 45 kDa, as well as a high molecular smear, and is ubiquitinated [16, 61, 62, 63]. The quantity of these modified TDP-43 species detected by biochemistry [62, 63] may be variable, but correlates with the amount of pathology detected by IHC. Additionally, the 45 kDa species collapse into a 43 kDa band upon dephosphorylation with alkaline phosphatase, indicating that TDP-43 is abnormally phosphorylated, with features paralleling the biochemical changes seen in the tauopathies. Hippo-campal sclerosis (HS) may be found as a coexisting pathology in FTLD-U with or without MND, and small numbers of ubiquitin- and TDP-43-positive inclusions may be seen exclusively in the dentate granule cells. Preliminary data indicate that some, or perhaps most, cases of HS are TDP-43 proteinopathies [2], but further studies on larger samples of “pure HS” and biochemical studies are required to determine the nosologic status of HS.
TDP-43-negative inclusions
In those cases that have ubiquitin- or P62-positive, TDP-43-, tau-, and α-internexin-negative NCIs in the frontal and temporal lobes and dentate gyrus, the most likely diagnosis is FTLD with CHMP2B mutation [16]. Mutations in the CHMP2B gene are the cause of FTD-linked to chromosome 3 in a large Danish pedigree [68]. Human CHMP2B is a component of the endosomal secretory complex, which becomes dysregulated by the gene defect. However, the absence of DN and the presence of granular, ubiquitin-positive structures within the neocortex of these cases distinguish this FTLD-U subtype from the types 1–4 described above. Thus, based on the small number of cases studied to date, familial FTLD with CHMP2B mutation appears to be a distinctive pathologic subtype of FTLD-U and is not a TDP-43 proteinopathy on the basis of IHC.
In cases of FTLD where there is frontotemporal neuron loss and gliosis, α-internexin- or neurofilament-positive, TDP-43-, α-synuclein-, and tau-negative, and variably ubiquitinated but P62-positive neuronal inclusions, the most likely diagnosis is neuronal intermediate filament inclusion disease [14]. Where there is FTLD and P62-positive, variably ubiquitin-positive, TDP-43-, α-internexin-, α-synuclein-, and tau-negative basophilic inclusions, the most likely neuropathologic diagnosis is basophilic inclusion body disease (BIBD) [58]. If all of the antibodies listed above and routine histological stains such as hematoxylin and eosin fail to reveal signature lesions, and if prion disease has been excluded by IHC or molecular genetics, the remaining FTLD diagnosis is dementia lacking distinctive histologic features (DLDH) [42]. However, it should be borne in mind that in many of the earlier histopathologic surveys, a relatively high proportion of cases of DLDH were encountered [32, 67]. Re-evaluation of such cases using either more sensitive ubiquitin IHC methodologies [25, 45, 51] or TDP-43 IHC [21] shows DLDH to be an uncommon cause of FTLD and, indeed, it still remains to be proven as a discrete entity with diagnostic criteria other than default characteristics being employed when all IHC and other methods have failed to reveal signature lesions.
Conclusions
The proposed criteria for the neuropathologic diagnosis of FTLD described here are an evolution of the previous criteria described by McKhann et al. [53]. When formulating these proposed criteria, we acknowledged revised staging schemes for other disorders (e.g., AD and DLB), which recommend the use of IHC for the diagnosis of neurodegenerative diseases, replacing silver impregnation methods and averting the lack of reproducibility between centers when these stains are used. We have incorporated the recent identification of new entities into the nosology of FTLD. Most significantly, the discovery of TDP-43 as the major pathologic protein of most forms of FTLD identifies a novel molecular pathology, TDP-43 proteinopathy, and this is now included in the revised criteria. We have also considered the great progress in determining the molecular genetics of FTLD. In addition to FTLD with MAPT mutation, other familial subtypes are now recognized on the basis of the neuropathology of ubiquitin and/or TDP-43 IHC, biochemistry, and molecular genetics (FTLD with PGRN, CHMP2B, and VCP mutations, and cases linked to chromosome 9p), which reveal a strong correlation between genotype and neuropathologic phenotype. To facilitate neuropathologic diagnoses of FTLD at research and other centers, commercially available antibodies are now readily available for the identification of the underlying molecular pathologies (e.g., TDP-43 proteinopathy and tauopathy) and the rational diagnosis and nosology of a case of FLTD that comes to autopsy. The neuropathologic diagnostic algorithm described here is based on the current level of knowledge, but the consortium members appreciate that further study of TDP-43 proteinopathy may reveal new subtypes and that other FTLD entities may yet be identified. The proposed neuropathologic algorithm will facilitate efforts to improve the diagnosis of FTLD and encourage multi-center clinico-pathologic studies. Together, these efforts should improve the early and reliable neuropatho-logic diagnosis of the FTLD, raise the awareness of possible coexisting pathologies, and facilitate research efforts into pathogenesis and potential treatments where none currently exists.
Acknowledgments
We thank the clinical, genetic, pathology, and technical staff of the collaborating centers for making information and tissue samples available for this study and we thank the families of patients whose generosity made this research possible. Support for this work was provided by grants from the National Institute on Aging of the National Institutes of Health (P50-AG05681, P01-AG03991, U01-AG16976, P30-AG13854, P30-NS057105, AG10124 and AG17586), the Buchanan Fund, the Winspear Family Center for Research on the Neuropathology of Alzheimer Disease, and the Charles and Joanne Knight Alzheimer Research Initiative.
References
- 1.Alafuzoff I, Pikkarainen M, Al Sarraj S, Arzberger T, Bell J, Bodi I, Bogdanovic N, Budka H, Bugiani O, Ferrer I, Gelpi E, Giaccone G, Graeber MB, Hauw JJ, Kamphorst W, King A, Kopp N, Korkolopoulou P, Kovacs GG, Meyronet D, Parchi P, Patsouris E, Preusser M, Ravid R, Roggendorf W, Seilhean D, Streichenberger N, Thal DR, Kretzschmar H. Interlaboratory comparison of assessments of Alzheimer disease-related lesions: a study of the BrainNet Europe Consortium. J Neuropathol Exp Neurol. 2006;65:740–757. doi: 10.1097/01.jnen.0000229986.17548.27. [DOI] [PubMed] [Google Scholar]
- 2.Amador-Ortiz C, Lin W-L, Ahmed Z, Personett D, Davies P, Duara R, Graff-Radford NR, Hutton ML, Dickson DW. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol. 2007;61:435–445. doi: 10.1002/ana.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 4.Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 5.Behrens MI, Mukherjee O, Tu PH, Liscic RM, Grinberg LT, Carter D, Paulsmeyer K, Taylor-Reinwald L, Gitcho M, Norton JB, Chakraverty S, Goate AM, Morris JC, Cairns NJ. Neuropathologic heterogeneity in HDDD1: a familial frontotemporal lobar degeneration with ubiquitin-positive inclusions and progranulin mutation. Alzheimer Dis Assoc Disord. 2007;21:1–7. doi: 10.1097/WAD.0b013e31803083f2. [DOI] [PubMed] [Google Scholar]
- 6.Bigio EH, Lipton AM, Yen SH, Hutton ML, Baker M, Nacharaju P, White CL, III, Davies P, Lin W, Dickson DW. Frontal lobe dementia with novel tauopathy: sporadic multiple system tauopathy with dementia. J Neuropathol Exp Neurol. 2001;60:328–341. doi: 10.1093/jnen/60.4.328. [DOI] [PubMed] [Google Scholar]
- 7.Braak H, Braak E. Cortical and subcortical argyrophilic grains characterize a disease associated with adult onset dementia. Neuropathol Appl Neurobiol. 1989;15:13–26. doi: 10.1111/j.1365-2990.1989.tb01146.x. [DOI] [PubMed] [Google Scholar]
- 8.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 9.Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K. Stages in the development of Parkinson's disease-related pathology. Cell Tissue Res. 2004;318:121–134. doi: 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
- 10.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Broe M, Hodges JR, Schofield E, Shepherd CE, Kril JJ, Halliday GM. Staging disease severity in pathologically confirmed cases of frontotemporal dementia. Neurology. 2003;60:1005–1011. doi: 10.1212/01.wnl.0000052685.09194.39. [DOI] [PubMed] [Google Scholar]
- 12.Brun A, Englund B, Gustafson L, Passant U, Mann DMA, Neary D, Snowden JS. Clinical and neuropathological criteria for frontotemporal dementia. J Neurol Neurosurg Psychiatry. 1994;57:416–418. doi: 10.1136/jnnp.57.4.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buee L, Delacourte A. Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP-17 and Pick's disease. Brain Pathol. 1999;9:681–693. doi: 10.1111/j.1750-3639.1999.tb00550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cairns NJ, Grossman M, Arnold SE, Burn DJ, Jaros E, Perry RH, Duyckaerts C, Stankoff B, Pillon B, Skullerud K, Cruz-Sanchez FF, Bigio EH, Mackenzie IR, Gearing M, Juncos JL, Glass JD, Yokoo H, Nakazato Y, Mosaheb S, Thorpe JR, Uryu K, Lee VM, Trojanowski JQ. Clinical and neuropathologic variation in neuronal intermediate filament inclusion disease. Neurology. 2004;63:1376–1384. doi: 10.1212/01.wnl.0000139809.16817.dd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cairns NJ, Lee VM, Trojanowski JQ. The cytoskeleton in neurodegenerative diseases. J Pathol. 2004;204:438–449. doi: 10.1002/path.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ, Foong C, White CL, III, Schneider JA, Kretzschmar H, Carter D, Taylor-Reinwald L, Paulsmeyer K, Strider J, Gitcho M, Goate AM, Morris JC, Mishra M, Kwong LK, Stieber A, Xu Y, Forman MS, Lee VMY, Trojanowski JQ, Mackenzie IR. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol. 2007 doi: 10.2353/ajpath.2007.070182. (in press). doi:10.2353/ajpath. 2007.070182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron. 2003;40:427–446. doi: 10.1016/s0896-6273(03)00606-8. [DOI] [PubMed] [Google Scholar]
- 18.Consensus recommendations for the postmortem diagnosis of Alzheimer's disease The National Institute on Aging, eagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging. 1997;18:S1–S2. [PubMed] [Google Scholar]
- 19.Cooper PN, Jackson M, Lennox G, Lowe J, Mann DM. Tau, ubiquitin, and alpha B-crystallin immunohistochemistry define the principal causes of degenerative frontotemporal dementia. Arch Neurol. 1995;52:1011–1015. doi: 10.1001/archneur.1995.00540340103019. [DOI] [PubMed] [Google Scholar]
- 20.Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- 21.Davidson Y, Kelley T, Mackenzie IR, Pickering-Brown S, Du PD, Neary D, Snowden JS, Mann DM. Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol. 2007;113:521–33. doi: 10.1007/s00401-006-0189-y. [DOI] [PubMed] [Google Scholar]
- 22.de Silva R, Lashley T, Strand C, Shiarli AM, Shi J, Tian J, Bailey KL, Davies P, Bigio EH, Arima K, Iseki E, Murayama S, Kretzschmar H, Neumann M, Lippa C, Halliday G, Mackenzie J, Ravid R, Dickson D, Wszolek Z, Iwatsubo T, Pickering-Brown SM, Holton J, Lees A, Revesz T, Mann DM. An immunohistochemical study of cases of sporadic and inherited frontotemporal lobar degeneration using 3R- and 4R-specific tau monoclonal antibodies. Acta Neuropathol. 2006;111:329–340. doi: 10.1007/s00401-006-0048-x. [DOI] [PubMed] [Google Scholar]
- 23.Dickson DW, Wertkin A, Kress Y, Ksiezak-Reding H, Yen SH. Ubiquitin immunoreactive structures in normal human brains. Distribution and developmental aspects. Lab Invest. 1990;63:87–99. [PubMed] [Google Scholar]
- 24.Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K, Jellinger K, Lantos PL, Lippa CF, Mirra SS, Tabaton M, Vonsattel JP, Wakabayashi K, Litvan I. Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol. 2002;61:935–946. doi: 10.1093/jnen/61.11.935. [DOI] [PubMed] [Google Scholar]
- 25.Forman MS, Farmer J, Johnson JK, Clark CM, Arnold SE, Coslett HB, Chatterjee A, Hurtig HI, Karlawish JH, Rosen HJ, Van Deerlin V, Lee VM, Miller BL, Trojanowski JQ, Grossman M. Frontotemporal dementia: clinicopathological correlations. Ann Neurol. 2006;59:952–962. doi: 10.1002/ana.20873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forman MS, Mackenzie IR, Cairns NJ, Swanson E, Boyer PJ, Drachman DA, Jhaveri BS, Karlawish JH, Pestronk A, Smith TW, Tu PH, Watts GD, Markesbery WR, Smith CD, Kimonis VE. Novel ubiquitin neuropathology in frontotemporal dementia with valosin-containing protein gene mutations. J Neuropathol Exp Neurol. 2006;65:571–581. doi: 10.1097/00005072-200606000-00005. [DOI] [PubMed] [Google Scholar]
- 27.Gibb WR, Luthert PJ, Marsden CD. Corticobasal degeneration. Brain. 1989;112:1171–1192. doi: 10.1093/brain/112.5.1171. [DOI] [PubMed] [Google Scholar]
- 28.Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. 1992;8:159–168. doi: 10.1016/0896-6273(92)90117-v. [DOI] [PubMed] [Google Scholar]
- 29.Graham A, Davies R, Xuereb J, Halliday G, Kril J, Creasey H, Graham K, Hodges J. Pathologically proven frontotemporal dementia presenting with severe amnesia. Brain. 2005;128:597–605. doi: 10.1093/brain/awh348. [DOI] [PubMed] [Google Scholar]
- 30.Guyant-Marechal L, Laquerriere A, Duyckaerts C, Dumanchin C, Bou J, Dugny F, Le Ber I, Frebourg T, Hannequin D, Campion D. Valosin-containing protein gene mutations: clinical and neuropathologic features. Neurology. 2006;67:644–651. doi: 10.1212/01.wnl.0000225184.14578.d3. [DOI] [PubMed] [Google Scholar]
- 31.Hauw JJ, Daniel SE, Dickson D, Horoupian DS, Jellinger K, Lantos PL, McKee A, Tabaton M, Litvan I. Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy) Neurology. 1994;44:2015–2019. doi: 10.1212/wnl.44.11.2015. [DOI] [PubMed] [Google Scholar]
- 32.Hodges JR, Davies RR, Xuereb JH, Casey B, Broe M, Bak TH, Kril JJ, Halliday GM. Clinicopathological correlates in frontotemporal dementia. Ann Neurol. 2004;56:399–406. doi: 10.1002/ana.20203. [DOI] [PubMed] [Google Scholar]
- 33.Ironside JW, Head MW, Bell JE, McCardle L, Will RG. Laboratory diagnosis of variant Creutzfeldt–Jakob disease. Histopathology. 2000;37:1–9. doi: 10.1046/j.1365-2559.2000.00946.x. [DOI] [PubMed] [Google Scholar]
- 34.Iseki E, Yamamoto R, Murayama N, Minegishi M, Togo T, Katsuse O, Kosaka K, Akiyama H, Tsuchiya K, de Silva R, Lees A, Arai H. Immunohistochemical investigation of neurofibrillary tangles and their tau isoforms in brains of limbic neurofibrillary tangle dementia. Neurosci Lett. 2006;405:29–33. doi: 10.1016/j.neulet.2006.06.036. [DOI] [PubMed] [Google Scholar]
- 35.Jellinger KA, Bancher C. Senile dementia with tangles (tangle predominant form of senile dementia. Brain Pathol. 1998;8:367–376. doi: 10.1111/j.1750-3639.1998.tb00160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Josephs KA, Holton JL, Rossor MN, Godbolt AK, Ozawa T, Strand K, Khan N, Al Sarraj S, Revesz T. Frontotemporal lobar degeneration and ubiquitin immunohistochemistry. Neuropathol Appl Neurobiol. 2004;30:369–373. doi: 10.1111/j.1365-2990.2003.00545.x. [DOI] [PubMed] [Google Scholar]
- 37.Josephs KA, Ahmed Z, Katsuse O, Parisi JF, Boeve BF, Knopman DS, Petersen RC, Davies P, Duara R, Graff-Radford NR, Uitti RJ, Rademakers R, Adamson J, Baker M, Hutton ML, Dickson DW. Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. J Neuropathol Exp Neurol. 2007;66:142–151. doi: 10.1097/nen.0b013e31803020cf. [DOI] [PubMed] [Google Scholar]
- 38.Katsuse O, Dickson DW. Ubiquitin immunohistochemistry of frontotemporal lobar degeneration differentiates cases with and without motor neuron disease. Alzheimer Dis Assoc Disord. 2005;19(Suppl 1):S37–S43. doi: 10.1097/01.wad.0000183889.61421.a8. [DOI] [PubMed] [Google Scholar]
- 39.Kersaitis C, Halliday GM, Kril JJ. Regional and cellular pathology in frontotemporal dementia: relationship to stage of disease in cases with and without Pick bodies. Acta Neuropathol. 2004;108:515–523. doi: 10.1007/s00401-004-0917-0. [DOI] [PubMed] [Google Scholar]
- 40.Kertesz A, Davidson W, McCabe P, Takagi K, Munoz D. Primary progressive aphasia: diagnosis, varieties, evolution. J Int Neuropsychol Soc. 2003;9:710–719. doi: 10.1017/S1355617703950041. [DOI] [PubMed] [Google Scholar]
- 41.Kipps CM, Davies RR, Mitchell J, Kril JJ, Halliday GM, Hodges JR. Clinical significance of lobar atrophy in frontotemporal dementia: application of an MRI visual rating scale. Dement Geriatr Cogn Disord. 2007;23:334–342. doi: 10.1159/000100973. [DOI] [PubMed] [Google Scholar]
- 42.Knopman DS, Mastri AR, Frey WH, Sung JH, Rustan T. Dementia lacking distinctive histologic features: a common non-Alzheimer degenerative dementia. Neurology. 1990;40:251–256. doi: 10.1212/wnl.40.2.251. [DOI] [PubMed] [Google Scholar]
- 43.Knopman DS, Boeve BF, Petersen RC. Essentials of the proper diagnoses of mild cognitive impairment, dementia, and major subtypes of dementia. Mayo Clin Proc. 2003;78:1290–1308. doi: 10.4065/78.10.1290. [DOI] [PubMed] [Google Scholar]
- 44.Lantos PL, Cairns NJ, Khan MN, King A, Revesz T, Janssen JC, Morris H, Rossor MN. Neuropathologic variation in frontotemporal dementia due to the intronic tau 10(+16) mutation. Neurology. 2002;58:1169–1175. doi: 10.1212/wnl.58.8.1169. [DOI] [PubMed] [Google Scholar]
- 45.Lipton AM, White CL, III, Bigio EH. Frontotemporal lobar degeneration with motor neuron disease-type inclusions predominates in 76 cases of frontotemporal degeneration. Acta Neuropathol. 2004;108:379–385. doi: 10.1007/s00401-004-0900-9. [DOI] [PubMed] [Google Scholar]
- 46.Liscic RM, Storandt M, Cairns NJ, Morris JC. Clinical and psychometric distinction of frontotemporal and Alzheimer dementias. Arch Neurol. 2007;64:535–540. doi: 10.1001/archneur.64.4.535. [DOI] [PubMed] [Google Scholar]
- 47.Lowe J. Establishing a pathological diagnosis in degenerative dementias. Brain Pathol. 1998;8:403–406. doi: 10.1111/j.1750-3639.1998.tb00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lowe J, Hand N, Mayer RJ. Application of ubiquitin immunohistochemistry to the diagnosis of disease. Methods Enzymol. 2005;399:86–119. doi: 10.1016/S0076-6879(05)99007-1. [DOI] [PubMed] [Google Scholar]
- 49.Mackenzie IR, Baborie A, Pickering-Brown S, Plessis DD, Jaros E, Perry RH, Neary D, Snowden JS, Mann DM. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol. 2006;112:539–549. doi: 10.1007/s00401-006-0138-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mackenzie IR, Baker M, Pickering-Brown S, Hsiung GY, Lindholm C, Dwosh E, Gass J, Cannon A, Rademakers R, Hutton M, Feldman HH. The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain. 2006;129:3081–3090. doi: 10.1093/brain/awl271. [DOI] [PubMed] [Google Scholar]
- 51.Mackenzie IR, Shi J, Shaw CL, Duplessis D, Neary D, Snowden JS, Mann DM. Dementia lacking distinctive histology (DLDH) revisited. Acta Neuropathol. 2006;112:551–559. doi: 10.1007/s00401-006-0123-3. [DOI] [PubMed] [Google Scholar]
- 52.McKeith IG, Dickson DW, Lowe J, Emre M, O'Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, Aarsland D, Arai H, Ballard CG, Boeve B, Burn DJ, Costa D, Del Ser T, Dubois B, Galasko D, Gauthier S, Goetz CG, Gomez-Tortosa E, Halliday G, Hansen LA, Hardy J, Iwatsubo T, Kalaria RN, Kaufer D, Kenny RA, Korczyn A, Kosaka K, Lee VM, Lees A, Litvan I, Londos E, Lopez OL, Minoshima S, Mizuno Y, Molina JA, Mukaetova-Ladinska EB, Pasquier F, Perry RH, Schulz JB, Trojanowski JQ, Yamada M. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65:1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 53.McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ. Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick's Disease. Arch Neurol. 2001;58:1803–1809. doi: 10.1001/archneur.58.11.1803. [DOI] [PubMed] [Google Scholar]
- 54.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 55.Momeni P, Schymick J, Jain S, Cookson MR, Cairns NJ, Greggio E, Greenway MJ, Berger S, Pickering-Brown S, Chio A, Fung HC, Holtzman DM, Huey ED, Wassermann EM, Adamson J, Hutton ML, Rogaeva E, George-Hyslop P, Rothstein JD, Hardiman O, Grafman J, Singleton A, Hardy J, Traynor BJ. Analysis of IFT74 as a candidate gene for chromosome 9p-linked ALS-FTD. BMC Neurol. 2006 doi: 10.1186/1471-2377-6-44. doi:10.1186/1471-2377-6-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morita M, Al Chalabi A, Andersen PM, Hosler B, Sapp P, Englund E, Mitchell JE, Habgood JJ, de Belleroche J, Xi J, Jongjaroenprasert W, Horvitz HR, Gunnarsson LG, Brown RH., Jr A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology. 2006;66:839–844. doi: 10.1212/01.wnl.0000200048.53766.b4. [DOI] [PubMed] [Google Scholar]
- 57.Mukherjee O, Pastor P, Cairns NJ, Chakraverty S, Kauwe JS, Shears S, Behrens MI, Budde J, Hinrichs AL, Norton J, Levitch D, Taylor-Reinwald L, Gitcho M, Tu PH, Tenenholz GL, Liscic RM, Armendariz J, Morris JC, Goate AM. HDDD2 is a familial frontotemporal lobar degeneration with ubiquitin-positive, tau-negative inclusions caused by a missense mutation in the signal peptide of progranulin. Ann Neurol. 2006;60:314–322. doi: 10.1002/ana.20963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Munoz DG. The pathology of pick complex. In: Munoz DG, editor. Pick's disease and pick complex. Wiley-Liss; New York: 1998. pp. 211–241. [Google Scholar]
- 59.Neary D, Snowden JS, Mann DM, Northen B, Goulding PJ, Macdermott N. Frontal lobe dementia and motor neuron disease. J Neurol Neurosurg Psychiatry. 1990;53:23–32. doi: 10.1136/jnnp.53.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J, Benson DF. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 61.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 62.Neumann M, Kwong LK, Truax AC, Vanmassenhove B, Kretzschmar HA, Van Deerlin VM, Clark CM, Grossman M, Miller BL, Trojanowski JQ, Lee VM. TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J Neuropathol Exp Neurol. 2007;66:177–183. doi: 10.1097/01.jnen.0000248554.45456.58. [DOI] [PubMed] [Google Scholar]
- 63.Neumann M, Mackenzie IR, Cairns NJ, Boyer PJ, Markesbery WR, Smith CD, Taylor JP, Kretzschmar HA, Kimonis VE, Forman MS. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol. 2007;66:152–157. doi: 10.1097/nen.0b013e31803020b9. [DOI] [PubMed] [Google Scholar]
- 64.Okamoto K, Hirai S, Yamazaki T, Sun XY, Nakazato Y. New ubiquitin-positive intraneuronal inclusions in the extra-motor cortices in patients with amyotrophic lateral sclerosis. Neurosci Lett. 1991;129:233–236. doi: 10.1016/0304-3940(91)90469-a. [DOI] [PubMed] [Google Scholar]
- 65.Rossor MN, Revesz T, Lantos PL, Warrington EK. Semantic dementia with ubiquitin-positive tau-negative inclusion bodies. Brain. 2000;123:267–276. doi: 10.1093/brain/123.2.267. [DOI] [PubMed] [Google Scholar]
- 66.Sampathu DM, Neumann M, Kwong LK, Chou TT, Micsenyi M, Truax A, Bruce J, Grossman M, Trojanowski JQ, Lee VM. Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol. 2006;169:1343–1352. doi: 10.2353/ajpath.2006.060438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shi J, Shaw CL, Du PD, Richardson AM, Bailey KL, Julien C, Stopford C, Thompson J, Varma A, Craufurd D, Tian J, Pickering-Brown S, Neary D, Snowden JS, Mann DM. Histopathological changes underlying frontotemporal lobar degeneration with clinicopathological correlation. Acta Neuropathol. 2005;110:501–512. doi: 10.1007/s00401-005-1079-4. [DOI] [PubMed] [Google Scholar]
- 68.Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, Nielsen JE, Hodges JR, Spillantini MG, Thusgaard T, Brandner S, Brun A, Rossor MN, Gade A, Johannsen P, Sorensen SA, Gydesen S, Fisher EM, Collinge J. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37:806–808. doi: 10.1038/ng1609. [DOI] [PubMed] [Google Scholar]
- 69.Snowden JS, Neary D, Mann DM. Frontotemporal dementia. Br J Psychiatry. 2002;180:140–143. doi: 10.1192/bjp.180.2.140. [DOI] [PubMed] [Google Scholar]
- 70.Spillantini MG, van Swieten JC, Goedert M. Tau gene mutations in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) Neurogenetics. 2000;2:193–205. doi: 10.1007/pl00022972. [DOI] [PubMed] [Google Scholar]
- 71.Srinivasan R, Davidson Y, Gibbons L, Payton A, Richardson AM, Varma A, Julien C, Stopford C, Thompson J, Horan MA, Pendleton N, Pickering-Brown SM, Neary D, Snowden JS, Mann DM. The apolipoprotein E epsilon 4 allele selectively increases the risk of frontotemporal lobar degeneration in males. J Neurol Neurosurg Psychiatry. 2006;77:154–158. doi: 10.1136/jnnp.2005.063966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Steele JC, Richardson JC, Olszewski J. Progressive supranuclear palsy. A heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol. 1964;10:333–359. doi: 10.1001/archneur.1964.00460160003001. [DOI] [PubMed] [Google Scholar]
- 73.Taniguchi S, McDonagh AM, Pickering-Brown SM, Umeda Y, Iwatsubo T, Hasegawa M, Mann DM. The neuropathology of frontotemporal lobar degeneration with respect to the cytological and biochemical characteristics of tau protein. Neuropathol Appl Neurobiol. 2004;30:1–18. doi: 10.1046/j.0305-1846.2003.00481.x. [DOI] [PubMed] [Google Scholar]
- 74.Togo T, Sahara N, Yen SH, Cookson N, Ishizawa T, Hutton M, de Silva R, Lees A, Dickson DW. Argyrophilic grain disease is a sporadic 4-repeat tauopathy. J Neuropathol Exp Neurol. 2002;61:547–556. doi: 10.1093/jnen/61.6.547. [DOI] [PubMed] [Google Scholar]
- 75.Vance C, Al Chalabi A, Ruddy D, Smith BN, Hu X, Sreedharan J, Siddique T, Schelhaas HJ, Kusters B, Troost D, Baas F, de Jong V, Shaw CE. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2-21.3. Brain. 2006;129:868–876. doi: 10.1093/brain/awl030. [DOI] [PubMed] [Google Scholar]
- 76.van der Knaap MS, Naidu S, Kleinschmidt-Demasters BK, Kamphorst W, Weinstein HC. Autosomal dominant diffuse leukoencephalopathy with neuroaxonal spheroids. Neurology. 2000;54:463–468. doi: 10.1212/wnl.54.2.463. [DOI] [PubMed] [Google Scholar]
- 77.Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, Kimonis VE. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36:377–381. doi: 10.1038/ng1332. [DOI] [PubMed] [Google Scholar]
- 78.Weintraub S, Rubin NP, Mesulam MM. Primary progressive aphasia. Longitudinal course, neuropsychological profile, and language features. Arch Neurol. 1990;47:1329–1335. doi: 10.1001/archneur.1990.00530120075013. [DOI] [PubMed] [Google Scholar]
- 79.Wightman G, Anderson VER, Martin J, Swash M, Anderton BH, Neary D, Mann D, Luthert P, Leigh PN. Hippocampal and neocortical ubiquitin-immunoreactive inclusions in amyotrophic-lateral-sclerosis with dementia. Neurosci Lett. 1992;139:269–274. doi: 10.1016/0304-3940(92)90569-s. [DOI] [PubMed] [Google Scholar]
- 80.Zhukareva V, Mann D, Pickering-Brown S, Uryu K, Shuck T, Shah K, Grossman M, Miller BL, Hulette CM, Feinstein SC, Trojanowski JQ, Lee VM. Sporadic Pick's disease: a tauopathy characterized by a spectrum of pathological tau isoforms in gray and white matter. Ann Neurol. 2002;51:730–739. doi: 10.1002/ana.10222. [DOI] [PubMed] [Google Scholar]