

Abstract
The human epidermis serves 2 crucial barrier functions: it protects against water loss and prevents penetration of infectious agents and allergens. The physiology of the epidermis is maintained by a balance of protease and antiprotease activities, as illustrated by the rare genetic skin disease Netherton syndrome (NS), in which impaired inhibition of serine proteases causes severe skin erythema and scaling. Here, utilizing mass spectrometry, we have identified elastase 2 (ELA2), which we believe to be a new epidermal protease that is specifically expressed in the most differentiated layer of living human and mouse epidermis. ELA2 localized to keratohyalin granules, where it was found to directly participate in (pro-)filaggrin processing. Consistent with the observation that ELA2 was hyperactive in skin from NS patients, transgenic mice overexpressing ELA2 in the granular layer of the epidermis displayed abnormal (pro-)filaggrin processing and impaired lipid lamellae structure, which are both observed in NS patients. These anomalies led to dehydration, implicating ELA2 in the skin barrier defect seen in NS patients. Thus, our work identifies ELA2 as a major new epidermal protease involved in essential pathways for skin barrier function. These results highlight the importance of the control of epidermal protease activity in skin homeostasis and designate ELA2 as a major protease driving the pathogenesis of NS.
Introduction
As the most external organ of the human body, the main function of the epidermis is to provide a protective barrier against water loss and the penetration of infectious agents and allergens. The stratum corneum (SC) is the outermost layer of the epidermis and is largely responsible for this vital barrier function. The SC arises from a specific differentiation program of keratinocytes from the basal layer to the spinous and granular layers of the epidermis (1). Two key structures of the SC underlie its barrier properties. The first is the cornified envelope (CE), a reinforcement of the corneocyte plasma membrane by an insoluble protein scaffold comprising involucrin, loricrin, and filaggrin (2). Prior to their covalent attachment to the CE, loricrin and (pro-)filaggrin accumulate as keratohyalin granules, non–membrane-bound cytoplasmic aggregates characteristic of the granular layer, or stratum granulosum (SG) (2). The second barrier component is the intercellular lipid matrix, which holds near-equimolar amounts of cholesterol, FFA, and ceramide (Cer), a ratio which is imperative for lipid organization into competent lamellar structures (3). These structures result from the extracellular processing of lipid precursors into mature lipids, which arrange themselves spatially into lipid bilayers filling the entire SC extracellular space (3). Lamellae are correctly aligned through the contact with the corneocyte-bound lipid envelope (CLE), a specific extracellular lipid monolayer that is covalently bound to the CE (4). In granular keratinocytes, lipid precursors and lipases involved in their maturation are sequestered into lamellar bodies (LB). These specialized vesicles of the SG fuse to the plasma membrane at the SG-SC interface to extrude lipid precursors and a variety of enzymes with their inhibitors, including proteases, which contribute to the desquamation process.
In epidermal biology, the importance of the protease-antiprotease balance is illustrated by rare genetic skin diseases including Papillon-Lefèvre syndrome (OMIM 245000) (5), autosomal recessive ichthyosis with hypotrichosis (OMIM 610765) (6), and Netherton syndrome (NS) (OMIM 256500) (7).
NS is a severe ichthyosis arising from a genetic deficiency of lymphoepithelial kazal-type-related inhibitor (LEKTI), a serine protease inhibitor (7). This rare autosomal recessive skin disorder is characterized by congenital ichthyosiform erythroderma, a specific hair shaft defect, and severe atopic manifestations (8–10). Frequent complications such as bacterial infection, hypernatremic dehydration, and nutritional defects are probably caused by the severe alteration of the skin barrier function and compromise vital prognosis in neonates. Histologically, NS skin shows epidermal hyperplasia (acanthosis) with the persistence of nuclei in the SC (parakeratosis). The SC is thick (hyperkeratosis), often detached from the underlying epidermis, while the SG is reduced or absent (agranulosis) (11). We have previously identified serine protease inhibitor kazal–type 5 (SPINK5) as the defective gene in NS (7). The vast majority of causative mutations reported to date lead to the complete absence of the encoded protein, LEKTI (7, 12–14). LEKTI is expressed in the SG of the epidermis, and we showed that bioactive LEKTI fragments inhibit 3 major proteases involved in SC desquamation, kallikreins (KLK) 5, 7, and 14 (15).
Spink5-knockout mice mimic key features of NS, including premature desquamation through deep SC detachment, a profound skin barrier defect, impaired terminal differentiation of keratinocytes, and hair malformation (16). We have shown that lack of LEKTI led to kallikrein 5 (KLK5) and KLK7 proteolytic hyperactivity, resulting in desmoglein-1 degradation and, in turn, to abnormal corneodesmosome cleavage below the SC (16). Furthermore, this mouse model revealed the presence of a third protease of 28 kDa, which was hyperactive in the absence of LEKTI (16).
Using tandem mass spectrometry, we have now identified this protease as elastase 2 (ELA2), whose expression in the skin has not previously been reported. In this work, we demonstrate ELA2 expression and activity in the granular layer of mouse and human epidermis. We show that ELA2 colocalizes with (pro-)filaggrin, and we establish that ELA2 directly degrades (pro-)filaggrin. To investigate the role of ELA2 in vivo and its contribution to NS pathophysiological pathways, we mimicked ELA2 hyperactivity by engineering transgenic mice overexpressing murine ELA2 in the SG. Transgenic animals display severe skin barrier dysfunction due to excessive degradation of (pro-)filaggrin and altered intercorneocyte lipid metabolism. This study identifies ELA2 for the first time, to our knowledge, as a major protease involved in the molecular pathways of skin barrier function, a central process of epidermal biology, and in the pathogenesis of NS.
Results
ELA2 is a new epidermal protease which is hyperactive in LEKTI-deficient epidermis.
We previously reported increased proteolytic activities of KLK5, KLK7, and an unknown 28-kDa protease in Spink5–/– murine epidermis (16). Since classical biochemical approaches were unable to identify this protease, we used a strategy combining enrichment and peptide assignment by mass spectrometry (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI41440DS1). The occurrence of 3 high-confidence peptides, covering 8% of the sequence, allowed the unambiguous identification of this protease as murine ELA2 (Swiss-Prot P05208; EC 3.4.21.71), encoded by the Ela2a gene (Figure 1A). This protease is entirely different from murine neutrophil elastase (33% amino acid identity) and was hitherto known as a pancreas-specific protein.
Figure 1. Identification and characterization of ELA2, a new epidermal protease.

(A) Sequence of murine Ela2 precursor. The peptides identified by mass spectrometry are shown in bold red, corresponding to 8% sequence coverage. The score of each peptide is indicated. (B) RT-PCR analysis showing ELA2 mRNA expression in murine and human epidermis, but not in dermis. RT-PCR experiments performed from cultured cells with human ELA2 (UTR) primers, showing ELA2 mRNA expression in differentiated human keratinocytes, but not in undifferentiated keratinocytes nor fibroblasts. As a control, the hypoxanthine phosphoribosyl transferase (HPRT) gene was amplified in all samples. Lanes were run on the same gel but were noncontiguous (white lines). (C) Expression of ELA2 by immunohistochemistry on WT and Spink5–/– (KO) mouse skin as well as human healthy and NS skin sections. ELA2 is detected in the granular layer in each sample. The asterisk indicates the loss of ELA2 signal in an area where the granular layer is absent. BL, basal layer; SP, spinous layer. Scale bar: 75 μm. (D) In situ zymography analysis of normal and LEKTI-deficient mouse and human skins. Elastolytic activity is mainly found in the granular layer as well as in the SC of normal and LEKTI-deficient epidermis. The signal intensity is strongly increased and extended to the outermost layers of the spinous layer in LEKTI-deficient epidermis. Activity intensity is indicated in a pseudo-color gradient ranging from 0 (dark) to 255 (white). The dotted line represents the basal membrane of the epidermis. Scale bar: 50 μm.
RT-PCR analysis revealed a single transcript of Ela2a restricted to the murine epidermal compartment (Figure 1B), and sequencing confirmed 100% identity to the full-length pancreatic cDNA. We also showed transcriptional expression of its ortholog ELA2A (Swiss-Prot accession number P08217) in human epidermis (Figure 1B) and more specifically in human-differentiated keratinocytes in culture, but not in undifferentiated keratinocytes nor in fibroblasts (Figure 1B).
In agreement with these results, ELA2 was immunodetected in the SG of WT and Spink5–/– newborn mouse epidermis as well as in the epidermis of NS patients and healthy human controls (Figure 1C). In situ zymography detected elastolytic activity in the SG and the SC of normal mouse and human epidermis, concordant with ELA2 immunolocalization (Figure 1D). In LEKTI-deficient murine and human epidermis, elastolytic activity was strongly enhanced and extended to the spinous compartment of the epidermis (Figure 1D and Supplemental Figure 2A). In Spink5–/– epidermal extracts, a time-course analysis revealed a 5.3-fold increase in elastolytic activity compared with the WT (Supplemental Figure 2B), which was not due to a higher Ela2a expression level, as shown by quantitative PCR analysis (Supplemental Figure 2C). As ELA2 is the first described elastase physiologically expressed in the suprabasal epidermis, it could be the main protease responsible for the increased elastolytic activity observed in the absence of LEKTI. However, activity tests showed no inhibition of ELA2 activity by LEKTI fragments (15) (data not shown), which suggested an indirect control mechanism. Previous studies have shown pancreatic ELA2 zymogen activation by trypsin cleavage (17). As KLK5, a trypsin-like protease directly inhibited by LEKTI, is hyperactive in LEKTI-deficient epidermis (15, 18–20), we tested the effect of KLK5 on pro-ELA2 activation. Coincubation of pro-ELA2 with KLK5 led to a 4-fold increase in elastolytic activity compared with pro-ELA2 basal activity (Figure 2A). These data, together with localization of both partners in the SG, strongly suggest that LEKTI inhibits ELA2 activity indirectly, by controlling KLK5-mediated cleavage of pro-ELA2.
Figure 2. KLK5, SLPI, and SKALP regulate ELA2 activity.

(A) Activation of pro-ELA2 by KLK5 and KLK7. The degradation velocity of BODIPY-FL-elastin by activated ELA2 was measured. In the proteolytic assay, KLK5 leads to an elastolytic activity increase of 4 times compared with the basal activity level of pro-ELA2 (–). No increase in elastolytic activity was observed with KLK7. Charts represent each individual value of 3 independent experiments (dots) and average (histograms). (B) Inhibition properties of SLPI and SKALP toward ELA2. ELA2 was incubated with increased concentrations of inhibitors. The curves represent the percentage of resulting elastolytic activity according to the ratio (inhibitor)/(enzyme) (I/E). Ki values are indicated. SKALP displays a better inhibitory capacity toward ELA2 compared with SLPI.
We next investigated possible ELA2 physiological inhibitors, the secretory leukoprotease inhibitor (SLPI) and the skin-derived antileukoprotease (SKALP), 2 elastase inhibitors also expressed in the SG of the epidermis (21, 22). Both SLPI and SKALP displayed high inhibitory capacity against ELA2, as indicated by their Ki values of 29.9 nM and 14.3 nM, respectively (Figure 2B). The inhibition strength of SLPI and SKALP was well correlated with the Ki values determined in vitro for each inhibitor.
Altogether, our results demonstrate that ELA2 is expressed and active in murine and human epidermis and identify KLK5, SLPI, and SKALP as potential regulators of its activity.
The epidermal barrier is impaired in Tg-ELA2 mice.
We developed transgenic mice (Tg-ELA2) overexpressing murine ELA2 fused to a FLAG tag (ELA2flag) under the control of the human involucrin promoter (23) to specifically target its expression to the SG (Figure 3, A and B). Western blot analysis using an anti-FLAG antibody confirmed the expression of the zymogen and the active forms of exogenous ELA2 in the epidermis of Tg-ELA2 animals (Figure 3C). At birth, transgenic mice were indistinguishable from the WT animals. However, they subsequently developed generalized scaling that was maximal between P4 and P6 and varied in severity within the litter. Surprisingly, the ichthyosiform phenotype regressed spontaneously in the following days concomitantly with hair growth, and the mice developed normally (Figure 3D).
Figure 3. Tg-ELA2 mice display an ichthyotic phenotype.

(A) Representation of the transgenic cassette. The murine Ela2a cDNA fused with the FLAG tag was cloned downstream of the human involucrin (INV) promoter. (B) PCR analysis of genomic DNA with transgene-specific primers indicated by arrowheads in A showed the presence of a specific 1 kb-band in transgenic animals. As a control, exon 1 of the SPINK5 gene was amplified in both samples (0.4 kb-band). (C) Western blot analysis of WT and Tg-ELA2 (Tg) epidermal and dermal extracts, using an anti-FLAG antibody, reveals exogenous ELA2flag as pro-forms and active forms in the epidermis of transgenic animals. (D) Tg-ELA2 animals (Tg), compared with WT littermates, developed generalized scaling a few days after birth, accompanied by growth retardation. This phenotype was maximal between P4 and P6, then spontaneously regressed concomitantly with the beginning of hair growth.
Interestingly, this transient scaly phenotype correlated over time with the levels of the active form of ELA2flag with a maximum at day 4 (Figure 4A). At this time point, FLAG immunodetection revealed patchy but high expression of exogenous ELA2 in the SG of Tg-ELA2 epidermis (Figure 4B), which correlated with increased in situ elastolytic activity (Figure 4C). Notably, the barrier function in Tg-ELA2 animals was defective, as measured by transepidermal water loss (TEWL) from days 4 to 7 (Figure 4D) and dye penetration assay at day 4 (Figure 4E), concordant with the scaly critical period and with the level of active ELA2 expression.
Figure 4. Skin barrier defect is linked to ELA2flag expression and activity.

(A) Immunodetection of ELA2flag (pro-form and active forms) in Tg-ELA2 epidermal extracts. At birth (P0), only the pro-ELA2flag was detected. Active ELA2flag is detected from P2 to P6. Lanes were run on the same gel but were noncontiguous (white lines). (B) Immunofluorescence analysis showing expression of ELA2flag in the granular layer of the epidermis of Tg-ELA2 at P4. The dotted line represents the basal membrane of the epidermis. Scale bar: 125 μm. (C) In situ zymography analysis showing a strong elastolytic activity in suprabasal layers of the epidermis (brackets) and in the dermis of Tg-ELA2 mice. Activity intensity is indicated in a pseudocolor gradient ranging from 0 (dark) to 255 (white). The dotted line represents the basal membrane of the epidermis. Scale bar: 200 μm. (D) TEWL measurements. Between day 4 and day 8, the TEWL values were higher in Tg-ELA2 animals compared with the WT (up to 3.2 times on day 5). After day 8, TEWL values of WT and transgenic mice were similar. Charts represent each individual value (dot) and average (line). (E) Whole-mount dye penetration assay performed at day 4. Transgenic animals displayed skin areas with blue staining revealing an outside-in skin barrier defect compared with uncolored WT mice.
We next investigated the proliferation/differentiation program in the transgenic epidermis. Histological analysis revealed marked acanthosis and compact and hyperkeratotic SC (Figure 5), associated with focal agranulosis. Immunohistochemical analysis revealed increased epidermal proliferation in Tg-ELA2 animals as shown by strong keratin 6 staining (Figure 5). Keratin 14, a differentiation marker normally restricted to the basal layer, was extended to the suprabasal compartment of Tg-ELA2 epidermis (Figure 5). In contrast, the suprabasal keratin 10 was slightly reduced and the terminal differentiation marker involucrin was markedly decreased (Figure 5). Detection of CD45-positive cells revealed the presence of an inflammatory infiltrate in the Tg-ELA2 dermis (Figure 5).
Figure 5. Alteration of the epidermal differentiation program in Tg-ELA2 animals.
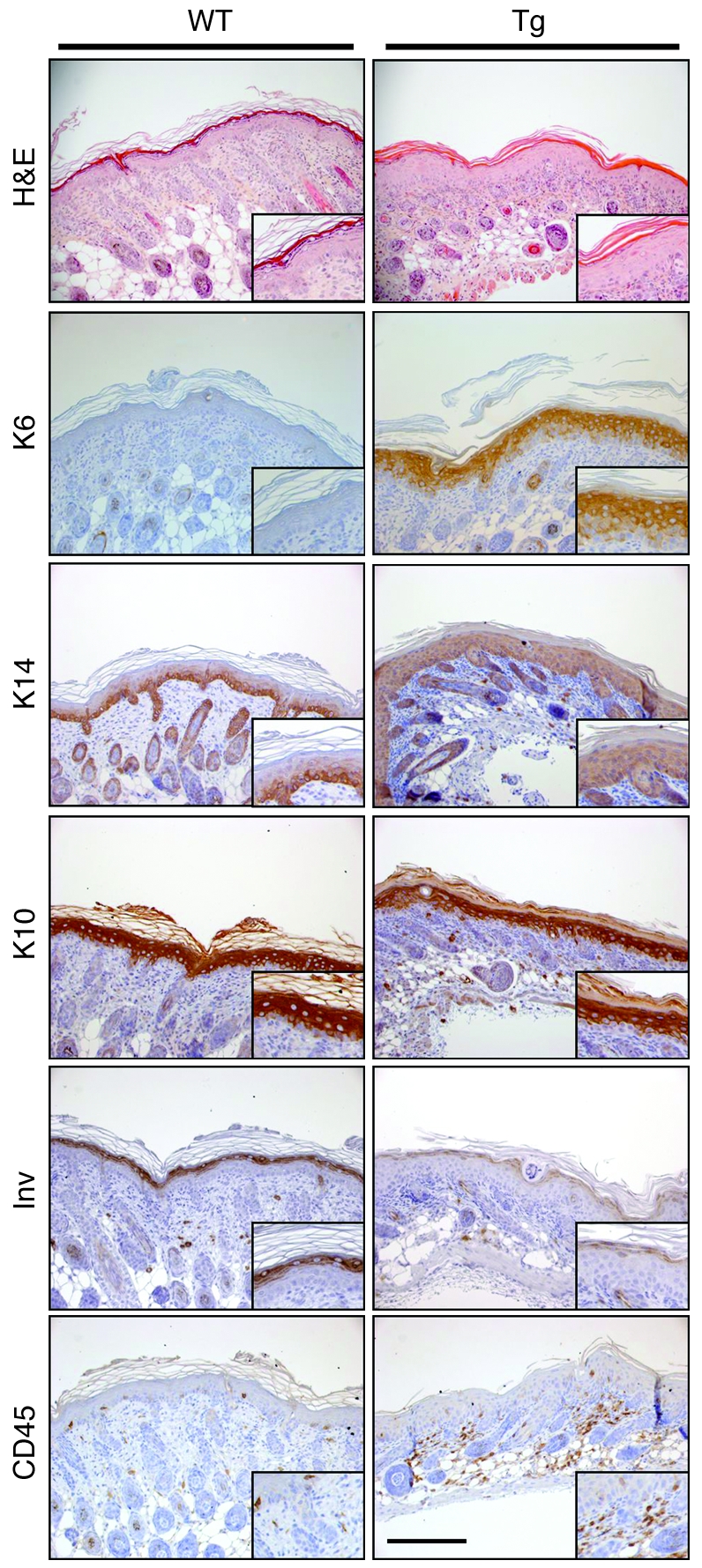
Histological and immunohistochemical analysis of 4-day-old Tg-ELA2 epidermis. H&E staining of 4-day-old WT and Tg-ELA2 (Tg) epidermis shows thickening of the epidermis (acanthosis) with hypogranulosis and a compact SC in Tg-ELA2 mice. The hyperproliferation marker keratin 6 (K6) is not detectable in the WT epidermis, whereas it is strongly expressed in the suprabasal layers of Tg-ELA2 epidermis. The basal marker keratin 14 (K14) is extended to the suprabasal compartment in the transgenic epidermis. Keratin 10 (K10) is detected in all suprabasal layers of WT epidermis but is slightly reduced in intensity in the granular layer of transgenic animal. Involucrin (Inv) staining is reduced in intensity in Tg-ELA2 epidermis. The number of CD45-positive cells is significantly increased in the dermis of Tg-ELA2 mice. Scale bar: 250 μm. Higher magnifications of each panel are shown as insets.
A Tg-ELA2 mouse with a more severe phenotype was also obtained but died shortly after birth. This animal presented similar but more pronounced cutaneous defects associated with a higher elastolytic activity, consistent with a dose-effect of exogenous ELA2 (Supplemental Figure 3).
We concluded that ELA2 overactivity in murine SG results in defective skin barrier function associated with an abnormal epidermal differentiation pattern.
Lipid lamellae formation is defective in Tg-ELA2 mice epidermis and in NS patients.
In newborn WT and Tg-ELA2 mice, Nile red staining revealed uniform, linear lipophilic structures corresponding to the intercellular spaces of corneocytes (Figure 6A; WTnb, Tgnb). In contrast, on day 4, numerous lipid droplets became evident in the Tg-ELA2 SC simultaneous with the development of the ichthyosiform phenotype (Figure 6A; WTP4, TgP4). Ultrastructural examination after ruthenium postfixation revealed an almost complete absence of lipid lamellae in the extracellular domain of the entire Tg-ELA2 SC. Instead, secreted lipid material remained globular and disorganized (Figure 6B).
Figure 6. Abnormal epidermal lipid organization and quantification.

(A) Nile red staining of mouse and human skin cryosections. In normal mouse (WT) and human (healthy) skin, extracellular lipid lamellae appear as yellow parallel lines. In the epidermis of newborn Tg-ELA2 mice (Tgnb), no abnormality is detectable, but numerous yellow lipid droplets are observed at P4 (TgP4). In KOnb mice, lipid droplets are visible in the central layers of the SC. In KOg, the staining reveals numerous lipid droplets and no extracellular lamellae. NS patient epidermis shows similar abnormal lipid droplets. Scale bar: 10 μm. (B) Structure of intercellular lipids revealed with RuO4 postfixation in 4-day-old WT and Tg-ELA2 epidermis. Note broad and compact lipid bilayers in WT SC (bracket). In contrast, unprocessed lipids are seen in the SC intercellular space in Tg-ELA2. At the SG-SC interface of WT epidermis, LB are observed. In Tg-ELA2, lipid material remains globular and disorganized (asterisk) and dilates the extracellular space. Scale bar: 10 nm. (C) Epidermal lipid content in 4-day-old Tg-ELA2 compared with WT animals. ω-hydroxy Cers, ω-OH Cer in CLE; ω-hydroxy fatty acids, ω-OH FA in CLE. Quantification shows a significant decrease in FFA, SM, and ω-OH FA amount in Tg-ELA2 epidermis, while GlcCer amount is significantly increased. Results represent the mean ± SD of 4 WT and 4 transgenic epidermis. Significant P values are indicated.
Lipid analysis showed FFA content to be decreased by 25% in Tg-ELA2 epidermis, while cholesterol and total Cer were unchanged (Figure 6C). Total glucosylceramide (GlcCer) had increased by 20% while sphingomyelin (SM) had decreased by 20%. Finally, catabolites of bound lipids (ω-OH Cer → ω-OH FA) had decreased by 45% in the cornified lipid envelope of Tg-ELA2. Consistent with Nile red staining, the epidermal lipid profile of newborn Tg-ELA2 mice did not differ from that of WT mice (data not shown). These results indicate that the lack of competent lamellae in transgenic epidermis is attributable to alterations of lipid composition secondary to ELA2 hyperactivity.
Remarkably, the Nile red staining pattern in Spink5–/– and NS patient skin was highly reminiscent of the disorganized lipid structures observed in Tg-ELA2. In Spink5–/– newborn mice, small lipid vesicles were visible in addition to lamellae, suggesting a minimal lipid defect at birth (Figure 6A; WTnb, KOnb). LEKTI deficiency exacerbated this lipid defect over time, as shown by the complete lack of lamellae in Spink5–/– grafted epidermis (Figure 6A; WTg, KOg). Interestingly, NS patient skin displayed similar anomalies (Figure 6A), consistent with the ultrastructural lipid defects previously reported in several patients (24, 25). Our results show that ELA2 hyperactivity is sufficient to cause the lipid lamellae defect in LEKTI-deficient skin.
ELA2 directly degrades (pro-)filaggrin, promoting skin barrier impairment.
Tg-ELA2 epidermis had fewer keratohyalin granules, as shown in electron microscopy (Figure 7A), which correlated with decreased (pro-)filaggrin immunostaining (Figure 7B). Filaggrin oligomers and monomers, resulting from profilaggrin processing, were detected in Western blots of (pro-)filaggrin–enriched epidermal extracts from 4-day-old WT animals (Figure 7C). In Tg-ELA2, the amount of filaggrin intermediates, and filaggrin monomers declined to nearly undetectable levels (Figure 7C). Excessive degradation appeared on day 2 and persisted until day 6 (Supplemental Figure 4A), concomitant with the detection of active ELA2flag (Figure 4A). Since no significant difference was observed in profilaggrin transcriptional expression between WT and Tg animals (Supplemental Figure 4B), we hypothesized that ELA2 could affect the (pro-)filaggrin protein level. Indeed, when (pro-)filaggrin–enriched murine epidermal extracts were incubated with purified human ELA2, all forms of (pro-)filaggrin were progressively degraded (Figure 7D), demonstrating the ability of ELA2 to directly proteolyse both profilaggrin and filaggrin monomers.
Figure 7. (Pro-)filaggrin is a target of ELA2.

(A) Ultrastructural analysis of 4-day-old Tg-ELA2 (Tg) shows hyperkeratosis and a reduction in size and number of keratohyalin granules (blue arrowheads). SP, spinous layer. Scale bar: 5 μm. Each panel is a composite of 2 images. (B) Filaggrin immunostaining is markedly decreased in 4-day-old Tg-ELA2 epidermis compared with littermate WT animals. Scale bar: 250 μm. (C) Immunodetection of profilaggrin (proFlg), filaggrin (Flg), and filaggrin-processing intermediate dimers (2xFlg) and trimers (3xFlg) in epidermal extract from a 4-day-old litter composed of 2 WT and 2 Tg-ELA2 animals (Tg). In Tg-ELA2 epidermis, profilaggrin and filaggrin intermediates are decreased in intensity and filaggrin monomers are not detectable. (D) In vitro degradation of (pro-)filaggrin by purified ELA2. Epidermal extracts from WT animals were incubated with (+) or without (–) purified ELA2 for different time periods (0, 15, 30, and 60 minutes) before Western blotting analyses using an anti-filaggrin antibody. In the absence of ELA2, no degradation of either filaggrin form is observed. In the presence of ELA2, profilaggrin and filaggrin intermediates are digested progressively with incubation time.
To investigate the interaction between ELA2 and filaggrin, we next analyzed the fine localization of ELA2 and (pro-)filaggrin by immunofluorescence on human skin sections. ELA2, mainly expressed in the granular layers of healthy human skin, colocalized with (pro-)filaggrin in the SG, supporting the interaction of these 2 proteins in vivo (Figure 8A). In NS patient epidermis, colocalization persisted despite agranulosis and reduced (pro-)filaggrin levels (Figure 8A).
Figure 8. ELA2 colocalizes with filaggrin and KLK5 in differentiated keratinocytes and in vivo.

(A) Colocalization between ELA2 and SG-specific proteins in normal and NS skin sections. ELA2 staining (green) and FLG or KLK5 staining (red) were analyzed in confocal laser scanning microscopy. Graphs, representing the signal intensity from each protein along the arrow, show a perfect colocalization of both signals in normal as well as in NS epidermis in spite of the decreased filaggrin signal level. Scale bars: 25 μm. (B) Colocalization experiment of ELA2 with FLG or KLK5 in normal human keratinocytes differentiated in culture, analyzed by confocal laser scanning microscopy. ELA2 staining is quasi-exclusively localized around the (pro-)filaggrin–containing keratohyalin granules. KLK5 staining spreads in the entire cytoplasmic compartment, with concentrated areas at the periphery of the nucleus and around keratohyalin granules, where KLK5 and ELA2 colocalized. Note the presence of some intense yellow dots at a distance from the keratohyalin granules. Dotted outlines define the nucleus of the cells. The 2 far-right panels (ELA/FLG and ELA/KLK5) represent higher magnifications of the regions delineated by the dotted squares. Scale bar: 10 μm.
We further examined the subcellular distribution of this newly identified protease in human cultured primary keratinocytes. Ring-shaped ELA2 immunostaining was observed in the cytoplasm of the most differentiated keratinocytes (Figure 8B) and was absent in undifferentiated cells (data not shown). This particular ELA2 staining pattern exactly overlapped with that of (pro-)filaggrin detected in small rounded granules of different size, previously reported as keratohyalin granules (26) (Figure 8B). This colocalization was detected at the periphery of these granules concordant with a potential interaction and probably a direct degradation of (pro-)filaggrin by ELA2.
To confirm our earlier result showing activation of pro-ELA2 by KLK5 (Figure 2A), we performed similar double-labeling experiments. ELA2 and KLK5 colocalized in the SG of human epidermis, as shown by immunofluorescence analysis (Figure 8A). In differentiated human primary keratinocytes, KLK5 staining was observed throughout the cytoplasm, with areas of accumulation near the nucleus and, interestingly, around keratohyalin granules. The ELA2 and KLK5 stainings overlapped at the outside edge of the granules as well as in isolated dots (Figure 8B), supporting a direct interaction between both proteases in keratinocytes and substantiating pro-ELA2 activation by KLK5.
Taken together, these data demonstrate that ELA2 hyperactivity plays a key role in the skin barrier impairment observed in Tg-ELA2 mice and in NS patients through (pro-)filaggrin degradation.
Discussion
We brought to light what we believe to be a novel epidermal serine protease, ELA2, previously thought to be restricted to the pancreas (27). We demonstrated that ELA2 is constitutively expressed and active in the SG and SC of murine and human epidermis, supporting a role for this protease in skin biology. Low ELA2 expression levels in the skin may have precluded earlier detection in keratinocyte cDNA libraries and during transcriptomic analysis of the SG (28). This new epidermal protease is clearly different from the very well-characterized neutrophil elastase, and its role is not restricted to inflammation, since ELA2 is expressed in epidermis under normal physiological conditions. Cell-culture analysis confirmed that ELA2 is specifically expressed in differentiated keratinocytes. Analysis for predicted transcription factor–binding sites in the ELA2 promoter revealed the presence of AP1, SP1, and C/EBP motifs (data not shown), which are known to activate gene transcription in the most differentiated layer of the epidermis (29, 30) and could therefore drive ELA2 expression.
We showed that ELA2 is hyperactive in LEKTI-deficient epidermis from NS patients and Spink5–/– mice. Like most proteases, the ELA2 precursor contains a propeptide, which must be cleaved to release the active enzyme. KLK5 interacts with ELA2, as shown by our immunofluorescence experiments and by the observation that KLK5 cleaves pro-ELA2 into its active form in vitro. As expected from its known inhibitory properties (15), we found LEKTI to be ineffective against ELA2. Therefore, the lack of KLK5 inhibition in LEKTI-deficient epidermis probably leads to ELA2 hyperactivity. Our data revealed that SLPI and SKALP inhibited ELA2 in vitro and that these inhibitors were also able to control native epidermal ELA2 activity, as evidenced by in situ zymography (Supplemental Figure 5). SLPI and SKALP are thus potential physiological inhibitors of ELA2 in the skin, which is in agreement with their specific capacity to inhibit members of the elastase family (31, 32). High expression of SKALP has been reported in NS patient skin in response to skin inflammation and recurrent infections (14, 33). However, SKALP overexpression is not sufficient to counteract ELA2 hyperactivity, as revealed by in situ zymography on patient skin sections.
Elastases are characterized by their capacity to interact with very insoluble proteins like elastin fibers. This feature is in agreement with our observation of ELA2 immunodetection in differentiated keratinocytes, in close contact with keratohyalin granules, which result from the aggregation of highly insoluble proteins such as (pro-)filaggrin and loricrin. More precisely, we showed that ELA2 colocalized with (pro-)filaggrin. ELA2 retention in the cytoplasm of keratinocytes was unexpected, since epidermal and pancreatic ELA2 carry the same signal peptide, which drives secretion of the pancreatic form. However, Western blot analysis revealed that ELA2 is secreted in the cultured medium of differentiated transgenic mouse keratinocytes (Supplemental Figure 4C). Therefore, ELA2 displays a dual, cytoplasmic and extracellular, localization in epidermal cells.
Our data demonstrate the capacity of ELA2 to process (pro-)filaggrin. Indeed, ELA2 progressively degrades all forms of (pro-)filaggrin in vitro, from the precursor to the monomer form. This cleavage pattern appears to be different from that of caspase-14, which preferentially degrades the filaggrin monomer (34). In vivo, transgenic ELA2 mice displayed excessive processing of profilaggrin and filaggrin monomers. This abnormality, together with the skin barrier impairment and the transient ichthyosiform phenotype of Tg-ELA2 animals, was time correlated with the presence of active exogenous protease. ELA2 is thus a new protease directly involved in (pro-)filaggrin maturation, in addition to the proposed enzymes, PEP-1, μ-calpain, furin, cap1, matriptase, and caspase-14 (34–39).
(Pro-)filaggrin anomalies have been described in several murine models with a skin barrier defect (Spink5–/–, flaky tail, matriptase–/–, and caspase14–/– mice) (34, 38, 40) as well as in patients with ichthyosis vulgaris and atopic dermatitis involving stop mutations in the filaggrin (FLG) gene (41, 42). NS patient skin also shows a decrease in keratohyalin granules and (pro-)filaggrin levels (25). Therefore, ELA2 hyperactivity could be a major molecular mechanism underlying the skin barrier defect observed in NS through excessive (pro-)filaggrin processing.
In addition to excessive degradation of (pro-)filaggrin, lipid abnormalities contribute to the epidermal barrier dysfunction in our animal model. The present study revealed changes in the lipid contents of Tg-ELA2 epidermis. Notably the amount of FFAs was significantly decreased, disrupting the equimolar ratio among Cer, cholesterol, and FFAs, which is necessary for efficient skin barrier function. FFAs are generated mainly from esterified lipids, suggesting a phospholipase A2 (sPLA2) dysregulation in Tg-ELA2 epidermis. Since the sPLA2 transcriptional level was similar between WT and Tg-ELA2 mice (data not shown), ELA2 hyperactivity could impair sPLA2 function and/or its activators. Our results show that a pool of ELA2 is secreted by keratinocytes (Supplemental Figure 4C) and could therefore directly regulate secreted lipases. The amount of total Cer was unchanged in contrast to their immediate precursors (GlcCer and SM). However, detailed Cer analysis revealed independent variations among Cer species levels (data not shown), indicating that specific step(s) of Cer synthesis rather than global lipid processing are affected by ELA2 hyperactivity. In addition, CLE maturation was also altered in Tg-ELA2 epidermis, as indicated by decreased ω-hydroxy FA despite there being no change in the amount of their immediate precursors (ω-hydroxy Cer). This observation highlights the complexity of ELA2 involvement in lipid metabolism. Alteration of lipid profiles is thus likely to give rise to the abnormal lamellae observed in Tg-ELA2 epidermis. Interestingly, lamellae were well-formed in newborn Spink5–/– pups, but few small lipid droplets were already present, in agreement with the observation of small intracorneocyte lipid droplets at the ultrastructural level (16), which could account to some extent for the intrinsic barrier impairment reported in these animals (16). Spink5–/– skin grafts displayed a severe ichthyosic phenotype and a prominent defect in lipid organization, illustrating the consequences of chronic ELA2 hyperactivity. We observed the same lipid structural anomalies in the skin of NS patients, which could be related to the abnormal morphology and secretion of LB also described in NS epidermis (24). The mechanisms of LB secretion are not well understood, but proteases seem to play an important role in controlling this process, since topical application of serine protease inhibitors after barrier disruption has been shown to improve barrier recovery through enhanced LB secretion (43). Interestingly, among these inhibitors, PMSF displayed inhibitory capacity toward ELA2. These results strongly support the hypothesis that ELA2 hyperactivity could prevent the formation of a protective skin barrier in NS through impaired LB secretion associated with defective lipid metabolism.
In NS patients, the SC is hyperkeratotic and is often detached from the underlying cell layers (16, 25). Loss of SC adhesion arises from asymmetrical corneodesmosome cleavage through DSG1 and desmoplakin degradation by hyperactive proteases (16, 25). In scaly Tg-ELA2 mice, neither Dsg-1 nor desmoplakin were degraded (data not shown), and corneodesmosomes remained assembled, suggesting that ELA2 is not involved in this specific NS feature. Nevertheless, epidermal lipids, besides their established role as major components of the skin barrier, also influence desquamation, as illustrated in Harlequin ichthyosis, recessive X-linked ichthyosis, and Refsum disease as well as Sjögren-Larsson syndrome. This suggests that the ichthyosiform phenotype of Tg-ELA2 mice could result from abnormal cutaneous lipid composition.
Barrier disruption by itself stimulates keratinocyte proliferation as well as cytokine and chemokine production (IL-1α, IL-1β, TNF-α, GM-CSF), which are features of skin inflammatory diseases (44, 45). The active form of Il-1β was overexpressed in Lekti-deficient mouse epidermis (46). In addition, proteases with elastolytic activity such as cathepsin G and neutrophil elastase activate IL-1β through proteolytic cleavage of its pro-form in vitro (47). It is thus possible that ELA2 likewise activates IL-1β produced by keratinocytes in NS epidermis, resulting in the accumulation of neutrophils and other IL-1β–chemoattracted inflammatory cells. These observations support the possibility that ELA2 may also participate in the initiation of epidermal inflammation in NS skin.
Epidermal hyperproliferation is a general compensatory mechanism for impaired skin barrier function, and we provided evidence that ELA2 hyperactivity affects the skin proliferation/differentiation program. Lipid abnormalities could also contribute to the dysregulated proliferation/differentiation balance observed in Tg-ELA2 epidermis, since various Cer species and metabolites have well-recognized roles in signaling different cell fates (48, 49).
In conclusion, we demonstrated that this new epidermal protease has a major role in (pro-)filaggrin maturation and in lipid metabolism, although its precise role in the intricate lipid pathway requires further investigation. The unexpected discovery of the key role of ELA2 in cutaneous biology highlights the importance of the control of epidermal protease activity for the maintenance of skin barrier function.
ELA2 emerges as a new target, together with hyperactive proteases in LEKTI-deficient epidermis (16), with the prospect of a specific therapy for NS based on protease inhibitors. This would represent a substantial advance over the symptomatic treatment available at present.
Methods
Affinity chromatography and mass spectrometry analysis.
The skin from twenty euthanized newborn mice was heated for 30 minutes at 56°C for epidermis/dermis separation. The epidermis was crushed in 1 M acetic acid with Ultra-Turrax, and epidermal proteins were prepared and purified on an sbTI column as described (50) (see Supplemental Methods). The 28-kDa protease-containing fractions were pooled and concentrated on a Vivaspin 20 MW 3 kDa (Vivasciences). The concentrated eluate was submitted to MS/MS analysis (technical details in Supplemental Methods).
Antibodies.
A polyclonal rabbit anti-ELA2 antibody raised against a peptide highly conserved between human and murine pancreatic ELA2 was produced by Interchim. Note that this antibody was unsuccessful in Western blot application in detecting endogenous protein. Primary antibodies directed against FLAG (Sigma-Aldrich), CD45 (BD Biosciences — Pharmingen), keratin 6 (Abcam), keratin 14 (Covance), keratin 10 (Covance), involucrin (Santa Cruz Biotechnology Inc.), KLK5 (Abcam), filaggrin (Covance and Interchim), and actin (Sigma-Aldrich) were used. FITC-conjugated swine anti-rabbit and TRITC-coupled rabbit anti-mouse were from Dako secondary antibodies. Alexa Fluor 546–coupled goat anti-rabbit and Alexa Fluor 546–coupled goat anti-mouse IgG1 secondary antibodies were from Invitrogen. Coupling of rabbit anti-ELA2 antibody to Alexa Fluor 488 was performed with the Zenon Rabbit IgG Labeling Kit (Molecular Probes) according to manufacturer’s recommendations.
Cell culture.
Normal human primary keratinocytes (NHK) were cocultured with γ-irradiated 3T3-J2 mouse fibroblasts in Green medium (51), at 37°C, under 10% CO2. For RT-PCR analysis, NHK were harvested before confluence (undifferentiated state) or after 7 days postconfluence (differentiated state).
For immunocytofluorescence experiments, NHK were grown onto glass coverslips in EpiLife 0.06 mM CaCl2, supplemented with Human Keratinocyte Growth Supplement (Cascade Biologics), 10 μg/ml streptomycin, and 10 U/ml penicillin until confluence. NHK were differentiated during 7 days after confluence in Epilife 1.2 mM CaCl2.
Human primary fibroblasts were grown in DMEM supplemented with 10% fetal calf serum, penicillin, and streptomycin (Invitrogen).
WT and Tg-ELA2 murine primary keratinocytes were isolated from skin biopsies of newborn mice as previously described for human biopsy (52). Murine keratinocytes were expanded on γ-irradiated 3T3-J2 mouse fibroblasts in a modified green medium (DMEM without Ca2+ Invitrogen) but with 2 mM l-glutamine (PAA), and 1 mM pyruvate sodium (Gibco; Invitrogen).
RT-PCR analysis.
RNA was isolated using the RNeasy kit (QIAGEN). 1.5 to 5 μg RNA were reverse transcribed following the recommendations of the Superscript first strand synthesis system. PCR was performed with GoTaq polymerase (Promega), using 1/10 to undiluted cDNA as a matrix, with primers indicated in Supplemental Table 1.
If needed, amplicons were cloned into pGEMT-easy (Promega) and then sequenced using Big Dye Terminator.
Construction and genotyping of ELA2 transgenic mice.
All animal studies were approved by the French Genetic Engineering Commission (01-10-08, agreement number 4858). All experiments were performed in accordance with the relevant guidelines and regulations. The involucrin promoter vector (pH3700-pL2) was a gift from J. Segre (Genetics and Molecular Biology Branch, National Human Genome Research Institute, Bethesda, Maryland) (53). Mouse cDNA encoding Ela2 was amplified by RT-PCR, allowing the introduction of the FLAG octapeptide epitope at the 3′ end of the cDNA as well as NotI restriction sites at each end. The final construct was introduced in pH3700-pL2 using NotI sites and confirmed by sequencing.
Transgenic mice were produced by injecting the transgene (containing the involucrin promoter and mouse ELA2FLAG cDNA excised with SalI) into the pronuclei of fertilized oocytes from (C57BL/6 × CBA) F1 mice. Injected oocytes were implanted in pseudopregnant females. Mouse-tail DNA, extracted with the High Pure PCR template preparation kit (Roche) was used for PCR to establish genotypes, using “transgene” primers (Supplemental Table 1).
The founder mouse was crossed with WT C57BL/6 animals to establish the transgenic line named Tg(INVO-Ela2aFLAG)55CPTP in agreement with the International Committee on Standardized Genetic Nomenclature for Mice. Transgenic mice produced were named Tg-ELA2 for the following study.
Immunohistochemical and immunofluorescence analysis.
The anti-ELA2 antibody was diluted 1:50. For commercially available primary antibodies, we followed the manufacturer’s recommendations.
Coimmunolocalization experiments were performed on paraffin human and mouse skin sections. We performed sequential incubations of rabbit anti-ELA2, FITC-conjugated swine anti-rabbit, and mouse anti-filaggrin, followed by TRITC-coupled rabbit anti-mouse. Coimmunolocalization between KLK5 and ELA2 was performed with overnight incubation with rabbit anti-KLK5 at 4°C and sequentially with Alexa Fluor 546–coupled goat anti-rabbit and Alexa Fluor 488–coupled rabbit anti-ELA2 antibodies. Fluorescence was analyzed using an inverted Zeiss LSM 510 confocal microscope.
In situ zymography.
Frozen sections (5-μm thickness) were rinsed with a washing solution (2% Tween 20 in deionized water) and incubated at 37°C overnight with 100 μl of FITC-conjugated elastin using the EnzChek-elastase Assay kit (Invitrogen) in 50 mM Tris-HCl, pH 8, in order to visualize elastolytic activity. All sections were rinsed with PBS solution and visualized with the Axiovert 200 inverted high-end microscope (Zeiss). Images were analyzed with Metamorph Imaging system software, version 3.6 (Universal Imaging Corp.).
Western blotting.
Epidermis, dermis, or cultured cells were crushed in a protein extraction buffer (PEB) containing 50 mM Tris-HCl, pH 8, 150 mM NaCl, 5 mM EDTA, pH 8, 0.1% Nonidet-P40, and 1X Protease Inhibitor Cocktail Tablets (Complete; Roche) with Ultra-Turrax. Lysates were clarified from insoluble material by centrifugation at 13,000 g, 4°C for 30 minutes. Western blotting was performed as previously described (15). (pro-)filaggrin–enriched fractions were prepared as previously described prior to anti-filaggrin immunodetection (16).
Degradation tests in vitro.
For proteolysis experiments, 20 μg of (pro-)filaggrin–enriched epidermal fraction was mixed with 7.5 ng of recombinant human ELA2 (gift from C. Largman, University of California School of Medicine, Hematology Research [151H], Veterans Affairs [VA] Medical Center, San Francisco, California) in a reaction buffer (100 mM Tris-HCl, pH 8, 0.02 mM azide, 15 mM EDTA, 50 μM leupeptin, 1 μM E64). Mixtures were incubated at 37°C for 0, 15, 30, and 60 minutes. At each time-step, 4 μg of filaggrin-enriched protein extracts were removed and reactions were interrupted by boiling for 5 minutes in Laemmli buffer. Samples were analyzed by immunoblotting, as described above.
Nile red staining.
Frozen sections were immersed for 20 minutes in a buffer containing 50 mM glycine, 50 mM NaCl, and 50 mM NaOH, pH 12.5, as described (54). Nile red (Sigma-Aldrich) working solution (12.5 μg/ml in 75% glycerol) was applied to each tissue section and immediately covered with a coverslip. Nile red fluorescence was captured with an inverted Zeiss LSM 510 microscope.
Lipid quantification.
Epidermis was separated from dermis and total epidermal lipids were extracted, as previously described (4). The individual lipid species were separated by high-performance thin-layer chromatography (HPTLC), followed by quantitation by scanning densitometry, as described previously (4). Covalently bound lipids were isolated using the method of Wertz et al. (55), and resultant Cer fractions were quantified by HPTLC-scanning densitometry, as above.
Barrier function assay.
We performed whole-mount dye penetration assay on euthanized 4-day-old mice as described (16). TEWL measurements were recorded daily with the EP1 evaporimeter (ServoMed).
Transmission electron microscopy.
For classical electron microscopy, skin tissues were prepared as described previously (16). For the ruthenium tetroxide post-fixation experiment, the following protocol was used: 1 mm pieces were fixed overnight in 2% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.4, at 4°C. The samples were post-fixed with 0.25% ruthenium tetroxide for 1 hour in the dark at 4°C. This was followed by dehydration and embedding using Embed 812 resin. The blocks were polymerized at 60°C for 30 hours. 80- to 90-nm “near-surface” sections were mounted on copper grids and examined with a transmission electron microscope.
Immunocytofluorescence.
Cells were fixed in 4% formaldehyde solution and permeabilized with 0.2% Triton X-100.
Double immunofluorescence staining was performed with overnight incubation of cells with rabbit anti-KLK5 or mouse anti-filaggrin antibodies at 4°C and sequentially at room temperature with Alexa Fluor 546–coupled goat anti-rabbit, Alexa Fluor 546–coupled goat anti-mouse IgG1, and Alexa Fluor 488–coupled rabbit anti-ELA2 antibodies. Cells were imaged using a Zeiss 510 confocal microscope (Zeiss).
Statistics.
We determined P values by a 2-tailed Student’s t test, assuming equal variance. We considered differences between groups significant at P < 0.05.
Supplementary Material
Acknowledgments
We are indebted to Corey Largman for providing us with the purified human pancreatic ELA2. We are grateful to Julia Segre for providing us with the pH3700-pL2 vector. We thank Sophie Allart from the imagery platform, Florence Capilla and Talal Al Saati from the experimental histopathology platform, and Anne Huchenq-Champagne and Sylvie Appolinaire-Pilipenko from the transgenesis and cryoconservation platform (IFR150, Institut Claude de Préval, Toulouse, F-31300 France). We are grateful to Isabelle Fourquaux and Nacer Benmeradi from the electron microscopy platform of IFR109 (Toulouse), Sam Moradian for technical assistance in lipid analysis (Northern California Institute For Research and Education and Veterans Affairs Medical Center, San Francisco, California), and Florine Oualid (INSERM U563) for experiment assistance. We thank José Mejia (INSERM U563) for helpful discussion and critical readings. This work was supported by grants from the French Ministry of Research and Technology, the European GENESKIN Coordination Action Project, the Génopole Toulouse Midi-Pyrénées, the Région Midi-Pyrénées, the National Institute of Health Grants AR 051077, the French Foundation for Medical Research (FRM), and the National Agency for Research (Netherlink: ANR-05-MRAR-009-01/NS2AD:ANR-08-GENO-033).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2010;120(3):871–882. doi:10.1172/JCI41440.
References
- 1.Fuchs E. Scratching the surface of skin development. Nature. 2007;445(7130):834–842. doi: 10.1038/nature05659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sandilands A, Sutherland C, Irvine AD, Irwin McLean WH. Filaggrin in the fronline: role in skin barrier function and disease. J Cell Sci. 2009;122(Pt 9):1285–1294. doi: 10.1242/jcs.033969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swartzendruber DC, Wertz PW, Kitko DJ, Madison KC, Downing DT. Molecular models of the intercellular lipid lamellae in mammalian stratum corneum. J Invest Dermatol. 1989;92(2):251–257. doi: 10.1111/1523-1747.ep12276794. [DOI] [PubMed] [Google Scholar]
- 4.Uchida Y, et al. Epidermal sphingomyelins are precursors for selected stratum corneum ceramides. J Lipid Res. 2000;41(12):2071–2082. [PubMed] [Google Scholar]
- 5.Hart TC, et al. Mutations of the cathepsin C gene are responsible for Papillon-Lefevre syndrome. J Med Genet. 1999;36(12):881–887. [PMC free article] [PubMed] [Google Scholar]
- 6.Basel-Vanagaite L, et al. Autosomal recessive ichthyosis with hypotrichosis caused by a mutation in ST14, encoding type II transmembrane serine protease matriptase. Am J Hum Genet. 2007;80(3):467–477. doi: 10.1086/512487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chavanas S, et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet. 2000;25(2):141–142. doi: 10.1038/75977. [DOI] [PubMed] [Google Scholar]
- 8.Comel M. Ichtyosis Linearis circumflexa. Dermatologica. 1949;98(3):133–136. doi: 10.1159/000257290. [DOI] [PubMed] [Google Scholar]
- 9.Netherton EW. A unique case of Trichorrexis Invaginata. Arch Dermatol. 1958;78:483–487. doi: 10.1001/archderm.1958.01560100059009. [DOI] [PubMed] [Google Scholar]
- 10. Ong C, and Harper J. Netherton’s Syndrome. In: Harper J, Oranje A, Prose N, eds.Textbook of Pediatric Dermatology . Oxford, United Kingdom: Wiley/Blackwell. 2006:1359–1366. [Google Scholar]
- 11.Hausser I, Anton-Lamprecht I. Severe congenital generalized exfoliative erythroderma in newborns and infants: a possible sign of Netherton syndrome. Pediatr Dermatol. 1996;13(3):183–199. doi: 10.1111/j.1525-1470.1996.tb01202.x. [DOI] [PubMed] [Google Scholar]
- 12.Bitoun E, et al. Netherton syndrome: disease expression and spectrum of SPINK5 mutations in 21 families. J Invest Dermatol. 2002;118(2):352–361. doi: 10.1046/j.1523-1747.2002.01603.x. [DOI] [PubMed] [Google Scholar]
- 13.Sprecher E, et al. The spectrum of pathogenic mutations in SPINK5 in 19 families with Netherton syndrome: implications for mutation detection and first case of prenatal diagnosis. J Invest Dermatol. 2001;117(2):179–187. doi: 10.1046/j.1523-1747.2001.01389.x. [DOI] [PubMed] [Google Scholar]
- 14.Raghunath M, et al. SPINK5 and Netherton syndrome: novel mutations, demonstration of missing LEKTI, and differential expression of transglutaminases. J Invest Dermatol. 2004;123(3):474–483. doi: 10.1111/j.0022-202X.2004.23220.x. [DOI] [PubMed] [Google Scholar]
- 15.Deraison C, et al. LEKTI fragments specifically inhibit KLK5, KLK7, and KLK14 and control desquamation through a pH-dependent interaction. Mol Biol Cell. 2007;18(9):3607–3619. doi: 10.1091/mbc.E07-02-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Descargues P, et al. Spink5-deficient mice mimic Netherton syndrome through degradation of desmoglein 1 by epidermal protease hyperactivity. Nat Genet. 2005;37(1):56–65. doi: 10.1038/ng1493. [DOI] [PubMed] [Google Scholar]
- 17.Largman C, Brodrick JW, Geokas MC, Johnson JH, Fassett M. Demonstration of a pancreatic proelastase 2-alpha 1-protease inhibitor complex in normal human plasma. Am J Physiol. 1980;238(3):G177–G182. doi: 10.1152/ajpgi.1980.238.3.G177. [DOI] [PubMed] [Google Scholar]
- 18.Egelrud T, et al. hK5 and hK7, two serine proteinases abundant in human skin, are inhibited by LEKTI domain 6. Br J Dermatol. 2005;153(6):1200–1203. doi: 10.1111/j.1365-2133.2005.06834.x. [DOI] [PubMed] [Google Scholar]
- 19.Borgono CA, et al. A potential role for multiple tissue kallikrein serine proteases in epidermal desquamation. J Biol Chem. 2007;282(6):3640–3652. doi: 10.1074/jbc.M607567200. [DOI] [PubMed] [Google Scholar]
- 20.Schechter NM, et al. Inhibition of human kallikreins 5 and 7 by the serine protease inhibitor lympho-epithelial Kazal-type inhibitor (LEKTI). Biol Chem. 2005;386(11):1173–1184. doi: 10.1515/BC.2005.134. [DOI] [PubMed] [Google Scholar]
- 21.Wingens M, et al. Induction of SLPI (ALP/HUSI-I) in epidermal keratinocytes. J Invest Dermatol. 1998;111(6):996–1002. doi: 10.1046/j.1523-1747.1998.00425.x. [DOI] [PubMed] [Google Scholar]
- 22.Alkemade JA, et al. SKALP/elafin is an inducible proteinase inhibitor in human epidermal keratinocytes. J Cell Sci. 1994;107(Pt 8):2335–2342. doi: 10.1242/jcs.107.8.2335. [DOI] [PubMed] [Google Scholar]
- 23.Carroll JM, Albers KM, Garlick JA, Harrington R, Taichman LB. Tissue- and stratum-specific expression of the human involucrin promoter in transgenic mice. Proc Natl Acad Sci U S A. 1993;90(21):10270–10274. doi: 10.1073/pnas.90.21.10270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fartasch M, Williams ML, Elias PM. Altered lamellar body secretion and stratum corneum membrane structure in Netherton syndrome: differentiation from other infantile erythrodermas and pathogenic implications. Arch Dermatol. 1999;135(7):823–832. doi: 10.1001/archderm.135.7.823. [DOI] [PubMed] [Google Scholar]
- 25.Descargues P, et al. Corneodesmosomal cadherins are preferential targets of stratum corneum trypsin- and chymotrypsin-like hyperactivity in Netherton syndrome. J Invest Dermatol. 2006;126(7):1622–1632. doi: 10.1038/sj.jid.5700284. [DOI] [PubMed] [Google Scholar]
- 26.Ishida-Yamamoto A, Hashimoto Y, Manabe M, O’Guin WM, Dale BA, Iizuka H. Distinctive expression of filaggrin and trichohyalin during various pathways of epithelial differentiation. Br J Dermatol. 1997;137(1):9–16. doi: 10.1111/j.1365-2133.1997.tb03694.x. [DOI] [PubMed] [Google Scholar]
- 27.Kawashima I, Tani T, Shimoda K, Takiguchi Y. Characterization of pancreatic elastase II cDNAs: two elastase II mRNAs are expressed in human pancreas. DNA. 1987;6(2):163–172. doi: 10.1089/dna.1987.6.163. [DOI] [PubMed] [Google Scholar]
- 28.Toulza E, et al. Large-scale identification of human genes implicated in epidermal barrier function. Genome Biol. 2007;8(6):R107. doi: 10.1186/gb-2007-8-6-r107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Banks EB, Crish JF, Welter JF, Eckert RL. Characterization of human involucrin promoter distal regulatory region transcriptional activator elements-a role for Sp1 and AP1 binding sites. Biochem J. 1998;331(Pt 1):61–68. doi: 10.1042/bj3310061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oh HS, Smart RC. Expression of CCAAT/enhancer binding proteins (C/EBP) is associated with squamous differentiation in epidermis and isolated primary keratinocytes and is altered in skin neoplasms. J Invest Dermatol. 1998;110(6):939–945. doi: 10.1046/j.1523-1747.1998.00199.x. [DOI] [PubMed] [Google Scholar]
- 31.Thompson RC, Ohlsson K. Isolation, properties, and complete amino acid sequence of human secretory leukocyte protease inhibitor, a potent inhibitor of leukocyte elastase. Proc Natl Acad Sci U S A. 1986;83(18):6692–6696. doi: 10.1073/pnas.83.18.6692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schalkwijk J, de Roo C, de Jongh GJ. Skin-derived antileukoproteinase (SKALP), an elastase inhibitor from human keratinocytes. Purification and biochemical properties. Biochim Biophys Acta. 1991;1096(2):148–154. doi: 10.1016/0925-4439(91)90053-c. [DOI] [PubMed] [Google Scholar]
- 33.Shimomura Y, Sato N, Kariya N, Takatsuka S, Ito M. Netherton syndrome in two Japanese siblings with a novel mutation in the SPINK5 gene: immunohistochemical studies of LEKTI and other epidermal molecules. Br J Dermatol. 2005;153(5):1026–1030. doi: 10.1111/j.1365-2133.2005.06900.x. [DOI] [PubMed] [Google Scholar]
- 34.Denecker G, et al. Caspase-14 protects against epidermal UVB photodamage and water loss. Nat Cell Biol. 2007;9(6):666–674. doi: 10.1038/ncb1597. [DOI] [PubMed] [Google Scholar]
- 35.Resing KA, Thulin C, Whiting K, al-Alawi N, Mostad S. Characterization of profilaggrin endoproteinase 1. A regulated cytoplasmic endoproteinase of epidermis. J Biol Chem. 1995;270(47):28193–28198. doi: 10.1074/jbc.270.47.28193. [DOI] [PubMed] [Google Scholar]
- 36.Yamazaki M, et al. Cytoplasmic processing of human profilaggrin by active mu-calpain. Biochem Biophys Res Commun. 1997;235(3):652–656. doi: 10.1006/bbrc.1997.6809. [DOI] [PubMed] [Google Scholar]
- 37.Pearton DJ, Nirunsuksiri W, Rehemtulla A, Lewis SP, Presland RB, Dale BA. Proprotein convertase expression and localization in epidermis: evidence for multiple roles and substrates. Exp Dermatol. 2001;10(3):193–203. doi: 10.1034/j.1600-0625.2001.010003193.x. [DOI] [PubMed] [Google Scholar]
- 38.List K, et al. Loss of proteolytically processed filaggrin caused by epidermal deletion of Matriptase/MT-SP1. J Cell Biol. 2003;163(4):901–910. doi: 10.1083/jcb.200304161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leyvraz C, et al. The epidermal barrier function is dependent on the serine protease CAP1/Prss8. J Cell Biol. 2005;170(3):487–496. doi: 10.1083/jcb.200501038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Presland RB, Boggess D, Lewis SP, Hull C, Fleckman P, Sundberg JP. Loss of normal profilaggrin and filaggrin in flaky tail (ft/ft) mice: an animal model for the filaggrin-deficient skin disease ichthyosis vulgaris. J Invest Dermatol. 2000;115(6):1072–1081. doi: 10.1046/j.1523-1747.2000.00178.x. [DOI] [PubMed] [Google Scholar]
- 41.Smith FJ, et al. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat Genet. 2006;38(3):337–342. doi: 10.1038/ng1743. [DOI] [PubMed] [Google Scholar]
- 42.Palmer CN, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38(4):441–446. doi: 10.1038/ng1767. [DOI] [PubMed] [Google Scholar]
- 43.Hachem JP, et al. Serine protease signaling of epidermal permeability barrier homeostasis. J Invest Dermatol. 2006;126(9):2074–2086. doi: 10.1038/sj.jid.5700351. [DOI] [PubMed] [Google Scholar]
- 44.Nickoloff BJ, Naidu Y. Perturbation of epidermal barrier function correlates with initiation of cytokine cascade in human skin. J Am Acad Dermatol. 1994;30(4):535–546. doi: 10.1016/S0190-9622(94)70059-1. [DOI] [PubMed] [Google Scholar]
- 45.Wood LC, Jackson SM, Elias PM, Grunfeld C, Feingold KR. Cutaneous barrier perturbation stimulates cytokine production in the epidermis of mice. J Clin Invest. 1992;90(2):482–487. doi: 10.1172/JCI115884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Briot A, et al. Kallikrein 5 induces atopic dermatitis-like lesions through PAR2-mediated thymic stromal lymphopoietin expression in Netherton syndrome. J Exp Med. 2009;206(5):1135–1147. doi: 10.1084/jem.20082242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hazuda DJ, Strickler J, Kueppers F, Simon PL, Young PR. Processing of precursor interleukin 1 beta and inflammatory disease. J Biol Chem. 1990;265(11):6318–6322. [PubMed] [Google Scholar]
- 48.Bieberich E. Integration of glycosphingolipid metabolism and cell-fate decisions in cancer and stem cells: review and hypothesis. Glycoconj J. 2004;21(6):315–327. doi: 10.1023/B:GLYC.0000046274.35732.47. [DOI] [PubMed] [Google Scholar]
- 49.Teichgraber V, et al. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat Med. 2008;14(4):382–391. doi: 10.1038/nm1748. [DOI] [PubMed] [Google Scholar]
- 50.Egelrud T. Purification and preliminary characterization of stratum corneum chymotryptic enzyme: a proteinase that may be involved in desquamation. J Invest Dermatol. 1993;101(2):200–204. doi: 10.1111/1523-1747.ep12363804. [DOI] [PubMed] [Google Scholar]
- 51.Rheinwald JG, Green H. Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell. 1975;6(3):331–343. doi: 10.1016/S0092-8674(75)80001-8. [DOI] [PubMed] [Google Scholar]
- 52.Watt FM, Boukamp P, Hornung J, Fusenig NE. Effect of growth environment on spatial expression of involucrin by human epidermal keratinocytes. Arch Dermatol Res. 1987;279(5):335–340. doi: 10.1007/BF00431227. [DOI] [PubMed] [Google Scholar]
- 53.Carroll JM, Taichman LB. Characterization of the human involucrin promoter using a transient beta-galactosidase assay. J Cell Sci. 1992;103(Pt 4):925–930. doi: 10.1242/jcs.103.4.925. [DOI] [PubMed] [Google Scholar]
- 54.Talreja P, Kleene NK, Pickens WL, Wang TF, Kasting GB. Visualization of the lipid barrier and measurement of lipid pathlength in human stratum corneum. AAPS PharmSci. 2001;3:E13. doi: 10.1208/ps030213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wertz PW, Madison KC, Downing DT. Covalently bound lipids of human stratum corneum. J Invest Dermatol. 1989;92(1):109–111. doi: 10.1111/1523-1747.ep13071317. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.