

Abstract
Background and purpose:
Sphingosine-1-phosphate and its receptors may be involved in vascular smooth muscle cell (VSMC) proliferation following vascular injury. Here, we evaluate the effect of d-erythro-N,N-dimethylsphingosine (DMS), a sphingosine kinase (SK) inhibitor, on VSMC proliferation, apoptosis and neointimal formation.
Experimental approach:
Growth responses in vitro to fetal calf serum (FCS) were measured by [3H]-thymidine incorporation and extracellular signal-regulated kinase-1/2 (ERK-1/2) activation in quiescent primary cultures of porcine VSMC in the presence and absence of various concentrations of the SK inhibitor DMS. In vivo treatment with DMS was delivered with a local endoluminal catheter, following balloon injury of coronary arteries. The artery intimal formation was investigated by angiography, myography and histomorphometry.
Key results:
In vitro experiments indicated that DMS induced a dose-dependent reduction in [3H]-thymidine incorporation and ERK-1/2 activation via a protein kinase C (PKC) independent mechanism with an IC50 value of 12 ± 6 and 15 ± 10 µM respectively. DMS also reduced Akt signalling. Four weeks following in vivo delivery of DMS, complete functional endothelial regeneration was observed in all treatment groups, with significant reduction of intimal formation (vehicle 23.7 ± 4.6% vs. DMS infusion 8.92 ± 2.9%, P < 0.05).
Conclusions and implications:
Taken together, these results demonstrate that local administration of the SK inhibitor, DMS, reduced neointimal formation, and this effect could involve inhibition of ERK-1/2 and Akt signalling, and modulation of smooth muscle growth.
Keywords: anti-proliferative, smooth muscle cells (vascular), intimal thickening, extracellular signal-regulated protein kinase, protein kinase C, restenosis, sphingosine, sphingosine kinase
Introduction
The introduction of drug-eluting stents has greatly reduced the incidence of restenosis and the need for revascularization following percutaneous coronary interventions (Steffel and Tanner, 2007; Windecker and Juni, 2008). Nevertheless, there remain concerns about the long-term safety of drug-eluting stents due to a slight increased risk of late thrombosis (Steffel and Tanner, 2007; Windecker and Juni, 2008). The drug-eluting stents in current use inhibit endothelial regeneration and function, and therefore there remains a need for novel inhibitors of cell proliferation, with improved pharmacological profiles (Steffel and Tanner, 2007).
Several sphingolipids act as important signalling molecules (Hannun and Obeid, 2008). In particular, ceramide has a second messenger role in growth arrest and apoptosis, whereas sphingosine-1-phosphate (S1P) promotes cell survival and cell growth through an intracellular action, and also acts as an agonist at several S1P-specific G protein-coupled receptors (Chun et al., 2002). Ceramide and S1P can be interconverted, via sphingosine, which inhibits protein kinase C (PKC), a cell survival protein, in vitro by competing with its effectors, diacylglycerol and phorbol esters (Hannun and Bell, 1987). Various ceramidase isoforms deacylate ceramide to form sphingosine, which can be phosphorylated to S1P by sphingosine kinase (SK). Conversely, S1P can be dephosphorylated (by S1P phosphatase or lipid phosphate phosphatases), and the resulting sphingosine acylated by ceramide synthase to produce ceramide (Hannun and Obeid, 2008). Indeed, a sphingolipid ‘rheostat’ has been proposed where the relative amounts of ceramide/sphingosine:S1P determine cellular fate (Cuvillier et al., 1996). The therapeutic potential of manipulating this ‘rheostat’ is well recognized (Kolesnick, 2002). In the context of restenosis, Kester and colleagues demonstrated that in vivo application of C6-ceramide on a balloon catheter prevented subsequent neointimal hyperplasia upon injury of rabbit carotid arteries (Charles et al., 2000). An alternative approach is to apply SK inhibitors to shift the ‘rheostat’ towards sphingosine and ceramide, and thereby to promote growth arrest and prevent restenosis.
Two isoforms of SK, termed SK1 and SK2, have been cloned and of which there are N-terminal variants (reviewed by Alemany et al., 2007). N,N-dimethyl-sphingosine (DMS) has been shown to inhibit SK activity in several cell types (Yatomi et al., 1996; Edsall et al., 1998). DMS has also been reported to inhibit PKC (Kim et al., 2005). However, in PC12 cells, DMS did not inhibit PKC activity nor membrane translocation of PKCα or PKCδ even at concentrations higher than those required to inhibit SK1 (Edsall et al., 1998). DMS has also been found to inhibit basic fibroblast growth factor-stimulated proliferation of human coronary and rat arterial smooth muscle cells (SMCs) (Xu et al., 2002), and to inhibit platelet-derived growth factor (PDGF)-stimulated proliferation of mesangial cells (Katsuma et al., 2002). ‘Inside out’ signalling has been reported where S1P formed from SK1-catalysed phosphorylation of sphingosine is released from cells or partitions into the lipid bilayer in close proximity with S1P receptors (Takabe et al., 2008). In this regard, S1P1/S1P3 receptors have been reported to enhance, while S1P2 receptors reduce, vascular smooth muscle proliferation in vitro in response to S1P, or in vivo after vascular injury (Wamhoff et al., 2008). The aim of this study was to evaluate the effect of SK1 inhibition with DMS on intracellular signalling pathways such as extracellular signal-regulated kinase-1/2 (ERK-1/2), DNA synthesis, and SMC remodelling after vascular injury, and to test whether local application of DMS was capable of inhibiting neointimal hypertrophy in vivo.
Methods
Cell culture
Hearts from White/Welsh Landrace pigs were obtained from the local abattoir. The primary cultures of porcine vascular smooth muscle cells (PVSMCs) were obtained by explantation from segments of the left anterior descending (LAD) coronary artery over a 14 day period. These arteries were maintained in a 1:1 (v/v) mixture of Waymouth's medium MB 752/1 and nutrient mixture Ham's F12, containing 1% (v/v) penicillin/streptomycin with 10% (v/v) Australian fetal calf serum (FCS). These PVSMCs were passaged and used between passages 3 and 4 for experiments (Work et al., 2001).
[3H]-thymidine cell proliferation assay
Incorporation of [3H]-thymidine was used to measure DNA synthesis (a marker of proliferation). Cultured cells were made quiescent for 24 h in media containing 0.1% (v/v) (FCS). The quiescent cells were stimulated for 24 h using 10% (v/v) FCS with [3H]-thymidine added for the final 5 h. The sphingolipid derivatives (0.1–100 µM) were added 15 min before serum stimulation, and remained present thereafter. At the completion of serum stimulation, cells were washed with ice-cold phosphate-buffered saline followed by 10% trichloroacetic acid (3 × 10 min at 4°C). Nuclear material was solubilized with lauryl sulphate (10% w/v) plus sodium hydroxide (0.3 M), and radioactivity was quantified by liquid scintillation (Work et al., 2001).
Phosphorylated ERK-1/2 detection
ERK-1/2 activation was measured because of the well-defined role of ERK-1/2 in regulating cyclin D1 activation, cell cycle progression and subsequent mitogenesis of SMC (Sherr, 1996). Sub-confluent layers of vascular PVSMC were made quiescent for 48 h, pretreated with DMS for 10 min, and then stimulated with FCS for 15 min.
The phosphorylated forms of ERK-1/2 were detected by Western blotting of cell lysates with anti-phospho-ERK-1/2 antibody. Anti-ERK-2 and anti-alpha-actin antibodies were used for Western blotting to establish equal loading of protein in each sample and SMC identity. Immunoblotting was performed as described previously (Work et al., 2001).
SK assay
Lysates from HEK 293 cells expressing recombinant SK1 were prepared in SK assay buffer [20 mM Tris (pH 7.4), 1 mM EDTA, 1 mM Na3VO4, 40 mM β-glycerophosphate, 1 mM NaF, 10 µg·mL−1 soya bean trypsin inhibitor, 10 µg·mL−1 aprotinin, 1 mM phenylmethylsulphonyl fluoride, 0.5 mM 4-deoxypyridoxine, 0.007% (v/v) β-mercaptoethanol and 20% (v/v) glycerol. The incubation mixture consisted of 15 µg protein, 50 µM sphingosine, 250 µM ATP and 250 µM phosphatidylserine in 200 µL final volume in the presence of Triton X-100 (0.06%) and [γ-32P] ATP (1 µCi, 44 400 dpm·nmol−1); incubation was for 15 min at 30°C. The resulting [32P] S1P was extracted using n-butanol, which was washed twice using 2 M KCl, before quantification of radioactivity and calculation of SK activity.
RT-PCR
RNA was extracted from PVSMC using Qiagen preparation kit (Crawley, UK). cDNA was synthesized in a 20 µL reaction using 5 µg of RNA template, Superscript II reverse transcriptase (200 U), 1 µL of oligo(dT)12–18 (500 µg·mL−1), 0.5 µL of dNTP Mix (20 mM), 4 µL of 5× first-strand buffer, 2 µL of 0.1 M dithiothreitol and up to 12 µL of distilled water. The mixture was incubated at 42°C for 50 min, and inactivated at 72°C for 15 min. Then, 2 µg of the resulting cDNA was used as a template for amplification by PCR with 1.5 µL of 50 mM MgCl2, 5 µL of 10× PCR buffer [200 mM Tris–HCl (pH 8.4), 500 mM KCl], 0.5 µL of 20 mM dNTP Mix, 2 µL of forward primer (10 µM), 2 µL reverse primer (10 µM) and 0.4 µL of Taq DNA polymerase (5 U·µL−1). Negative controls contained 2 µL of water. The primers used for SK1 were FWD: CTG TCA CCC ATG AAC CTG CTG TC; REV: CAT GGC CAG GAA GAG GCG CAG CA. The PCR parameters were as follows: 25 cycles at 94°C for 1 min, 50°C for 1 min and 60°C for 2 min with a final primer extension at 68°C for 10 min. A single product was obtained, with the predicted size of 477 base pairs.
In vitro balloon injury
Porcine hearts were obtained from a local abattoir, and the LAD coronary artery was dissected from the surrounding fat and connective tissue in a class II culture hood, and cut into 3–4 mm segments that were divided into three groups: group 1, arteries were immediately fixed to determine baseline morphology; group 2, placed in culture medium containing 10% serum (v/v); group 3, balloon injured with a 3.5 mm catheter at 9 atm for 60 s, and placed in a culture medium containing 10% serum (v/v) for a period of 14 days. This culture medium was replaced every 48 h. These artery segments were cultured in the presence and absence of varying concentrations of DMS 0.1–100 µM for 14 days, after which they were fixed in formal saline, embedded in wax and 3 µM sections cut on a microtome for histological analysis (Voisard et al., 1995; Work et al., 2001).
Angioplasty procedure
All animal care and experimental procedures conformed to the requirements of the UK Animal (Scientific Procedures) Act 1986. Male Large White/Welsh Landrace pigs (n = 14; 17–27 kg) were premedicated with aspirin (325 mg orally), sedated with azaperone (5 mg·kg−1 IM) and anaesthetized with a mixture of O2/halothane/nitrous oxide. An arteriotomy was performed on the femoral artery, and a 7F introducer sheath was inserted into the artery to allow a 6F guide catheter to be advanced to the coronary ostium. Immediately before the coronary arterial advancement of the balloon catheter, a 50 mg intravenous dose of the anti-fibrillatory and anti-arrhythmic agent bretylium tosylate was given, and an intracoronary injection of nitroglycerin (50–150 ug) to alleviate post-PTCA vasospasm. A 3.5 mm balloon catheter was then passed along over the guide wire to engage the midpoint between the first and second diagonal branches of the LAD coronary artery. After a coronary angiogram to confirm the position of the balloon catheter, the balloon was inflated three times to 10 atm, providing an approximate balloon-to-artery ratio of 2:1. Immediately after withdrawal of the balloon catheter, a 3.5 mm Dispatch catheter (SCIMED, Boston Scientific, Natick, MA, USA) with a double lumen was advanced to the site of balloon angioplasty to deliver either saline (vehicle: n = 6) or DMS (25 µmol·L−1; n = 6) over a 15 min infusion period at a rate of 200 uL·min−1 (total injected volume: 5 mL containing 25 µM DMS). After removal of all catheters, the femoral artery was permanently ligated, and the wound was sutured. All animals were given antibiotic (ampicillin 50 mg·kg−1) and analgesic (buprenorphine 0.5 mg) cover before recovery from the anaesthetic. Four weeks after surgery, the pigs were given an overdose of sodium pentobarbitone (70 mg·kg−1). A sternotomy was performed to excise the heart, and both the LAD and left circumflex (LCx) coronary arteries were dissected free. The injured section of the LAD and a segment of the LCx of equivalent diameter were each cut into four artery rings. Two of the rings were used immediately for functional studies, with the remaining two rings placed into phosphate-buffered formol saline for subsequent histological analysis (Work et al., 2001).
The concentration of DMS to be used in the Dispatch catheter was determined from the organ and cell culture experiments, as 25 µM caused substantial inhibition of cell proliferation and neointimal formation, and near complete inhibition of ERK phosphorylation. The rather constant inhibition of proliferation across a range of concentrations was advantageous since a relatively wide range of concentrations would have efficacy without toxicity.
Functional analysis
For functional experiments, artery rings were suspended between two parallel stainless steel wires, one fixed and the other connected to an isometric transducer (FT03 Grass Instrument Division, WestRI, USA), in a 10 mL organ bath containing Krebs solution (37°C) of the following composition (mM): NaCl, 118.3; NaHCO3, 25; KCl, 4.7; CaCl2, 2.5; MgSO4, 1.2; KH2PO4, 1.2; and glucose, 11.1. Coronary artery rings were placed under an optimum resting force of 4 g, and were allowed to equilibrate for 1 h. All rings were subjected to repeated exposure to 40 mM KCl to ensure adequate contractility before commencing the experimental protocol. Cumulative dose–response curves were conducted with KCl (5–100 mM). In the vasorelaxant studies, coronary arteries were contracted with the thromboxane mimetic 9,11-dideoxy-1α,9α-epoxymethano-prostaglandin F2α (U46619; 5 × 10−7 M). Once the level of contraction had stabilized, relaxation to cumulative addition of the endothelium-dependent vasodilator, calcimycin (10−9 to 10−5 M) was measured (Work et al., 2001).
Histology
The fixed LAD and LCx artery rings were processed for histological analysis, and embedded in paraffin wax. Serial sections were cut (3 µm) from each artery using a rotary microtome (Leitz GmbH, Wetzler, Germany). These artery segments were stained with: (i) haematoxylin and eosin for morphological analysis; and (ii) alcian blue to determine the presence of proteoglycans (Kennedy et al., 2006).
Measurement of neointimal formation
Neointimal formation was quantified from sections (n = 7–14 per group) stained with haematoxylin and eosin. For each section, the neointimal, luminal surface and external elastic lamina were traced from digital images (Image-Pro Plus version 3, Media Cybernetics, Bethesda, MD, USA), and neointimal and medial areas were calculated by subtraction of the relevant profile area. Neointimal area was expressed as a percentage of total medial and neointimal area.
TUNEL staining
Cell viability was determined by in situ end labelling as described previously (George et al., 1998). Briefly, artery sections were rehydrated and incubated at room temperture for 15 min with 5 µg·mL−1 proteinase K in 1 × TE buffer [10 mM Tris–HCl (pH 8), 1 mM EDTA], and washed twice in 1 × TE. As a positive control, one section from each artery was incubated with DNAse I [1500 U·mL−1 in 1 × TE buffer with 1 mg·mL−1 of bovine serum albumin (BSA)] for 10 min to induce DNA strand breaks. Again, sections were washed in TE buffer and then incubated with labelling mix [50 mM Tris–HCl (pH 7.2), 10 mM MgSO4, 0.1 mM biotin-dUTP and 8 units of DNA polymerase 1 (Klenow, Promega, Southampton, UK)] for 60 min. Sections were rinsed in TE buffer before endogenous peroxidase activity was inhibited in 3% H202 for 5 min. Following further washing, biotin was labelled with extravidin peroxidase (Sigma, Poole, UK; 1 in 200 dilution) in 10% BSA, incubated in diaminobenzidine and subsequently counterstained with haematoxylin.
Statistics
For the in vitro biochemical studies, values shown are the mean ± SEM of n experiments performed on separate cell preparations obtained from different LAD coronary arteries. For the in vivo studies, values shown are the mean ± SEM of n animals. Means were compared by analysis of variance or t-test with P ≤ 0.05 indicating statistical significance.
Materials
Unless otherwise stated, chemicals were purchased from Sigma Chemicals Co. Cell culture materials were purchased from Life Technologies, Inc. (Carlsbad, CA, USA) Anti-phospho-ERK-1/2 and ERK-2 antibodies were from Transduction Laboratories (Lexington, KY, USA). Anti-phospho Akt1 (Thr 308) and anti-Akt1 were from Upstate Cell Signalling Technologies (Charlottesville, VA, USA). Reporter horseradish peroxidase–anti rabbit, mouse and goat antibodies were from the Scottish Antibody Production Unit (Carluke, Scotland). N,N-dimethylsphingosine, d-erthyro-sphingosine, C2-ceramide and C2-dihydroceramide were from Biomol (Exeter, UK). d-erythro-S1P was from TCS Biosciences (Botolph Claydon, Bucks, UK). Bisindolylmaleimide I was from Calbiochem (Beeston, Notts, UK). Halothane was obtained from Mallinckrodt Veterinary (Hazelwood, MO, USA) and Amfipen LA from Mycofarm UK Ltd. (Braintree, Essex, UK) Hexabrix was from May & Baker (Dagenham, UK). Bretylium tosylate MgSO4, nitroglycerin and sterile saline were supplied by Baxter Healthcare Ltd. (Deerfield, IL, USA)
Results
Effect of DMS and bisindolylmaleimide I on cell proliferation and ERK-1/2 activation
DMS induced a concentration-dependent inhibition of FCS-stimulated [3H]-thymidine incorporation into PVSMC with an IC50 of 12 ± 6 µM (Figure 1A). DMS (0.1–100 µM) reduced serum-stimulated ERK-1/2 activation in a concentration-dependent manner with an IC50 of 15 ± 10 µM (Figure 1B), in close agreement with inhibition of DNA synthesis (Figure 1A). Moreover, another SK1 inhibitor, d,l,-threo-dihydrosphingosine (DHS) also inhibited FCS-stimulated ERK-1/2 activation and [3H]-thymidine incorporation with similar sensitivity compared with DMS (Figure 1C). These data suggest a possible role for SK1 in regulating serum-dependent activation of ERK-1/2 and DNA synthesis. DMS also reduced FCS-stimulated Akt (Mr = 65 kDa) phosphorylation (Figure 1D).
Figure 1.
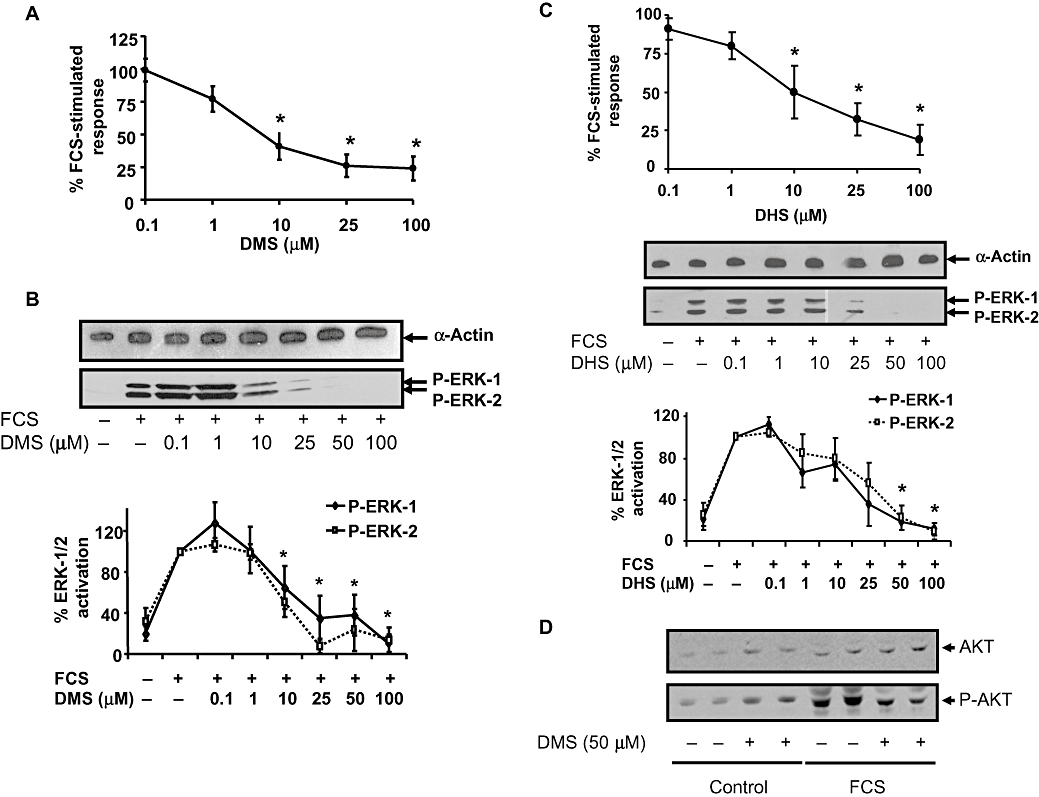
Effect of DMS and DHS on FCS-stimulated DNA synthesis and ERK-1/2 activation. (A) and (C) PVSMC were pretreated with vehicle or (A) DMS (0.1–100 µM) or (C) DMS (0.1–100 µM) for 15 min before FCS stimulation for 24 h. [3H]-thymidine was added for the last 5 h, and its incorporation into DNA quantified. Results are expressed as a percentage of FCS-stimulated vehicle-treated controls. The * indicates P ≤ 0.05 compared to FCS-stimulated control using one-way anova followed by a Bonferroni test, n = 6. (B) and (C) PVSMCs were pretreated with vehicle or (B) DMS (0.1–100 µM) or (C) DHS (0.1–100 µM) for 15 min before FCS stimulation for 10 min. Samples were Western blotted for phospho-ERK-1/2 (P-ERK-1 /2). Blots were stripped and re-probed with anti-α-actin antibody to ensure equal protein loading. Representative Western blots are shown. Blots were quantified by densitometry, and results are presented as % maximal FCS-stimulated phospho-ERK-1/2 (mean ± SEM, n = 5, b and n = 5, c). (D) PVSMCs were pretreated with vehicle or DMS (50 µM) for 15 min before FCS stimulation for 10 min. Samples were Western blotted for phospho-Akt. Blots were stripped and re-probed with anti-Akt antibody to ensure equal protein loading. The Western blot shown is representative of two cell preparations with each treatment performed in duplicate wells.
The effects of DMS observed here are dependent on the expression of SK1 in PVSMC. In this regard, SK1 mRNA was detected in PVSMC by RT-PCR (Figure 2A), and DMS inhibited recombinant SK1 activity in vitro (Figure 2B).
Figure 2.
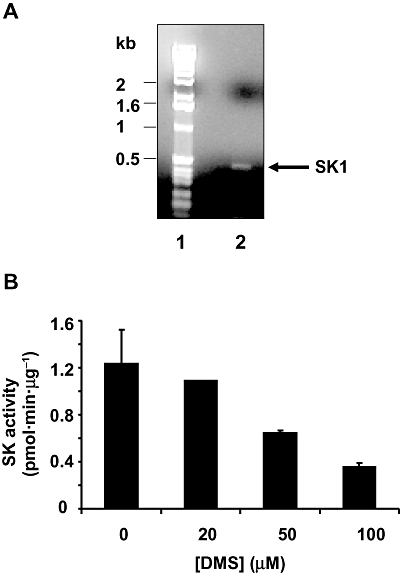
Detection of SK1 and effect of DMS on SK1 activity. (A) RNA was extracted from PVSMC and subjected to RT-PCR with SK1 gene-specific primers. A single amplicon of the expected size (480 bp) was obtained. Lane 1 – size markers; lane 2 – RT-PCR of SK1 amplicon. (B) DMS was confirmed to inhibit recombinant SK1 activity in vitro (expressed as pmol·min·µg−1 protein) in a concentration-dependent manner. Recombinant SK1 activity was assayed in triplicate using cell lysates derived from HEK 293 cells over-expressing hSK1.
We also assessed whether the inhibition of PKC by DMS might account for its effect on the stimulation of ERK-1/2 by FCS. In this regard, the PKC inhibitor, bisindolylmaleimide I failed to inhibit FCS-stimulated activation of ERK-1/2 (Figure 3A and B). These findings contrast with phorbol 12-myristate 13-acetate-stimulated activation of ERK-1/2, which involves PKC and is sensitive to inhibition by bisindolylmaleimide I and DMS (Figure 3A and B). This suggests that DMS does not reduce basal or FCS-stimulated ERK-1/2 signalling via inhibition of typical PKC isoforms. The inhibition of SK1 by DMS may increase intracellular sphingosine (the substrate of SK1) and/or ceramide, which might itself have anti-proliferative activity, rather than this being due to a reduction in S1P formation. To test this possibility, we treated PVSMC with sphingosine or C2-ceramide. Sphingosine inhibited both FCS-stimulated DNA synthesis and ERK-1/2 activation in a concentration-dependent manner (Figure 4A and B) as did C2-ceramide (Figure 4C and D). As a negative control, the inactive sphingolipid, C2-dihydroceramide was without effect on FCS-stimulated DNA synthesis (Figure 4D). We also found that S1P is a rather weak stimulator of ERK-1/2 activation (Figure 4E), suggesting that the involvement of SK1/S1P in ‘inside-out’ signalling is unlikely to contribute significantly to FCS-stimulated activation of ERK-1/2, which is substantial. In addition, S1P was without effect on FCS-stimulated ERK-1/2 activation (Figure 4F).
Figure 3.
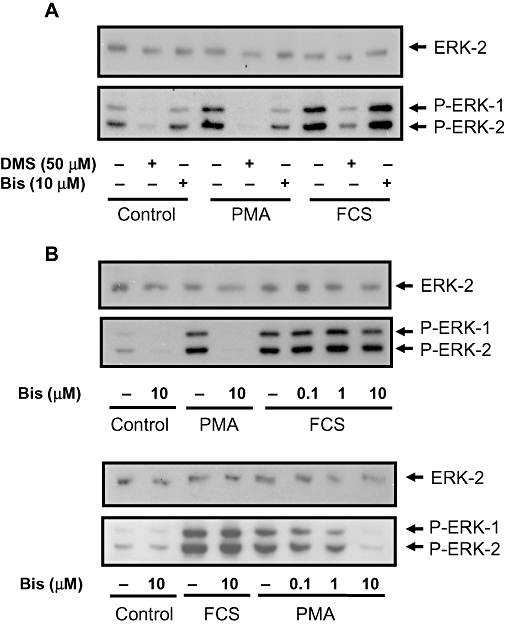
Effect of bisindolylmaleimide I and DMS on FCS- and PMA-stimulated ERK-1/2 activation. PVSMCs were pretreated with and without bisindolylmaleimide I (Bis; 0.1–10 µM or DMS (50 µM for 10 min, and then stimulated with either PMA (1 µM or FCS (10%, v/v) for 10 min. (A) Western blot showing the comparative effect of DMS and bisindolylmaleimide I on PMA- and FCS-stimulated ERK-1/2 activation; (B) Western blots showing the concentration-dependent effect of bisindolylmaleimide I on PMA, but not FCS-stimulated ERK-1/2 activation. Phosphorylated ERK-1/2 (P-ERK-1/2) was detected on Western blots probed with anti-phospho ERK-1/2 antibody. Blots were also probed with anti-ERK-2 antibody to ensure equal protein loading. Representative blots are shown. DMS/PMA (n = 18), DMS/FCS (n = 10), Bis/PMA (n = 14), Bis/FCS (n = 11).
Figure 4.
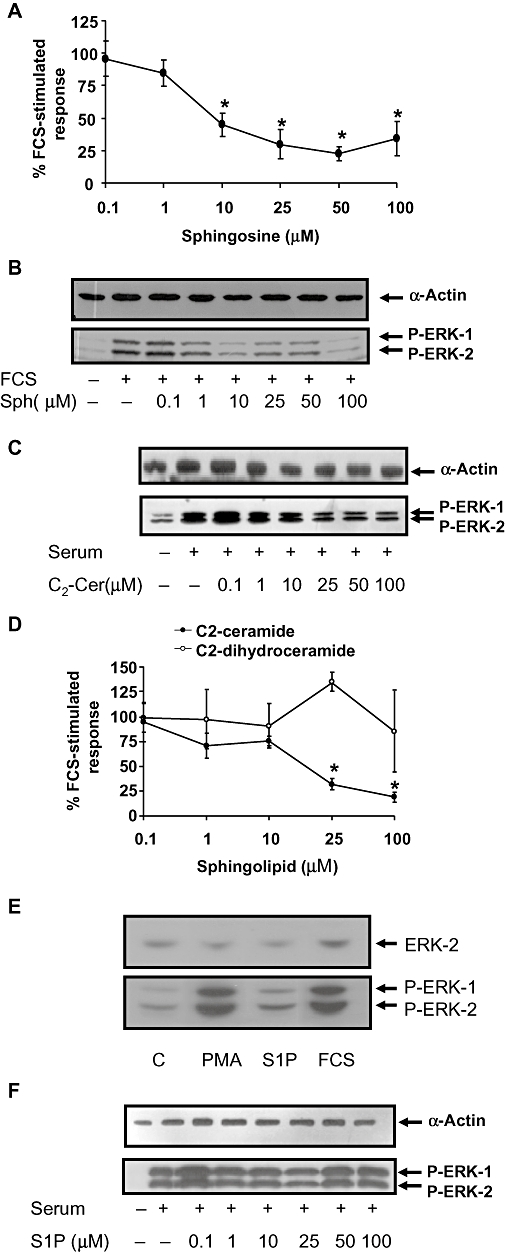
Effect of sphingosine, C2-ceramide, C2-dihydroceramide and S1P on FCS-stimulated DNA synthesis or ERK-1/2 activation. PVSMCs were pretreated with vehicle or (A, B) sphingosine or (C, D) C2-ceramide (0.1–100 µM) or (D) C2-dihydroceramide (0.1–100 µM) or (E) S1P (5 µM) or PMA (1 µM or (F) 0.1–100 µM S1P for 15 min before FCS stimulation for 10 min (ERK-1/2 phosphorylation) or 24 h (DNA synthesis). (A, D) [3H]-thymidine was added for the last 5 h, and its incorporation into DNA was quantified. Results are expressed as a percentage of FCS-stimulated vehicle-treated controls. The * indicates P ≤ 0.05 compared to FCS-stimulated control using one-way anova followed by a Bonferroni test, n = 6. (B, C, E, F) Samples were Western blotted for phospho-ERK-1/2. Blots were stripped and re-probed with anti-α-actin or anti-ERK-2 antibody to ensure equal protein loading. Western blots representative of at least three experiments are shown.
Effect of DMS on in vitro balloon injury
Organ culture of pig coronary artery rings caused a low level of neointimal formation (Figure 5B) compared to non-cultured control arteries (Figure 5A). However, much greater neointimal formation occurred when the internal elastic lamina (IEL) was ruptured by balloon injury (Figure 5C), as described previously (Work et al., 2001). In order to determine if DMS could inhibit PVSMC proliferation and reduce neointimal formation, we incubated balloon-injured arterial segments with DMS (10–100 µM) or vehicle. A significant neointima was formed in response to balloon injury both in the absence and in the presence of vehicle (DMSO) during the 14 day culture period. However, the continued presence of DMS between 10 and 100 µM resulted in a significant attenuation of neointimal formation compared to both control (70–84% inhibition) and vehicle-treated (59–79% inhibition) balloon-injured arteries, although there was no evidence of a concentration-dependent effect over a range of concentrations (10–100 µM) (Figure 5D). To establish whether DMS affects growth or apoptosis, we performed TUNEL staining on isolated injured artery rings incubated with DMS for 14 days in culture. We did not find any apoptosis in response to DMS, while DNAse I treatment of these DMS-treated arteries produced TUNEL staining (Figure 5E).
Figure 5.
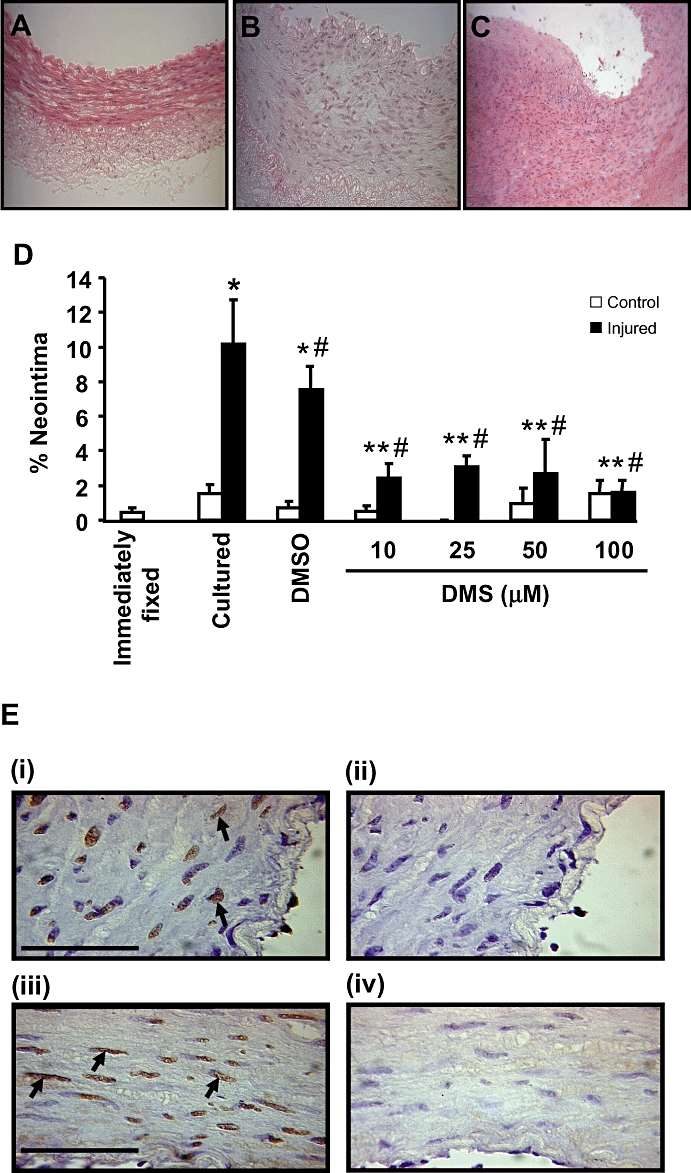
Effect of DMS on neointimal formation in vitro. Haematoxylin and eosin-stained sections showing the extent of neointimal formation in arterial segments that were (A) immediately fixed, (B) control cultured or (C) balloon injured and cultured. (D) Quantification of neointimal formation in balloon-injured LAD coronary arteries cultured in the absence and presence of DMS, 10–100 µM, n = 7–14). Values shown are mean + SEM, which *indicates P ≤ 0.05 compared to control, non injured cultured arteries using a Student's t-test; # indicates P ≤ 0.05 compared to injured cultured LAD coronary arteries using one-way anova followed by Tukey's test; ** indicates P ≤ 0.05 compared to injured, vehicle-treated cultured LAD coronary arteries using one-way anova followed by Tukey's test. (E) The effect of DMS on apoptosis. Apoptosis is marked by TUNEL staining (brown, marked with arrows) and counterstained (blue) with haematoxylin. Panels i and ii are arteries treated with 25 µM DMS. Panels iii and iv are artery sections which have been treated with 50 µM DMS. Panel i and iii have been treated with DNAse I to induce DNA strand breaks. All arterial sections received TUNEL staining. The extent of apoptosis with DNAse I + DMS was not different from DNAse I alone. Bar represents 50 µm.
Effect of DMS on in vivo balloon injury
The non-injured LCx coronary arteries showed normal morphology with occasional small areas of spontaneous neointima (Figure 6B and E). Following angioplasty injury, there was disruption of the IEL with significant neointimal hyperplasia at the site of the IEL break (Figure 6C and F), and neointimal thickening that was usually eccentric. In DMS-infused animals, the non-injured LCx coronary arteries appeared identical to non-injured arteries. DMS infusion (25 µM) markedly reduced cell hyperplasia and neointimal formation (Figure 6A, D and G). Although there was evidence of neointima in DMS-infused animals, this was not significantly different from that found in LCx arteries, which had not received balloon injury. In addition to the reduction in neointimal formation, there was a substantial reduction in the level of proteoglycan deposition at the site of IEL rupture and neointimal formation, which corresponds with the reduction in VSMC deposition at the site of neointimal formation (Figure 6E to G).
Figure 6.
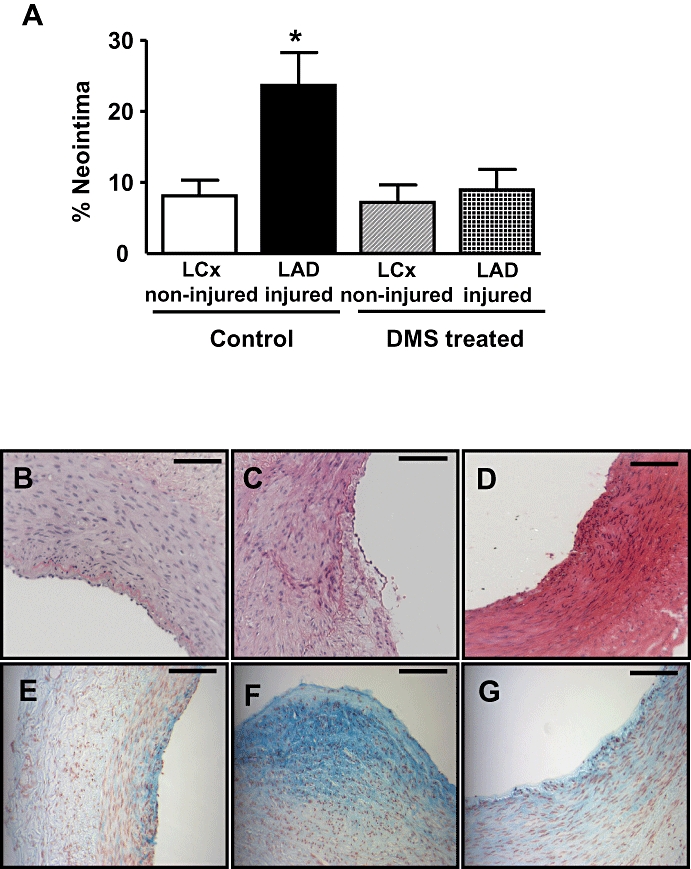
Effect of DMS on neointimal formation in vivo. (A) Neointimal formation was quantified in sections of coronary arteries isolated from non-injured LCx and balloon-injured LAD arteries 28 days after surgery. Neointima was calculated as a percentage of the total neointima and medial area. Local infusion of DMS (5 mL of 25 mM solution, where indicated) following balloon injury significantly reduced neointimal formation. (Values shown are mean + SEM; n = 6–7. *P ≤ 0.05 compared to the other three groups of arteries using one-way anova followed by Tukey's test.) (b–g) Photomicrographs of coronary arteries removed 28 days after angioplasty. Sections B–D were stained with haematoxylin and eosin, and sections E–G were stained with alcian blue. Scale bar represents 100 mm. Panels (B) and (E) represent a small spontaneous lesion in the non-injured LCx (vehicle-infused animals). Panels (C) and (D) represent sections isolated from balloon-injured LAD coronary artery (vehicle-infused animals). Panels (D) and (G) show that neointimal formation was markedly reduced in sections isolated from balloon-injured LAD coronary arteries in DMS-infused animals.
Artery contraction and endothelium-dependent relaxation
Twenty-eight days after balloon injury, DMSO-infused arteries were hyper-responsive to KCl, with significantly enhanced maximum contraction and no change in EC50 (Figure 7A). Similarly, the contractile response to KCl was also increased following injury and endoluminal infusion of DMS (Figure 7B). Endothelium-dependent relaxation was also assessed in arteries 28 days after balloon injury. Following pre-contraction with the thromboxane mimetic U46619, endothelium-dependent relaxation in response to calcimycin was unaffected by vehicle or DMS when compared to that observed in non-injured LCx from the same animals. These results indicate that local infusion of DMS did not inhibit the regeneration of a functionally active endothelial layer 28 days after injury (Figure 7C and D).
Figure 7.
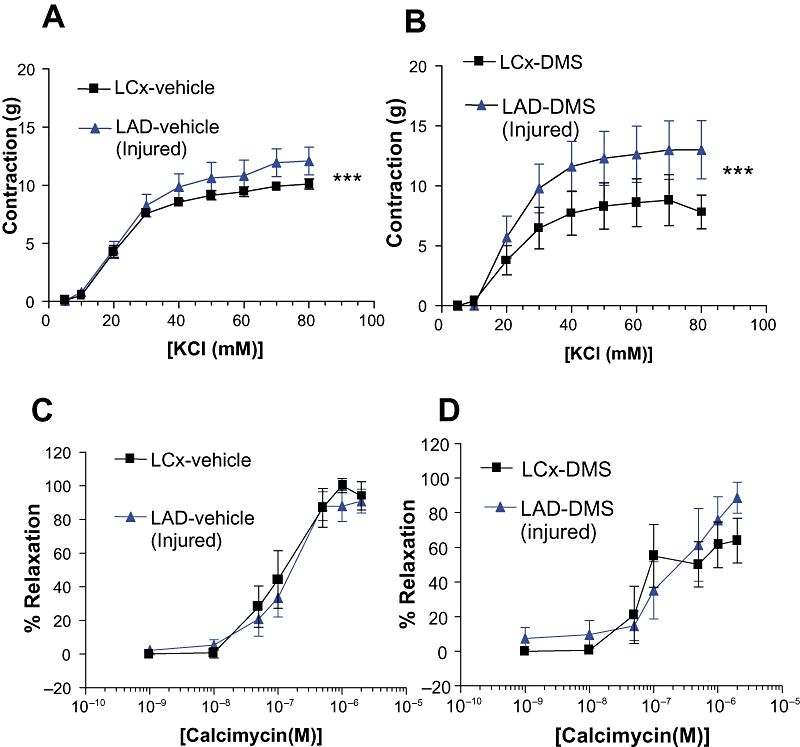
Effect of DMS on vascular function. Isolated rings derived from non-injured or injured coronary arteries 28 days after surgery, and local infusion of vehicle or DMS (5 mL of 25 µM solution, where indicated) were assessed for vascular function. (A) and (B) Contractile responses to KCl (n = 6). (C) and (D) Endothelial-dependent relaxation induced by calcimycin after pre-contraction with U46619 (5 × 10−7 M). n = 6; P < 0.05 vs. control.
Discussion
The in vitro studies suggest that DMS mediated its effects by reducing DNA synthesis and SMC growth rather than inducing apoptosis through inhibition of SK1. Indeed, DMS reduced FCS-stimulated ERK-1/2 activation, an effect which is independent of typical PKC. In addition, a similar inhibition of ERK-1/2 signalling and DNA synthesis was achieved with a different SK1 inhibitor, DHS. DMS also inhibited FCS-stimulated Akt phosphorylation. Akt signalling is involved in regulating both cell growth and survival. We did not detect apoptosis in response to DMS in isolated injured arteries, and therefore inhibition of both ERK-1/2 and Akt signalling by DMS appears linked with inhibition of cell growth.
Our findings are compatible with the rheostat model proposed to explain the differential effects of ceramide, sphingosine and S1P (Pyne and Pyne, 2000; Spiegel and Milstien, 2003). In this regard, ceramide and sphingosine are defined as reducing cell survival, while conversion of sphingosine to S1P by SK1 promotes cell survival and proliferation. Thus, accumulation of sphingosine and ceramide as a consequence of SK1 inhibition by DMS may account for the effect of DMS on ERK-1/2, Akt and DNA synthesis. Indeed, we demonstrate here that C2-ceramide and sphingosine also inhibited FCS-stimulated ERK-1/2 activation and DNA synthesis. DMS-induced perturbation of the ceramide–sphingosine–S1P rheostat, and the subsequent inhibitory action of ceramide/sphingosine on ERK-1/2 and Akt signalling are supported by the findings of Kester et al. These workers have demonstrated that C6-ceramide administration at the site of surgical intervention inhibits neointimal hyperplasia via a mechanism involving block of SMC cell cycle progression and inhibition of ERK-1/2 and Akt signalling (Charles et al., 2000). Further characterization demonstrated that C6-ceramide activated PKCζ, which in turn associates with Akt and inhibits activation of Akt by PDGF, leading to growth arrest of SMC (Bourbon et al., 2002). The effect of ceramide on growth arrest is therefore in line with our findings where we demonstrate that C2-ceramide inhibits DNA synthesis and growth, rather than inducing apoptosis.
The question arises as to whether there would be sufficient sphingosine and ceramide formed after inhibition of SK1. In this respect, there are numerous reports demonstrating that SK1 is localized in different subcellular compartments. Moreover, such compartmentalization of SK1 can be linked with different functions of this enzyme. Compartmentalization of SK1 suggests that it is highly probable that multiple sphingosine pools are accessed by SK1 (Pyne and Pyne, 2008; Pyne et al., 2009). Therefore, only relatively small changes in the amounts of S1P, sphingosine and ceramide in response to DMS might be sufficient to elicit the profound changes in proliferation pathways.
In the current study, 10 µM DMS induced a maximal inhibitory effect on hyperplasia in isolated vessels with little, if any, further inhibition with higher concentrations (Figure 5D). In addition, 10 µM DMS induced about 50% inhibition of FCS-stimulated ERK-1/2 activation (Figure 1B). Our findings with DMS in isolated arteries might therefore suggest that there is a threshold requirement for ERK-1/2 activation in terms of stimulating neointimal formation.
In addition, the sensitivity of arteries to DMS might be defined by mechanisms that do not operate in cultured PVSMC. Wamhoff et al. (2008) have also demonstrated that S1P1/S1P3 receptors enhance vascular smooth muscle proliferation in vitro in response to S1P or in vivo after vascular injury, suggesting a role for ‘inside-out’ signalling by S1P (Takabe et al., 2008). Thus, inhibition of SK1 by DMS might also reduce S1P ‘inside-out signalling’in vivo. Indeed, S1P1 receptor expression is also induced in neointimal lesions of human in-stent restenosis (Zohlnhofer et al., 2001). However, in our in vitro studies using PVSMC, exogenous S1P was a very poor stimulator of ERK-1/2, and is therefore, unlikely to account for the stimulation of ERK-1/2 by FCS. We therefore do not consider a significant role for ‘inside-out’ signalling by SK1 in PVSMC stimulated with FCS. Therefore, the inhibition of FCS stimulation of ERK-1/2 by DMS is unlikely to be due to effects on inside-out signalling by SK1/S1P.
DMS also inhibits a second isoform of SK – SK2. However, SK2 promotes apoptosis, by sequestration of Bcl2 via a BH3 domain in SK2 (Maceyka et al., 2005). Therefore, inhibition of this isoform of SK by DMS might be expected to inhibit apoptosis and increase neointima formation, and which is the opposite of the effects of DMS observed here.
The functional data obtained from coronary arteries 28 days following balloon angioplasty demonstrated that local infusion of DMS had no effect on artery contraction. Contraction to KCl was enhanced by the earlier balloon injury itself, as has been previously observed (Work et al., 2001; Kennedy et al., 2006), but was not further changed by DMS. Endothelial-dependent relaxation was the same in injured arteries as in parallel non-injured arteries, in keeping with the histological findings that the endothelium had re-grown at this time. As DMS did not affect endothelium-dependent relaxation, it may be inferred that DMS had no deleterious actions on endothelium regrowth or function. This is consistent with the results we have obtained following local infusion with other anti-proliferative drugs (Work et al., 2001; Kennedy et al., 2006). This observation is important as S1P is said to be involved in endothelial migration and regeneration (Kimura et al., 2003; Limaye, 2008).
When administered as an intra-arterial irrigation directly after angioplasty injury, 25 µM DMS was remarkably effective at inhibiting neointimal growth. This method of administration involved a brief application of DMS to the artery wall (infusion duration: 15 min), showing that DMS inhibited the first wave of cell proliferation in the immediate aftermath of balloon dilation and that this determined the course of neointimal growth over the subsequent 28 days. The organ culture results showed that even at maximal concentrations of DMS, cell growth was not completely prevented. This is a favourable profile for an anti-restenotic intervention following stent deployment, as it would be expected to permit a minimal amount of cell proliferation leading to satisfactory strut coverage.
In conclusion, the results presented here suggest that an SK1 inhibitor, presumably by causing accumulation of sphingosine and ceramide reduced neointimal hyperplasia both in vitro and possibly in vivo. Therefore, we suggest that DMS represents a class of sphingoid compounds that offer potential as therapeutic agents to alleviate arterial remodelling as a consequence of angioplasty-induced injury.
Acknowledgments
This research was supported by the British Heart Foundation (FS/99071).
Glossary
Abbreviations:
- bFGF
basic fibroblast growth factor
- BSA
bovine serum albumin
- DDT
dithiothreitol
- DHS
d,l-threo-dihydrosphingosine
- DMS
d-erythro-N,N-dimethylsphingosine
- DMSO
dimethyl sulphoxide
- ERK-1/2
extracellular signal-regulated kinase-1/2
- FCS
fetal calf serum
- IEL
internal elastic lamina
- IM ratio
intima/media ratio
- PBS
phosphate-buffered saline
- PDGF
platelet-derived growth factor
- PKC
protein kinase C
- PMA
phorbol 12-myristate 13-acetate
- PMSF
phenylmethylsulphonyl fluoride
- S1P
sphingosine-1-phosphate
- SDS–PAGE
sodium dodecylsulphate–polyacrylamide gel electrophoresis
Conflict of interest
None.
References
- Alemany R, van Koppen CJ, Danneberg K, Ter Braak M, Meyer zu Heringdorf D. Regulation and functional roles of sphingosine kinases. Naunyn Schmiedebergs Arch Pharmacol. 2007;374:413–428. doi: 10.1007/s00210-007-0132-3. [DOI] [PubMed] [Google Scholar]
- Bourbon NA, Sandirasegarane L, Kester M. Ceramide-induced inhibition of Akt is mediated through protein kinase Cζ: implications for growth arrest. J Biol Chem. 2002;277:3286–3292. doi: 10.1074/jbc.M110541200. [DOI] [PubMed] [Google Scholar]
- Charles R, Sandirasegarane L, Yun J, Bourbon N, Wilson R, Rothstein RP, et al. Ceramide-coated balloon catheters limit neointimal hyperplasia after stretch injury in carotid arteries. Circ Res. 2000;87:282–288. doi: 10.1161/01.res.87.4.282. [DOI] [PubMed] [Google Scholar]
- Chun J, Goetzl EG, Hla T, Igarashi Y, Lynch KR, Moolenaar W, et al. IUPHAR XXXIV Lysophospholipid receptor nomenclature. Pharmacol Rev. 2002;54:265–269. doi: 10.1124/pr.54.2.265. [DOI] [PubMed] [Google Scholar]
- Cuvillier O, Pirianov G, Kleuser B, Vanek PG, Coso OA, Gutkind S, et al. Suppression of ceramide-mediated programmed cell death by sphingosine 1-phosphate. Nature. 1996;381:800–803. doi: 10.1038/381800a0. [DOI] [PubMed] [Google Scholar]
- Edsall LC, Van Brocklyn JR, Cuvillier O, Kleuser B, Spiegel S. N,N-dimethylsphingosine is a potent competitive inhibitor of sphingosine kinase but not of protein kinase C: Modulation of cellular levels of sphingosine 1-phosphate and ceramide. Biochemistry. 1998;37:12892–12898. doi: 10.1021/bi980744d. [DOI] [PubMed] [Google Scholar]
- George SJ, Baker AH, Angelini AB, Newby AC. Gene transfer of tissue inhibitor of metalloproteinase 2 inhibits metalloproteinase activity and neointima formation in human saphenous vein. Gene Ther. 1998;5:1552–1560. doi: 10.1038/sj.gt.3300764. [DOI] [PubMed] [Google Scholar]
- Hannun YA, Bell R. Lysosphingolipids inhibit protein kinase C: implications for the sphingolipidoses. Science. 1987;235:670–674. doi: 10.1126/science.3101176. [DOI] [PubMed] [Google Scholar]
- Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- Katsuma S, Hada Y, Ueda T, Shiojima S, Hirasawa A, Tanoue A, et al. Signalling mechanisms in sphingosine 1-phosphate-promoted mesangial cell proliferation. Genes Cells. 2002;7:1217–1230. doi: 10.1046/j.1365-2443.2002.00594.x. [DOI] [PubMed] [Google Scholar]
- Kennedy S, Wadsworth RM, Wainwright CL. Locally administered anti-proliferative drugs inhibit hypercontractility to serotonin in balloon-injured pig coronary artery. Vascul Pharmacol. 2006;44:363–371. doi: 10.1016/j.vph.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Kim JW, Kim YW, Inagaki Y, Hwang YA, Mitsutake S, Ryu YW, et al. Synthesis and evaluation of sphingoid analogs as inhibitors of sphingosine kinases. Bioorg Med Chem. 2005;13:3475–3485. doi: 10.1016/j.bmc.2005.02.053. [DOI] [PubMed] [Google Scholar]
- Kimura T, Sato K, Malchinkhuu E, Tomura H, Tamama K, Kuwabara A, et al. High-density lipoprotein stimulates endothelial cell migration and survival through sphingosine 1-phosphate and its receptors. Arterioscler Thromb Vasc Biol. 2003;23:1283–1288. doi: 10.1161/01.ATV.0000079011.67194.5A. [DOI] [PubMed] [Google Scholar]
- Kolesnick R. The therapeutic potential of modulating the ceramide/sphingomyelin pathway. J Clin Invest. 2002;110:3–8. doi: 10.1172/JCI16127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limaye V. The role of sphingosine kinase and sphingosine-1-phosphate in the regulation of endothelial cell biology. Endothelium. 2008;15:101–112. doi: 10.1080/10623320802125342. [DOI] [PubMed] [Google Scholar]
- Maceyka M, Sankala H, Hait NC, Le Stunff H, Liu H, Toman R, et al. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J Biol Chem. 2005;280:37118–37129. doi: 10.1074/jbc.M502207200. [DOI] [PubMed] [Google Scholar]
- Pyne S, Pyne NJ. Sphingosine 1-phosphate signalling in mammalian cells. Biochem J. 2000;349:385–402. doi: 10.1042/0264-6021:3490385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyne NJ, Pyne S. Sphingosine 1-phosphate, lysophosphatidic acid and growth factor signalling and termination. Biochim Biophys Acta. 2008;1781:467–476. doi: 10.1016/j.bbalip.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Pyne S, Lee SC, Long J, Pyne NJ. Role of sphingosine kinases and lipid phosphate phosphatases in regulating spatial sphingosine 1-phosphate signalling in health and disease. Cell Signal. 2009;21:14–21. doi: 10.1016/j.cellsig.2008.08.008. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003;4:397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- Steffel J, Tanner FC. Biological effects of drug-eluting stents in the coronary circulation. Herz. 2007;32:268–273. doi: 10.1007/s00059-007-3000-5. [DOI] [PubMed] [Google Scholar]
- Takabe K, Paugh SW, Milstien S, Spiegel S. ‘Inside-out’ signalling of sphingosine-1-phosphate: therapeutic targets. Pharmacol Rev. 2008;60:181–195. doi: 10.1124/pr.107.07113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voisard R, Jensch V, Baur R, Hoher M, Hombach V. A coronary porcine organ culture system for studies of post-angioplasty cell proliferation. Coron Artery Dis. 1995;6:657–665. doi: 10.1097/00019501-199508000-00011. [DOI] [PubMed] [Google Scholar]
- Wamhoff BR, Lynch KR, Macdonald TL, Owens GK. Sphingosine-1-phosphate receptor subtypes differentially regulate smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol. 2008;8:1454–1461. doi: 10.1161/ATVBAHA.107.159392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windecker S, Juni P. Safety of drug-eluting stents. Nat Clin Pract Cardiovasc Med. 2008;5:316–328. doi: 10.1038/ncpcardio1189. [DOI] [PubMed] [Google Scholar]
- Work LM, McPhaden AR, Pyne NJ, Pyne S, Wadsworth RM, Wainwright CL. Short-term local delivery of an inhibitor of Ras farnesyltransferase prevents neointima formation in vivo after porcine coronary balloon angioplasty. Circulation. 2001;104:1538–1543. doi: 10.1161/hc3801.095661. [DOI] [PubMed] [Google Scholar]
- Xu CB, Zhang YP, Stenman E, Edvinsson L. d-erythro-N,N-dimethylsphingosine inhibits bFGF-induced proliferation of cerebral, aortic and coronary smooth muscle cells. Atherosclerosis. 2002;164:237–243. doi: 10.1016/s0021-9150(02)00100-4. [DOI] [PubMed] [Google Scholar]
- Yatomi Y, Ruan F, Megidish T, Toyokuni T, Hakomori S, Igarashi Y. N,N-dimethylsphingosine inhibition of sphingosine kinase and sphingosine 1-phosphate activity in human platelets. Biochemistry. 1996;35:626–633. doi: 10.1021/bi9515533. [DOI] [PubMed] [Google Scholar]
- Zohlnhofer D, Richter T, Neumann F, Nuhrenberg T, Wessely R, Brandl R, et al. Transcriptome analysis reveals a role of interferon gamma in human neointima formation. Mol Cell. 2001;7:1059–1069. doi: 10.1016/s1097-2765(01)00239-8. [DOI] [PubMed] [Google Scholar]