

Abstract
Platelet inhibitors are the mainstay treatment for patients with vascular diseases. The current ‘gold standard’ antiplatelet agent clopidogrel has several pharmacological and clinical limitations that have prompted the search for more effective platelet antagonists. The candidates include various blockers of the purinergic P2Y12 receptor such as prasugrel, an oral irreversible thienopyridine; two adenosine triphosphate analogues that bind reversibly to the P2Y12 receptor: ticagrelor (oral) and cangrelor (intravenous); elinogrel, a direct-acting reversible P2Y12 receptor inhibitor (the only antiplatelet compound that can be administered both intravenously and orally); BX 667, an orally active and reversible small-molecule P2Y12 receptor antagonist; SCH 530348, SCH 205831, SCH 602539 and E5555, highly selective and orally active antagonists on the protease-activated receptor 1. A number of drugs also hit new targets: terutroban, an oral, selective and specific inhibitor of the thromboxane receptor; ARC1779, a second-generation, nuclease resistant aptamer which inhibits von Willebrand factor-dependent platelet aggregation; ALX-0081, a bivalent humanized nanobody targeting the GPIb binding site of von Willebrand factor and AJW200, an IgG4 monoclonal antibody of von Willebrand factor. The pharmacology and clinical profiles of new platelet antagonists indicate that they provide more consistent, more rapid and more potent platelet inhibition than agents currently used. Whether these potential advantages will translate into clinical advantages will require additional comparisons in properly powered, randomized, controlled trials.
Keywords: prasugrel, ticagrelor, cangrelor, elinogrel, BX 667, SCH 530348, E5555, SCH 602539, terutroban, ARC1779
Introduction
Platelet activation plays a pivotal role in the pathogenesis of atherothrombotic events, such as acute coronary syndrome (ACS). Rupture of an atherosclerotic plaque exposes collagen and von Willebrand factor (vWF). The vWF binds with its A1 domain to the platelets Gp-Ib receptor. The initial binding is followed by further platelet recruitment, platelet activation and release of adenosine diphosphate (ADP) (Siller-Matula et al., 2007). ADP binds to the P2Y12 receptor and amplifies response to other agonists such as thrombin (Raju et al., 2008). Thrombin mediates its effect largely through the protease-activated receptor 1 (PAR-1), augmenting platelet activation and aggregation. Thromboxane A2 (TXA2) binds to the thromboxane receptor (TP), amplifying platelet aggregation (Figure 1). Activated platelets undergo conformational change and the platelet membrane glycoprotein IIa/IIIb binds to fibrinogen, which leads to fibrinogen-platelet cross-linking and the formation of a hemostatic plug at sites of vascular injury (McNicol and Israels, 2003).
Figure 1.
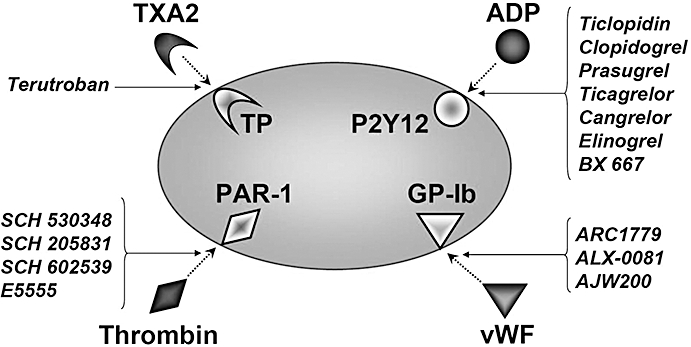
Platelet receptors, their agonists and antagonists. ADP, adenosine diphosphate; PAR, protease-activated receptor; TP, thromboxane receptor; TXA2, thromboxane A2; vWF, von Willebrand factor.
Antiplatelet agents such as aspirin and clopidogrel are effective in the prevention of atherothrombotic events. However, due to the clinical limitations of clopidogrel such as high inter-individual variability in platelet inhibition and potential for drug–drug interactions (Siller-Matula et al., 2008; Siller-Matula et al., 2009a,b,c;), new antiplatelet drugs are currently under development.
Previous reviews in this journal have described the evidence for platelet activation in various inflammatory diseases, the mechanisms by which this pathogenesis occurs and potential candidate target molecules for future anti-platelet drug development (Pitchford, 2007; Barrett et al., 2008). This review focuses on the novel antiplatelet drugs, which have already progressed to clinical trials. These include P2Y12 receptor antagonists (prasugrel, ticagrelor, cangrelor, elinogrel, BX 667), PAR-1 antagonists (SCH 530348, SCH 205831, SCH 602539, E5555), the TP antagonist terutroban and vWF antagonists ARC1779, ALX-0081 and AJW200. The pharmacokinetic/pharmacodynamic properties of those compounds are summarized in Table 1; a summary of clinical trials is shown in Table 2. Drug/molecular target nomenclature in this review conforms to BJP's Guide to Receptors and Channels (Alexander et al., 2008).
Table 1.
Properties of novel antiplatelet drugs
| Drug | Antagonism on | Structure | Prodrug | Half-life | Steady state | Reversible | Route of administration | Frequency of administration | Time to peak platelet inhibition | Phase | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Prasugrel | CS-747 | P2Y12 receptor | Thienopyridine | Yes | 8 h | 2–4 days | No | Oral | Once daily | 2 h | Approved |
| Cangrelor | ARC-69931MX | P2Y12 receptor | ATP analog | No | 3 min | – | Yes | Intravenous | Continuous | 30 min | 3 |
| Ticagrelor | AZD6140 | P2Y12 receptor | ATP analog; cyclopentyl-triazolo-pyrimidine | No | 6–12 h | 2–3 days | Yes | Oral | Twice daily | 2 h | 3 |
| Elinogrel | PRT060128 | P2Y12 receptor | – | No | – | – | Yes | Intravenous/oral | Bolus/twice daily | 20 min | 2 |
| – | BX 667 | P2Y12 receptor | – | Yes | – | – | Yes | Oral | – | – | Pre-clinical |
| – | SCH 530348 | PAR-1 receptor | Himbacine derivative | No | 126–269 h | – | Yes | Oral | Once daily | 0.5–1 h | 3 |
| – | E5555 | PAR-1 receptor | – | – | – | – | – | Oral | Once daily | – | 2 |
| – | SCH 205831 | PAR-1 receptor | Himbacine derivative | No | – | – | Yes | Oral | – | – | Pre-clinical |
| – | SCH 602539 | PAR-1 receptor | Himbacine derivative | No | – | – | Yes | Oral | – | – | Pre-clinical |
| Terutroban | S18886 | TP receptor | – | No | 6–10 h | – | Yes | Oral | Once daily | 1 h | 3 |
| – | ARC1779 | von Willebrand factor | Aptamer | No | 2 h | – | Yes | Intravenous | Continuous | 10 min | 2 |
| – | ALX-0081 | von Willebrand factor | Nanobody | No | – | – | – | Intravenous | – | immediately | 2 |
| – | AJW200 | von Willebrand factor | Monoclonal antibody | No | – | – | – | Intravenous | – | immediately | 2 |
ATP, adenosine triphosphate; PAR, protease-activated receptor.
Table 2.
Clinical trials examining novel antiplatelet drugs
| Drug | Trial | Phase | Type | Patients | n | Dose of drug tested | Dose of comparator drug | Primary outcome | Status | Comment |
|---|---|---|---|---|---|---|---|---|---|---|
| Prasugrel CS-747 | JUMBO-TIMI 26 | 2 | Randomized, multi-centre, parallel group | urgent or Elective PCI | 904 | (i) LD: 40 mg, MD: 7.5 mg; (ii) LD: 60 mg, MD: 10 mg; (iii) LD: 60 mg, MD: 15 mg | Clopidogrel LD: 300 mg, MD: 75 mg | TIMI major and minor hemorrhage | Completed | Bleedings were not significantly more frequent in the prasugrel group than in the clopidogrel group |
| PRINCIPLE-TIMI 44 | 2 | Double-blind, active-comparator-controlled | Planned PCI | 201 | LD: 60 mg, MD: 10 mg | Clopidogrel LD: 600 mg, MD: 150 mg | Inhibition of platelet aggregation | Completed | Inhibition of platelet aggregation was greater in prasugrel group compared with clopidogrel | |
| TRIGGER-PCI | 2 | Randomized, double-blind, active-comparator-controlled, parallel assigned | Elective PCI | 2 150 | LD: 60 mg, MD:10 mg | Clopidogrel MD: 75 mg | Time to first occurrence of heart attack or cardiovascular death | Ongoing | Anticipated study completion date: May 2011 | |
| TRITON-TIMI 38 | 3 | Randomized, double-blind, parallel group | ACS scheduled PCI | 13 608 | LD: 60 mg, MD:10 mg | Clopidogrel LD: 300 mg, MD: 75 mg | Death from CV causes, nonfatal MI and stroke | Completed | MACE occurred more frequently in the clopidogrel group; major bleeding was more frequent in the in the prasugrel group | |
| TRILOGY ACS | 3 | Randomized, double-blind, active-comparator-controlled, parallel assigned | UA/NSTEMI who are medically managed | 10 300 | LD: 30 mg, MD: 5 or 10 mg | Clopidogrel LD: 300 mg, MD: 75 mg | Reduction in risk of the composite end point of CV death, MI or stroke | Ongoing | Anticipated study completion date: October 2011 | |
| Cangrelor ARC-69931MX | STEP-AMI | 2 | Randomized, parallel group, open-label | STEMI | 92 | (i) 280 µg·min−1; (ii) 35 µg·min−1; (iii) 140 µg·min−1; (iv) 280 µg·min−1+ half-dose t-PA | Full dose t-PA alone | TIMI grade 3 flow at 60 min | Completed | TIMI grade 3 was reached in 61% patients receiving combination therapy versus 50% in full-dose t-PA |
| BRIDGE | 2 | Randomized, double-blind, parallel assigned | ACS undergoing non-emergent CABG | 220 | Infusion 4 µg·kg−1·min−1 before CABG | Placebo | Platelet inhibition | Ongoing | Anticipated study completion date: October 2009 | |
| CHAMPION-PCI | 3 | Randomized, double-blind, parallel assigned | ACS requiring PCI | 9 000 | Bolus 30 µg·kg−1+ infusion 4 µg·kg−1·min−1 within 30 min of start of PCI | Clopidogrel 600 mg 30 min prior to PCI | MACE | Terminated | Discontinued because no convincing clinical efficacy needed for approval was seen | |
| CHAMPION-PLATFORM | 3 | Randomized, double-blind, parallel assigned | ACS requiring PCI | 6 400 | Bolus 30 µg·kg−1+ infusion 4 µg·kg−1·min−1 within 30 min of start of PCI | Placebo 30 min of start of PCI | MACE | Terminated | Discontinued because no convincing clinical efficacy needed for approval was seen | |
| Ticagrelor AZD6140 | DISPERSE | 2 | Randomized, double-blind, double-dummy, parallel group | CAD | 200 | (i) 50 mg, (ii) 100 mg, (iii) 200 mg twice daily, (iv) 400 mg daily | Clopidogrel 75 mg | Platelet inhibition and bleeding events | Completed | Greater inhibition of platelets compared with clopidogrel |
| DISPERSE II | 2 | Randomized, multi-centre, double-blind, double-dummy | NSTEMI | 990 | (i) 90 mg twice daily, (ii) 180 mg twice daily | Clopidogrel LD: 300 mg, MD: 75 mg | Total bleeding events | Completed | No statistical difference in the bleeding rates | |
| PLATO | 3 | Randomized, double-blind, parallel group, safety and efficacy | STEMI scheduled for PCI or NSTEMI | >18 000 | LD: 180 mg, MD: 90 mg twice daily | Clopidogrel LD: 300 mg, MD: 75 mg | Reduction in the relative risk of vascular death, nonfatal MI and stroke; safety variable is major bleeding | Completed | MACE occurred more frequently in the clopidogrel group; no difference in overall major bleeding; increase in non-procedure-related bleeding with ticagrelor | |
| Elinogrel PRT060128 | ERASE-MI | 2 | Randomized, double blind, placebo-controlled, parallel assigned | STEMI prior to primary PCI | 70 | i.v. bolus before angiography at doses of 10, 20, 40, 60 mg | Placebo | Safety and tolerability | Completed | Study was completed in July 2008 |
| INNOVATE-PCI | 2 | Randomized, double-blind, triple-dummy | ACS scheduled for non-urgend PCI | 800 | 80 mg iv bolus prior to PCI following (i) 50 mg, (ii) 100 mg or (iii) 150 mg twice daily | Clopidogrel LD 300 or 600 mg, MD: 75 mg | No pre-specified end point | Ongoing | Anticipated study completion date September 2009 | |
| SCH 530348 | TRA-PCI | 2 | Multi-centre, randomized, double-blind, placebo-controlled | ACS undergoing non-urgent PCI | 1 030 | (i) LD: 10 mg, MD: 0.5 mg, (ii) LD: 20 mg, MD: 1.0 mg, (iii) LD: 40 mg, MD: 2.5 mg | Placebo | TIMI major and minor bleeding | Completed | No statistical difference in the bleeding rates |
| TRA-CER | 3 | Randomized, double-blind, placebo-controlled, parallel assigned | NSTEMI | 10 000 | LD: 40 mg, MD: 2.5 mg once daily | Placebo | First occurrence of MACE | Ongoing | Anticipated study completion date July 2011 | |
| TRA 2P-TIMI 50 | 3 | Randomized, double-blind, placebo-controlled, parallel assigned | Patients with a history of atherosclerosis (MI, stroke, peripheral arterial disease) | 25 000 | MD: 2.5 mg once daily for 1 year | Placebo | First occurrence of MACE | Ongoing | Anticipated study completion date September 2010 | |
| E5555 | LANCELOT | 2 | Randomized, double-blind, placebo-controlled | NSTEMI | 600 | (i)50 mg, (ii)100 mg, (iii)200 mg | Placebo | Safety and tolerability | Ongoing | Anticipated study completion date: October 2009 |
| Terutroban S18886 | PERFORM | 3 | Randomized, double-blind, parallel group | History of ischemic strokes or transient ischemic attacks | 19 000 | 30 mg | Aspirin 100 mg | Ischemic stroke, MI or other vascular death | Ongoing | Anticipated study completion date: 2011 |
| ARC 1779 | vITAL-1 | 2 | Randomized, double blind, active-comparator-controlled, parallel assigned | Acute MI undergoing PCI | 300 | (i)0.1 mg·kg−1, (ii)0.3 mg·kg−1, (iii)1.0 mg·kg−1 | Abciximab 0.25 mg·kg−1 bolus + 0.125 mg·kg−1·min−1 infusion | Reperfusion within 48 h | Terminated |
ACS, acute coronary syndrome; CAD, coronary artery disease; MACE, major adverse cardiovascular events; NSTEMI, none ST-elevation myocardial infarction; PCI, percutaneous coronary intervention; STEMI, ST-elevation myocardial infarction; TIMI, thrombolysis in myocardial infarction; t-PA, tissue plasminogen activator; UA, unstable angina.
P2Y12 receptor antagonists
Prasugrel (CS-747)
Prasugrel is an oral third generation thienopyridine, which acts as a selective and irreversible oral inhibitor of the P2Y12 receptor (Figure 1).
Preclinical studies
In preclinical studies with rats, prasugrel showed a more potent, a longer-lasting antiaggregatory effect and a faster onset to peak-action compared with clopidogrel and ticlopidin (Sugidachi et al., 2000; Niitsu et al., 2005). Prasugrel inhibited ADP-induced platelet aggregation by 50% (effective dose 50%, ED50) at a dose of 1.2 mg·kg−1. At 30 min, prasugrel inhibited platelets by 80%. The antiaggregatory effect after a single oral dose of prasugrel was 10-fold higher compared with clopidogrel. The antithrombotic property was shown through reduced thrombus-formation in a dose-related manner with an ED50 of 0.68 mg·kg−1 for prasugrel (Sugidachi et al., 2000).
The in vitro antiplatelet effect of prasugrel/clopidogrel active metabolites is almost identical, but when aggregation inhibition following a single oral dose was tested ex vivo, prasugrel was 13 times more effective than clopidogrel 4 h after intake (clopidogrel ED50: 16 mg·kg−1, prasugrel ED50: 1.2 mg·kg−1) (Sugidachi et al., 2007) in rats. In addition, the plasma concentration of active metabolites showed a higher Cmax and AUC for prasugrel than for clopidogrel (Sugidachi et al., 2007). In a thrombosis model in rabbits, prasugrel produced a strong antithrombotic effect with ED50 of 1.2 mg·kg−1·day−1 and resulted in dose-dependent prolongation of bleeding time (ED50: 1.9 mg·kg−1·day−1) (Wong et al., 2009).
Prasugrel's active metabolite effectively blocked the platelet P2Y12 receptor and inhibited procoagulant and pro-inflammatory platelet responses (Judge et al., 2008).
Clinical studies
Pharmacokinetic/pharmacodynamic
Prasugrel is an oral prodrug that requires conversion into an active metabolite (R-138727) by the hepatic cytochrome P450 (CYP) system (Farid et al., 2007a). Prasugrel is transformed through hydrolization of esterases into the thiolactone, R-95913 and then further metabolized via oxidation of intestinal and hepatic CYP450 into active metabolite R-138727 (Jakubowski et al., 2007b). The greatest contribution to this conversion is made by CYP3A4 and CYP2B6 (Farid et al., 2007a). Neither the intermediate nor the final active metabolite of prasugrel is expected to inhibit the P450-mediated metabolism of co-administered drugs, even though a weak inhibition of various CYPs was shown in vitro (Rehmel et al., 2006) and a weak inhibition of CYP2B6 in vivo (Farid et al., 2008). Due to the difference in the metabolic pathway, prasugrel is more efficiently generated in vivo and better able to produce a higher concentration of its effective metabolite, which results in more predictable pharmacodynamic responses and a faster onset of action compared with clopidogrel (Farid et al., 2007b).
The active metabolite of prasugrel appears rapidly in the blood after ingestion, showing a significant effect after 15 min with a median time to reach a 20% inhibition of platelet aggregation 30 min after administration (Brandt et al., 2007). The maximum effect of prasugrel shows a plateau after 1 h. Cmax is about 500 ng·mL−1 after a 60 mg loading dose. Inhibition of platelet aggregation reaches a steady-state after 2–4 days (Jakubowski et al., 2007a). Prasugrel's metabolites are eliminated with a median half-life ranging from 3 to 9 h. Two-thirds of metabolites are excreted in the urine, one-third in feces (Farid et al., 2007b).
Prasugrel inhibited platelet aggregation better than clopidogrel after single dose (84% vs. 49%). In this crossover study subjects switched study medication after a 2 weeks wash-out-period. Those subjects who first responded poorly to clopidogrel showed a greater inhibition in response to prasugrel (Brandt et al., 2007).
Prasugrel-treated subjects also reached higher levels of platelet inhibition in a multiple dosing study (Jakubowski et al., 2007a). Further, a consistent response over time was obtained, which did not occur with clopidogrel, where half of the subjects showed widely variable platelet inhibition over 10 days (Jakubowski et al., 2007a).
In a three-way crossover study with 41 healthy subjects, who were treated for 7 days, prasugrel more effectively inhibited platelets after 30 min, 1 h and 2 h post-loading compared with clopidogrel (Payne et al., 2007). Also during maintenance dose regimen, platelet inhibition by prasugrel exceeded clopidogrel (78% vs. 56%), showing a 2.2-fold higher AUC of 60 mg prasugrel as compared with 600 mg clopidogrel.
In a phase Ib study, prasugrel (loading dose 60 mg, maintenance dose 10 mg) was compared with clopidogrel (loading dose 600 mg, maintenance dose 75 mg) in 110 stable coronary artery disease (CAD) patients (Wallentin et al., 2008). Patients received study medication for 28 days. Although clopidogrel doses were doubled, the maximal platelet aggregation was significantly lower with prasugrel as compared with clopidogrel at all time-points after the loading dose (34% vs. 55%). Through maintenance treatment this trend continued: 43% for prasugrel and 54% for clopidogrel measured on day 29 (Wallentin et al., 2008). The pharmacokinetic analysis showed a higher peak-concentration of prasugrel, which occurred earlier. Similarly the peak concentrations during maintenance regimen were consistently higher with prasugrel. The mean P2Y12 receptor blockade was significantly greater with prasugrel, which was shown by a lower platelet reactivity index in the vasodilator-stimulated phosphoprotein assay (Wallentin et al., 2008).
An integrated analysis confirmed that prasugrel achieves faster onset, greater extent and more consistent platelet inhibition compared with the approved or even higher loading doses of clopidogrel. Gender, race, body weight and age were identified as statistically significant covariates impacting platelet inhibition (Li et al., 2009).
Phase II: JUMBO-thrombolysis in myocardial infarction (TIMI) 26: joint utilization of medications to block platelets optimally
JUMBO-TIMI 26 was a dose ranging safety trial of prasugrel versus clopidogrel in 904 patients undergoing percutaneous coronary intervention (PCI) (Wiviott et al., 2005). The primary hypothesis for this study was that prasugrel is at least as safe as clopidogrel with respect to bleeding events after PCI. Major adverse cardiac events occurring through the 30-day visit after PCI were assessed. After diagnostic catheterization approximately 900 patients were randomized to three different doses of prasugrel versus standard dose of clopidogrel. The major bleeding rate was overall very low (0.5% for prasugrel vs. 0.8% for clopidogrel). Combined major and minor bleedings were more frequent in the prasugrel group than in the clopidogrel group: 1.7% versus 1.2%, but without statistical significance. Minimal bleeding was more frequent in the high-dose prasugrel group (3.6%) compared with the low dose (2.0%), the intermediate dose (1.5%) and the clopidogrel group (2.4%). Major adverse cardiac events occurred more frequently in the clopidogrel group as compared with prasugrel (9.4% vs. 7.2%).
Phase II: PRINCIPLE-TIMI 44: the prasugrel in comparison with clopidogrel for inhibition of platelet activation and aggregation-TIMI
The PRINCIPLE-TIMI 44 trial compared prasugrel to a 600 mg loading dose of clopidogrel in patients undergoing cardiac catheterization for PCI (Wiviott et al., 2007b). Patients received a 60 mg loading dose of prasugrel or a 600 mg of clopidogrel 1 h before PCI. After PCI patients received 10 mg prasugrel or 150 mg clopidogrel once daily for 14 days. Subjects directly switched to the alternate maintenance therapy for an additional 14 days without a wash-out period in between. A total of 201 patients were randomized with finally 112 patients receiving PCI. Greater platelet inhibition was observed with prasugrel compared with clopidogrel at 6 h (75% vs. 32%). The greater antiplatelet effect was apparent as early as 30 min and maintained through 18–24 h. No major bleedings were observed in either treatment arm. More patients experienced a bleeding event in the prasugrel period (19%) compared with the clopidogrel period (14%). After the switch, four subjects in the clopidogrel period, versus one subject in the prasugrel period had a hemorrhagic event. The frequency of major adverse cardiac events was low: one clopidogrel-receiving subject had an acute stent thrombosis requiring urgent revascularization therapy versus two subjects in the prasugrel group, who experienced periprocedural myocardial infarctions. After the crossover one patient with prasugrel had a myocardial infarction.
Phase II: TRIGGER-PCI: testing platelet reactivity in patients undergoing elective stent placement on clopidogrel to guide alternative therapy with prasugrel
The TRIGGER-PCI is an ongoing trial, which aims to determine the efficacy of prasugrel versus clopidogrel for the reduction of adverse cardiovascular outcomes in patients with high platelet reactivity on clopidogrel after successful implantation of coronary drug-eluting stents. The primary end point is the time to first occurrence of heart attack or cardiovascular death.
Phase III: TRITON-TIMI 38: trial to assess improvement in therapeutic outcomes by optimizing platelet inhibition with prasugrel
The TRITON-TIMI 38 trial was a double-blind, parallel group study comparing prasugrel (LD 60 mg, MD 10 mg) with clopidogrel (LD 300 mg, MD 75 mg) in >13 000 patients with ACS undergoing PCI (Wiviott et al., 2007a; Murphy et al., 2008; Montalescot et al., 2009). More than 10 000 patients with unstable angina (UA) or none ST-elevation myocardial infarction (NSTEMI) (74%) and more than 3500 patients with ST-elevation myocardial infarction (STEMI) (26%) were enrolled. The median duration of therapy was 14.5 months. A total of 781 patients of the clopidogrel group (12.1%) suffered the primary end point compared with 643 patients in the prasugrel-treated group (9.9%) with an overall relative reduction of 19%. This effect was apparent within three days (prasugrel: 4.7% vs. clopidogrel: 5.6%). The reduction of the primary end points was largely due to a significant reduction of nonfatal myocardial infarctions, whereas no significant difference was shown for cardiovascular death or nonfatal stroke. The secondary end point of death from cardiovascular causes, nonfatal MI, stroke or rehospitalization due to recurrent ischemia was also in favour of prasugrel (12.4% vs. 14.6%). Stent thrombosis occurred more often in the clopidogrel group (2.4% vs. 1.1%) (Wiviott et al., 2007a; Murphy et al., 2008; Montalescot et al., 2009).
In diabetic patients the primary end point occurred in 12.2% under prasugrel compared with 17.0% in the clopidogrel group. A significant risk reduction was shown with prasugrel in both groups (diabetics vs. non-diabetics), but with a greater risk reduction for cardiovascular death, myocardial infarction and stroke in the diabetics group. The risk reduction was independent of diabetes type (non-insulin vs. insulin), but leading to the highest level of relative risk reduction for diabetics treated with insulin (37%). The combination of a relatively greater reduction in ischemic events together with a similar risk of major hemorrhage compared with non-diabetics yielded an improved statistical net benefit of prasugrel in diabetic patients (Wiviott et al., 2008).
In the prasugrel group, 2.4% of patients had at least one major hemorrhagic event that was not related to coronary artery bypass graft compared with 1.8% in the clopidogrel group. More patients in the prasugrel group suffered from life-threatening bleedings (1.4% vs. 0.9%), where 0.4% ended fatally with prasugrel versus 0.1% with clopidogrel (Wiviott et al., 2007a; Murphy et al., 2008; Montalescot et al., 2009).
In a prespecified analysis of net clinical benefit, where the major efficacy end points of reduced rates of ischemic events and safety problems were included, findings still favoured prasugrel. Three groups did not benefit from the treatment with prasugrel: patients, who had a history of cerebrovascular events, patients being older than 75 years and those patients that weighed less than 60 kg (Wiviott et al., 2007a; Murphy et al., 2008; Montalescot et al., 2009).
However, the TRITON-TIMI 38 trial has also been criticized for its study design, definition of events, interpretation of the results and suitability of the high maintenance prasugrel dose for chronic use in the viewpoint article by Serebruany et al. (2009a).
Phase III: TRILOGY ACS: a comparison of prasugrel and clopidogrel in ACS subjects
TRILOGY ACS is an ongoing trial, which will evaluate the relative efficacy and safety of prasugrel and clopidogrel in a medically managed UA or NSTEMI population (i.e. patients who are not managed with acute coronary revascularization). The primary end point is a reduction in risk of the composite end point of first occurrence of cardiovascular death, myocardial infarction or stroke.
Ticagrelor (AZD6140)
Ticagrelor is an adenosine triphosphate analog, which belongs to a new chemical class cyclopentyl-triazolo-pyrimidines. Ticagrelor is the first reversibly binding oral ADP receptor (P2Y12) antagonist, which is highly selective for the receptor (Figure 1).
Preclinical studies
Ticagrelor was found to have an advantageous profile with a beneficial separation of antithrombotic effect and increase of bleeding time in dogs (van Giezen and Humphries, 2005). The peak plasma level occurred between 1.5 and 3 h after ingestion of the drug with a steady-state concentration after 2 to 3 days.
Clinical studies
Pharmacokinetic/pharmacodynamic
When ticagrelor was first tested in a phase I study in healthy volunteers, single doses from 100 to 400 mg were administered orally (Tantry et al., 2007; Angiolillo et al., 2009). After rapid oral absorption there was a linear and dose-dependent pharmacokinetic profile with complete platelet inhibition 2 h after dosing, at the same time when peak-plasma levels of the drug were present in the blood (Tantry et al., 2007). Higher doses produced maximal platelet inhibition over a period of 24 h. The elimination half-life was 6–12 h (Tantry et al., 2007).
Phase II: DISPERSE
The DISPERSE trial aimed to assess the pharmacokinetics, pharmacodynamics and safety profile of ticagrelor in 200 patients with atherosclerotic disease (Husted et al., 2006). Patients were either allocated to a 50, 100 or 200 mg dose of ticagrelor twice daily, 400 mg once daily or clopidogrel 75 mg. The doses >100 mg daily of ticagrelor achieved rapid and near complete inhibition of platelet aggregation. Platelet inhibition was greater with ticagrelor compared with clopidogrel. On day 1, clopidogrel did not inhibit platelet aggregation by more than 15%, whereas the higher doses of ticagrelor yielded 80–90% platelet inhibition. The concentration of ticagrelor and its active metabolite AR-C124910XX increased dose-proportionally and was stable at steady-state until day 14. The AUC was independent from age or sex. The most common adverse event reported was minor or moderate bleeding with one major gastrointestinal bleeding with a drop in haemoglobin reported in the ticagrelor 400 mg group. There was a dose–response relationship in the incidence of bleeding events. Adverse events other than bleeding that were reported in at least 10% of patients were dyspnoea, dizziness, headache and red blood cells in the urine.
Phase II: DISPERSE II: dose confirmation study assessing anti-platelet effects of azd6140 versus clopidogrel in non-st segment elevation myocardial infarction
The DISPERSE II trial was performed in 990 patients. Patients received either clopidogrel (LD 300 mg, MD 75 mg) or twice-daily ticagrelor (90 or 180 mg) for up to 3 months (Cannon et al., 2007). Patients in the ticagrelor group were subrandomized to receive or not receive a loading dose of 270 mg ticagrelor. Inhibition of platelet aggregation was greater with all ticagrelor doses than maximum levels of inhibition seen with clopidogrel and more stable over a period of 4 weeks (Storey et al., 2007). No statistical difference was seen between the three treatment groups according to bleeding events despite the higher level of antiplatelet effect yielded with ticagrelor: 8.1% in the clopidogrel group, 9.8% in the 90 mg-ticagrelor group and 8.0% in the 180 mg ticagrelor group. Among patients undergoing coronary artery bypass graft surgery, a lower incidence of bleeding was seen with ticagrelor compared with clopidogrel (36% vs. 64%) 1–5 days after the treatment had been stopped. This indicates more rapid recovery of platelet function with ticagrelor. There was a numerical trend towards lower rates of MI in the ticagrelor group compared with clopidogrel (3.8% vs. 5.6%). Dyspnoea was more common in the ticagrelor groups compared with clopidogrel (11% with 90 mg ticagrelor; 16% with 80 mg ticagrelor vs. 6% with clopidogrel). Half of the patients that were bothered by dyspnoea experienced prolonged symptoms during the treatment, whereas the other half of patients had a fast resolution of symptoms.
One explanation for the respiratory symptoms could be that ticagrelor damages platelets because of its reversible nature and that the substance still has features of the adenosine triphosphate parent compound, which acts as bronchial irritant causing bronchoconstriction, inflammation and coughs (Doggrell, 2009). A greater number of asymptomatic ventricular pauses were found post-hoc in the ticagrelor groups, although no plausible explanation could be found. It is discussed, if ticagrelor somehow modulates and affects the adenosine metabolism (Cannon et al., 2007).
Phase III: PLATO trial: a study of platelet inhibition and patient outcomes
PLATO was an international, randomized, double-blind, event-driven trial involving 18 624 patients hospitalized for ST-elevation ACS with scheduled primary PCI or for non-ST-elevation ACS. After loading doses of ticagrelor 180 mg or clopidogrel 300 mg, patients received ticagrelor 90 mg twice daily or clopidogrel 75 mg once daily for 6–12 months. The median duration of exposure to the study drug was 277 days.
The primary efficacy end point (death from vascular causes, myocardial infarction or stroke) occurred in 9.8% of patients receiving ticagrelor as compared with 11.7% of those receiving clopidogrel (hazard ratio, 0.84) (Wallentin et al., 2009). The difference in the treatment effect was apparent within the first 30 days of therapy and persisted throughout the study period. Similarly, the study showed significant differences in the rates of other composite end points, as well as myocardial infarction alone (5.8% in the ticagrelor group vs. 6.9% in the clopidogrel group), death from vascular causes (4.0% vs. 5.1%) but not stroke alone (1.5% vs. 1.3%). The rate of death from any cause was also reduced with ticagrelor (4.5%, vs. 5.9%). The results regarding the primary end point did not show significant heterogeneity in analyses of the 33 subgroups, with three exceptions. The benefit of ticagrelor appeared to be attenuated in patients with low body weight, those not taking lipid-lowering drugs at randomization and those enrolled in North America.
The primary safety end point (major bleeding) did not differ between the ticagrelor and clopidogrel groups (11.6% and 11.2%) but ticagrelor was associated with a higher rate of major bleeding not related to coronary-artery bypass grafting (4.5% vs. 3.8%), including more instances of fatal intracranial bleeding and fewer of fatal bleeding of other types (Wallentin et al., 2009).
New adverse effects, not seen with clopidogrel or prasugrel, were seen with the use of ticagrelor. These include dyspnea, bradyarrhythmia and increased serum levels of uric acid and creatinine (Schomig, 2009; Wallentin et al., 2009).
Cangrelor (ARC-69931MX)
Cangrelor is a reversible, short-acting, potent, competitive P2Y12 inhibitor that has the advantage of being an active drug not requiring metabolic conversion, although it is not orally available (Figure 1).
Preclinical studies
Cangrelor has been tested in several models of arterial thrombosis in dogs and rabbits (Ingall et al., 1999; Huang et al., 2000; van Gestel et al., 2003; Wang et al., 2003). A 98-fold separation between antithrombotic effect and increase in bleeding time for cangrelor has been shown as compared with GpIIb/IIIa antagonists in a thrombosis model in dog (Ingall et al., 1999). The full inhibition of platelet aggregation was achieved at doses which extend bleeding times less than twofold. That is in contrast to GpIIb/IIIa antagonists, which achieved full inhibition of aggregation at dose levels that increased bleeding times six- to sevenfold (Ingall et al., 1999).
In another study, placebo or cangrelor (4 µg·kg−1·min−1 for 6 h) were administered intravenously before vessel wall injury (Huang et al., 2000). Whereas each dog from the placebo-treated group developed a thrombus (n= 5), carotid artery blood flow was sustained in five out of six cangrelor treated dogs. Thrombus weight was smaller, buccal and tongue bleeding time longer in the cangrelor group, but bleeding times returned to baseline within 60 min. ADP-induced platelet aggregation was clearly inhibited within the first 75 min, maintained for the duration of the protocol, returned to normal values after cessation of cangrelor infusion (Huang et al., 2000).
In another canine coronary electrolytic injury thrombosis model, cangrelor was administered additionally to tissue plasminogen activator (t-PA) (Wang et al., 2003). Myocardial tissue perfusion and the incidence of reocclusion were measured. The combination therapy of t-PA and cangrelor led to a significantly better blood flow and a decreased number of reocclusion events in the antagonist group than t-PA and saline alone (Wang et al., 2003).
Clinical studies
Pharmacokinetic/pharmacodynamic
Because the triphosphate chain of cangrelor results in a short half-life in vivo (3 min), is has to be infused intravenously (van Giezen and Humphries, 2005). Thus, cangrelor achieves a rapid steady-state concentration with a clearance of approximately 50 L·h−1. The pharmacokinetic profile is similar in men and women. Cangrelor is not dependent on hepatic or renal mechanisms of activation or clearance (Angiolillo et al., 2009).
In a pharmacokinetic study, 33 patients with a diagnosis of UA or non-Q-wave myocardial infarction were assigned to three different groups: stepwise dose increments over 3 h to a plateau of either 2 µg·kg−1·min−1 for 21 h or the same dose up to 69 h or 4 µg·kg−1·min−1 for up to 69 h (Storey et al., 2001). Elimination of plasma cangrelor was biphasic, clearance was rapid (44.3 L·h−1), the initial volume of distribution was small (5.1 L), indicating the restriction to the blood compartment. Estimated mean population half-life was less than 5 min. The mean volume of distribution at steady state was 13.37 L (Storey et al., 2001). Cangrelor dose-dependently inhibited platelet aggregation by 100% and a rapid return of normal platelet function has been demonstrated after withdrawal of treatment (Storey et al., 2001). The complete reversibility of platelet inhibition appears within 20–50 min after administration (Storey et al., 2001; Greenbaum et al., 2006; Steinhubl et al., 2008; Angiolillo et al., 2009).
A very interesting study investigated the effect on platelet aggregation when clopidogrel was administered concurrently with or immediately upon termination of cangrelor infusion (Steinhubl et al., 2008). Ten healthy subjects received an i.v. bolus (30 µg·kg−1) and 4 µg·kg−1·min−1 infusion of cangrelor for 1 h, followed by oral administration of clopidogrel (600 mg). In subjects receiving clopidogrel simultaneously with cangrelor the effect of co-administered clopidogrel was attenuated and the degree of platelet inhibition reduced, suggesting that there is a competitive effect between these two substances. A possible explanation is that cangrelor's high affinity to the receptor might prevent clopidogrel's active metabolite, which is unstable, from binding to the P2Y12 receptor.
Similar results have been reported in another study. Preincubation of blood with cangrelor before adding active metabolites of clopidogrel or prasugrel reduced the ability of the active metabolites of clopidogrel/prasugrel to irreversibly antagonize the P2Y12 receptor. In contrast, addition of cangrelor after preincubation with active metabolites of clopidogrel/prasugrel led to a sustained platelet inhibition (Dovlatova et al., 2008).
In contrast, another study reported that cangrelor and clopidogrel have an additive effect on inhibition of platelet aggregation (Storey et al., 2002).
Phase II
Cangrelor's safety and tolerability profile have been tested in a multi-centre phase 2 study (Jacobsson et al., 2002). Ninety-one patients with UA or non-Q-wave-myocardial infarction received cangrelor (n= 45) or placebo (n= 46). Cangrelor's plasma concentration reached 401 ng·mL−1 at steady state. There was no sign of accumulation; interindividual variability in clearance (41.0 L·h−1) was not distinct (22%). The half-life was <5 min (Jacobsson et al., 2002).
In another phase 2 study, patients with ACS received an infusion of cangrelor (2 µg·kg−1·min−1 or 4 µg·kg−1·min−1; n= 13) or a standard regimen of clopidogrel (Storey et al., 2002). Cangrelor was able to inhibit platelet aggregation after 30 s: both doses yielded in a highly effective inhibition, which was maintained during the course of the infusion.
In a cohort of 200 patients undergoing PCI, platelet aggregation was rapid and reversible (Greenbaum et al., 2006). In the first part, patients received either placebo or cangrelor (1, 2 or 4 µg·kg−1·min−1). In the second part, patients received cangrelor (4 µg·kg−1·min−1) or abciximab before PCI. Major or minor bleedings occurred in 13% of patients receiving cangrelor versus 8% in these receiving placebo (part 1) and in 7% receiving cangrelor compared with 10% randomized to abciximab (part 2). Not surprisingly, thrombocytopenia was less frequent during cangrelor infusion compared with abciximab (1% vs. 7%). Both study medications increased steady-state bleeding times, but with a tendency toward longer bleeding times with abciximab compared with cangrelor (Greenbaum et al., 2006). The 30-day composite incidence of major adverse coronary events was similar between those receiving cangrelor and abciximab (7.6% vs. 5.3%). Platelet inhibition was maximal after 15 min and returned to baseline within 15 min after cessation of infusion, except for the 4 µg·kg−1·min−1-dose where it remained significantly inhibited. Cangrelor at a dose of 4 µg·kg−1·min−1 completely inhibited platelets in 95% of subjects within minutes (Greenbaum et al., 2006).
Phase II: STEP-AMI trial: safety, tolerability and effect on patency in acute myocardial infarction
In the STEP-AMI trial 92 patients with ST-segment elevation ACS received either cangrelor alone, t-PA (100 mg) alone, or one to three doses of cangrelor together with half-dose t-PA (Greenbaum et al., 2007). The goal was to evaluate the coronary patency and occurrence of adverse events and bleeding under cangrelor in combination with a fibrinolytic antithrombotic therapy. The combination therapy led to similar results in 60-min patency (TIMI flow grade 3) as full-dose t-PA alone (55% vs. 50%) and greater patency than with cangrelor alone (55% vs. 18%). Myocardial perfusion grade 3 was reached in 61% versus 50% in patients receiving combination therapy and full-dose t-PA, but only in 18% in those receiving cangrelor monotherapy. Safety and adverse events were comparable between groups (Greenbaum et al., 2007).
Phase II: BRIDGE trial: maintenance of platelet inhibition with cangrelor after discontinuation of thienopyridines in patients undergoing surgery
The BRIDGE trial has been designed to demonstrate that patients receiving cangrelor infusion before coronary artery bypass surgery have an acceptable safety profile and can undergo surgery without excessive bleeding peri-operatively. This ongoing trial will enrol 220 patients.
Phase III
CHAMPION-PCI (a clinical trial comparing cangrelor to clopidogrel in subjects who require PCI) has been designed to evaluate if cangrelor is superior, or at least non-inferior to the standard therapy of clopidogrel in patients in need for PCI as measured by a composite end point of all-cause mortality, myocardial infarction and ischemia-driven revascularization in a time frame of 48 h (primary) and 30 days (secondary).
CHAMPION-PLATFORM [a clinical trial comparing treatment with cangrelor (in combination with usual care) to usual care, in subjects who require PCI] has been designed to assess the efficacy of cangrelor compared with placebo in patients undergoing PCI as measured by a composite end point of all-cause mortality, myocardial infarction and ischemia-driven revascularization in a time frame of 48 h (primary outcome measure) and 30 days (secondary outcome measure).
In May 2009 both CHAMPION trials have been stopped, following a decision by the interim analysis review committee that the studies would not show the persuasive clinical efficacy needed for approval, although 98% of planned 9000 patients for CHAMPION-PCI and 83% of planned 6000 patients for CHAMPION-PLATFORM had already been enrolled.
Elinogrel (PRT060128)
Elinogrel is a direct-acting reversible P2Y12 receptor inhibitor that is in phase 2 clinical trials (Figure 1). Elinogrel is the only antiplatelet compound that can be administered both intravenously and orally. This allows an immediate high-level platelet inhibition with the i.v. formulation in the acute setting and a smooth transition to predictable platelet inhibition with the oral formulation in the chronic setting (Michelson, 2009).
Preclinical studies
Elinogrel demonstrated dose-proportional antithrombotic activity in vivo at plasma concentrations which had minimal effect on tail bleeding times in mice (Andre et al., 2007). At the highest plasma concentrations of elinogrel, inhibition of thrombosis was superior to that observed with doses of clopidogrel which achieved full blockade of ADP-induced aggregation (Andre et al., 2007).
Clinical studies
Pharmacokinetic/pharmacodynamic
In a pharmacodynamic study, single oral dose of elinogrel has been shown to overcome high platelet reactivity in patients undergoing PCI who were non-responsive to clopidogrel therapy (Gurbel et al., 2008).
Following a 50 mg oral dose of 14C-elinogrel, mean total radioactivity Cmax and AUC were 3895 ng eq·mL−1 and 28985 ng eq*h·mL−1 respectively (Hutchaleelaha et al., 2008). Approximately 56% of the total dose administered was recovered in urine and 48% in feces. Unchanged elinogrel was the dominant circulating radioactivity in plasma and the major radioactive component in urine and feces, accounting for 66.2% of the total administered dose in 0–36 h urine and 0–120 h in feces (Hutchaleelaha et al., 2008). The major metabolic route was demethylation to form PRT060301, which was determined to be the only prominent circulating metabolite in plasma (AUC approximately 10% of elinogrel) and the only major metabolite in urine and feces (22.4% of the dose) (Hutchaleelaha et al., 2008).
Phase II: INNOVATE-PCI: a randomized trial to evaluate the effect of adjunctive antiplatelet therapy with intravenous PRT060128, a selective P2Y12-receptor inhibitor, before primary percutaneous intervention in patients with STEMI
INNOVATE-PCI is a multi-centre, randomized, double-blind, triple-dummy, clopidogrel-controlled trial of i.v. and oral elinogrel compared with clopidogrel in patients undergoing non-urgent PCI. After diagnostic angiography, patients scheduled for non-urgent PCI will be randomized to clopidogrel or to one of three dose levels of elinogrel. The study is ongoing and will enrol 800 patients.
Phase II: ERASE-MI: safety and efficacy study of adjunctive antiplatelet therapy prior to primary PCI in patients with STEMI
ERASE-MI trial investigated the safety and efficacy of i.v. elinogrel at doses of 10, 20, 40 and 60 mg as an adjunctive antiplatelet therapy before primary PCI in 70 patients with STEMI. The results are not published yet.
BX 667
BX 667 is an orally active and reversible small-molecule P2Y12 receptor antagonist (Figure 1) (Wang et al., 2007).
Preclinical studies
BX 667 is metabolized by esterases to the carboxylic acid form, BX 048, without a significant change in binding affinity and platelet inhibitory potency [inhibitory concentration 50% (IC50) = 97 nM, IC50 = 290 nM respectively] (Wang et al., 2007; Bryant et al., 2008; Post et al., 2008). Administration of BX 667 results in a rapid and sustained inhibition of platelet aggregation where the extent and duration of platelet inhibition is directly proportional to circulating plasma levels (Post et al., 2008).
In a rat arteriovenous-shunt model, both intravenous BX 048 and oral BX 667 administrations resulted in a similar efficacy with a similar pharmacodynamic relationship between the plasma concentration of BX 048 and thrombus inhibition for both compounds even given by different routes of administration (Wang et al., 2007).
In dogs, both compounds have been measured in circulating blood because the conversion from BX 667 to BX 048 was gradual and incomplete. In the dog cyclic flow variation model, the EC50 (0.34 µM) for thrombus inhibition was comparable with the EC50 (0.71 µM) in the rat model of platelet-rich thrombosis (Wang et al., 2007). Even when given with aspirin in the dog model, BX 667 still maintained a wider therapeutic index than clopidogrel (Wang et al., 2007).
PAR-1 antagonists
SCH 530348
SCH 530348 is a highly selective and orally active PAR-1 antagonist, which inhibits the thrombin-mediated platelet activation, without interfering with the thrombin-mediated cleavage of fibrinogen (Chintala et al., 2008; Becker et al., 2009).
Preclinical studies
Oral administration of SCH 530348 at doses ≥0.1 mg·kg−1 completely inhibited platelet aggregation for 24 h in cynomolgus monkeys (Chackalamannil et al., 2008). In human platelet-rich plasma, SCH 530348 inhibited thrombin-induced and thrombin receptor activating peptide-induced platelet aggregation with IC50 of 47 and 25 nM, respectively, without affecting the aggregation induced by ADP, the TXA2 mimetic U46619 or collagen (Chackalamannil et al., 2008; Chintala et al., 2008). SCH 530348 did not affect prothrombin time or activated partial thromboplastin time, suggesting that the potential for bleeding events may not be increased. SCH 530348 also demonstrated a bioavailability of 86% in monkeys (Chintala et al., 2008). Treatment of cynomolgus monkeys with SCH 530348, either alone or in combination with aspirin plus clopidogrel, did not increase surgical blood loss or bleeding times versus placebo and aspirin plus clopidogrel respectively (Chintala et al., 2008).
Clinical studies
Pharmacokinetic/pharmacodynamic
The route of administration is oral and the route of elimination is through the gastrointestinal and biliary tract. SCH 530348 shows a high bioavailability and a half-life of 126–269 h (Kasoglou et al., 2005; Becker et al., 2009).
SCH 530348 given to healthy volunteers in a single dose (5–40 mg) caused a mean inhibition of >90% of thrombin receptor activating peptide-induced platelet aggregation for more than 72 h (Kasoglou et al., 2005; Angiolillo et al., 2009; Becker et al., 2009). A 40 mg loading dose followed by a 2.5 mg once-daily dose of SCH 530348 effectively inhibited thrombin receptor activating peptide-induced platelet aggregation throughout 28 days (Chintala et al., 2008).
Phase II: TRA-PCI
In the TRA-PCI trial SCH 530348 was administered to 1030 patients who were scheduled non-urgently for angiography and possible stenting (Becker et al., 2009). Patients received SCH 530348 (10, 20 or 40 mg) or placebo. Those patients who underwent primary PCI received the standard therapy of clopidogrel (300 or 600 mg) and heparin/bivalirudin and continued taking an oral dose of SCH 530348 (0.5, 1.0 or 2.5 mg) for 60 days. Those patients who did not undergo PCI, but CABG or medical management continued in a separate, secondary evaluable cohort. Primary end points were the incidence of clinically significant minor or major bleedings. Secondary end points were death or major adverse coronary events.
SCH 530348 was not associated with an increase in major and minor bleedings versus placebo: the primary end point occurred in 2%, 3% and 4% of patients, taking 10, 20 or 40 mg of SCH 530348 versus 3% of patients in the placebo group. During maintenance dose regimen 2%, 4% and 3% of patients receiving 0.5, 1.0 or 2.5 mg SCH 530348 suffered a primary end point versus 2% of patients receiving placebo. The combined end point of death/major adverse cardiovascular events/stroke occurred less frequently in the SCH 530348 group compared with placebo: 6% versus 9% (Becker et al., 2009).
Phase II
Two additional trials in patients with ACS or ischemic stroke have confirmed the favourable safety profile of SCH 530348 observed in the TRA-PCI study (Goto et al., 2008; Shinohara et al., 2008).
Phase III: TRA-CER: thrombin receptor antagonist for clinical event reduction in ACS
The TRA-CER trial has been designed to determine whether SCH 530348, when added to the existing standard of care prevents heart attack and stroke in patients with ACS. The primary efficacy end point of the study is the first occurrence of any component of the composite of cardiovascular death, myocardial infarction, stroke, recurrent ischemia with rehospitalization and urgent coronary revascularization. The study is also designed to assess the risk of bleeding with SCH 530348 added to the standard of care versus the standard of care alone. TRA-CER study evaluates a 40 mg loading dose and 2.5 mg maintenance dose of SCH 530348 in approximately 10 000 patients with ACS for >1 year of follow-up. The study is ongoing.
Phase III: TRA 2P-TIMI 50: thrombin receptor antagonist in secondary prevention of atherothrombotic ischemic events
The TRA 2P-TIMI 50 trial has been designed to determine whether SCH 530348, when added to the existing standard of care for preventing heart attack and stroke in patients with a known history of atherosclerosis, will yield additional benefit over the existing standard of care without SCH 530348 in preventing heart attack and stroke. The primary efficacy end point of the study is the first occurrence of any component of the composite of cardiovascular death, MI, stroke and urgent coronary revascularization. TRA 2P-TIMI 50 study is an ongoing trial, which will enrol 19 000 patients with a history of cardiovascular disease receiving a maintenance dose of 2.5 mg of SCH 530348 or placebo.
SCH 205831
SCH 205831 is a potent, selective and orally active PAR-1 antagonist derived from the natural product himbacine (Figure 1) (Chintala et al., 2005a).
Preclinical studies
SCH 205831 competes with the thrombin receptor activating peptide for the PAR-1 receptor with a Ki of 2.7 nM. SCH 205831 inhibits thrombin receptor activating peptide-induced platelet aggregation in washed human platelet with an IC50 of 65 nM (Chintala et al., 2005a). Oral administration of SCH 205831 to cynomolgus monkeys caused a dose-dependent and complete inhibition of thrombin receptor activating peptide-induced platelet aggregation for 6 h post-dosing (Chintala et al., 2005a).
SCH 205831 was also evaluated in two models of thrombosis in baboons. SCH 205831 inhibited platelet deposition by 50–70% onto uncoated stents in an arteriovenous-shunt model of thrombosis in baboons (Chintala et al., 2005a). SCH 205831 at 10 mg·kg−1, inhibited platelet deposition by 90% in an endarterectomy model of arteriovenous-shunt thrombosis in baboons. Platelet aggregation responses to thrombin receptor activating peptide but not to ADP were inhibited by SCH 205831. Template bleeding times and coagulation parameters were unchanged (Chintala et al., 2005a,b;).
SCH 602539
SCH 602539 is another potent, selective and orally active PAR-1 antagonist (Figure 1) (Chintala et al., 2007).
Preclinical studies
SCH 602539 inhibited thrombosis in a dose-dependent manner in the Folts model of thrombosis in anesthetized cynomolgus monkeys (Chintala et al., 2007). SCH 602539 and cangrelor exhibited additive and synergistic antithrombotic effects (Chintala et al., 2007).
E5555
E5555 is a novel PAR-1 antagonist targeting the G-coupled receptor and modulating thrombin-platelet-endothelial interactions (Kogushi et al., 2007; Serebruany et al., 2009b). The drug is currently being tested in phase 2 trials in patients with CAD and has antithrombotic and anti-inflammatory effects (Serebruany et al., 2009b).
Preclinical studies
E5555 showed anti-thrombotic effect in a thrombosis model in guinea pigs and inhibitory effects on neointima hyperplasia in a rat balloon injury model (Kogushi et al., 2007). Moreover, E5555 inhibits the release of the inflammatory markers: sCD40L, IL-6 and expression of P-selectin (Kogushi et al., 2007).
E5555 moderately but significantly inhibited platelet activity beyond PAR-1 blockade in blood from healthy volunteers and CAD patients treated with aspirin with or without clopidogrel (Serebruany et al., 2009b). Platelet inhibition was present already at 20 ng·mL−1, and was not dose-dependent without thrombin receptor activating peptide stimulation. E5555 caused a 10–15% inhibition of ADP- and collagen-induced platelet aggregation. As expected, thrombin receptor activating peptide-induced aggregation was inhibited dose-dependently at 20–50 ng·mL−1 (Serebruany et al., 2009b).
Phase II: LANCELOT ACS: a randomized, double-blind, placebo-controlled study of the safety and tolerability of E5555 and its effects on clinical events and biomarkers in patients with non-ST-segment elevation ACS
The primary objective of this ongoing study is to investigate the safety and tolerability of E5555 at three dose levels in patients with ACS. The secondary objectives are to determine the effect of E5555 on major adverse coronary events and transient ischemia, and explore the pharmacokinetics of E5555 and effects of E5555 on inflammatory markers.
Thromboxan receptor (TP receptor) antagonist
Terutroban (S18886)
Terutroban is a selective oral and specific antagonist on the TP (Figure 1). It blocks thromboxane-induced platelet aggregation and vasoconstriction, improves endothelial function and has an antiatherosclerotic effect (Chamorro, 2009).
Preclinical studies
Terutroban has a high antithrombotic efficacy with a favourable bleeding risk profile in an experimental porcine model of stent-induced thrombosis (Vilahur et al., 2007).
Terutroban dose-dependently prolonged the time to primary occlusive coronary artery thrombosis, whereas terutroban plus clopidogrel were effective in preventing occlusive thrombus formation with only a moderate increase of tongue-bleeding time in coronary arterial thrombosis and myocardial ischemia-reperfusion model in canines (Hong et al., 2006).
Terutroban attenuated renal damage in the double transgenic rat model of hypertension, indicating that TP inhibition ameliorates angiotensin II-induced nephropathy (Sebekova et al., 2008). Additionally, it has been reported that terutroban has renoprotective properties in an experimental model of type 2 diabetes: it prevented mesangiolysis, reduced the urinary transforming growth factor-beta and 2,3-dinor-thromboxane B excretion and enhanced the antioxidative defense (Sebekova et al., 2007). In another study terutroban attenuated renal oxidative stress and proteinuria in diabetic apolipoprotein E-deficient mice (Xu et al., 2006).
Clinical studies
Pharmacokinetic/pharmacodynamic
A pharmacokinetic/pharmacodynamic study was conducted in 30 patients with peripheral artery disease, who were randomized to receive five different oral doses of terutroban (1, 2.5, 5, 10 or 30 mg) for 12 weeks (Gaussem et al., 2005). Pharmacokinetics of terutroban was linear, with peak plasma levels being reached between 30 min and 2 h and a terminal half-life of 5.8–10 h. No significant accumulation of terutroban in plasma was observed after repeated dosing. There was a predictable relation between the plasma drug concentration and the degree of platelet inhibition. Maximal inhibition of platelet aggregation was achieved within 1 h with all oral doses of terutroban, and this effect was maintained for at least 12 h. The pharmacokinetic/pharmacodynamic relationship was direct and platelet aggregation was strongly inhibited by terutroban plasma concentrations above 10 ng·mL−1. The safety profile of terutroban was excellent with no attributable adverse events (Gaussem et al., 2005).
Phase III: PERFORM: the prevention of cerebrovascular and cardiovascular events of ischemic origin with terutroban in patients with a history of ischemic stroke or transient ischemic attack
PERFORM is an ongoing, international, double-blind, randomized controlled trial designed to investigate the superiority of the terutroban (30 mg·day−1) over aspirin (100 mg·day−1), in reducing cerebrovascular and cardiovascular events in patients with a recent history of ischemic stroke or transient ischemic attack in 19 000 patients (Hennerici, 2009).
vWF antagonists
AJW200
AJW200 is an IgG4 humanized monoclonal antibody to human vWF derived from Sp2/0 mouse myeloma cells.
Preclinical studies
AJW200 specifically inhibited high-shear-stress-induced platelet aggregation in a concentration-dependent manner (IC50: 1 µg·mL−1) in blood from volunteers. In contrary, low-shear-stress-induced platelet aggregation was not affected, even at a concentration of 80 µg·mL−1 (Kageyama et al., 2002).
Sustained inhibition of platelet aggregation was observed over 24 h after a single bolus injection of 0.3 mg·kg−1 AJW200 in cynomolgus monkeys (Kageyama et al., 2002). Moderate prolongation of the bleeding time was observed at the doses of 1 and 3 mg·kg−1 (Kageyama et al., 2002).
Clinical studies
Pharmacokinetic/pharmacodynamic
In the pharmacokinetic/pharmacodynamic study healthy volunteers received placebo or AJW200 (0.01, 0.03 or 0.05 mg·kg−1) intravenously. Cmax (205, 586 and 833 ng·mL−1), Tmax (1.0, 0.92 and 0.57 h) and T1/2? (23.5, 24.3, 27.2 h) were dose-dependent. The maximum vWF occupancy by AJW200 was 19, 51 and 62% respectively. AJW200 prolonged closure times in the Platelet Function Analyzer 100, which lasted for 3–6 h with the lower and up to 12 h for the higher dose. No clinically significant adverse events were recorded and there was no evidence of immunogenicity.
ARC1779
ARC1779 is a second-generation, nuclease resistant aptamer, which has been conjugated to a 20-kDA polyethylene glycol. Aptamers are a novel therapeutic class of oligonucleotides with drug-like properties that share some of the attributes of monoclonal antibodies (resulting in the class being referred to as chemical antibodies), as well as some of those of low molecular weight, chemically synthesized drugs (Gilbert et al., 2007).
ARC1779 binds with high affinity (KD∼2 nM) to the activated vWF A1-domain and inhibits vWF dependent platelet aggregation through preventing Gp-Ib receptor to interact with Willebrand factor (Figure 1) (Gilbert et al., 2007). It represents both, a new therapeutic class and a novel therapeutic mechanism.
As vWF is a mediator of atherosclerotic cardiovascular disease due to its causal role in arterial thrombogenesis (Paulinska et al., 2009), Willebrand factor antagonists might act specifically in the setting of ACS (Gilbert et al., 2007). A further advantage is that vWF is only active in the presence of high shear forces seen in the arterial side of circulation (Spiel et al., 2008).
Preclinical studies
ARC1779 inhibited vWF dependent platelet aggregation measured by botrocetin-induced platelet aggregation and the Platelet Function Analyzer 100 closure time (Diener et al., 2009). Moreover, ARC1779 reduced adhesion of platelets to collagen-coated matrices and formation of platelet thrombi on denuded porcine arteries (Diener et al., 2009). In a cynomolgus monkey carotid electrical injury thrombosis model, ARC1779 inhibited the formation of occlusive thrombi (Diener et al., 2009). An infusion of 0.5 mg·kg−1 body weight achieved plasma Cmax of 1 µM (13 µg·mL−1) (Diener et al., 2009).
ARC1779 inhibited vWF activity (IC90: 3–4 µg·mL−1) and shear dependent platelet function assessed by the Platelet Function Analyzer 100 (IC50: 0.5–0.9 µg·mL−1) and the Cone and Plate Analyzer (IC50: 0.1–0.4 µg·mL−1) in blood from patients with ACS and volunteers (Spiel et al., 2009).
Clinical studies
Pharmacokinetic/pharmacodynamic
In the pharmacokinetic/pharmacodynamic study, the concentration-time curve of ARC1779 was monophasic with a dose-dependent and linear increase in mean Cmax and AUC when administered as i.v. bolus, 15 min slow i.v. bolus with 0.05–1.0 mg·kg−1 or as i.v. bolus followed by a 4-h infusion. The maximal exposure to ARC1779 was produced by the highest concentration through the slow i.v. bolus and was 21.2 µg·mL−1 (Cmax) with a mean AUC of 80.9 µg·mL−1·h−1. Tmax was 7–10 min after the bolus and 30 min after the slow bolus administration. The mean elimination half-life was 2 h, the mean volume of distribution was 74.3 mL·kg−1 (Gilbert et al., 2007).
A dose-dependent effect on closure time in the Platelet Function Analyzer 100 (PFA-100) was seen up to 0.3 mg·kg−1. With doses >0.3 mg·kg−1 closure time was maximally prolonged with complete inhibition of platelet function for up to 4 h and recovery of platelet function after 8–12 h. VWF activity was inhibited dose-dependently, with a maximum 1–2 h after the 15-min-bolus. Inhibition ranged from 60% with the lowest dose until 95% with 1.0 mg·kg−1 ARC1779 (Gilbert et al., 2007).
EC50 and EC90 values of ARC1779 were 0.2 µg·mL−1 and 2.0 µg·mL−1 for inhibition of vWF activity after slow i.v. bolus administration. EC50 and EC90 values for inhibition of platelet function as measured by PFA-100 closure time were 0.75 µg·mL−1 and 2.6 µg·mL−1 respectively (Gilbert et al., 2007).
Phase II
ARC1779 effectively increased platelet counts in critically ill thrombotic thrombocytopenic purpura patients through preventing platelet aggregation and loss of platelets. Cessation of ARC1779 infusion was associated with a drop in platelets and a progression in thrombotic thrombocytopenic purpura-related organ damage (Jilma et al., 2009; Knobl et al., 2009).
In an open-labelled clinical trial, the safety and efficacy of the ARC1779 added to plasma exchange therapy in seven patients with acute thrombotic thrombocytopenic purpura were tested. Continuous infusions of ARC1779 (0.001–0.002 mg·kg−1·min−1) effectively blocked vWF – platelet binding in thrombotic thrombocytopenic purpura patients and this was associated with a rapid increase in platelet counts (Jilma et al., 2009).
In a prospective open label trial ARC1779 was administered in three periods in patients with familial thrombotic thrombocytopenic purpura. In the first period 0.002 mg·kg−1·min−1 of ARC1779 was given for 24 h. In the second period, 50 mg of ARC1779 was administered s.c. once daily for 7 days, then tapered in two steps (25 mg on day 8, 12.5 mg day 9). Finally, the patient received a bolus primed continuous infusion of 0.004 mg·kg−1 of ARC1779 for 48 h and 0.006 mg·kg−1 up to a total of 72 h. Median steady state ARC1779 concentrations were approximately 10 µg·mL−1 (24 h i.v.) in period I, 0.6 µg·mL−1 (daily s.c.) in period II and 64 µg·mL−1 (high dose i.v.) in period III. High dose ARC1779 i.v. blocked the GPIb binding site of the vWF A1 domain by >95%, the lower i.v. dose by 85%, and s.c. ARC1779 decreased vWF activity to 57% of normal activity; this was mirrored by maximal prolongation of collagen/ADP closure times with PFA-100 when the drug was infused i.v., but not when ARC1779 was injected s.c. ARC1779 injected s.c. did not reach therapeutically effective plasma concentrations and did not prevent the periodic drop in platelet counts observed in this patient. In striking contrast, ARC1779 raised the platelet count by 46% and by 106% when given for 24 h or 72 h i.v. respectively (Firbas et al., 2009).
Another study showed that the ARC1779 can prevent desmopressin-induced thrombocytopenia in patients with von Willebrand disease type 2B (Schranz et al., 2009).
ALX-0081
ALX-0081 is a bivalent humanized Nanobody specifically targeting the GPIb binding site of vWF. Due to its bivalency, ALX-0081 is able to avidly interact with vWF resulting in an increased potency compared with its monovalent analogue (Ulrichts et al., 2009).
Preclinical studies
ALX-0081 completely inhibited platelet adhesion in collagen perfusion studies using blood obtained from patients undergoing PCI. In contrast, when aspirin and clopidogrel were used only an incomplete platelet inhibition was observed (Ulrichts et al., 2009).
In a combined baboon efficacy and safety model measuring acute thrombosis and surgical bleeding, ALX-0081 showed a superior therapeutic window compared with aspirin, clopidogrel and abciximab. The half-life of ALX-0081 was prolonged by vWF binding, enabling predictable drug levels in cynomolgus monkey (Ulrichts et al., 2009).
Clinical studies
Pharmacokinetic/pharmacodynamic
Treatment with ALX-0081 was well tolerated and safe, no signs of bleeding were reported and no immunogenic response was detected after intravenous infusions of ALX-0081 for 1 h at doses 0.5–12 mg in 40 male healthy volunteers (Holz et al., 2009). ALX-0081 displayed non-linear pharmacokinetic properties, following a two-compartment model. Full inhibition of ristocetin-induced platelet aggregation was observed at ALX-0081 concentrations of 400 ng·mL−1. A total dose of ALX-0081 >2 mg caused complete inhibition of ristocetin-induced platelet aggregation 1 h post-dosing with a maximal duration of 12 h. Mild and transient laboratory changes in the reduction of factor VIII and vWF plasma levels were observed, all events were fully reversible within 24 h (Holz et al., 2009).
Conclusion
Novel platelet inhibitors are currently in advanced clinical testing. The pharmacology and clinical profiles of new platelet antagonists indicate that they have the potential to provide more consistent, more rapid and more potent platelet inhibition than agents currently used. In addition, as most new platelet antagonists are reversible and do not require hepatic bioactivation, more flexibility in their use is ensured if rapid onset of action is needed before PCI or when cessation is required before coronary artery bypass graft surgery. On the other hand, enhanced efficacy of antiplatelet drugs is often associated with an increased risk of major bleedings. Additionally, as the current knowledge of safety of any new drug is limited, the occurrence of adverse events such as thrombotic thrombocytopenic purpura or cancer risk are unknown. Whether the potential advantages of platelet inhibitors will translate into clinical advantages will require additional comparisons in properly powered, randomized, controlled trials.
Glossary
Abbreviations:
- ACS
acute coronary syndrome
- ADP
adenosine diphosphate
- ATP
adenosine triphosphate
- CAD
coronary artery disease
- CYP
cytochrome P
- ED50
effective dose 50%
- IC50
inhibitory concentration 50%
- MACE
major adverse cardiovascular events
- NSTEMI
none ST-elevation myocardial infarction
- PAR
protease-activated receptor
- PCI
percutaneous coronary intervention
- STEMI
ST-elevation myocardial infarction
- TIMI
thrombolysis in myocardial infarction
- TP
thromboxane receptor
- t-PA
tissue plasminogen activator
- TXA2
thromboxane A2
- UA
unstable angina
- vWF
von Willebrand factor
References
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edn. Br J Pharmacol. 2008;153(Suppl. 2):S1–209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre P, Jurek M, Sim D, Deguzman F, Hollenbach S, Phillips DR, et al. PRT060128, a novel, direct-acting orally available P2Y12 antagonist, confers superior antithrombotic activity over clopidogrel in a mice thrombosis model. J Thromb Haemost. 2007;5:O-W-031. [Google Scholar]
- Angiolillo DJ, Bhatt DL, Gurbel PA, Jennings LK. Advances in antiplatelet therapy: agents in clinical development. Am J Cardiol. 2009;103:40A–51A. doi: 10.1016/j.amjcard.2008.11.023. [DOI] [PubMed] [Google Scholar]
- Barrett NE, Holbrook L, Jones S, Kaiser WJ, Moraes LA, Rana R, et al. Future innovations in anti-platelet therapies. Br J Pharmacol. 2008;154:918–939. doi: 10.1038/bjp.2008.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker RC, Moliterno DJ, Jennings LK, Pieper KS, Pei J, Niederman A, et al. Safety and tolerability of SCH 530348 in patients undergoing non-urgent percutaneous coronary intervention: a randomised, double-blind, placebo-controlled phase II study. Lancet. 2009;373:919–928. doi: 10.1016/S0140-6736(09)60230-0. [DOI] [PubMed] [Google Scholar]
- Brandt JT, Payne CD, Wiviott SD, Weerakkody G, Farid NA, Small DS, et al. A comparison of prasugrel and clopidogrel loading doses on platelet function: magnitude of platelet inhibition is related to active metabolite formation. Am Heart J. 2007;153:66 e9-16. doi: 10.1016/j.ahj.2006.10.010. [DOI] [PubMed] [Google Scholar]
- Bryant J, Post JM, Alexander S, Wang YX, Kent L, Schirm S, et al. Novel P2Y12 adenosine diphosphate receptor antagonists for inhibition of platelet aggregation (I): in vitro effects on platelets. Thromb Res. 2008;122:523–532. doi: 10.1016/j.thromres.2008.03.026. [DOI] [PubMed] [Google Scholar]
- Cannon CP, Husted S, Harrington RA, Scirica BM, Emanuelsson H, Peters G, et al. Safety, tolerability, and initial efficacy of AZD6140, the first reversible oral adenosine diphosphate receptor antagonist, compared with clopidogrel, in patients with non-ST-segment elevation acute coronary syndrome: primary results of the DISPERSE-2 trial. J Am Coll Cardiol. 2007;50:1844–1851. doi: 10.1016/j.jacc.2007.07.053. [DOI] [PubMed] [Google Scholar]
- Chackalamannil S, Wang Y, Greenlee WJ, Hu Z, Xia Y, Ahn HS, et al. Discovery of a novel, orally active himbacine-based thrombin receptor antagonist (SCH 530348) with potent antiplatelet activity. J Med Chem. 2008;51:3061–3064. doi: 10.1021/jm800180e. [DOI] [PubMed] [Google Scholar]
- Chamorro A. TP receptor antagonism: a new concept in atherothrombosis and stroke prevention. Cerebrovasc Dis. 2009;27(Suppl. 3):20–27. doi: 10.1159/000209262. [DOI] [PubMed] [Google Scholar]
- Chintala M, Ahn H, Foster C, Xia Y, Chackalamannil S. Antithrombotic effects of SCH 205831: a potent, selective and orally active antagonist of the PAR-1 thrombin receptor. J Thromb Haemost. 2005a;3 Abstract OR286. [Google Scholar]
- Chintala M, Ahn HS, Foster C, Xia Y, Chackalamannil S. Chintala M, Ahn HS, Foster C, Xia Y, Chackalamannil S. J Thromb Haemost. 2005b;3(Suppl.):OR286. [Google Scholar]
- Chintala M, Kurowski S, Vemulapalli S, Li Q, Brown A, Strony J. Efficacy of SCH 602539, a selective thrombin receptor antagonist alone and in combination with cangrelor in a Folts model of thrombosis in nesthetized monkeys. Eur Heart J. 2007;28(Suppl. 1):188. [Google Scholar]
- Chintala M, Shimizu K, Ogawa M, Yamaguchi H, Doi M, Jensen P. Basic and translational research on proteinase-activated receptors: antagonism of the proteinase-activated receptor 1 for thrombin, a novel approach to antiplatelet therapy for atherothrombotic disease. J Pharmacol Sci. 2008;108:433–438. doi: 10.1254/jphs.08r06fm. [DOI] [PubMed] [Google Scholar]
- Diener JL, Daniel Lagasse HA, Duerschmied D, Merhi Y, Tanguay JF, Hutabarat R, et al. Inhibition of von Willebrand factor-mediated platelet activation and thrombosis by the anti-von Willebrand factor A1-domain aptamer ARC1779. J Thromb Haemost. 2009;7:1155–1162. doi: 10.1111/j.1538-7836.2009.03459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doggrell SA. Ticagrelor, a platelet aggregation inhibitor for the potential prevention and treatment of arterial thrombosis and acute coronary syndromes. IDrugs. 2009;12:309–317. [PubMed] [Google Scholar]
- Dovlatova NL, Jakubowski JA, Sugidachi A, Heptinstall S. The reversible P2Y antagonist cangrelor influences the ability of the active metabolites of clopidogrel and prasugrel to produce irreversible inhibition of platelet function. J Thromb Haemost. 2008;6:1153–1159. doi: 10.1111/j.1538-7836.2008.03020.x. [DOI] [PubMed] [Google Scholar]
- Farid NA, Payne CD, Small DS, Winters KJ, Ernest CS, 2nd, Brandt JT, et al. Cytochrome P450 3A inhibition by ketoconazole affects prasugrel and clopidogrel pharmacokinetics and pharmacodynamics differently. Clin Pharmacol Ther. 2007a;81:735–741. doi: 10.1038/sj.clpt.6100139. [DOI] [PubMed] [Google Scholar]
- Farid NA, Smith RL, Gillespie TA, Rash TJ, Blair PE, Kurihara A, et al. The disposition of prasugrel, a novel thienopyridine, in humans. Drug Metab Dispos. 2007b;35:1096–1104. doi: 10.1124/dmd.106.014522. [DOI] [PubMed] [Google Scholar]
- Farid NA, Payne CD, Ernest CS, 2nd, Li YG, Winters KJ, Salazar DE, et al. Prasugrel, a new thienopyridine antiplatelet drug, weakly inhibits cytochrome P450 2B6 in humans. J Clin Pharmacol. 2008;48:53–59. doi: 10.1177/0091270007309709. [DOI] [PubMed] [Google Scholar]
- Firbas C, Jilma B, Gilbert JC, Schranz S, Knöbl P. The anti von Willebrand factor aptamer ARC1779 increases platelet counts in a patient with familial thrombotic thrombocytopenic purpura (TTP) J Thromb Haemost. 2009;7(Suppl. 1):OC-WE-011. [Google Scholar]
- Gaussem P, Reny JL, Thalamas C, Chatelain N, Kroumova M, Jude B, et al. The specific thromboxane receptor antagonist S18886: pharmacokinetic and pharmacodynamic studies. J Thromb Haemost. 2005;3:1437–1445. doi: 10.1111/j.1538-7836.2005.01468.x. [DOI] [PubMed] [Google Scholar]
- van Gestel MA, Heemskerk JW, Slaaf DW, Heijnen VV, Reneman RS, oude Egbrink MG. In vivo blockade of platelet ADP receptor P2Y12 reduces embolus and thrombus formation but not thrombus stability. Arterioscler Thromb Vasc Biol. 2003;23:518–523. doi: 10.1161/01.ATV.0000057809.32354.22. [DOI] [PubMed] [Google Scholar]
- van Giezen JJ, Humphries RG. Preclinical and clinical studies with selective reversible direct P2Y12 antagonists. Semin Thromb Hemost. 2005;31:195–204. doi: 10.1055/s-2005-869525. [DOI] [PubMed] [Google Scholar]
- Gilbert JC, DeFeo-Fraulini T, Hutabarat RM, Horvath CJ, Merlino PG, Marsh HN, et al. First-in-human evaluation of anti von Willebrand factor therapeutic aptamer ARC1779 in healthy volunteers. Circulation. 2007;116:2678–2686. doi: 10.1161/CIRCULATIONAHA.107.724864. [DOI] [PubMed] [Google Scholar]
- Goto S, Yamaguchi T, Ikeda Y, Yamaguchi H, Shimizu K, Jensen P. Phase II trial of the novel antiplatelet agent, SCH 530348, in Japanese patients with non-ST segment elevation acute coronary syndromes (NSTE ACS) Eur Heart J. 2008;29(Suppl.) Abstract: P4767. [Google Scholar]
- Greenbaum AB, Grines CL, Bittl JA, Becker RC, Kereiakes DJ, Gilchrist IC, et al. Initial experience with an intravenous P2Y12 platelet receptor antagonist in patients undergoing percutaneous coronary intervention: results from a 2-part, phase II, multicenter, randomized, placebo- and active-controlled trial. Am Heart J. 2006;151:689. e1–689. e10. doi: 10.1016/j.ahj.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Greenbaum AB, Ohman EM, Gibson CM, Borzak S, Stebbins AL, Lu M, et al. Preliminary experience with intravenous P2Y12 platelet receptor inhibition as an adjunct to reduced-dose alteplase during acute myocardial infarction: results of the Safety, Tolerability and Effect on Patency in Acute Myocardial Infarction (STEP-AMI) angiographic trial. Am Heart J. 2007;154:702–709. doi: 10.1016/j.ahj.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Gurbel PA, Conley PB, Andre P, Stephens G, Gretler DD, Jurek MM, et al. Oral Dosing of PRT060128, a Novel Direct-acting, Reversible P2Y12 Antagonist Overcomes High Platelet Reactivity in Patients Non-responsive to Clopidogrel Therapy. Circulation. 2008;118:S_972. [Google Scholar]
- Hennerici MG. Rationale and design of the Prevention of Cerebrovascular and Cardiovascular Events of Ischemic Origin with Terutroban in Patients with a History of Ischemic Stroke or Transient Ischemic Attack (PERFORM) Study. Cerebrovasc Dis. 2009;27(Suppl. 3):28–32. doi: 10.1159/000209263. [DOI] [PubMed] [Google Scholar]
- Holz J, Bartunek J, Barbato E, Vercruysse K, Pullan S, Heyndrickxs G. ALX-0081 a novel anti-thrombotic: first results of a multiple dose phase 1 study in patients with stable angina undergoing PCI. J Thromb Haemost. 2009;(Suppl.) abstract PP-WE-416. [Google Scholar]
- Hong TT, Huang J, Driscoll E, Lucchesi BR. Preclinical evaluation of S18886 in an experimental model of coronary arterial thrombosis. J Cardiovasc Pharmacol. 2006;48:239–248. doi: 10.1097/01.fjc.0000248234.08277.7e. [DOI] [PubMed] [Google Scholar]
- Huang J, Driscoll EM, Gonzales ML, Park AM, Lucchesi BR. Prevention of arterial thrombosis by intravenously administered platelet P2T receptor antagonist AR-C69931MX in a canine model. J Pharmacol Exp Ther. 2000;295:492–499. [PubMed] [Google Scholar]
- Husted S, Emanuelsson H, Heptinstall S, Sandset PM, Wickens M, Peters G. Pharmacodynamics, pharmacokinetics, and safety of the oral reversible P2Y12 antagonist AZD6140 with aspirin in patients with atherosclerosis: a double-blind comparison to clopidogrel with aspirin. Eur Heart J. 2006;27:1038–1047. doi: 10.1093/eurheartj/ehi754. [DOI] [PubMed] [Google Scholar]
- Hutchaleelaha A, Ye C, Lambing JL, Desai M, Gu ZM, Kissling J, et al. Metabolism and excretion of PRT060128, a new direct-acting, reversible P2Y12 inhibitor, in healthy human subjects. AAPS J. 2008 http://www.aapsj.org/abstracts/AM_2008/AAPS2008-001676.PDF. [Google Scholar]
- Ingall AH, Dixon J, Bailey A, Coombs ME, Cox D, McInally JI, et al. Antagonists of the platelet P2T receptor: a novel approach to antithrombotic therapy. J Med Chem. 1999;42:213–220. doi: 10.1021/jm981072s. [DOI] [PubMed] [Google Scholar]
- Jacobsson F, Swahn E, Wallentin L, Ellborg M. Safety profile and tolerability of intravenous AR-C69931MX, a new antiplatelet drug, in unstable angina pectoris and non-Q-wave myocardial infarction. Clin Ther. 2002;24:752–765. doi: 10.1016/s0149-2918(02)85149-9. [DOI] [PubMed] [Google Scholar]
- Jakubowski JA, Matsushima N, Asai F, Naganuma H, Brandt JT, Hirota T, et al. A multiple dose study of prasugrel (CS-747), a novel thienopyridine P2Y12 inhibitor, compared with clopidogrel in healthy humans. Br J Clin Pharmacol. 2007a;63:421–430. doi: 10.1111/j.1365-2125.2006.02792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubowski JA, Winters KJ, Naganuma H, Wallentin L. Prasugrel: a novel thienopyridine antiplatelet agent. A review of preclinical and clinical studies and the mechanistic basis for its distinct antiplatelet profile. Cardiovasc Drug Rev. 2007b;25:357–374. doi: 10.1111/j.1527-3466.2007.00027.x. [DOI] [PubMed] [Google Scholar]
- Jilma B, Jilma P, Gilbert JC, Hutabarat R, Siller J, Spiel A, et al. Safety, pharmacokinetics and pharmacodynamics of the anti von Willebrand factor aptamer ARC1779 in patients with acute thrombotic thrombocytopenic purpura TTP. J Thromb Haemost. 2009;7(Suppl. 1):AS-WE-014. [Google Scholar]
- Judge HM, Buckland RJ, Sugidachi A, Jakubowski JA, Storey RF. The active metabolite of prasugrel effectively blocks the platelet P2Y12 receptor and inhibits procoagulant and pro-inflammatory platelet responses. Platelets. 2008;19:125–133. doi: 10.1080/09537100701694144. [DOI] [PubMed] [Google Scholar]
- Kageyama S, Yamamoto H, Nakazawa H, Matsushita J, Kouyama T, Gonsho A, et al. Pharmacokinetics and pharmacodynamics of AJW200, a humanized monoclonal antibody to von Willebrand factor, in monkeys. Arterioscler Thromb Vasc Biol. 2002;22:187–192. doi: 10.1161/hq0102.101520. [DOI] [PubMed] [Google Scholar]
- Kasoglou T, Reyderman L, Robert R. Pharmacodynamics and pharmacocinetics of a novel protease-activated receptor (PAR-1) antagonist SCH 530348. Circulation. 2005;112:1132. [Google Scholar]
- Knobl P, Jilma B, Gilbert JC, Hutabarat RM, Wagner PG, Jilma-Stohlawetz P. Anti-von Willebrand factor aptamer ARC1779 for refractory thrombotic thrombocytopenic purpura. Transfusion. 2009 doi: 10.1111/j.1537-2995.2009.02232.x. doi: 10.1111/j.1537-2995.2009.02232.x. [DOI] [PubMed] [Google Scholar]
- Kogushi M, Yokohama H, Kitamura S, Hishinuma I. Effects of E5555, a protease-activated receptor-1 antagonist, on the inflammatory markers in vitro. J Thromb Haemost. 2007;5 Abstract: P-M-059. [Google Scholar]
- Li YG, Ni L, Brandt JT, Small DS, Payne CD, Ernest CS, et al. Inhibition of platelet aggregation with prasugrel and clopidogrel: an integrated analysis in 846 subjects. Platelets. 2009;20:316–327. doi: 10.1080/09537100903046317. [DOI] [PubMed] [Google Scholar]
- McNicol A, Israels SJ. Platelets and anti-platelet therapy. J Pharmacol Sci. 2003;93:381–396. doi: 10.1254/jphs.93.381. [DOI] [PubMed] [Google Scholar]
- Michelson AD. New P2Y12 antagonists. Curr Opin Hematol. 2009;16:371–377. doi: 10.1097/MOH.0b013e32832ea2f2. [DOI] [PubMed] [Google Scholar]
- Montalescot G, Wiviott SD, Braunwald E, Murphy SA, Gibson CM, McCabe CH, et al. Prasugrel compared with clopidogrel in patients undergoing percutaneous coronary intervention for ST-elevation myocardial infarction (TRITON-TIMI 38): double-blind, randomised controlled trial. Lancet. 2009;373:723–731. doi: 10.1016/S0140-6736(09)60441-4. [DOI] [PubMed] [Google Scholar]
- Murphy SA, Antman EM, Wiviott SD, Weerakkody G, Morocutti G, Huber K, et al. Reduction in recurrent cardiovascular events with prasugrel compared with clopidogrel in patients with acute coronary syndromes from the TRITON-TIMI 38 trial. Eur Heart J. 2008;29:2473–2479. doi: 10.1093/eurheartj/ehn362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niitsu Y, Jakubowski JA, Sugidachi A, Asai F. Pharmacology of CS-747 (prasugrel, LY640315), a novel, potent antiplatelet agent with in vivo P2Y12 receptor antagonist activity. Semin Thromb Hemost. 2005;31:184–194. doi: 10.1055/s-2005-869524. [DOI] [PubMed] [Google Scholar]
- Paulinska P, Spiel A, Jilma B. Role of von Willebrand factor in vascular disease. Hamostaseologie. 2009;29:32–38. [PubMed] [Google Scholar]
- Payne CD, Li YG, Small DS, Ernest CS, 2nd, Farid NA, Jakubowski JA, et al. Increased active metabolite formation explains the greater platelet inhibition with prasugrel compared to high-dose clopidogrel. J Cardiovasc Pharmacol. 2007;50:555–562. doi: 10.1097/FJC.0b013e3181492209. [DOI] [PubMed] [Google Scholar]
- Pitchford SC. Novel uses for anti-platelet agents as anti-inflammatory drugs. Br J Pharmacol. 2007;152:987–1002. doi: 10.1038/sj.bjp.0707364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post JM, Alexander S, Wang YX, Vincelette J, Vergona R, Kent L, et al. Novel P2Y12 adenosine diphosphate receptor antagonists for inhibition of platelet aggregation (II): pharmacodynamic and pharmacokinetic characterization. Thromb Res. 2008;122:533–540. doi: 10.1016/j.thromres.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Raju NC, Eikelboom JW, Hirsh J. Platelet ADP-receptor antagonists for cardiovascular disease: past, present and future. Nat Clin Pract Cardiovasc Med. 2008;5:766–780. doi: 10.1038/ncpcardio1372. [DOI] [PubMed] [Google Scholar]
- Rehmel JL, Eckstein JA, Farid NA, Heim JB, Kasper SC, Kurihara A, et al. Interactions of two major metabolites of prasugrel, a thienopyridine antiplatelet agent, with the cytochromes P450. Drug Metab Dispos. 2006;34:600–607. doi: 10.1124/dmd.105.007989. [DOI] [PubMed] [Google Scholar]
- Schomig A. Ticagrelor – is there need for a new player in the antiplatelet-therapy field? N Engl J Med. 2009;361:1108–1111. doi: 10.1056/NEJMe0906549. [DOI] [PubMed] [Google Scholar]
- Schranz S, Jilma B, Firbas C, Gilbert JC, Hutabarat R, Knöbl P. Pharmacokinetics and pharmacodynamics of the aptamer ARC1779, a specific inhibitor of von Willebrand factor A1 domains. A pilot study in patients with type 2B von Willebrand disease. J Thromb Haemost. 2009;7(Suppl. 1):PP-MO-622. [Google Scholar]
- Sebekova K, Eifert T, Klassen A, Heidland A, Amann K. Renal effects of S18886 (Terutroban), a TP receptor antagonist, in an experimental model of type 2 diabetes. Diabetes. 2007;56:968–974. doi: 10.2337/db06-1136. [DOI] [PubMed] [Google Scholar]
- Sebekova K, Ramuscak A, Boor P, Heidland A, Amann K. The selective TP receptor antagonist, S18886 (terutroban), attenuates renal damage in the double transgenic rat model of hypertension. Am J Nephrol. 2008;28:47–53. doi: 10.1159/000108760. [DOI] [PubMed] [Google Scholar]
- Serebruany V, Shalito I, Kopyleva O. Prasugrel development – claims and achievements. Thromb Haemost. 2009a;101:14–22. [PubMed] [Google Scholar]
- Serebruany VL, Kogushi M, Dastros-Pitei D, Flather M, Bhatt DL. The in-vitro effects of E5555, a protease-activated receptor (PAR)-1 antagonist, on platelet biomarkers in healthy volunteers and patients with coronary artery disease. Thromb Haemost. 2009b;102:111–119. doi: 10.1160/TH08-12-0805. [DOI] [PubMed] [Google Scholar]
- Shinohara Y, Goto S, Shimizu K, Jensen P, Shinohara Y, Goto S, et al. A phase II safety study of novel antiplatelet agent, SCH 530348, in Japanese patients with prior ischemic stroke. Int J Stroke. 2008;3(Suppl. 1) Abstract PO01–193. [Google Scholar]
- Siller-Matula J, Schror K, Wojta J, Huber K. Thienopyridines in cardiovascular disease: focus on clopidogrel resistance. Thromb Haemost. 2007;97:385–393. [PubMed] [Google Scholar]
- Siller-Matula JM, Lang I, Christ G, Jilma B. Calcium-channel blockers reduce the antiplatelet effect of clopidogrel. J Am Coll Cardiol. 2008;52:1557–1563. doi: 10.1016/j.jacc.2008.07.055. [DOI] [PubMed] [Google Scholar]
- Siller-Matula JM, Gouya G, Wolzt M, Jilma B. Cross validation of the Multiple Electrode Aggregometry. A prospective trial in healthy volunteers. Thromb Haemost. 2009a;102:397–403. doi: 10.1160/TH08-10-0669. [DOI] [PubMed] [Google Scholar]
- Siller-Matula JM, Haberl K, Prillinger K, Panzer S, Lang I, Jilma B. The effect of antiplatelet drugs clopidogrel and aspirin is less immediately after stent implantation. Thromb Res. 2009b;123:874–880. doi: 10.1016/j.thromres.2008.11.005. [DOI] [PubMed] [Google Scholar]
- Siller-Matula JM, Spiel AO, Lang IM, Kreiner G, Christ G, Jilma B. Effects of pantoprazole and esomeprazole on platelet inhibition by clopidogrel. Am Heart J. 2009c;157:148. doi: 10.1016/j.ahj.2008.09.017. e1-5. [DOI] [PubMed] [Google Scholar]
- Spiel AO, Gilbert JC, Jilma B. von Willebrand factor in cardiovascular disease: focus on acute coronary syndromes. Circulation. 2008;117:1449–1459. doi: 10.1161/CIRCULATIONAHA.107.722827. [DOI] [PubMed] [Google Scholar]
- Spiel AO, Mayr FB, Ladani N, Wagner PG, Schaub RG, Gilbert JC, et al. The aptamer ARC1779 is a potent and specific inhibitor of von Willebrand Factor mediated ex vivo platelet function in acute myocardial infarction. Platelets. 2009;20:334–340. doi: 10.1080/09537100903085927. [DOI] [PubMed] [Google Scholar]
- Steinhubl SR, Oh JJ, Oestreich JH, Ferraris S, Charnigo R, Akers WS. Transitioning patients from cangrelor to clopidogrel: pharmacodynamic evidence of a competitive effect. Thromb Res. 2008;121:527–534. doi: 10.1016/j.thromres.2007.05.020. [DOI] [PubMed] [Google Scholar]
- Storey RF, Oldroyd KG, Wilcox RG. Open multicentre study of the P2T receptor antagonist AR-C69931MX assessing safety, tolerability and activity in patients with acute coronary syndromes. Thromb Haemost. 2001;85:401–407. [PubMed] [Google Scholar]
- Storey RF, Wilcox RG, Heptinstall S. Comparison of the pharmacodynamic effects of the platelet ADP receptor antagonists clopidogrel and AR-C69931MX in patients with ischaemic heart disease. Platelets. 2002;13:407–413. doi: 10.1080/0953710021000024402. [DOI] [PubMed] [Google Scholar]
- Storey RF, Husted S, Harrington RA, Heptinstall S, Wilcox RG, Peters G, et al. Inhibition of platelet aggregation by AZD6140, a reversible oral P2Y12 receptor antagonist, compared with clopidogrel in patients with acute coronary syndromes. J Am Coll Cardiol. 2007;50:1852–1856. doi: 10.1016/j.jacc.2007.07.058. [DOI] [PubMed] [Google Scholar]
- Sugidachi A, Asai F, Ogawa T, Inoue T, Koike H. The in vivo pharmacological profile of CS-747, a novel antiplatelet agent with platelet ADP receptor antagonist properties. Br J Pharmacol. 2000;129:1439–1446. doi: 10.1038/sj.bjp.0703237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugidachi A, Ogawa T, Kurihara A, Hagihara K, Jakubowski JA, Hashimoto M, et al. The greater in vivo antiplatelet effects of prasugrel as compared to clopidogrel reflect more efficient generation of its active metabolite with similar antiplatelet activity to that of clopidogrel's active metabolite. J Thromb Haemost. 2007;5:1545–1551. doi: 10.1111/j.1538-7836.2007.02598.x. [DOI] [PubMed] [Google Scholar]
- Tantry US, Bliden KP, Gurbel PA. Azd6140. Expert Opin Investig Drugs. 2007;16:225–229. doi: 10.1517/13543784.16.2.225. [DOI] [PubMed] [Google Scholar]
- Ulrichts H, Schoolmeester A, Hoefman S, Lauwereys M, Casteels P, Stanssens P, et al. Anti-thrombotic drug candidate ALX-0081 shows superior preclinical efficacy and safety compared to current marketed antiplatelet drugs. J Thromb Haemost. 2009;(Suppl.) doi: 10.1182/blood-2010-11-317859. abstract AS-TH-024. [DOI] [PubMed] [Google Scholar]
- Vilahur G, Casani L, Badimon L. A thromboxane A2/prostaglandin H2 receptor antagonist (S18886) shows high antithrombotic efficacy in an experimental model of stent-induced thrombosis. Thromb Haemost. 2007;98:662–669. [PubMed] [Google Scholar]
- Wallentin L, Varenhorst C, James S, Erlinge D, Braun OO, Jakubowski JA, et al. Prasugrel achieves greater and faster P2Y12receptor-mediated platelet inhibition than clopidogrel due to more efficient generation of its active metabolite in aspirin-treated patients with coronary artery disease. Eur Heart J. 2008;29:21–30. doi: 10.1093/eurheartj/ehm545. [DOI] [PubMed] [Google Scholar]
- Wallentin L, Becker RC, Budaj A, Cannon CP, Emanuelsson H, Held C, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361:1045–1057. doi: 10.1056/NEJMoa0904327. [DOI] [PubMed] [Google Scholar]
- Wang K, Zhou X, Zhou Z, Tarakji K, Carneiro M, Penn MS, et al. Blockade of the platelet P2Y12 receptor by AR-C69931MX sustains coronary artery recanalization and improves the myocardial tissue perfusion in a canine thrombosis model. Arterioscler Thromb Vasc Biol. 2003;23:357–362. doi: 10.1161/01.atv.0000052669.50791.0b. [DOI] [PubMed] [Google Scholar]
- Wang YX, Vincelette J, da Cunha V, Martin-McNulty B, Mallari C, Fitch RM, et al. A novel P2Y(12) adenosine diphosphate receptor antagonist that inhibits platelet aggregation and thrombus formation in rat and dog models. Thromb Haemost. 2007;97:847–855. [PubMed] [Google Scholar]
- Wiviott SD, Antman EM, Winters KJ, Weerakkody G, Murphy SA, Behounek BD, et al. Randomized comparison of prasugrel (CS-747, LY640315), a novel thienopyridine P2Y12 antagonist, with clopidogrel in percutaneous coronary intervention: results of the Joint Utilization of Medications to Block Platelets Optimally (JUMBO)-TIMI 26 trial. Circulation. 2005;111:3366–3373. doi: 10.1161/CIRCULATIONAHA.104.502815. [DOI] [PubMed] [Google Scholar]
- Wiviott SD, Braunwald E, McCabe CH, Montalescot G, Ruzyllo W, Gottlieb S, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007a;357:2001–2015. doi: 10.1056/NEJMoa0706482. [DOI] [PubMed] [Google Scholar]
- Wiviott SD, Trenk D, Frelinger AL, O'Donoghue M, Neumann FJ, Michelson AD, et al. Prasugrel compared with high loading- and maintenance-dose clopidogrel in patients with planned percutaneous coronary intervention: the Prasugrel in Comparison to Clopidogrel for Inhibition of Platelet Activation and Aggregation-Thrombolysis in Myocardial Infarction 44 trial. Circulation. 2007b;116:2923–2932. doi: 10.1161/CIRCULATIONAHA.107.740324. [DOI] [PubMed] [Google Scholar]
- Wiviott SD, Braunwald E, Angiolillo DJ, Meisel S, Dalby AJ, Verheugt FW, et al. Greater clinical benefit of more intensive oral antiplatelet therapy with prasugrel in patients with diabetes mellitus in the trial to assess improvement in therapeutic outcomes by optimizing platelet inhibition with prasugrel-Thrombolysis in Myocardial Infarction 38. Circulation. 2008;118:1626–1636. doi: 10.1161/CIRCULATIONAHA.108.791061. [DOI] [PubMed] [Google Scholar]
- Wong PC, Crain EJ, Watson CA, Hua J, Schumacher WA, Rehfuss R. Clopidogrel versus prasugrel in rabbits. Effects on thrombosis, haemostasis, platelet function and response variability. Thromb Haemost. 2009;101:108–115. [PubMed] [Google Scholar]
- Xu S, Jiang B, Maitland KA, Bayat H, Gu J, Nadler JL, et al. The thromboxane receptor antagonist S18886 attenuates renal oxidant stress and proteinuria in diabetic apolipoprotein E-deficient mice. Diabetes. 2006;55:110–119. [PubMed] [Google Scholar]