

Abstract
The 6-billion human population provides a vast reservoir of mutations, which, in addition to the opportunity of detecting very subtle defects, including specific cognitive dysfunctions as well as late appearing disorders, offers a unique background in which to investigate the roles of cell–cell adhesion proteins. Here we focus on inherited human disorders involving members of the cadherin superfamily. Most of the advances concern monogenic disorders. Yet, with the development of single nucleotide polymorphism (SNP) association studies, cadherin genes are emerging as susceptibility genes in multifactorial disorders. Various skin and heart disorders revealed the critical role played by desmosomal cadherins in epidermis, hairs, and myocardium, which experience high mechanical stress. Of particular interest in that respect is the study of Usher syndrome type 1 (USH1), a hereditary syndromic form of deafness. Studies of USH1 brought to light the crucial role of transient fibrous links formed by cadherin 23 and protocadherin 15 in the cohesion of the developing hair bundle, the mechanoreceptive structure of the auditory sensory cells, as well as the involvement of these cadherins in the formation of the tip-link, a key component of the mechano-electrical transduction machinery. Finally, in line with the well-established role of cadherins in synaptic formation, maintenance, strength, and plasticity, a growing number of cadherin family members, especially protocadherins, have been found to be involved in neuropsychiatric disorders.
Mutations in cadherins cause hereditary deafness by compromising development of mechanotransducers in the ear. They also contribute to skin, heart, and psychiatric disorders.
Cell–cell adhesion maintains the structural and functional integrity of multicellular organisms. It ensures cell recognition, sorting, and signaling in various tissues and organs. In line with the diversity of the structural and physiological properties of the cell–cell junctions (tight junctions, adherens junctions, desmosomes, and gap junctions), related disorders span a highly heterogeneous group of diseases. Abnormal or loss of cell–cell adhesion and/or associated signaling are hallmarks of tumor growth, malignant transformation, and metastases (see Berx and van Roy 2009). In addition, nonneoplastic diseases caused by defects in genes encoding proteins of the tight junctions, adherens junctions, desmosomes, and gap junctions have been identified (Table 1) (see also Lai-Cheong et al. 2007; Awad et al. 2008; Org et al. 2009). In this article, we summarize the current understanding of human inherited disorders because of defects in members of the cadherin superfamily, i.e., classical cadherins, desmosomal cadherins, protocadherins, and atypical cadherins.
Table 1.
Integral membrane proteins of cell–cell junctions involved in human diseases.
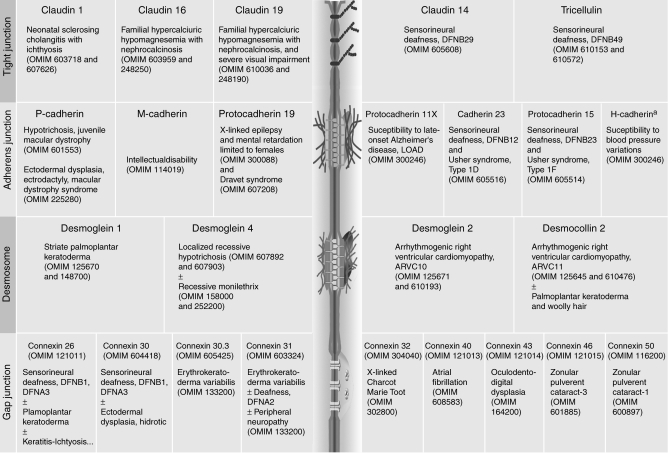 |
aH-cadherin (also referred to as cadherin 13) lacks transmembrane and cytoplasmic domains (see Org et al. 2009). OMIM numbers are listed for references and details on the indicated protein and/or associated human diseases.
Cadherins are characterized by the presence of 1–34 extracellular cadherin (EC) domains (comprised of ∼110 amino acids). Variations in their cytoplasmic domains impart functional specificity by conferring to each molecule the ability to interact with different ligands (see Shapiro and Weis 2009). These cadherins initiate and build up two types of specialized cell–cell contacts: (1) adherens junctions, and (2) desmosomes, where they tether the plasma membrane to actin microfilaments and intermediate filaments, respectively.
Clinical interest in cadherins stems from the discovery, about 25 years ago, of autoantibodies directed against desmosomal cadherins, desmoglein 1 and desmoglein 3, in the autoimmune blistering skin diseases pemphigus foliaceous and pemphigus vulgaris, respectively (Waschke 2008). Pemphigus-affected patients display a loss of intercellular adhesion between keratinocytes (acantholysis) caused by the binding of autoantibodies to desmosomal cadherins (Waschke 2008). Later on, when a dominantly inherited skin disease, striate palmoplantar keratoderma (SPPK; OMIM 148700), was mapped at chromosome 18q12.1, near the cluster of desmosomal cadherin genes (Hennies et al. 1995), the corresponding genes were considered as candidate causative genes for this disorder (Allen et al. 1996). However, it was not until 1999 that the first mutation—an in-frame deletion—was detected in the desmoglein 1 gene in SPPK patients (Rickman et al. 1999).
Genome sequence analysis has revealed an impressive diversity of the cadherin superfamily, with 113 cadherins identified in humans to date (Hulpiau and van Roy 2009). Despite functional redundancy among cell–cell junction proteins, several genetic defects affecting the skin, heart, nervous system, ear, or eye have been reported in mice and humans carrying mutations in some cadherin genes (Table 1). So far, mutations in genes encoding 11 cadherins (two classical, four desmosomal, three protocadherins, and two atypical) have been associated with hereditary human diseases (Table 1). These have highlighted a particular need for dynamic and positional cues for axon guidance during the establishment of neural circuits, or for high adhesive strength in tissues that experience mechanical stress, such as the epidermis and hairs, myocardium, and inner ear sensory epithelia.
CADHERINS IN SKIN AND HEART
Skin, Retinal, and Limb Defects Caused by P-Cadherin Deficiency
Sixteen members of the classical cadherin subfamily have been identified in the human genome (Hulpiau and van Roy 2009). P-cadherin (also referred to as cadherin 3) displays a wide tissue distribution. In 2001, mutations in P-cadherin were identified in a human disease exclusively affecting the hair follicle and the retina (Fig. 1B,C), termed hypotrichosis with juvenile macular dystrophy (HJMD syndrome; OMIM 601553). HJMD patients have short, sparse hair, and progressive blindness (Sprecher et al. 2001). Mutations in P-cadherin were later reported to cause another autosomal recessive disease, ectodermal dysplasia, ectrodactyly, and macular dystrophy (EEM syndrome; OMIM 225280) (Kjaer et al. 2005; Shimomura et al. 2008). The common features of the two diseases are early hair loss followed by progressive degeneration of the central retina, leading to blindness between the first and third decades. EEM affected patients, however, also display hypodontia and limb defects such as a split hand/foot malformation (see Fig. 1C). Genetic and clinical analyses revealed that HJMD patients carrying identical P-cadherin mutations display varying degrees of visual impairment (Kjaer et al. 2005; Shimomura et al. 2008). Whether modifier genes or possible digenic inheritance could explain the phenotype variability remains to be established.
Figure 1.

Mutations in P-cadherin, desmogleins (1 and 4), and desmocollin 2 cause hair, retina, and skin disorders. (A) Predicted structure of classical and desmosomal cadherins. These cadherins are composed of four highly conserved extracellular domains (EC1–4), a more variable extracellular anchor (EA, also referred to as EC5), a single transmembrane domain (TM), an intracellular anchor (IA), and additional cytoplasmic domains. The four desmogleins are unique in having extended tails beyond the ICS (intracellular catenin-binding site segment), namely an intracellular proline-rich linker domain (IPL), a variable number of repeated-unit domains (RUD; five in Dsg1, six in Dsg2, two in Dsg3, and three in Dsg4) and a desmoglein-specific terminal domain (DTD). Each of the three desmocollins (DSC1-3) have two spliced forms, a and b, with the “b” form lacking the ICS and therefore being unable to bind to plakoglobin. (B) P-cadherin mutations identified in HJMD and EEM affected patients are indicated. Note that a similar mutation (bold) can result either in EEM or HJMD disease (see Shimomura et al. 2008). (C, D) Clinical features. (C) HJMD patients show sparse, light and short hairs over the scalp and macular pigmentary abnormalities. EEM patients in addition show limb defects (adapted, with permission, from Indelman et al. 2007 [© Blackwell], and Shimomura et al. 2008). (D) SPPK patients show hyperkeratotic lesions on the palms and soles (adapted, with permission, from Dua-Awereh et al. 2009 [© Elsevier]).
Differences in the phenotypes caused by P-cadherin deficiency in humans and mice have slowed down the deciphering of mechanisms underlying skin, limb, and eye anomalies in HJMD and EEM syndromes. P-cadherin null mice show precocious mammary gland development, but surprisingly hairs, eyes, and limbs develop normally (Radice et al. 1997). Chromatin immunoprecipitation assays revealed that P-cadherin is a direct target for p63, a transcription factor known to be involved in hair follicle and limb development, and whose defects also cause split hand/foot malformations in humans. The p63 protein directly interacts with two distinct regions of the P-cadherin promoter, which could be crucial for hair follicle and limb bud development (Shimomura et al. 2008). Interestingly, one of the p63-target sites of the human P-cadherin gene is located within an intronic region not conserved in the mouse gene (Yang et al. 2006), thereby possibly accounting for the species-related phenotypic differences associated with mutations in the P-cadherin gene.
The strong expression of the P-cadherin gene that is observed in the hair follicle matrix, the apical ridge of limb buds, and the retinal pigment epithelium (RPE) correlate with the affected sites in humans carrying mutations in this gene (Xu et al. 2002). The changes in the spatio-temporal expression pattern of this cadherin during development may explain the restricted spectrum of clinical manifestations resulting from mutations in the P-cadherin gene (Indelman et al. 2007). In the developing epidermis, P-cadherin up-regulation correlated specifically with follicle downgrowth (Jamora et al. 2003). Also, when the outer layer of the optic cup differentiates into RPE cells, an increase in P-cadherin expression is observed concomitantly with a down-regulation of N-cadherin (the most abundant cadherin expressed by RPE cells) (Xu et al. 2002). Details on the subcellular defects caused by P-cadherin dysfunction in RPE cells and the underlying photoreceptor cells in HJMD patients are however still missing.
Desmosomal Cadherins in the Skin and Hair Follicles
Desmosomal cadherins form the core of desmosomes, adhesion structures often present in tissues under high mechanical stress, such as skin epidermis and hair follicles (see Delva et al. 2009). In humans, there are seven desmosomal cadherins: four desmogleins (DSG1–4) and three desmocollins (DSC1–3, each of them having two splice variants) (Fig. 1A). These cadherins resemble the classical cadherins, such as E-cadherin. They contain five tandemly repeated EC domains, a single transmembrane region, and a cytoplasmic domain. The desmogleins, however, differ from classical cadherins by the presence of several additional cytoplasmic domains that interact with the intermediate filament-based cytoskeleton through plakoglobin, plakophilins, and desmoplakin (Figs. 1A and 2).
Figure 2.

The desmosomal molecular network. The tissues that are affected in human individuals who carry mutations in genes encoding desmosomal proteins are indicated. (PKP) plakophilins 1 and 2; (PKG) plakoglobin; (DSP) desmoplakin.
Desmogleins and desmocollins are both needed for normal adhesive function. They are expressed in a tissue-specific and differentiation step-specific manner (Bazzi et al. 2006). Monolayered epithelia express only the desmoglein 2–desmocollin 2 pair, whereas stratified epithelia, such as the epidermis and the hair follicle, express combinations of the seven desmosomal cadherins (see Delva et al. 2009).
Desmoglein 1: Desmoglein 1 was the first cadherin found as a target of a human hereditary disease, striate palmoplantar keratoderma (Rickman et al. 1999). Sixteen different mutations have been identified in the DSG1 gene, most of which result in premature stop codons (Dua-Awereh et al. 2009; Hershkovitz et al. 2009). SPPK is likely caused by desmoglein 1 haploinsufficiency, even though a dominant-negative effect of mutant alleles cannot be excluded (Dua-Awereh et al. 2009).
SPPK patients display longitudinal hyperkeratotic lesions on the palms and soles, usually evident on the pressure areas that are under continuous mechanical stress and friction (see Fig. 1D). To date, no desmoglein 1 defective mice have been reported. Three Dsg1 genes, Dsg1α, Dsg1β, and Dsg1γ, have been identified in the mouse genome, whereas a single DSG1 gene is present in man. The murine Dsg1 genes display redundant expression patterns, suggesting that a targeted deletion of all three genes (technically difficult because of their genomic proximity) may be required to obtain a murine model of the human disease. Palmar skin analysis by EM in SPPK patients showed a reduced number of desmosomes in suprabasal layers that are abnormally small and poorly formed. At the molecular level, the expression of keratins K5, K14, and K10 was reduced, whereas that of K16 was increased (Wan et al. 2004). The up-regulation of K16, a known marker of the onset of epidermal proliferation and regeneration, may account for the incomplete differentiation of keratinocytes and skin thickening characteristic of the phenotype (Wan et al. 2004).
Desmoglein 4: Mutations in the DSG4 gene have been associated with an inherited human hair disorder, the Localized autosomal recessive hypotrichosis (LAH; OMIM 607903). The first identified mutation was a large, intragenic deletion, which leads to an in-frame deletion of the EC2 domain and part of the EC1 and EC3 domains (Kljuic et al. 2003). The frequent occurrence and high incidence of this mutation in Pakistani individuals from different geographical regions suggest that this is an ancestral mutation (Schweizer 2006). Additional mutations, missense, and null mutations have been reported (Wajid et al. 2007).
LAH affects the scalp, trunk, and extremities, largely sparing the facial, pubic, and axillary hairs. Hairs are fragile and break easily, leaving short sparse scalp hairs with a characteristic appearance (Kljuic et al. 2003). DSG4 is abundant in the hair follicle shaft cortex, where the abnormal phenotype is observed in LAH patients. Skin biopsy analysis from patients showed widening of spaces between keratinocytes throughout most of the epidermis (apart from the basal layer), with ultrastructural evidence of a reduced number of desmosomes that are abnormally small and poorly formed. A clinical overlap was observed between LAH and monilethrix, another human disease that also manifests a beaded hair phenotype (Schweizer 2006). Monilethrix is inherited either as a dominant or a recessive disorder. Homozygous mutations in DSG4 were identified in monilethrix-affected patients (Schaffer et al. 2006; Shimomura et al. 2006; Zlotogorski et al. 2006). The existence of potential modifying genes or different types/combinations of mutations may explain the interfamilial variations in phenotype among individuals carrying DSG4 mutations.
Desmoglein 2 and Desmocollin 2 in Heart Function
Myocardium desmosomes are essentially made of desmoglein 2 and desmocollin 2. Several studies have shown that heterozygous mutations in desmoglein 2 and desmocollin 2 in humans cause arrhythmogenic right ventricular cardiomyopathy (ARVC; OMIM 107970). ARVC is estimated to affect approximately one in 5000 individuals. The disease is frequently familial and typically involves autosomal dominant transmission with low penetrance and variable expressivity. Clinical features include right ventricular enlargement and dysfunction, characteristic electrocardiographic abnormalities with ventricular arrhythmia, and fibrofatty replacement of myocytes (see Awad et al. 2008 and references therein).
Desmoglein 2: Complete loss of desmoglein 2 in mice is embryonic lethal, shortly after implantation (Eshkind et al. 2002). Heterozygous mutations in the human DSG2 gene cause isolated ARVC without skin or hair abnormalities (Awad et al. 2006; Pilichou et al. 2006). Compensation by other DSG genes likely takes place in the epidermis but not in the myocardium of DSG2 mutation carriers. More than 22 mutations have been identified in ARVC patients, most of which are missense mutations located in the extracellular region of DSG2 (Awad et al. 2008). Histopathological analyses in several patients revealed a loss of cardiac cells and replacement with fibrofatty tissue, a decreased desmosome number and intercellular gap widening.
Desmocollin 2: A few families with autosomal dominant ARVC have been described to date (see Awad et al. 2008). Although inactivation of Dsc2 in the mouse has not yet been reported, knockdown of this gene in zebrafish results in dose-dependent impaired contractility and dilation of cardiac ventricles and bradycardia, associated with consistent loss of the desmosomal extracellular electron-dense midlines. The cardiac phenotype can be rescued with wild-type human DSC2 mRNA but not with the mutated DSC2 mRNA that carries the M211fs221 mutation (a frame shift mutation at amino acid position 211, leading to a premature stop codon at position 221) identified in ARVC patients. The markedly decreased levels of mRNA observed with the mutant DSC2 allele (M211fs221) led the authors to suggest that nonsense-mediated mRNA decay and consequent haploinsufficiency likely contribute to the disease mechanism (Heuser et al. 2006). Recently, sequence analysis of DSC2 revealed a homozygous single-base deletion in exon 12 (1841delG) in a family affected by autosomal recessive ARVC associated with palmoplantar keratoderma and woolly hair (Simpson et al. 2008), a phenotype similar to Naxos disease (OMIM 601214).
The Desmosomal Molecular Complex in Skin and Heart
Abnormal skin and heart phenotypes are not restricted to mutations of desmosomal cadherins. Twelve ARVC loci have been identified so far (Awad et al. 2008). In approximately 50% of ARVC patients, a mutation in one of the genes encoding the desmosomal cadherins and four major components of the cardiac desmosome have been identified (see Fig. 2). The four genes encode plakoglobin (skin, hair, heart; OMIM 601214), plakophilin 1 (skin; OMIM 604536), plakophilin 2 (heart; OMIM 609040), and desmoplakin (skin, hair, and heart; OMIM 605656) (see Fig. 2) (McGrath 2005; Syrris et al. 2006; Awad et al. 2008).
Two nonmutually exclusive models have been proposed to explain the phenotypes in skin, hair, and heart diseases. The first one involves a structural defect and suggests that the mechanical disruption of cell–cell adhesion underlies the observed anomalies. The second one involves signaling pathways, likely initiated by the translocation of the armadillo proteins, β-catenin and/or plakoglobin (γ-catenin), to the nucleus (Martin et al. 2009). It has been proposed that the translocation of plakoglobin to the nucleus decreases canonical Wnt signaling through Tcf/Lef1, and decreases the transcription of β-catenin target genes, e.g., c-Myc and cyclin D1 (see Fig. 2) (for details of the Wnt signaling pathway, see Cadigan and Peifer 2009). The resulting suppression of the Wnt pathway ultimately enhances the expression of adipogenic genes, thus promoting the transdifferentiation of cardiac myocytes to adipocytes, as observed in ARVC patients (Garcia-Gras et al. 2006).
The existence of crosstalk between desmosomes, adherens junctions, and gap junctions is well established (Fidler et al. 2008; Li et al. 2008). Consistently, abnormal expression of the gap junction protein connexin 43 has been reported in autosomal dominant forms of ARVC, Naxos, and Carvajal diseases, caused by homozygous mutations of desmosomal proteins (Fidler et al. 2008) (for details of gap junction structure and function, see Goodenough and Paul 2009). Connexin 43-containing gap junctions were significantly reduced in the heart both in number and in size, after N-cadherin depletion, which led the authors to suggest that germline mutations in the human N-cadherin gene may predispose patients to increased risk of cardiac arrhythmias (Li et al. 2008). Additional studies are thus necessary to discern how the interplay between the distinct specialized cell–cell junctions relates to the pathogenesis of the previously described diseases. Disentangling the respective contributions of adhesion and signaling functions of the desmosomal cadherins in tissue development and physiology as well as their respective contribution in the associated human diseases is an important issue regarding the development of appropriate therapeutic approaches.
CADHERINS IN THE CENTRAL NERVOUS SYSTEM
Synapses of the central nervous system are specialized asymmetric cell-adhesion junctions (Yamada and Nelson 2007) (for details of the neuronal synapse, see Giagtzoglou et al. 2009). In vitro experiments and analyses of spontaneous as well as engineered mutant mice have shed light on the roles played by members of the cadherin family in both synapse morphogenesis and plasticity (Morishita and Yagi 2007; Takeichi 2007; Arikkath and Reichardt 2008). Given their wide, differential, and combinatorial expression patterns and their critical roles in dendrite spine morphogenesis, synapse formation and remodeling, cadherins have over the past years been considered as possible targets for cognitive disorders (Bourgeron 2007; Arikkath and Reichardt 2008). Accordingly, causative mutations in several adhesion proteins (e.g., neuroligins and neurexins) have been identified in patients that manifest autistic spectrum disorders (Bourgeron 2007; Sudhof 2008). Nonetheless, it is only recently that causative mutations in some cadherin genes have been reported in cognitive disorders.
M-cadherin Deficiency Associated with Mild to Severe Intellectual Disability
In 2008, a balanced translocation in a female patient with severe intellectual disability (ID) was characterized (Bhalla et al. 2008). Intellectual disability, also referred to as mental retardation, is the most frequently reported developmental disability, affecting cognitive function in about 1%–3% of people worldwide. Whereas the origin of at least half of all ID cases is still unknown, chromosome abnormalities, single-gene mutations, and multifactorial interactions may account for approximately 30% of ID cases. The t(11;16) translocation truncates two genes encoding cell-adhesion molecules, M-cadherin (cadherin 15; OMIM 114019) and KIRREL3, Kin of irregular chiasm-like 3 (member of the immunoglobulin superfamily; OMIM 607761) (Bhalla et al. 2008). Subsequent analysis revealed four nonsynonymous variants in the M-cadherin gene, three of which (R60C, R92W, and A122V) were found to alter M-cadherin-mediated adhesiveness (Bhalla et al. 2008).
How defects in M-cadherin lead to ID in humans is still unknown. Expression studies in mice suggested that M-cadherin might play a critical role in skeletal muscle and cerebellum, where this cadherin is mainly expressed. Nevertheless, M-cadherin–deficient mice do not show defects in skeletal muscle development. Because adherens junctions in the cerebellum were normal in the mutant mice, it has been proposed that other cadherins, in particular N-cadherin, substitute for the mutated M-cadherin (Hollnagel et al. 2002). Whether these mice show impaired learning and memory performances remains to be determined.
The second gene affected by the translocation is KIRREL3, a mammalian homolog of the Drosophila gene kirre (kin of irregular chiasm C-roughest). KIRREL3 expression was detected exclusively in human fetal and adult brain. Interestingly, KIRREL3 interacts with CASK, calcium/calmodulin-dependent serine protein kinase, which is defective in an X chromosome-linked ID (OMIM 300172). CASK interacts with GluR6, which is defective in an autosomal recessive ID (OMIM 138244), and with δ-catenin, mutations of which are associated with the severe mental retardation in cri-du-chat syndrome (OMIM 116806). Thus, a cadherin-catenin-KIRREL3-CASK molecular network has been proposed to operate in cognitive functions.
Mutations in Protocadherin 19 Associated with Epilepsy and Mental Retardation
Protocadherins are predominantly expressed in the nervous system and constitute the largest of the cadherin subfamilies (see Giagtzoglou et al. 2009). Although most of these genes are found in three clusters (Pcdh-α, Pcdh-β, and Pcdh-γ) located at chromosome 5q31, others are found dispersed in the genome (see Morishita and Yagi 2007; Hulpiau and van Roy 2009). Protocadherin genes are characterized by the presence of a large first exon coding for extracellular ectodomain repeats (typically six or seven), a transmembrane domain, and a part of the cytoplasmic domain (Morishita and Yagi 2007).
Mutations in the protocadherin 19 gene, a member of the δ2 subclass of nonclustered protocadherins, cause an X chromosome-linked epilepsy and mental retardation limited to females (EFMR), which is characterized by seizure onset in infancy or early childhood and cognitive impairment (OMIM 300088). EFMR has a unique inheritance pattern. Indeed, disorders caused by defects in genes located on the X chromosome typically affect males and leave carrier females unaffected. In contrast, EFMR spares male carriers of the mutations and only affects females (Dibbens et al. 2008). To explain the sex-limited mode of inheritance of EFMR, Dibbens et al. proposed that males lacking functional PCDH19 are unaffected because a related but nonparalogous protocadherin gene specific to the Y chromosome, PCDH11Y, compensates for the lack of PCDH19. By contrast, females, as a result of the X-inactivation process (Heard et al. 2004), display mosaicism with some neurons expressing the mutated PCDH19 gene and others the wild-type gene. The resulting mosaics would scramble neuronal identity and interfere with cell–cell communication, thus causing EFMR (Dibbens et al. 2008). Interestingly, additional point mutations (missense and truncating mutations, all located in the sequence encoding the extracellular region of the protein) were identified in female patients displaying Dravet syndrome, a genetically determined epileptic encephalopathy that mainly affects females (Depienne et al. 2009). In this study, a hemizygous deletion encompassing the PCDH19 gene was, however, found also in one male patient. Analysis of fibroblasts from the affected male revealed cell mosaicism for the PCDH19 deletion, which further argues in favor of “cellular interference” being the pathogenic mechanism in PCDH19-linked diseases (Depienne et al. 2009).
Genetic Variations in Cadherins and the Susceptibility to Psychiatric Disorders
Besides causative mutations, sequence variants, such as SNPs, in cadherin genes (see the following discussion) have recently been associated with an increased risk of developing psychiatric disorders. Given the roles that the adhesion proteins play in the development and the connectivity between individual neurons, corresponding genes have been considered as good candidates for susceptibility to psychiatric disorders.
Over the years, several laboratories have tested and excluded the involvement of sequence variants in PCDH8, PCDH11Y, or NrCAM, in patients with cognitive disorders including autism, attention deficit hyperactivity disorder, obsessive-compulsive disorder, and schizophrenia. More recently, a preliminary report analyzed 32 polymorphisms in 30 genes, including protocadherin α3, β11, cluster α (PCDHα8 and PCDHα10), and protocadherin 12 genes, in schizophrenic patients with abnormal neurodevelopment. These studies revealed an association between a polymorphism in protocadherin 12 (S640 N) and cortical folding (asymmetry coefficient of gyrification index) (Gregorio et al. 2009). Yet, further studies are needed to confirm this finding. Moreover, a recent two-stage genome-wide association study conducted in large cohorts of patients affected by late-onset Alzheimer’s disease (a neurodegenerative disease characterized by senile plaques and neurofibrillary tangles in the brain) and control cases identified a significant association between the disease and SNPs located within a haplotype block that encompasses PCDH11X at Xq21.3 (Carrasquillo et al. 2009). One of the SNPs (rs2573905) is more likely to alter PCDH11X function, as it resides in a 100-bp region in intron 2 that is conserved (70% identity) between the human and mouse sequences. To date, no mutation affecting protocadherin 11 encoded by the X chromosome has been identified in the patients. Additional studies are therefore needed to determine whether risk for Alzheimer’s disease is mediated by specific PCDH11X variants.
CADHERINS IN THE INNER EAR AND RETINA
The cell–cell junctions of the mammalian inner ear that house two sensory organs, the cochlea, the auditory sensory organ, the vestibule, the balance organ (see Fig. 3A), have focused a long-lasting interest as they are continuously submitted to tensions evoked by sound or acceleration stimuli. Surprisingly, the presence of cadherins at the junctions between mature sensory cells and their supporting cells is still under debate (Leonova and Raphael 1997; Nunes et al. 2006). The identification of genes which cause nonsyndromic (isolated) forms of deafness in humans combined with the study of the corresponding mouse mutants (see the following discussion) has highlighted the roles of the auditory neuroepithelium cell–cell junctions in cochlear physiology and the underlying molecular mechanisms. In addition, the study of a human syndromic form of deafness, Usher syndrome type 1 (USH1), has uncovered the key role of two members of the cadherin superfamily, cadherin 23 and protocadherin 15, in the development and function of the hair bundle, the mechanosensitive structure operating the conversion of acoustic pressure signals into electrical signals by the auditory sensory hair cells (Petit 2001; Petit and Richardson 2009).
Figure 3.

Atypical tight-adherens junctions are found between outer hair cells and their supporting cells in the mammalian auditory organ (cochlea). (A) A cross section through the cochlea, which is the auditory organ, shows the three fluid-filled compartments, the scala vestibuli, scala media, and scala tympani. (B) Focus on the scala media that houses the auditory sensory epithelium, the organ of Corti (OC), which contains the mechanosensitive inner (IHC) and outer (OHCs) hair cells. sv, stria vascularis. (C) Top view of the organ of Corti, showing the highly organized hair bundles of IHCs and OHCs. (D) At the TAJ junction between OHCs and Deiters cell (DC), claudins partition into claudin 14- and claudin-9/6 containing subdomains that are distinguishable by their strand morphology and their associated cytoskeletal network. Note the colocalization of catenin complexes with claudin-containing domains (modified, with permission, from Nunes et al. 2006).
Typical and Atypical Cell–Cell Junctions in the Cochlea
The cochlea is made up of three fluid-filled compartments of different ionic compositions—the scala vestibuli (perilymph), the scala media (endolymph), and the scala tympani (perilymph) (Fig. 3A,B). The perilymph has an ion composition typical of extracellular fluids (high Na+ and low K+ concentrations), whereas the endolymph has high K+ and low Na+ concentrations, and thus displays characteristics of an intracellular fluid (see Wangemann 2006). In addition, a transepithelial electric potential difference of ∼100 mV exists between the endolymphatic and perilymphatic compartments of the cochlea. This so-called endocochlear potential drives the cationic transduction current through hair cells in response to sound stimulation.
The auditory sensory epithelium, the organ of Corti, which keeps apart the endolymphatic and the perilymphatic compartments, contains sensory hair cells of two types, the inner (IHC) and the outer (OHC) hair cells (Fig. 3C,D). Various typical and atypical junctions ensure both the ion barrier between the endolymph and perilymph, and the integrity of the sensory epithelium despite the continuous mechanical stress the organ of Corti experiences. The hair bundle, at the apical region of hair cells, is exposed to endolymph, whereas hair cell basolateral regions are bathed in perilymph. The junctions between outer hair cells and their supporting cells, the Deiters cells, have a unique organization that does not conform to canonical TJs and AJs (Fig. 3D). It has been first proposed that mosaics of several alternating TJs and AJs make up these junctions (Raphael and Altschuler 2003). Later on, freeze-fracture electron microscopy and immunolabeling studies showed that these junctions are instead made up of a single large (3–5 μm) occluding junction with AJ features (Nunes et al. 2006). Strikingly, at least three canonical AJ proteins, α-, β-, and p120-catenins, were present between OHCs and Deiters cells, and colocalizing with TJ proteins (ZO1, claudin 14, and claudins 6/9) (Fig. 3D). This hybrid junction is now referred to as a tight adherens junction, TAJ (Nunes et al. 2006). Defects in two TJ proteins, tricellulin and claudin 14, cause deafness in humans (Table 1). So far, there is no mutant animal model for the tricellulin defect. In the claudin 14-deficient mice, a disruption of the junctional barrier resulting in an elevated K+ concentration around the hair cell bodies, and thereby chronic cell depolarization has been proposed to lead to hair cell death (Ben-Yosef et al. 2003).
The sound-induced displacement amplitude of the organ of Corti measured in the apical region of the guinea pig cochlea is roughly 10 nm for a sound intensity of 80 dB SPL. This displacement is estimated to generate a force per unit surface of 30–90 Pascals in the radial direction of the neuroepithelium, with similar forces in the transversal and longitudinal directions (Tomo et al. 2007). How cell–cell junctions of the organ of Corti cope with this mechanical stress is poorly understood. However, the critical role that the junctions between hair cells and their supporting cells play in the resilience of the auditory epithelium to sound trauma has recently been pinpointed. Hair cell conditional inactivation of the gene encoding vezatin, a transmembrane ubiquitous AJ protein previously shown to associate with the E-cadherin-catenin complex (Kussel-Andermann et al. 2000), resulted in a delayed spontaneous hearing loss in the mutant mice (Bahloul et al. 2009). Moreover, a single short exposure (1 minute) of these mice to a loud broadband sound (106 dB SPL) was sufficient to cause irreversible hearing impairment, with TAJ disorganization and hair cell loss (Bahloul et al. 2009).
Dual Function of Cadherin 23 and Protocadherin 15 in Auditory Hair Cell Development and Mechanoelectrical Transduction
Our ability to perceive sound and maintain balance depends on mechanoelectrical transduction (MET), a process achieved in the hair bundle. The hair bundle is made up of 20–300 actin-filled, stiff microvilli—the stereocilia—and a single genuine cilium, the kinocilium (only transient in the cochlea) (Fig. 4A). The stereocilia are arranged in three to four rows of increasing heights toward the kinocilium. Altogether they form a “V”-shaped staircase, with the kinocilium located at the vertex of the V. The vertices of the hair bundles of all OHCs are aligned uniformly along the mediolateral axis of the cochlea, thereby defining the planar cell polarity axis of the organ of Corti (Fig. 4A,B). Two types of links interconnect adjacent stereocilia in the mature hair bundle, namely the tip link and the horizontal top connectors (Fig. 4A). The tip link is a single oblique apical link that connects the tip of each stereocilium to the side of the nearest taller stereocilium, whereas the top connectors are lateral links that couple adjacent stereocilia both within and between rows. During development, different types of transient lateral links connect the growing stereocilia to each other and the kinocilium to the adjacent stereocilia of the tallest row. These links are the early transient lateral links, shaft links, ankle links, and kinociliary links (Fig. 4A) (see Petit and Richardson 2009). The function of these links was unknown until several deafness genes, including those responsible for the USH1 syndrome, were found to encode components of the different subsets of stereocilia links.
Figure 4.

Various links are found between adjacent stereocilia and between stereocilia and the kinocilium in the growing and mature hair bundle of inner ear sensory cells. (A) Throughout development and in mature hair cells, the stereocilia are connected by different types of transient and persistent links, respectively. The early transient lateral links (ELL), shaft links (SL), ankle links (AL), and kinociliary links (KL) are progressively replaced by the apical top connectors (TC) found in mature hair cells. The tip links (TL) are already present in the growing hair bundles, and they persist in mature hair cells. The inset shows a scanning electron microscopy image of an OHC hair bundle. (B) Kinocilium migration steps and planar cell polarity (PCP) in the organ of Corti. The kinocilium directs the staircase pattern of the hair bundle. (C) Cadherin 23 and protocadherin 15 isoforms can form the early transient lateral links, kinociliary links, and the tip link. Note that only protocadherin 15, which forms the lower part of the tip link, may be tightly associated to the MET channels.
Usher syndrome is the most frequent cause of hereditary deaf-blindness in humans. It is a monogenic disorder, transmitted on an autosomal recessive mode. Of the three clinical forms (USH1–3), USH1 is the most severe. It is characterized by severe to profound congenital hearing impairment, balance deficiency, and prepubertal-onset retinitis pigmentosa leading to blindness. Five USH1 genes have been identified (El-Amraoui and Petit 2005; Leibovici et al. 2008). Two cadherins, cadherin 23 and protocadherin 15, underlie the USH1D (Bolz et al. 2001; Bork et al. 2001) and USH1F (Ahmed et al. 2001; Alagramam et al. 2001b) forms, respectively. The three other USH1 genes encode an actin-based motor, myosin VIIa (USH1B) (Weil et al. 1995), a PDZ-domain-containing protein, harmonin (USH1C) (Bitner-Glindzicz et al. 2000; Verpy et al. 2000), and a putative scaffolding protein with ankyrin repeats, sans (USH1G) (Weil et al. 2003).
Cadherin 23 and Protocadherin 15 Form Transient Links that are Essential for the Cohesion and Proper Orientation of the Growing Hair Bundle
Cadherin 23 and protocadherin 15 consist of several isoforms with distinct extracellular regions and cytoplasmic domains, which result from alternatively spliced CDH23 and PCDH15 transcripts. Three classes of cadherin 23 isoforms (a to c) and four classes of protocadherin 15 isoforms (CD1 to CD3, and SI) have been predicted (Lagziel et al. 2005; Michel et al. 2005; Ahmed et al. 2006). The longest cadherin 23 and protocadherin 15 isoforms contain 27 and 11 EC domains, respectively (Fig. 5A,B). Notably, the cytoplasmic domains of both cadherin 23 and protocadherin 15 isoforms show no sequence similarity between each other or with classical cadherins. In particular, they all lack the consensus motif for binding to β-catenin (Cavey et al. 2008; Nelson 2008), raising the question of the existence of a dynamic link between these two cadherins and actin filaments.
Figure 5.

Cadherin 23 and protocadherin 15 defects underlie two genetic forms of Usher type 1 (USH1) syndrome. (A, B) Three classes of cadherin 23 isoforms (a, b, and c in A), and four classes of protocadherin 15 isoforms (CD1, CD2, CD3, and SI in B) have been reported. The distinct PDZ domain-binding motifs in the different protocadherin 15 cytodomains (CD) are indicated. SI, secreted isoform (see Ahmed et al. 2008b). (C) Scanning electron micrographs of OHC hair bundles in wild-type and protocadherin 15-deficient mice at postnatal day 15 (P15). Note the fragmentation of the hair bundles in the mutant (Source: M. Leibovici, Institut Pasteur, France). (D) Summary of the interactions between USH1 proteins. Bar = 1 µm.
Animal models defective for these proteins (Leibovici et al. 2008) have revealed the roles of these two cadherins, i.e., the deaf waltzer mouse (Di Palma et al. 2001) and sputnik zebrafish (Sollner et al. 2004) mutants for cadherin23, and the Ames waltzer mouse (Alagramam et al. 2001a) and orbiter zebrafish (Seiler et al. 2005) mutants for protocadherin 15. Mutations in either of these genes, or in the genes encoding myosin VIIa, harmonin, or sans all lead to similar morphological anomalies in the mouse cochlea, which consist of fragmented and misoriented hair bundles (see Fig. 5C, Lefevre et al. 2008; Petit and Richardson 2009).
When stereocilia emerge at the apical surface of auditory hair cells at E15.5 in the mouse, the kinocilium is located at the center. It then migrates to the cell periphery, thereby initiating the establishment of hair bundle polarity. Then, the kinocilium undergoes a second migratory step that results in a reorientation of the hair bundle, which reaches its final location on the apical cell surface, pointing toward the cochlear lateral wall (Fig. 4B) (Kelly and Chen 2007; Jones and Chen 2008; Petit and Richardson 2009). Analysis of the various USH1 mouse models revealed that although the kinocilium migrated from its central position to the cell periphery, it failed to reach its final correct position and was frequently disconnected from the neighboring stereocilia (Lefevre et al. 2008). In addition, in all Ush1 mutants, the hair cells had undergone early fragmentation of their hair bundle, which was split into a few clumps of stereocilia (Fig. 5C).
How does the absence of any of the USH1 proteins lead to this common hair bundle phenotype? Over the past 10 years, evidence for colocalization in the hair bundle and direct in vitro interactions of these proteins (Fig. 5D) has led to the conclusion that a similar pathogenic mechanism causes deafness in USH1 patients (see Leibovici et al. 2008). This mechanism involves defective early transient stereocilia lateral links and kinociliary links in the growing hair bundles (see Fig. 5C). Myosin VIIa and sans are required for the targeting of harmonin-b into the stereocilia, where it binds to F-actin and thus most likely anchors the cadherin 23- and protocadherin 15-containing links to the actin core of the stereocilium (Boëda et al. 2002; Lefevre et al. 2008). However, because the phenotype of the null harmonin b mutant is less severe than that of the mutant lacking all three harmonin isoforms, additional roles can be predicted for myosin VIIa and sans in the establishment of a cohesive hair bundle (I. Foucher, A. Bahloul, and E. Caberlotto, unpubl.). Cohesive forces applied to the early transient interstereociliary and kinociliary links in the forming hair bundle are thus necessary for the formation of a unitary and properly oriented “V”-shaped hair bundles. Conversely, the hair bundle misorientation suggests that the kinocilium final position is controlled by the sum of the forces applied to the entire hair bundle. Whether these forces originate outside the hair cell, for instance from the concomitant convergent-extension process driving cell intercalation and elongation of the cochlear neuroepithelium, or within the hair cell, is still unknown. This calls for both qualitative and quantitative information regarding the movements of the cells and of the kinocilia in the developing auditory neuroepithelium, as well as of actin filaments and microtubules organization over that time period.
Cadherin 23 and Protocadherin 15 Form the Tip Link in Mature Hair Cells
A late onset form of deafness caused by CDH23 mutations provided the first indication for its role beyond early hair bundle development and possibly also in MET (Noben-Trauth et al. 2003). The prevailing theory for MET states that tension in “gating springs” controls the open probability of MET channels (Corey and Hudspeth 1983). The tip link (Pickles et al. 1984), or a more compliant link in series with the tip link, comprises the gating spring. Sound displacement of the hair bundle toward the tallest stereocilia promotes MET channel opening, leading to an influx of endolymphatic cations. The resulting depolarizing receptor potential ultimately leads to neurotransmitter release by IHCs, and thereby the transfer of the acoustic information to the auditory neurons. Under a 90 dB sound stimulation, a 100 nm displacement of the hair bundle tip occurs in the guinea pig, generating a tension of 3 to 6 pN on the tip link and the molecular complex that bridges it to the stereocilia actin-filaments (Fridberger et al. 2006). This corresponds to a roughly 10% modulation of its resting tension evaluated in the range of 40 pN (Jaramillo and Hudspeth 1993; Prost et al. 2007).
High-resolution ultrastructural studies have shown that tip links consist of two to three helical filaments, approximately 150–200 nm in length (Kachar et al. 2000). Antibodies to specific epitopes of cadherin 23 or protocadherin 15 labeled the tips of stereocilia at the tip link emplacement (Ahmed et al. 2006; Kazmierczak et al. 2007). A detailed subcellular analysis showed that cadherin 23 and protocadherin 15 localize to the upper and lower part of the tip link, respectively (Fig. 4C). Cadherin 23 homodimers have been shown to interact in trans in a Ca2+-dependent manner with protocadherin 15 homodimers, forming a filament with structural similarity to the tip link (Kazmierczak et al. 2007; Muller 2008). To date, functional evidence establishing that cadherin 23 and protocadherin 15 form the tip link is a major issue, especially because careful analysis of the hair bundles in mouse mutants with predicted null alleles of cadherin 23 has shown the presence of tip link-like filaments connecting adjacent stereocilia (Rzadzinska and Steel 2009). Yet, a cadherin 23 mouse mutant, salsa, with an E737V substitution within the Ca2+ binding motif (LDRE) in the EC7 domain of the a-isoform, the longest one, further supports the involvement of the cadherins in the formation of the tip link because only rare disorganized hair bundles were observed, whereas absence of tension on the stereocilia tips argues for a loss of the tip links (Schwander et al. 2009). Protocadherin 15 isoforms from the CD2 subgroup have been proposed to be components of the early transient lateral links, and isoforms from the CD3 subgroup to form the tip link (Ahmed et al. 2006).
Cadherin 23 and Protocadherin 15 in the Retina
So far, progress in understanding the critical roles of cadherin 23 and protocadherin 15 in the retina has been hampered by the absence of retinitis pigmentosa in Ush1 mutant mice, including cadherin 23 and protocadherin 15 double knockout mutants (Ahmed et al. 2008a, see Leibovici et al. 2008 for a review). The shorter lifespan of the mouse, interspecies difference in light exposure, existence of a functional redundancy for USH1 proteins in murine but not human retina, or an intrinsic difference between murine and human photoreceptor cell’s physiology may account for this phenotypic discrepancy. In regards to the last possibility, the underdevelopment of photoreceptor cells’ calycal processes in mouse compared to human is an attractive hypothesis (Petit 2001). Indeed, these microvilli-like extensions of the photoreceptor inner segment of yet unknown functions are bridged to the connecting cilium (Cohen 1963) by fibrous links and some of these links have been shown to share molecular components with a subset of hair bundle transient lateral links, the ankle links (Kremer et al. 2006; Leibovici et al. 2008). To clarify this point, comparative studies determining the subcellular localization of cadherin 23 and protocadherin 15 isoforms in mouse and in human retinas are required.
According to the mutations detected in USH1 patients, the protocadherin 15-CD1 subclass may be the only subclass critical for retinal functions (Ahmed et al. 2008b). Whereas the USH1D mutations that have been identified do affect the a-isoforms of cadherin 23 (see http://webh01.ua.ac.be/hhh/), whether the b- and c-isoforms also play an important role in the retina is still unknown. Results of the comparative analysis will be essential to determine the appropriate animal model to decipher the pathogenesis of retinitis pigmentosa as well as to test future therapeutic approaches. Of note in zebrafish, two orthologues of the mammalian protocadherin 15 gene, pcdh15a and pcdh15b, encode proteins with highly dissimilar intracellular domains. Pcdh15a defects result in the hair bundle disorganization without visual abnormalities, whereas pcdh15b defects only cause retinal anomalies, i.e., abnormal alignment of photoreceptor outer disks (Seiler et al. 2005).
CONCLUDING REMARKS
The human genome-wide association studies are likely to extend the list of human disorders involving cadherins in a near future, especially regarding neuropsychiatric disorders. Epigenetic modifications of these genes are likely to broaden their implication in these diseases. Mutation screening will not only allow further deciphering of the cadherin isoforms critically underlying phenotypic features, but should also help identify the role of some of their interacting partners, such as cytoskeletal regulators, protein kinases and phosphatases, and transcription cofactors. The putative correlation between phenotypic features and copy number variants (CNV) of the cadherin genes would be of particular interest regarding the role of adhesion strength. Furthermore, the finding of modifier genes accounting for the failure of phenotype-genotype correlation in cadherin genes should add to our present knowledge of the roles played by the cadherins in various signaling pathways. Generating appropriate animal models remains, however, essential to achieve an in-depth understanding of cadherin functions/dysfunctions and as regards neuropsychiatric disorders, genetically modified animals with more elaborated cognitive functions than those of the mouse are needed.
ACKNOWLEDGMENTS
We are grateful to Jean-Pierre Hardelin for his comments and suggestions, to Michel Leibovici for SEM images. We apologize for not citing all the relevant references because of space limitation. The work in the authors’ laboratory is supported by ANR-07-MRARE-009-01, and Fondation pour la Recherche Médicale (FRM) to AE, the R. and G. Strittmatter Foundation, FAUN-Stiftung (Suchert Foundation), Fondation Orange, and the European Commission FP6 Integrated Project EUROHEAR, LSHG-CT-2004-512063 to CP.
Footnotes
Editors: W. James Nelson and Elaine Fuchs
Additional Perspectives on Cell Junctions available at www.cshperspectives.org
REFERENCES
- Ahmed ZM, Riazuddin S, Bernstein SL, Ahmed Z, Khan S, Griffith AJ, Morell RJ, Friedman TB, Riazuddin S, Wilcox ER 2001. Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am J Hum Genet 69:25–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed ZM, Goodyear R, Riazuddin S, Lagziel A, Legan PK, Behra M, Burgess SM, Lilley KS, Wilcox ER, Griffith AJ, et al. 2006. The tip-link antigen, a protein associated with the transduction complex of sensory hair cells, is protocadherin-15. J Neurosci 26:7022–7034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed ZM, Kjellstrom S, Haywood-Watson RJ, Bush RA, Hampton LL, Battey JF, Riazuddin S, Frolenkov G, Sieving PA, Friedman TB 2008a. Double homozygous waltzer and Ames waltzer mice provide no evidence of retinal degeneration. Mol Vis 14:2227–2236 [PMC free article] [PubMed] [Google Scholar]
- Ahmed ZM, Riazuddin S, Aye S, Ali RA, Venselaar H, Anwar S, Belyantseva PP, Qasim M, Friedman TB 2008b. Gene structure and mutant alleles of PCDH15: Nonsyndromic deafness DFNB23 and type 1 Usher syndrome. Hum Genet 124:215–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alagramam KN, Murcia CL, Kwon HY, Pawlowski KS, Wright CG, Woychik RP 2001a. The mouse Ames waltzer hearing-loss mutant is caused by mutation of Pcdh15, a novel protocadherin gene. Nature Genet 27:99–102 [DOI] [PubMed] [Google Scholar]
- Alagramam KN, Yuan H, Kuehn MH, Murcia CL, Wayne S, Srisailpathy CRS, Lowry RB, Knaus R, Van Laer L, Bernier FP, et al. 2001b. Mutations in the novel protocadherin PCDH15 cause Usher syndrome type 1F. Hum Mol Genet 10:1709–1718 [DOI] [PubMed] [Google Scholar]
- Allen E, Yu QC, Fuchs E 1996. Mice expressing a mutant desmosomal cadherin exhibit abnormalities in desmosomes, proliferation, and epidermal differentiation. J Cell Biol 133:1367–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arikkath J, Reichardt LF 2008. Cadherins and catenins at synapses: Roles in synaptogenesis and synaptic plasticity. Trends Neurosci 31:487–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awad MM, Dalal D, Cho E, Amat-Alarcon N, James C, Tichnell C, Tucker A, Russell SD, Bluemke DA, Dietz HC, et al. 2006. DSG2 mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am J Hum Genet 79:136–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awad MM, Calkins H, Judge DP 2008. Mechanisms of disease: Molecular genetics of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Nat Clin Pract Cardiovasc Med 5:258–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahloul A, Simmler MC, Michel V, Leibovici M, Perfettini I, Roux I, Weil D, Nouaille S, Zuo J, Zadro C, et al. 2009. Vezatin, an integral membrane protein of adherens junctions, is required for the sound-resilience of cochlear hair cells. EMBO Mol Medicine 1:125–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzi H, Getz A, Mahoney MG, Ishida-Yamamoto A, Langbein L, Wahl JK 3rd, Christiano AM 2006. Desmoglein 4 is expressed in highly differentiated keratinocytes and trichocytes in human epidermis and hair follicle. Differentiation 7:129–140 [DOI] [PubMed] [Google Scholar]
- Ben-Yosef T, Belyantseva IA, Saunders TL, Hughes ED, Kawamoto K, Van Itallie CM, Beyer LA, Halsey K, Gardner DJ, Wilcox ER, et al. 2003. Claudin 14 knockout mice, a model for autosomal recessive deafness DFNB29, are deaf due to cochlear hair cell degeneration. Hum Mol Genet 12:2049–2061 [DOI] [PubMed] [Google Scholar]
- Berx G, van Roy F 2009. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb Perspect Biol 1:a003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla K, Luo Y, Buchan T, Beachem MA, Guzauskas GF, Ladd S, Bratcher SJ, Schroer RJ, Balsamo J, DuPont BR, et al. 2008. Alterations in CDH15 and KIRREL3 in patients with mild to severe intellectual disability. Am J Hum Genet 83:703–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitner-Glindzicz M, Lindley KJ, Rutland P, Blaydon D, Smith VV, Milla PJ, Hussain K, Furth-Lavi J, Cosgrove KE, Shepherd RM, et al. 2000. A recessive contiguous gene deletion causing infantile hyperinsulinism, enteropathy and deafness identifies the Usher type 1C gene. Nature Genet 26:56–60 [DOI] [PubMed] [Google Scholar]
- Boëda B, El-Amraoui A, Bahloul A, Goodyear R, Daviet L, Blanchard S, Perfettini I, Fath KR, Shorte S, Reiners J, et al. 2002. Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. EMBO J 21:6689–6699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolz H, von Brederlow B, Ramirez A, Bryda EC, Kutsche K, Nothwang HG, Seeliger M, del CSCM, Vila MC, Molina OP, et al. 2001. Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nature Genet 27:108–112 [DOI] [PubMed] [Google Scholar]
- Bork JM, Peters LM, Riazuddin S, Bernstein SL, Ahmed ZM, Ness SL, Polomeno R, Ramesh A, Schloss M, Srisailpathy CRS, et al. 2001. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am J Hum Genet 68:26–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeron T 2007. The possible interplay of synaptic and clock genes in autism spectrum disorders. Cold Spring Harb Symp Quant Biol 72:645–654 [DOI] [PubMed] [Google Scholar]
- Cadigan KM, Peifer M 2009. Wnt signaling from development to disease: Insights from model systems. Cold Spring Harb Perspect Biol 1:a002881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasquillo MM, Zou F, Pankratz VS, Wilcox SL, Ma L, Walker LP, Younkin SG, Younkin CS, Younkin LH, Bisceglio GD, et al. 2009. Genetic variation in PCDH11X is associated with susceptibility to late-onset Alzheimer’s disease. Nature Genet 41:192–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavey M, Rauzi M, Lenne PF, Lecuit T 2008. A two-tiered mechanism for stabilization and immobilization of E-cadherin. Nature 453:751–756 [DOI] [PubMed] [Google Scholar]
- Cohen AI 1963. Vertebrate retinal cells and their organization. Biol rev Cambridge Philos Soc 38:427–459 [Google Scholar]
- Corey DP, Hudspeth AJ 1983. Kinetics of the receptor current in bullfrog saccular hair cells. J Neurosci 3:962–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delva E, Tucker DK, Kowalczyk AP 2009. The desmosome. Cold Spring Harb Perspect Biol 1:a002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depienne C, Bouteiller D, Keren B, Cheuret E, Poirier K, Trouillard O, Benyahia B, Quelin C, Carpentier W, Julia S, et al. 2009. Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females. PLoS Genet 5:pe1000381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Palma F, Holme RH, Bryda EC, Belyantseva IA, Pellegrino R, Kachar B, Steel KP, Noben-Trauth K 2001. Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nature Genet 27:103–107 [DOI] [PubMed] [Google Scholar]
- Dibbens LM, Tarpey PS, Hynes K, Bayly MA, Scheffer IE, Smith R, Bomar J, Sutton E, Vandeleur L, Shoubridge C, et al. 2008. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nature Genet 40:776–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dua-Awereh MB, Shimomura Y, Kraemer L, Wajid M, Christiano AM 2009. Mutations in the desmoglein 1 gene in five Pakistani families with striate palmoplantar keratoderma. J Dermatol Sci 53:192–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Amraoui A, Petit C 2005. Usher I syndrome: Unravelling the mechanisms that underlie the cohesion of the growing hair bundle in inner ear sensory cells. J Cell Sci 118:4593–4603 [DOI] [PubMed] [Google Scholar]
- Eshkind L, Tian Q, Schmidt A, Franke WW, Windoffer R, Leube RE 2002. Loss of desmoglein 2 suggests essential functions for early embryonic development and proliferation of embryonal stem cells. Eur J Cell Biol 81:592–598 [DOI] [PubMed] [Google Scholar]
- Fidler LM, Wilson GJ, Liu F, Cui X, Scherer SW, Taylor GP, Hamilton RM 2008. Abnormal connexin43 in arrhythmogenic right ventricular cardiomyopathy caused by plakophilin-2 mutations. J Cell Mol Med PMID: 18662195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridberger A, Tomo I, Ulfendahl M, Boutet de Monvel J 2006. Imaging hair cell transduction at the speed of sound: Dynamic behavior of mammalian stereocilia. Proc Natl Acad Sci USA 103:1918–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury DS, Marian AJ 2006. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest 116:2012–2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giagtzoglou N, Ly CV, Bellen HJ 2009. Cell adhesion, the backbone of the synapse: “vertebrate” and “invertebrate” perspectives. Cold Spring Harb Perspect Biol 1:a003079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenough DA, Paul DL 2009. Gap junctions. Cold Spring Harb Perspect Biol 1:a002576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorio SP, Sallet PC, Do KA, Lin E, Gattaz WF, Dias-Neto E 2009. Polymorphisms in genes involved in neurodevelopment may be associated with altered brain morphology in schizophrenia: Preliminary evidence. Psychiatry Res 165:1–9 [DOI] [PubMed] [Google Scholar]
- Heard E, Chaumeil J, Masui O, Okamoto I 2004. Mammalian X-chromosome inactivation: An epigenetics paradigm. Cold Spring Harb Symp Quant Biol 69:89–102 [DOI] [PubMed] [Google Scholar]
- Hennies HC, Kuster W, Mischke D, Reis A 1995. Localization of a locus for the striated form of palmoplantar keratoderma to chromosome 18q near the desmosomal cadherin gene cluster. Hum Mol Genet 4:1015–1020 [DOI] [PubMed] [Google Scholar]
- Hershkovitz D, Lugassy J, Indelman M, Bergman R, Sprecher E 2009. Novel mutations in DSG1 causing striate palmoplantar keratoderma. Clin Exp Dermatol 34:224–228 [DOI] [PubMed] [Google Scholar]
- Heuser A, Plovie ER, Ellinor PT, Grossmann KS, Shin JT, Wichter T, Basson CT, Lerman BB, Sasse-Klaassen S, Thierfelder L, et al. 2006. Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet 79:1081–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollnagel A, Grund C, Franke WW, Arnold HH 2002. The cell adhesion molecule M-cadherin is not essential for muscle development and regeneration. Mol Cell Biol 22:4760–4770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulpiau P, van Roy F 2009. Molecular evolution of the cadherin superfamily. Int J Biochem Cell Biol 41:349–369 [DOI] [PubMed] [Google Scholar]
- Indelman M, Eason J, Hummel M, Loza O, Suri M, Leys MJ, Bayne M, Schwartz FL, Sprecher E 2007. Novel CDH3 mutations in hypotrichosis with juvenile macular dystrophy. Clin Exp Dermatol 32:191–196 [DOI] [PubMed] [Google Scholar]
- Jamora C, DasGupta R, Kocieniewski P, Fuchs E 2003. Links between signal transduction, transcription and adhesion in epithelial bud development. Nature 422:317–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo F, Hudspeth AJ 1993. Displacement-clamp measurement of the forces exerted by gating springs in the hair bundle. Proc Natl Acad Sci USA 90:1330–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C, Chen P 2008. Primary cilia in planar cell polarity regulation of the inner ear. Curr Top Dev Biol 85:197–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachar B, Parakkal M, Kurc M, Zhao Y, Gillespie PG 2000. High-resolution structure of hair-cell tip links. Proc Natl Acad Sci USA 97:13336–13341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmierczak P, Sakaguchi H, Tokita J, Wilson-Kubalek EM, Milligan RA, Muller U, Kachar B 2007. Cadherin 23 and protocadherin 15 interact to form tip-link filaments in sensory hair cells. Nature 449:87–91 [DOI] [PubMed] [Google Scholar]
- Kelly M, Chen P 2007. Shaping the mammalian auditory sensory organ by the planar cell polarity pathway. Int J Dev Biol 51:535–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjaer KW, Hansen L, Schwabe GC, Marques-de-Faria AP, Eiberg H, Mundlos S, Tommerup N, Rosenberg T 2005. Distinct CDH3 mutations cause ectodermal dysplasia, ectrodactyly, macular dystrophy (EEM syndrome). J Med Genet 42:292–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kljuic A, Bazzi H, Sundberg JP, Martinez-Mir A, O’Shaughnessy R, Mahoney MG, Levy M, Montagutelli X, Ahmad W, Aita VM, et al. 2003. Desmoglein 4 in hair follicle differentiation and epidermal adhesion: Evidence from inherited hypotrichosis and acquired pemphigus vulgaris. Cell 113:249–260 [DOI] [PubMed] [Google Scholar]
- Kremer H, van Wijk E, Marker T, Wolfrum U, Roepman R 2006. Usher syndrome: Molecular links of pathogenesis, proteins and pathways. Hum Mol Genet 15:R262–270 [DOI] [PubMed] [Google Scholar]
- Kussel-Andermann P, El-Amraoui A, Safieddine S, Nouaille S, Perfettini I, Lecuit M, Cossart P, Wolfrum U, Petit C 2000. Vezatin, a novel transmembrane protein, bridges myosin VIIA to the cadherin-catenins complex. EMBO J 19:6020–6029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagziel A, Ahmed ZM, Schultz JM, Morell RJ, Belyantseva IA, Friedman TB 2005. Spatiotemporal pattern and isoforms of cadherin 23 in wild type and waltzer mice during inner ear hair cell development. Dev Dyn 280:295–306 [DOI] [PubMed] [Google Scholar]
- Lai-Cheong JE, Arita K, McGrath JA 2007. Genetic diseases of junctions. J Invest Dermatol 127:2713–2725 [DOI] [PubMed] [Google Scholar]
- Lefevre G, Michel V, Weil D, Lepelletier L, Bizard E, Wolfrum U, Hardelin JP, Petit C 2008. A core cochlear phenotype in USH1 mouse mutants implicates fibrous links of the hair bundle in its cohesion, orientation and differential growth. Development 135:1427–1437 [DOI] [PubMed] [Google Scholar]
- Leibovici M, Safieddine S, Petit C 2008. Mouse models for human hereditary deafness. Curr Top Dev Biol 84:385–429 [DOI] [PubMed] [Google Scholar]
- Leonova EV, Raphael Y 1997. Organization of cell junctions and cytoskeleton in the reticular lamina in normal and ototoxically damaged organ of Corti. Hear Res 113:14–28 [DOI] [PubMed] [Google Scholar]
- Li J, Levin MD, Xiong Y, Petrenko N, Patel VV, Radice GL 2008. N-cadherin haploinsufficiency affects cardiac gap junctions and arrhythmic susceptibility. J Mol Cell Cardiol 44:597–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin ED, Moriarty MA, Byrnes L, Grealy M 2009. Plakoglobin has both structural and signalling roles in zebrafish development. Dev Biol 327:83–96 [DOI] [PubMed] [Google Scholar]
- McGrath JA 2005. Inherited disorders of desmosomes. Australas J Dermatol 46:221–229 [DOI] [PubMed] [Google Scholar]
- Michel V, Goodyear RJ, Weil D, Marcotti W, Perfettini I, Wolfrum U, Kros CJ, Richardson GP, Petit C 2005. Cadherin 23 is a component of the transient lateral links in the developing hair bundles of cochlear sensory cells. Dev Dyn 280:281–294 [DOI] [PubMed] [Google Scholar]
- Morishita H, Yagi T 2007. Protocadherin family: Diversity, structure, and function. Curr Opin Cell Biol 19:584–592 [DOI] [PubMed] [Google Scholar]
- Muller U 2008. Cadherins and mechanotransduction by hair cells. Curr Opin Cell Biol 20:557–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson WJ 2008. Regulation of cell-cell adhesion by the cadherin-catenin complex. Biochem Soc Trans 36:149–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noben-Trauth K, Zheng QY, Johnson KR 2003. Association of cadherin 23 with polygenic inheritance and genetic modification of sensorineural hearing loss. Nature Genet 35:21–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes FD, Lopez LN, Lin HW, Davies C, Azevedo RB, Gow A, Kachar B 2006. Distinct subdomain organization and molecular composition of a tight junction with adherens junction features. J Cell Sci 119:4819–4827 [DOI] [PubMed] [Google Scholar]
- Org E, Eyheramendy S, Juhanson P, Gieger C, Lichtner P, Klopp N, Veldre G, Doring A, Viigimaa M, Sober S, et al. 2009. Genome-wide scan identifies CDH13 as a novel susceptibility locus contributing to blood pressure determination in two European populations. Hum Mol Genet 18:2288–2296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit C 2001. Usher syndrome: From genetics to pathogenesis. Annu Rev Genomics Hum Genet 2:271–297 [DOI] [PubMed] [Google Scholar]
- Petit C, Richardson G 2009. Linking deafness genes to hair-bundle development and function. Nature Neurosci 12:703–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickles JO, Comis SD, Osborne MP 1984. Cross-links between stereocilia in the guinea pig organ of Corti, and their possible relation to sensory transduction. Hear Res 15:103–112 [DOI] [PubMed] [Google Scholar]
- Pilichou K, Nava A, Basso C, Beffagna G, Bauce B, Lorenzon A, Frigo G, Vettori A, Valente M, Towbin J, et al. 2006. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation 113:1171–1179 [DOI] [PubMed] [Google Scholar]
- Prost J, Barbetta C, Joanny JF 2007. Dynamical control of the shape and size of stereocilia and microvilli. Biophys J 93:1124–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radice GL, Ferreira-Cornwell MC, Robinson SD, Rayburn H, Chodosh LA, Takeichi M, Hynes RO 1997. Precocious mammary gland development in P-cadherin-deficient mice. J Cell Biol 139:1025–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raphael Y, Altschuler RA 2003. Structure and innervation of the cochlea. Brain Res Bull 60:397–422 [DOI] [PubMed] [Google Scholar]
- Rickman L, Simrak D, Stevens HP, Hunt DM, King IA, Bryant SP, Eady RA, Leigh IM, Arnemann J, Magee AI, et al. 1999. N-terminal deletion in a desmosomal cadherin causes the autosomal dominant skin disease striate palmoplantar keratoderma. Hum Mol Genet 8:971–976 [DOI] [PubMed] [Google Scholar]
- Rzadzinska AK, Steel KP 2009. Presence of interstereocilial links in waltzer mutants suggests Cdh23 is not essential for tip link formation. Neuroscience 158:365–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer JV, Bazzi H, Vitebsky A, Witkiewicz A, Kovich OI, Kamino H, Shapiro LS, Amin SP, Orlow SJ, Christiano AM 2006. Mutations in the desmoglein 4 gene underlie localized autosomal recessive hypotrichosis with monilethrix hairs and congenital scalp erosions. J Invest Dermatol 126:1286–1291 [DOI] [PubMed] [Google Scholar]
- Schwander M, Xiong W, Tokita J, Lelli A, Elledge HM, Kazmierczak P, Sczaniecka A, Kolatkar A, Wiltshire T, Kuhn P, et al. 2009. A mouse model for nonsyndromic deafness (DFNB12) links hearing loss to defects in tip links of mechanosensory hair cells. Proc Natl Acad Sci USA (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer J 2006. More than one gene involved in monilethrix: Intracellular but also extracellular players. J Invest Dermatol 126:1216–1219 [DOI] [PubMed] [Google Scholar]
- Seiler C, Finger-Baier KC, Rinner O, Makhankov YV, Schwarz H, Neuhauss SC, Nicolson T 2005. Duplicated genes with split functions: Independent roles of protocadherin15 orthologues in zebrafish hearing and vision. Development 132:615–623 [DOI] [PubMed] [Google Scholar]
- Shapiro L, Weis WI 2009. Structure and biochemistry of cadherins and catenins. Cold Spring Harb Perspect Biol 1:a003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura Y, Sakamoto F, Kariya N, Matsunaga K, Ito M 2006. Mutations in the desmoglein 4 gene are associated with monilethrix-like congenital hypotrichosis. J Invest Dermatol 126:1281–1285 [DOI] [PubMed] [Google Scholar]
- Shimomura Y, Wajid M, Shapiro L, Christiano AM 2008. P-cadherin is a p63 target gene with a crucial role in the developing human limb bud and hair follicle. Development 135:743–753 [DOI] [PubMed] [Google Scholar]
- Simpson MA, Mansour S, Ahnood D, Kalidas K, Patton MA, McKenna WJ, Behr ER, Crosby AH 2008. Homozygous Mutation of Desmocollin-2 in Arrhythmogenic Right Ventricular Cardiomyopathy with Mild Palmoplantar Keratoderma and Woolly Hair. Cardiology 113:28–34 [DOI] [PubMed] [Google Scholar]
- Sollner C, Rauch GJ, Siemens J, Geisler R, Schuster SC, Muller U, Nicolson T 2004. Mutations in cadherin 23 affect tip links in zebrafish sensory hair cells. Nature 428:955–959 [DOI] [PubMed] [Google Scholar]
- Sprecher E, Bergman R, Richard G, Lurie R, Shalev S, Petronius D, Shalata A, Anbinder Y, Leibu R, Perlman I, et al. 2001. Hypotrichosis with juvenile macular dystrophy is caused by a mutation in CDH3, encoding P-cadherin. Nature Genet 29:134–136 [DOI] [PubMed] [Google Scholar]
- Sudhof TC 2008. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455:903–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syrris P, Ward D, Evans A, Asimaki A, Gandjbakhch E, Sen-Chowdhry S, McKenna WJ 2006. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am J Hum Genet 79:978–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeichi M 2007. The cadherin superfamily in neuronal connections and interactions. Nat Rev Neurosci 8:11–20 [DOI] [PubMed] [Google Scholar]
- Tomo I, Boutet de Monvel J, Fridberger A 2007. Sound-evoked radial strain in the hearing organ. Biophys J 93:3279–3284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verpy E, Leibovici M, Zwaenepoel I, Liu X-Z, Gal A, Salem N, Mansour A, Blanchard S, Kobayashi I, Keats BJB, et al. 2000. A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nature Genet 26:51–55 [DOI] [PubMed] [Google Scholar]
- Wajid M, Bazzi H, Rockey J, Lubetkin J, Zlotogorski A, Christiano AM 2007. Localized autosomal recessive hypotrichosis due to a frameshift mutation in the desmoglein 4 gene exhibits extensive phenotypic variability within a Pakistani family. J Invest Dermatol 127:1779–1782 [DOI] [PubMed] [Google Scholar]
- Wan H, Dopping-Hepenstal PJ, Gratian MJ, Stone MG, Zhu G, Purkis PE, South AP, Keane F, Armstrong DK, Buxton RS, et al. 2004. Striate palmoplantar keratoderma arising from desmoplakin and desmoglein 1 mutations is associated with contrasting perturbations of desmosomes and the keratin filament network. Br J Dermatol 150:878–891 [DOI] [PubMed] [Google Scholar]
- Wangemann P 2006. Supporting sensory transduction: Cochlear fluid homeostasis and the endocochlear potential. J Physiol 576:11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waschke J 2008. The desmosome and pemphigus. Histochem Cell Biol 130:21–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil D, Blanchard S, Kaplan J, Guilford P, Gibson F, Walsh J, Mburu P, Varela A, Levilliers J, Weston MD, et al. 1995. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature 374:60–61 [DOI] [PubMed] [Google Scholar]
- Weil D, El-Amraoui A, Masmoudi S, Mustapha M, Kikkawa Y, Laine S, Delmaghani S, Adato A, Nadifi S, Zina ZB, et al. 2003. Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Hum Mol Genet 12:463–471 [DOI] [PubMed] [Google Scholar]
- Xu L, Overbeek PA, Reneker LW 2002. Systematic analysis of E-, N- and P-cadherin expression in mouse eye development. Exp Eye Res 74:753–760 [DOI] [PubMed] [Google Scholar]
- Yamada S, Nelson WJ 2007. Synapses: Sites of cell recognition, adhesion, and functional specification. Annu Rev Biochem 76:267–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Zhu Z, Kapranov P, McKeon F, Church GM, Gingeras TR, Struhl K 2006. Relationships between p63 binding, DNA sequence, transcription activity, and biological function in human cells. Mol Cell 24:593–602 [DOI] [PubMed] [Google Scholar]
- Zlotogorski A, Marek D, Horev L, Abu A, Ben-Amitai D, Gerad L, Ingber A, Frydman M, Reznik-Wolf H, Vardy DA, et al. 2006. An autosomal recessive form of monilethrix is caused by mutations in DSG4: Clinical overlap with localized autosomal recessive hypotrichosis. J Invest Dermatol 126:1292–1296 [DOI] [PubMed] [Google Scholar]