

Abstract
Background and purpose:
α7 Nicotinic receptors have been suggested to play an important role in hippocampal learning and memory. However, the direct action of this receptor subtype on hippocampal pyramidal neurones in vivo has not yet been fully investigated. The availability of selective agonists for α7 receptors [AR-R17779 and (R)-(-)-5′-phenylspiro[1-azabicyclo[2.2.2] octane-3,2′-(3′H)furo[2,3-b]pyridine (PSAB-OFP)] has now allowed this role to be investigated.
Experimental approach:
Single-cell extracellular recordings were made from hippocampal CA3 pyramidal neurones in anaesthetized rats. The effects of nicotine, AR-R17779 and PSAB-OFP, applied either systemically or iontophoretically, were studied on the activity of these neurones.
Key results:
Intravenous injection of cumulative doses of nicotine and PSAB-OFP induced dose-related, significant increases in neuronal firing in the majority of neurones tested. This excitation could be inhibited by intravenous administration of methyllycaconitine (MLA), a selective α7 nicotinic receptor antagonist. Furthermore, iontophoretic application of nicotine, AR-R17779 and PSAB-OFP each evoked current-dependent excitation of most CA3 pyramidal neurones studied, and this excitation was antagonized by co-iontophoretic application of MLA. In addition, the excitation induced by iontophoretic application of nicotine, AR-R17779 or PSAB-OFP was also blocked by co-iontophoretic application of either 6,7-dinitroquinoxaline-2,3-dione (DNQX) or D(2)-2-amino-5-phosphonopentanoate (D-AP5), selective N-methyl-D-aspartic acid (NMDA) and non-NMDA receptor antagonists respectively.
Conclusions and implications:
CA3 pyramidal neurones are modulated by activation of presynaptic α7 nicotinic receptors, which, at least in part, enhances glutamate release onto post-synaptic (RS)-α-amino-3-hydroxy-5-methyl-4-isoxazole proprionic acid and NMDA receptors on these CA3 neurones. This mechanism probably contributes to the effects of nicotine on hippocampal learning and memory.
Keywords: presynaptic α7 nAChR, nicotine, AR-R17779, PSAB-OFP, hippocampus, CA3, iontophoresis, rat
Introduction
Nicotinic acetylcholine receptors [nAChRs; see Alexander et al. (2008) for the nomenclature used here] are widely expressed throughout the central nervous system and are known to be involved in various complex cognitive functions such as attention, learning, memory consolidation, arousal and sensory perception (Levin, 1992). The hippocampus, a key structure in learning and memory processes, receives a large cholinergic innervation from the septo-hippocampal pathway (Kasa, 1986). Post-synaptic nAChRs localized mainly on gamma aminobutyric acid (GABA)-ergic interneurones in the hippocampus mediate rapidly desensitizing nicotine currents (Jones and Yakel, 1997; Frazier et al., 1998), while presynaptic nAChRs present on both GABAergic and glutamatergic terminals regulate transmitter release (Radcliffe and Dani, 1998; Alkondon et al., 1999; Alkondon and Albuquerque, 2001; Maggi et al., 2001).
The predominant functional nAChR subtypes in the hippocampus are the homomeric α7 and the heteromeric α4β2 receptors (Alkondon and Albuquerque, 2004). The α7 subtype is distinguished by its high permeability to calcium, its affinity for the antagonists α-bungarotoxin and methyllycaconitine (MLA), and its rapid desensitization (Couturier et al., 1990; Seguela et al., 1993); in the hippocampus, α7 nAChRs are mainly localized on presynaptic nerve terminals to control transmitter release (Gray et al., 1996; Fabian-Fine et al., 2001), on the soma of GABAergic interneurones (Alkondon et al., 1996) or on the dendrites of CA1 pyramidal cells (Ji et al., 2001). Several studies have shown that α7 nAChRs can modulate the release of various neurotransmitters including glutamate, GABA, dopamine and noradrenaline and, thus, have the potential to participate in a range of neurological functions (Alkondon et al., 1997; 1999; Summers et al., 1997; Li et al., 1998; Schilstrom et al., 1998; Maggi et al., 2001). For example, activation of α7 nAChRs on hippocampal glutamatergic neurones is known to enhance the release of glutamate (Gray et al., 1996). Desensitization of α7 nAChRs on GABAergic inhibitory interneurones can cause disinhibition of glutamatergic neurones (Alkondon et al., 2000). Activation of α7 nAChRs by nicotine or by endogenously released acetylcholine has been shown to convert silent synapses into functional ones (Maggi et al., 2003) and to facilitate long-term potentiation (LTP) (Buccafusco et al., 2005). It is also known that activation of α7 nAChRs not only facilitates the induction of LTP in the rat hippocampus (Hunter et al., 1994) but also induces LTP of nicotinic receptors in the mouse dentate gyrus (Matsuyama et al., 2000). Animal behavioural studies have shown that selective activation of the α7 nAChRs improves sensory processing and cognition in animal models (Levin et al., 1999). However, the direct action of nicotinic receptors and particularly of the α7 nAChRs on the hippocampal pyramidal neurones in vivo has not yet been fully investigated.
Here, extracellular recordings from hippocampal CA3 pyramidal neurones obtained from anaesthetized rats were used to study the effects of nicotine and of two selective α7 nAChR agonists, AR-R17779 (Mullen et al., 2000) and PSAB-OFP (Astles et al., 2002; Broad et al., 2002), on the activity of these neurones. We found that both nicotine and the selective α7 nAChR agonists significantly enhanced pyramidal neuronal activity in CA3, an effect that was inhibited by both the selective α7 antagonist MLA and by the selective non-NMDA and NMDA receptor antagonists 6,7-dinitroquinoxaline-2,3-dione (DNQX) and D(2)-2-amino-5-phosphonopentanoate (D-AP5) respectively. This indicates that the excitation of CA3 pyramidal neurones induced by the activation of α7 nAChRs is, at least partially, mediated by presynaptically localized α7 nAChRs that enhance glutamate release.
Methods
Ethical information
All experiments were carried out under the approval of either the local committees of Laboratory Animals, Fudan University, and in accordance with Chinese authority regulation or of the Eli Lilly & Co. Ethics Committee, and in accordance with the Animals (Scientific Procedures) Act, 1986, UK. Stock animals were kept in normal animal house conditions within the animal facility, on a 12 h light–dark schedule. At the end of the experiment, the animals were killed by an overdose of anaesthetic followed by exsanguination.
General preparation
Experiments were carried out on 36 male Sprague Dawley rats (280–340 g), anaesthetized with choral hydrate (400 mg kg−1, i.p.). The level of anaesthesia was assessed by the absence of a withdrawal reflex and of a cardiovascular response to paw pinch and by the stability of resting blood pressure (BP) and heart rate. Additional anaesthetic (chloral hydrate, 100–150 mg kg−1, i.v.) was administered as necessary.
Rectal temperature was monitored and maintained between 37.0 ± 0.5°C with a Harvard homeothermic blanket. When surgical anaesthesia had been established, the femoral artery was cannulated for recording BP using a pressure transducer (Gould Instruments, Oxnard, CA) connected to a Grass Model 7D polygraph (Grass Medical Instruments, Quincy, MA, USA), and a lateral tail vein was cannulated for administration of drugs/fluids. The animals were then placed in a stereotaxic frame. A hole was drilled in the skull above the hippocampus approximately 3.8–4.4 mm caudal to bregma and 3.6–4.2 mm lateral to the midline.
Electrophysiology experiments
Either single or seven-barrelled glass microelectrodes pulled from a starbore glass capillary (Radnoti Glass Technology, Inc, Monrovia, CA, USA), filled with a solution containing 2% pontamine sky blue in 2 M NaCl, and with an in vitro impedance of 4–10 MΩ, were lowered into the brain using a Burleigh 6000ULN Controller (Burleigh Instrument, Burleigh Park, Fishers, NY, USA). A single-unit activity of CA3 neurones was found within the coordinates: 3.9–4.4 mm posterior to bregma, 3.7–4.2 mm lateral to midline and 3.6–4.2 mm below the brain surface. Hippocampal CA3 pyramidal neurones were identified by their characteristic electrophysiological properties, including action potential duration and complex spike discharges consisting of two to five action potentials, as described in detail previously (Frazier et al., 1996). At the end of the experiments, current deposition of pontamine sky blue dye with subsequent histology was used to obtain final confirmation of the location of the recording site, and any cells outside the CA3 pyramidal layer, as marked histologically, were not included in the analysis. An example is shown in Figure 1A,B.
Figure 1.
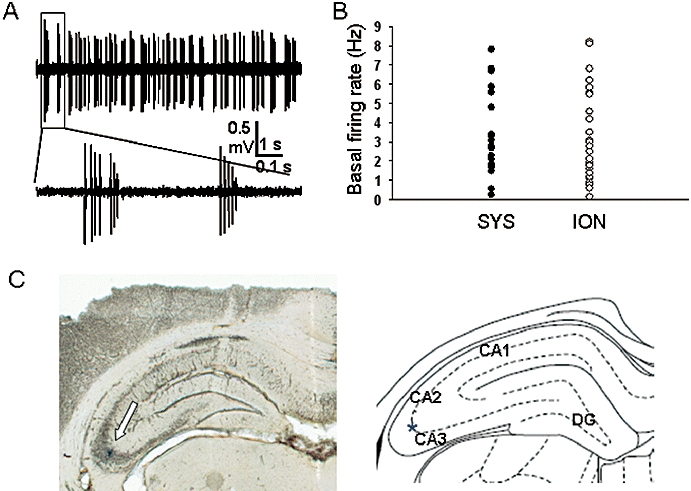
Identification of CA3 pyramidal neurones. (A) A typical CA3 pyramidal neurone recording trace showing the characteristic spontaneous firing pattern including complex spike discharges consisting of multiple action potentials. (B) Scatter plot of the basal firing rate of the neurones in systemic (SYS) study and iontophoretic (ION) study groups respectively. (C) Photograph of a brain section showing a pontamine sky blue-stained recording site within the CA3 pyramidal cell layer indicated by the arrow (and indicated by the star in the right panel diagram; DG, dentate gyrus).
Experimental protocol
Systemic study
Physiological saline of the same volume as that used for drug administration was injected i.v., at least 3–5 min after a stable baseline neuronal response had been recorded. Then, a further 3 min later, cumulative doses of either nicotine or PSAB-OFP were applied i.v. with an interdose interval of 2–3 min. At the end of the cumulative dose regimen, any neuronal response was challenged by an i.v. injection of MLA 3–5 min after the last agonist dose. For the systemic study, recordings were obtained from only one cell in each animal.
Iontophoretic study
Extracellular recordings were made from hippocampal CA3 neurones using seven-barrelled glass microelectrodes (tip diameter 5–7 µm) as described previously (Wang et al., 2006a; Jeggo et al., 2007). The recording barrel contained 2 M sodium chloride, and the other barrels contained pontamine sky blue (2% in 2 M NaCl) for current balancing and dye ejection, and a selection of the following drugs: nicotine (20 mM in 150 mM NaCl, pH 4.5), AR-R17779 (20 mM in 150 mM NaCl, pH 4.5), PSAB-OFP (20 mM in 150 mM NaCl, pH 4.5), MLA (20 mM in 150 mM NaCl, pH 4.5), NMDA (20 mM, in 150 mM NaCl, pH 8.5), (RS)-α-amino-3-hydroxy-5-methyl-4-isoxazole proprionic acid (AMPA) (20 mM, in 150 mM NaCl, pH 8.5), D-AP5 (20 mM, in 150 mM NaCl, pH 8.5) and DNQX (2.5 mM, in 150 mM NaCl, pH 8.5). Drugs were administered in the vicinity of the neurones by iontophoresis (Neurophore, Medical System Inc., Greenvale, NY, USA). Test drugs were ejected using positive currents (with a retaining current of 15–20 nA applied between ejection periods) or negative currents (with a positive retaining current) depending on the drug polarity. Responses were classified as excitation or inhibition if, during the ejection period, activity was increased or decreased by at least 20% of the baseline.
Data capture and analysis
Neuronal activity was amplified ×2000 and filtered (0.3–3 kHz, Dagan Corporation, Minneapolis, MN, USA). Complex spikes of CA3 pyramidal neurones with a consistently similar shape and amplitude (at least twice the amplitude of the basal noise and of other neuronal spikes) were defined as single-unit recordings and were then counted using a window discriminator (Digitimer D130, Digitimer, Welwyn Garden City, UK), with the discrimination level chosen to pick up only the largest spike and the output displayed as a rate histogram. Arterial BP and neuronal activity were displayed on a computer using an AD interface (CED 1401 micro, Cambridge Electronic Design, Cambridge, UK) and Spike2 software (Cambridge Electronic Design), and were stored on the hard disc of a desktop computer and subsequently copied to CDs. Off-line analysis of the recorded data was made using Spike2 software.
Baseline values for neuronal firing were taken as the mean over 3 min before the administration of saline and/or drug and then were standardized to 100%. After drug administration, the firing rate was taken as the mean over 1 min epochs and was then compared to the baseline value and expressed as a per cent of the pre-drug control. Excitation or inhibition was defined as at least a 20% change from the baseline control firing rate.
All data are presented as mean ± standard error of the mean, and all comparisons of the mean were made using one-way analysis of variance (anova) with post hoc Dunnett's test and/or Student's t-test. Differences between means were taken as significant when P < 0.05.
Localization of recording sites
Recording sites were marked by iontophoretic ejection of pontamine sky blue at the end of the electrophysiological recordings. After the rats had been killed, their brains were removed and fixed in 10% formaldehyde saline, and serial frozen sections (50 µm) were cut and photographed to confirm the recording site. The marked recording sites were displayed on standard sections of brain taken from a stereotaxic atlas of the rat brain (Paxinos and Watson, 1986).
Drugs
Drugs were obtained from the following sources: chloral hydrate and NMDA from Sigma-Aldrich Chemical Co. (Poole, Dorset, UK); pontamine sky blue dye from BDH (Poole, Dorset, UK); nicotine tartrate and DNQX from Research Biochemicals (Semat Technical Ltd., St Albans, Hertfordshire, UK); MLA, AMPA and D-AP5 from Tocris Cookson (Bristol, UK); (-)-Spiro[1-azabicyclo[2.2.2] octane-3,5′-oxazolidin-2′-one (AR-R17779; Mullen et al., 2000) and (R)-(-)-5′-phenylspiro[1-azabicyclo[2.2.2] octane-3,2′-(3′H)furo[2,3-b]pyridine (PSAB-OFP of Broad et al., 2002); Compound 35 of Astles et al. (2002) were synthesized at the Lilly Research Centre (Windlesham, Surrey, UK).
For systemic studies, all the drugs were dissolved in 0.9% saline and were injected i.v. in a volume of 1 mL kg−1. The doses for the drugs are all related to the salt form. The solutions for iontophoresis were described in a previous section.
Results
A total of 21 CA3 neurones from 21 rats were tested with systemic administration of drugs, and 31 CA3 neurones from 16 rats were studied with iontophoretic application of drugs. The mean arterial BP during data acquisition for 37 rats was 79 ± 3 mmHg (systolic: 101 ± 4 mmHg and diastolic: 68 ± 3 mmHg). Forty recording sites marked with pontamine sky blue dye at the end of the experiments and recovered after histological processing were within the CA3 pyramidal cell layer. Among these 40 neurones, 21 were from systemic studies and 19 were from iontophoretic studies. The other 12 neurones included in this study from iontophoretic experiments were within 200 µm of one of the marked sites in CA3 and thus were also defined as within CA3. Given the nature of the location, firing pattern and the spike shape of the neurones, we are confident that the majority, if not all, of the neurones analysed here were hippocampal CA3 pyramidal neurones.
The mean spontaneous firing frequency for 21 CA3 pyramidal neurones tested with systemic drug administration was 3.35 ± 0.52 Hz and that for 31 neurones tested with iontophoretic drug application was 3.10 ± 0.40 Hz. There is no significant difference between these two groups in terms of the baseline firing frequency, and hence there is no significant leakage of excitatory agonists from the multibarrel electrodes.
Systemic studies
Effects of systemic injection of nicotine and MLA on hippocampal CA3 pyramidal neurones
The effect of intravenous injection of nicotine on neuronal firing probability was studied first on 11 CA3 pyramidal neurones. Cumulative intravenous doses of nicotine (2–512 µg kg−1) evoked a dose-related increase in spontaneous firing in 6 of the 11 neurones tested (Figure 2A). In this group of six neurones, nicotine increased their firing rate to 199 ± 28% (P < 0.05, n= 6), 208 ± 32% (P < 0.05, n= 6), 251 ± 48% (P < 0.01, n= 6) and 271 ± 50% (P < 0.01, n= 5) of the baseline rate at the cumulative doses of 64, 128, 256 and 512 µg kg−1, respectively, whereas the saline vehicle was without effect (Figure 2B). Of the remaining five neurones, nicotine-evoked inhibition on four and had no effect on the other neurone.
Figure 2.

Effect of cumulative doses of nicotine and subsequent doses of methyllycaconitine (MLA) on the firing rate of CA3 pyramidal neurones. (A) Rate histogram showing that cumulative doses of nicotine (2, 2, 4, 8, 16, 32, 64, 128 and 256 µg kg−1, i.v. at arrows) dose dependently increased neuronal firing on a CA3 pyramidal neurone in a chloral hydrate anaesthetized rat. The firing rate increases evoked by nicotine were reversed by a single dose of MLA (0.25 mg kg−1, i.v.); (a,b) original traces showing the raw recordings of the neuronal activity taken at the times indicated in (A). (B) Group data showing nicotine significantly (*P < 0.05, **P < 0.01) increased the CA3 pyramidal neuronal activity, which was subsequently reversed by MLA (#P < 0.05).
Because α7 nAChRs have been implicated in hippocampal neuronal excitability (see Discussion for references), we examined whether nicotine-induced excitation of CA3 pyramidal neurones was likely to be mediated via α7 nAChRs by using the selective α7 antagonist MLA at a dose of 0.25 mg kg−1 (i.v.); this is a lower dose than we previously found to be effective centrally and selective for α7 nAChRs in vivo (0.3 mg·kg−1; Wang et al. 2006a). At the end of the cumulative dosing with nicotine, the increased neuronal activity was challenged with MLA (0.25 mg kg−1, i.v.). On all five neurones tested with 512 µg kg−1 nicotine, MLA reduced the excitation evoked. Thus, before and after the MLA challenge, the firing rate of CA3 neurones was 271 ± 50% and 172 ± 18% (P < 0.05, n= 5), respectively, of the pre-nicotine baseline value (Figure 2B). This result suggests that nicotine-evoked excitation of CA3 pyramidal neurones is due, at least in part, to the activation of α7 nicotinic receptors.
Effects of systemic administration of the selective α7 agonist PSAB-OFP on hippocampal CA3 pyramidal neurones
Next, the effect of an i.v. injection of the selective α7 agonist PSAB-OFP (Astles et al., 2002; Broad et al., 2002; Wang et al., 2006a;Moore et al., 2008) on CA3 pyramidal neurones was studied. Cumulative doses of PSAB-OFP (4–256 µg kg−1, i.v.) induced a sustained increase in the firing rate in 7 out of 10 CA3 neurones tested, which was dose dependent (Figure 3A). The increased firing rates at cumulative doses of approximately 128 and 256 µg kg−1 (i.v.) were 223 ± 51% (n= 7, P < 0.05) and 252 ± 55% (n= 6, P < 0.01) of the base firing rate, respectively (Figure 3B). The remaining three neurones were inhibited by cumulative doses of PSAB-OFP.
Figure 3.
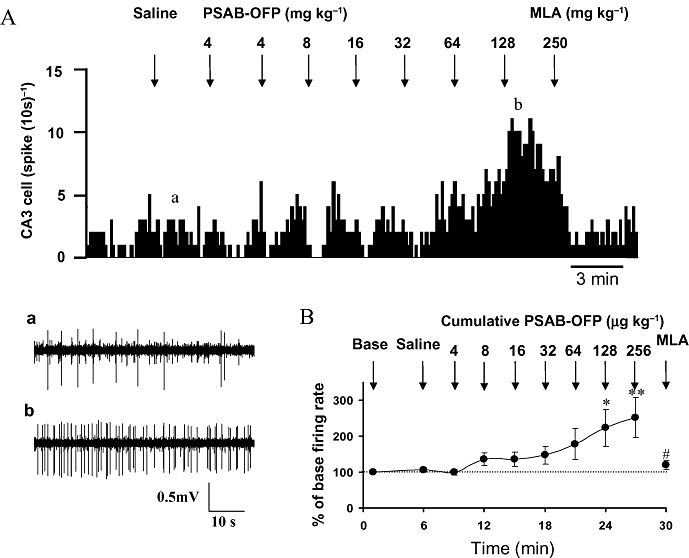
Effect of cumulative doses of (R)-(-)-5′-phenylspiro[1-azabicyclo[2.2.2] octane-3,2′-(3′H)furo[2,3-b]pyridine (PSAB-OFP) and subsequent doses of methyllycaconitine (MLA) on the firing rate of CA3 pyramidal neurones. (A) Rate histogram showing cumulative doses of PSAB-OFP (4, 4, 8, 16, 32, 64 and 128 µg kg−1, i.v. at arrows) dose dependently increased neuronal firing on a CA3 pyramidal neurone in a chloral hydrate anaesthetized rat. The firing rate increases evoked by PSAB-OFP were reversed by a single dose of MLA (0.25 mg kg−1, i.v.); (a,b) original traces showing the raw recordings of the neuronal activity taken at the times before and after PSAB-OFP injection indicated in (A). (B) Group data showing PSAB-OFP significantly (*P < 0.05, **P < 0.01) increased the CA3 pyramidal neurone activity, which was subsequently reversed by MLA (#P < 0.05).
At the end of the cumulative agonist test, an i.v. injection of MLA was applied 3 min after the last dose of PSAB-OFP to verify whether the excitation evoked by PSAB-OFP is mediated by α7 nAChRs. In the five neurones tested, MLA (0.25 mg kg−1, i.v.) significantly reversed PSAB-OFP (cumulative dose of 256 µg kg−1)-evoked excitation from 277 ± 61% to 120 ± 12% (P < 0.05) of the baseline firing rate (Figure 3B). This result indicates that PSAB-OFP-induced excitation of CA3 pyramidal neurones is indeed due to activation of α7 nicotinic receptors.
Iontophoretic studies
To investigate whether the excitation of CA3 pyramidal neurones by systemic nicotine and by PSAB-OFP is due to direct activation of the α7 nAChRs subtype within the CA3 itself, nicotine and two selective α7 receptor agonists, AR-R17779 and PSAB-OFP, were administered into the vicinity of single CA3 neurones by means of microiontophoresis.
Effect of iontophoretic application of nicotine, AR-R17779 and PSAB-OFP on the activity of CA3 pyramidal neurones
Cumulative ejection of nicotine (0–40 nA) was tested on 14 neurones, while 9 neurones showed current-related increases in neuronal firing (Figure 4A). Cumulative doses of nicotine increased the firing rate of CA3 pyramidal neurones to 253 ± 41% (n= 7, P < 0.05) and 421 ± 89% (n= 7, P < 0.01) of baseline firing rate at the ejection current of 20 and 40 nA, respectively (Figure 4B). In the five remaining CA3 neurones, nicotine inhibited three and had no effect on the two.
Figure 4.
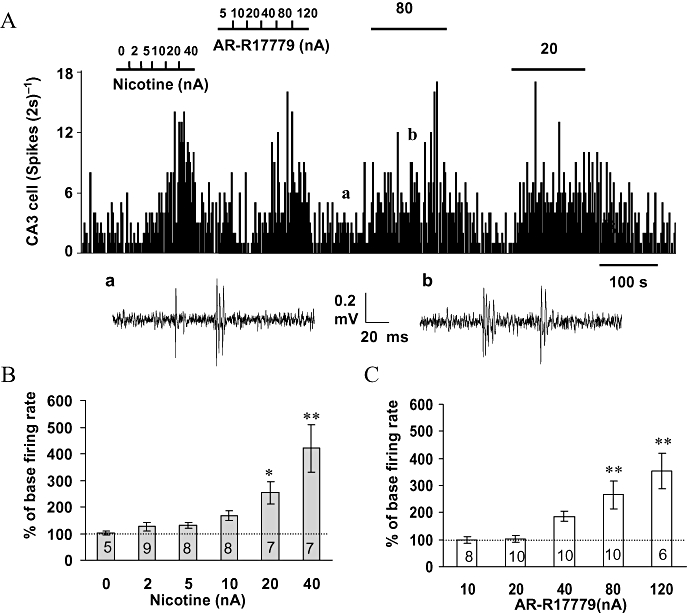
Effects of iontophoretic application of nicotine and AR-R17779 on CA3 pyramidal neurone activity. (A) Rate histogram showing nicotine (0–40 nA) and AR-R17779 (5–120 nA) dose dependently increased the firing rate. The bars above the rate histogram indicate the time when drugs were ejected by iontophoresis; (a,b) original traces showing the raw recordings of the neuronal activity taken at the times before and during AR-R17779 injection indicated in (A). (B) Histogram showing the group data for the excitation of CA3 pyramidal neurones evoked by (a) nicotine (0–40 nA) and (b) AR-R17779 (5–120 nA). *P < 0.05 and **P < 0.01 as compared to the baseline firing rate (n values are indicated in each column).
Similarly, cumulative ejection of AR-R17779 (10–120 nA) evoked excitation in 11 out of the 13 CA3 pyramidal neurones tested (Figure 4A). Of these 11 excited neurones, iontophoretic application of AR-R17779 increased the neuronal firing to 266 ± 52% (n= 10, P < 0.01) and 353 ± 65% (n= 6, P < 0.01) of the baseline firing rate at iontophoretic ejection currents of 80 and 120 nA, respectively (Figure 4C). Iontophoretic application of AR-R17779 had no effect on the remaining two neurones tested. In addition, in all 13 CA3 pyramidal neurones tested, iontophoretic application of PSAB-OFP (20 or 40 nA) increased the neuronal firing rate to 303 ± 59% (P < 0.01) of the baseline firing rate.
Among those neurones excited by iontophoretic application of nicotine, AR-R17779 and PSAB-OFP, four neurones were tested with three agonists, six neurones with nicotine and ARR-1779, four neurones with AR-R17779 and PSAB-OFP, and three neurones with nicotine and PSAB-OFP. All theseneurones gave similar responses, that is, clear excitation, to each of the three agonists (Figures 4A, 5A and 7A).
Figure 5.

Effects of methyllycaconitine (MLA) on the excitation evoked by nicotinic agonists. (A) Ratemeter records of the activity of a CA3 pyramidal neurone during iontophopretic application of nicotine, AR-R17779 and (R)-(-)-5′-phenylspiro[1-azabicyclo[2.2.2] octane-3,2′-(3′H)furo[2,3-b]pyridine (PSAB-OFP) with the stated iontophopretic currents for the times indicated by the bars. (B) Ratemeter records of the excitatory responses evoked by AR-R17779 before, during and after application of selective α7 receptor antagonist MLA with the stated iontophoretic currents for the times indicated by the bars. (C) Histogram showing the group data of the effect of MLA on the baseline firing rate and on the excitation evoked by nicotine, AR-R17779 and PSAB-OFP. *P < 0.05 and **P < 0.01 as compared to the baseline firing rate and #P < 0.05 as compared before and during MLA application.
Figure 7.
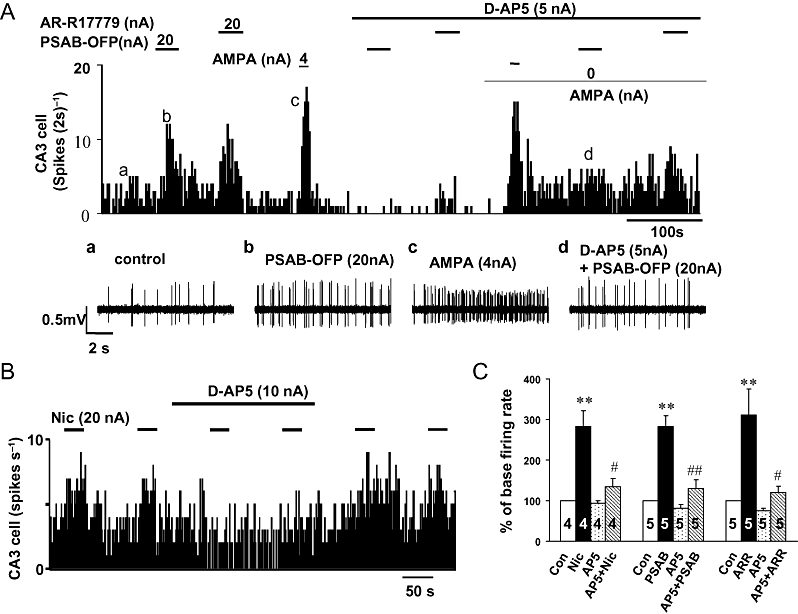
Effects of the selective NMDA receptor antagonist D(2)-2-amino-5-phosphonopentanoate (D-AP5) on responses evoked by nicotinic agonists. Continuous ratemeter records of the activity of two different CA3 pyramidal neurones during application of nicotine, AR-R17779 and (R)-(-)-5′-phenylspiro[1-azabicyclo[2.2.2] octane-3,2′-(3′H)furo[2,3-b]pyridine (PSAB-OFP) in the absence and presence of D-AP5, at the iontophoretic currents stated and for the times indicated by the bars. (A) Excitatory response evoked by application of AR-R17779, PSAB-OFP and (RS)-a-amino-3-hydroxy-5-methyl-4-isoxazole proprionic acid (AMPA) before, during and after application of low currents of the selective NMDA receptor antagonist D-AP5. Insets (a–d) show raw data at times indicated on the ratemeter record above. Note: co-application of D-AP5 attenuated the excitation evoked by AR-R17779 and PSAB-OFP but not that evoked by AMPA, even when the reduction in the baseline firing rate induced by D-AP5 was compensated back to control levels by a low ejecting current of AMPA. (B) Excitatory response, in another neurone, evoked by application of nicotine before, during and after application of low currents of D-AP5. (C) Histogram showing the group data for the effect of D-AP5 on the baseline firing rate and the excitation evoked by nicotine, AR-R17779 and PSAB-OFP. *P < 0.05 and **P < 0.01 as compared to the baseline firing rate and #P < 0.05, ##P < 0.01 as compared before and during D-AP5 application.
Effect of iontophoretic application of MLA on the excitation of CA3 pyramidal neurones evoked by nicotine, AR-R17779 and PSAB-OFP
The excitations of CA3 pyramidal neurones evoked by iontophoretic applications of nicotine, AR-R17779 and PSAB-OFP were challenged by the co-iontophoretic application of the selective α7 receptor antagonist MLA; this antagonist effectively reduced the excitatory responses to each of the three agonists in all the neurones tested (Figure 5B). Co-application of MLA (40–80 nA) attenuated the excitation evoked by nicotine from 317 ± 57% to 179 ± 30% (n= 7, P < 0.05), by AR-R17779 from 279 ± 40% to 138 ± 6% (n= 4, P < 0.05) and by PSAB-OFP from 307 ± 45% to 127 ± 12% (n= 6, P < 0.01) (Figure 5C).
Effect of DNQX and D-AP5 on the excitation of CA3 pyramidal neurones evoked by nicotine, AR-R17779 and PSAB-OFB
Presynaptic localization of α7 nAChRs has been implicated in the facilitatory effects of nicotine on hippocampal synaptic transmission (Gray et al., 1996; Fabian-Fine et al., 2001). To investigate whether the excitation of CA3 pyramidal neurones evoked by nicotine and selective α7 agonists is elicited pre- or post-synaptically, we used two glutamate receptor antagonists to test the hypothesis that the excitation of CA3 neurones following the activation of α7 nicotinic receptors is due to enhanced glutamate release. Firstly, the ejecting currents required to provide full antagonism and selectivity by the non-NMDA receptor antagonist DNQX and by the NMDA receptor antagonist D-AP5 were studied on excitations evoked by NMDA and AMPA. Among the three CA3 pyramidal neurones tested, iontophoretic application of DNQX (10–20 nA) or D-AP5 (5–10 nA) selectively blocked the excitations evoked by AMPA and NMDA respectively. These data (not illustrated) indicate that at this current range in our experimental conditions, DNQX and D-AP5 are selective for AMPA and NMDA receptors respectively.
DNQX, iontophoretically applied, blocked the excitation evoked by nicotine and its analogues in all the neurones tested (Figure 6A,B). A typical example is illustrated in Figure 6B in which iontophoretic application of DNQX (20 nA) attenuated PSAB-OFP (40 nA)-evoked excitations but without changing NMDA (3 nA)-induced excitations of the same neurone. Thus, for the whole group, co-iontophoretic application of DNQX (10–20 nA) attenuated the excitation evoked by nicotine from 282 ± 38% to 148 ± 21% (n= 4, P < 0.05), by PSAB-OFP from 282 ± 27% to 137 ± 21% (n= 4, P < 0.05) and by AR-R17779 from 311 ± 64% to 120 ± 15% (n= 5, P < 0.05) (Figure 6C).
Figure 6.

Effects of the selective non-NMDA receptor antagonist 6,7-dinitroquinoxaline-2,3-dione (DNQX) on the response evoked by nicotinic agonists. Continuous ratemeter records of the activity of two different CA3 pyramidal neurones during the application of nicotine and (R)-(-)-5′-phenylspiro[1-azabicyclo[2.2.2] octane-3,2′-(3′H)furo[2,3-b]pyridine (PSAB-OFP) in the absence and presence of DNQX, at the iontophoretic currents stated and for the times indicated by the bars. (A) Excitatory response evoked by application of nicotine before, during and after application of low currents of the selective non-NMDA receptor antagonist DNQX. Insets (a–c) show raw data at the time indicated in the ratemeter record above. (B) Excitatory response evoked by application of the selective a7 receptor agonist PSAB-OFP and NMDA before, during and after application of low currents of DNQX. Note: DNQX selectively attenuated the excitation evoked by PSAB-OFP but not that evoked by NMDA. (C) Histogram showing the group data of the effect of DNQX on the baseline firing rate and on the excitation evoked by nicotine, AR-R17779 and PSAB-OFP. *P < 0.05 and **P < 0.01 as compared to the baseline firing rate and #P < 0.05 as compared before and during DNQX application.
Iontophoretic co-application of D-AP5, using the predetermined selective currents of 5 or 10 nA significantly reduced excitation of CA3 pyramidal neurones evoked by nicotine, AR-R17779 and PSAB-OFP. A typical example as in Figure 7A demonstrated that iontophoretic application of D-AP5 (5 nA) attenuated the excitation evoked by AR-R17779 (20 nA) or PSAB-OFP (20 nA) but without affecting AMPA (4 nA)-induced excitation of the same neurone. Because ejection of D-AP5 alone caused a decrease in baseline firing rate, the retaining current on the barrel containing AMPA was reduced from 2 to 0 nA in order to compensate for the baseline change; under this condition, D-AP5 still significantly inhibited the excitation evoked by AR-R17779 or PSAB-OFP (Figure 7A). With D-AP5 co-application (5–10 nA), the excitation evoked by nicotine was reduced from 263 ± 28% to 133 ± 9% (n= 4, P < 0.05), by PSAB-OFP from 295 ± 44% to 130 ± 11% (n= 5, P < 0.01) and by AR-R17779 from 256 ± 41% to 120 ± 8% (n= 5, P < 0.05) (Figure 7C). These data demonstrate that activation of α7 nicotinic receptors in the vicinity of the CA3 pyramidal neurones results in excitation mediated by both NMDA and non-NMDA receptors.
Discussion
This study demonstrates that systemic application of nicotine increases the activity of CA3 pyramidal neurones in vivo, and this effect is at least partly due to the activation of α7 nAChRs because the α7 nAChR antagonist MLA reduced the effect of nicotine. The iontophoretic study demonstrated that this excitatory action of nicotine is due to local activation of α7 nicotinic receptors. Furthermore, the results with the glutamate antagonists DNQX and D-AP5 indicate that the excitation is mediated via glutamate receptors, presumably explained by presynaptically located nAChRs on glutamatergic terminals (see below). This is the first time, it has been directly demonstrated, in vivo, that activation of α7 nAChR in the hippocampal CA3 region excites CA3 pyramidal neurones via a presynaptic mechanism involving enhanced release of glutamate.
Compound selectivity
These conclusions, however, depend on the effectiveness and selectivity of the compounds used. The drug doses used in this study were carefully chosen, with reference either to the previously published papers or to the aid of pharmacokinetic studies, so as to be (i) high enough to stimulate the receptors of interest but (ii) low enough not to affect other receptor subtypes. In the systemic study, both nicotine and PSAB-OFP were injected i.v. at similar doses to those used in our previous publication (Wang et al., 2006a). Furthermore, the pharmacokinetic study, also published in that paper, demonstrated that PSAB-OFP given at 256 µg kg−1 (i.v.), which is also the highest dose given in the current study, induced an initial brain concentration of ∼1 µM for the first 15 min, and this declined by 50% at 30 min after drug injection (Wang et al., 2006a). This concentration of PSAB-OFP is selective for the α7 nAChR subtype relative to any other known nAChR subtypes (Astles et al., 2002; Broad et al., 2002). As for the antagonist MLA used in this study, we tried a lower dose than that previously used, which was known to be selective for α7 receptors (Wang et al., 2006a), and demonstrated again the effective attenuation of agonist-evoked excitation on CA3 neurones. Thus, we are confident that both the nicotinic agonists PSAB-OFP and ARR-1779 and the antagonist MLA used in the current systemic study are selective for the α7 nAChR subtype. With regard to the iontophoretic experiments, however, one limitation of the technique is a lack of knowledge of the exact concentration achieved at the receptors under study. For this reason, we performed careful control experiments with both agonists and antagonists. Firstly, for each neurone, we carried out cumulative current ejection experiments for the agonists to determine the lowest but stable effective current and used that current subsequently. Secondly, we tested the current range needed for DNQX and D-AP5 to selectively inhibit excitation evoked by AMPA and NMDA, respectively, and used this information to subsequently challenge the effects of the nicotinic agonists. Thirdly, we used the excitation evoked by either AMPA or NMDA, as appropriate, to serve as a neuronal excitability control during the subsequent experiments that tested the effects of the glutamate antagonists on the excitation evoked by the nicotinic agonists. Under such conditions, we are confident that the doses of the drugs used in this study are selective for the intended receptors.
Desensitization leading to disinhibition as an explanation of the excitatory effects?
The α7 nAChR displays a distinctive range of features including high Ca2+ permeability, high single-channel conductance and rapid desensitization upon the application of agonists (Seguela et al., 1993; Sudweeks and Yakel, 2000). Notably, the latter property of α7 nAChR complicates the explanation of the effects of α7 nAChR agonists because their actions may include both activation and desensitization mechanisms. Activation of α7 nAChRs on hippocampal glutamatergic neurones has been known to enhance the release of glutamate (Gray et al., 1996). Desensitization of tonically active α7 nAChRs on GABAergic inhibitory interneurones could cause disinhibition of glutamatergic neurones (Alkondon et al., 2000). In the present study, the finding that the α7 nAChR-selective antagonist MLA reduced the effect of the α7 nAChR-selective agonists AR-R17779 and PSAB-OFP and of nicotine itself on the activity of pyramidal neurones in the CA3 region suggests that this effect is mediated by the activation of α7 nAChRs rather than by desensitization leading to disinhibition of GABAergic function. MLA applied alone did not mimic the effect of nicotine or of the α7 agonists in the iontophoretic study (Figure 5), further suggesting that desensitization-induced inactivation of α7 nAChRs is not involved in the enhanced activity of pyramidal neurones seen in our study. Although we did not observe any evidence for desensitization, we cannot rule out the possibility of some desensitization of the α7 receptors occurring in our in vivo experiments. Relatively short periods (20–30 s) of agonist ejection may limit desensitization. Alternatively, desensitization may occur very rapidly, and desensitized receptors may be associated with the results from both our iontophoretic and systemic studies.
Inhibitory effects of nicotinic agonists
In addition to the excitation, either nicotine or the α7-selective agonists also induced inhibition in some of the hippocampal CA3 neurones tested. When nicotine or PSAB-OFP were applied systemically, around 36 and 30% of the neurones, respectively, showed inhibition, but in the iontophoretic studies, inhibition was seen in only around 21% of the neurones for nicotine and in none for either of the two α7-selective agonists. The difference between the systemic and iontophoretic effects of the α7 agonists suggests that their inhibitory effects are elicited at some distance from the CA3 neurones and are probably multisynaptic via inhibitory interneurones. The inhibitory effects of nicotine itself can be explained similarly, but, in addition, the local effects may be due to activation of receptor subtypes other than α7. Thus, it is well known that α7 nAChRs in the hippocampus exist not only on glutamatergic nerve terminals regulating transmitter release (Radcliffe and Dani, 1998; Alkondon et al., 1999; Alkondon and Albuquerque, 2001; Maggi et al., 2001; Sharma and Vijayaraghavan, 2003; Sharma et al., 2008) but also on interneurones in the hippocampus (Jones and Yakel, 1997; Frazier et al., 1998). α7 nAChRs are abundantly expressed on interneurones and GABAergic interneurones have been shown to be the major target of cholinergic inputs to the hippocampus (Yoshida and Oka, 1995). Stimulation of glutamatergic terminals on GABAergic interneuronal α7 nAChRs in the hippocampus will produce strong inhibitory currents in pyramidal cells (Fujii et al., 2000; Ji and Dani, 2000), which might be expected to result in a generalized depression of hippocampal activity. Desensitization of α7 nAChRs on interneurones in turn produces disinhibition of pyramidal cells, and activation of α7 nAChRs has also been reported to induce depression of interneurone activity via post-synaptic mechanisms (Wanaverbecq et al., 2007); this could also result from disynaptic inhibitions, which could disinhibit pyramidal neurones. Hence, systemic administration of nicotine can cause either excitation or inhibition, depending on the overall effects in a multisynaptic system. However, in our iontohoretic study, α7 receptor-selective agonists did not induce any inhibition in all the neurones tested, indicating that in the vicinity of the CA3 pyramidal neurones, stimulation of α7 nicotinic receptors causes only excitation.
The inhibition caused by iontophoretic application of nicotine is thus probably due to activation receptors other than α7 receptors. Various nicotinic receptor subtypes are localized in the hippocampus but the predominant functional nAChR subtypes in the hippocampus are the homomeric α7 and the heteromeric α4β2 receptors (Alkondon and Albuquerque, 2004). In our previous study (Wang et al., 2006b), we demonstrated that activation of α4β2 receptors inhibited LTP of the dentate gyrus. However, in the present study, we did not carry out further pharmacological testing of the inhibition of CA3 pyramidal neurones and, hence, to determine the subtype involved will require further study.
Do pre- or post-synaptic α7 nAChRs mediate excitation of CA3 pyramidal neurones?
In view of the above discussion and the abundance of literature supporting the location of α7 nAChRs on glutamatergic terminals (Fabian-Fine et al., 2001), the most parsimonious explanation of the present data, particularly the results obtained with the glutamate receptor antagonists, is that the nicotinic agonists activate these receptors to induce glutamate release, which in turn depolarizes the post-synaptic CA3 pyramidal neurones.
Direct excitation via post-synaptic α7 nAChRs on CA3 pyramidal neurones cannot be discounted but is unlikely to be a major factor because both DNQX and D-AP5 markedly inhibited the excitation induced by the nicotinic agonists. This suggests that both NMDA and AMPA receptors mediate much of the excitatory effect, with direct activation of putative post-synaptic α7 nAChRs having a minimal contribution. Our data do not accord with results from in vitro studies, which have demonstrated that the activation of pre-terminal α7 nAChRs in hippocampal CA3 enhances transmitter release to an extent that is sufficient to drive the post-synaptic pyramidal cell above its firing threshold (Sharma and Vijayaraghavan, 2003; Sharma et al., 2008).
The finding that the sum of the individual reductions of the nicotinic excitation by DNQX and D-AP5 is greater than 100% can be explained by the non-linearity between receptor occupation and the ensuing firing rate. In addition, the inhibition of AMPA receptor-mediated depolarization by DNQX will, because of the voltage sensitivity of the NMDA receptor channel complex, reduce the effect of glutamate released via NMDA receptor-mediated currents.
However, it is still possible that the nicotinic receptors are located on the soma of presynaptic glutamatergic neurones rather than on their terminals. It is generally assumed that the actions of iontophoretically applied drugs are limited to the immediate location of the electrode tip as small movements of the electrode produce dramatic changes in the effects on the neurone under study (personal observations), and hence polysynaptic mechanisms are unlikely. The major glutamatergic inputs to CA3 come from distal parts of the hippocampal formation, for example, the dentate gyrus and the entorhinal cortex. However, as CA3 pyramidal neurones have extensive recurrent collaterals, the iontophoretically applied drug may affect the closely packed cells, but two findings make this an unlikely occurrence. Firstly, the major recurrent collaterals from a given CA3 pyramidal neurone innervate regions of the CA3 more than 100 µm distant (Wittner et al., 2007). Secondly, it would imply considerable heterogeneity within CA3 neurones, some having a much higher concentration of α7 nAChRs, and there is no evidence for this in the literature.
Functional implications of these data
α7 nAChRs are one of the two major types of functional nAChRs in the brain, the other one being α4β2 nAChR. Both of these nAChRs appear to play important roles in cognitive function, particularly with regard to hippocampal involvement in learning and memory. Many studies have reported that nicotine induced facilitation or enhancement of LTP, and synaptic plasticity involves the activation or desensitization of α7 nAChRs or α4β2 nAChRs (Levin, 1992; Fujii et al., 1999; Matsuyama et al., 2000; Rezvani and Levin, 2001; Yamazaki et al., 2005; Wang et al., 2006b; Nakauchi et al., 2007), but the mechanism of this effect is still under discussion. This problem is especially complicated because nAChRs are widely expressed in the mammalian hippocampus. It is known that nAChRs exist pre- and post-synaptically on interneurones and pyramidal neurones in the hippocampus (Alkondon et al., 1996; Gray et al., 1996; Fabian-Fine et al., 2001; Ji et al., 2001). So the effect of nicotine on synaptic plasticity is likely to be an integrated effect. However, our results suggest that the predominant effect of α7 nAChRs in the CA3 region is induced by the activation of presynaptically localized α7 nAChRs that enhance the release of glutamate, which, in turn, excites the CA3 pyramidal neurones by the activation of both post-synaptic NMDA and AMPA receptors. Because CA3 pyramidal cells connect with CA1 pyramidal cells through the Schaffer collateral path, it is possible that the enhancement of the spontaneous activity of CA3 pyramidal neurones, via stimulation of α7 nAChRs, will contribute to the influence of nicotine on CA1 LTP. In addition, CA3 pyramidal neurones receive synaptic projections from the dentate gyrus via the mossy fibre pathway. Thus, the enhancement of the release of glutamate from the presynaptic terminal induced by the activation of α7 receptors in the vicinity of the CA3 pyramidal neurones may facilitate the mossy fibre – CA3 LTP. Similar arguments can be applied to other synaptic inputs; that is, a nicotinic-induced increase in tone could facilitate plasticity.
Nicotinic receptor deficits have been proposed to be a key to ageing (Prendergast et al., 1997; White and Levin, 2004) and to be involved in several cognitive diseases such as Alzheimer's disease (Paterson and Nordberg, 2000) and schizophrenia (Durany et al., 2000; Leonard et al., 2000). Novel nicotinic agonists that are selective for particular nicotinic receptor subtypes may provide more specific therapeutic benefits for treating cognitive dysfunction with fewer adverse side effects. Our results from both the systemic and iontophoretic studies show that the α7 nAChR-selective agonists AR-R17779 and PSAB-OFP mimic the effect of nicotine, which suggests that the α7 nAChR is the major nAChR subtype involved in evoking the excitation of CA3 neurones, which are important components of the hippocampal circuitry involved in learning and memory.
Conclusion
In conclusion, our results demonstrate that nicotine has a predominantly excitatory action on hippocampal CA3 pyramidal neurones, most likely involving the activation of presynaptically localized α7 nicotinic receptors and the enhancement of glutamate release, which may underlie the cellular mechanisms of nicotinic function in hippocampal plasticity.
Acknowledgments
We would like to thank Eli Lilly & Co. Ltd. for supplying AR-R17779 and PSAB-OFP. Part of this study was supported by the grants from the Shanghai Pujiang Program (07PJ14015), STCSM (09JC1401900), The National Basic Research Program of China (973 Program, 2009CB522000) and the National Nature Science Foundation of China (30770672, 30828014) to YW.
Glossary
Abbreviations:
- AMPA
(RS)-α-amino-3-hydroxy-5-methyl-4-isoxazole proprionic acid
- D-AP5
D(2)-2-amino-5-phosphonopentanoate
- DNQX
6,7-dinitroquinoxaline-2,3-dione
- LTP
long-term potentiation
- MLA
methyllycaconitine
- nAChR
nicotinic acetylcholine receptor
- NMDA
N-methyl-D-aspartic acid
- PSAB-OFP
(R)-(-)-5′-phenylspiro[1-azabicyclo[2.2.2] octane-3,2′-(3′H)furo[2,3-b]pyridine
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon M, Albuquerque EX. Nicotinic acetylcholine receptor alpha 7 and alpha4beta2 subtypes differentially control GABAergic input to CA1 neurons in rat hippocampus. J Neurophysiol. 2001;86:3043–3055. doi: 10.1152/jn.2001.86.6.3043. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Albuquerque EX. The nicotinic acetylcholine receptor subtypes and their function in the hippocampus and cerebral cortex. Prog Brain Res. 2004;145:109–120. doi: 10.1016/S0079-6123(03)45007-3. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Albuquerque EX. Mapping the location of functional nicotinic and gamma-aminobutyric acidA receptors on hippocampal neurones. JPET. 1996;279:1491–1506. [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Barbosa CT, Albuquerque EX. Neuronal nicotinic acetylcholine receptor activation modulates gammaaminobutyric acid release from CA1 neurons of rat hippocampal slices. JPET. 1997;283:1396–1411. [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Eisenberg HM, Albuquerque EX. Choline and selective antagonists identify two subtypes of nicotinic acetylcholine receptors that modulate GABA release from CA1 interneurons in rat hippocampal slices. J Neurosci. 1999;19:2693–2705. doi: 10.1523/JNEUROSCI.19-07-02693.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon M, Pereira EFR, Almedia LEF, Randall WRR, Albuquerque EX. Nicotine at concentrations found in cigarette smokers activates and desensitizes nicotinic acetylcholine receptors in CA1 interneurons of rat hippocampus. Neuropharmacology. 2000;39:2726–2739. doi: 10.1016/s0028-3908(00)00156-8. [DOI] [PubMed] [Google Scholar]
- Astles PC, Baker SR, Boot JR, Broad LM, Dell CP, Keenan M. Recent progress in the development of subtype selective nicotinic acetylcholine receptor ligands. Curr Drug Targets CNS Neurol Disord. 2002;1:337–348. doi: 10.2174/1568007023339256. [DOI] [PubMed] [Google Scholar]
- Buccafusco JJ, Letchworth SR, Bencherif M, Lippiello PM. Long-lasting cognitive improvement with nicotinic receptor agonists: mechanisms of pharmacokinetic-pharmacodynamic discordance. TIPS. 2005;26:352–360. doi: 10.1016/j.tips.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Broad LM, Felthouse C, Zwart R, Mcphie GI, Pearson KH, Craig PJ, et al. PSAB-OFP, a selective α7 nicotinic receptor agonist, is also a potent agonist of the 5-HT3 receptor. Eur J Pharmacol. 2002;452:137–144. doi: 10.1016/s0014-2999(02)02273-2. [DOI] [PubMed] [Google Scholar]
- Couturier S, Bertrand D, Matter JM, Hernandez MC, Bertrand S, Millar N, et al. A neuronal nicotinic acetylcholine receptor subunit (alpha 7) is developmentally regulated and forms a homo-oligomeric channel blocked by alpha-BTX. Neuron. 1990;5:847–856. doi: 10.1016/0896-6273(90)90344-f. [DOI] [PubMed] [Google Scholar]
- Durany N, Zochling R, Boissl KW, Paulus W, Ransmayr G, Tatschner T, et al. Human post-mortem striatal alpha4beta2 nicotinic acetylcholine receptor density in schizophrenia and Parkinson's syndrome. Neurosci Lett. 2000;287:109–112. doi: 10.1016/s0304-3940(00)01144-7. [DOI] [PubMed] [Google Scholar]
- Fabian-Fine R, Skehel P, Errington ML, Davies HA, Sher E, Stewart MG, et al. Ultrastructural distribution of the alpha7 nicotinic acetylcholine receptor subunit in rat hippocampus. J Neurosci. 2001;21:7993–8003. doi: 10.1523/JNEUROSCI.21-20-07993.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier CJ, Rollins YD, Hall ME, Young DA, Rose GM. Cholinergic deafferentation enhances rat hippocampal pyramidal neuron responsiveness to locally applied nicotine. Brain Res. 1996;727:217–220. doi: 10.1016/0006-8993(96)00402-7. [DOI] [PubMed] [Google Scholar]
- Frazier CJ, Rollins YD, Breese CR, Leonard S, Freedman R, Dunwiddie TV. Acetylcholine activates an alpha-bungarotoxin-sensitive nicotinic current in rat hippocampal interneurons, but not pyramidal cells. J Neurosci. 1998;18:1187–1195. doi: 10.1523/JNEUROSCI.18-04-01187.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S, Ji Z, Morita N, Sumikawa K. Acute and chronic nicotine exposure differentially facilitate the induction of LTP. Brain Res. 1999;846:137–143. doi: 10.1016/s0006-8993(99)01982-4. [DOI] [PubMed] [Google Scholar]
- Fujii S, Jia YS, Yan AZ, Sumikawa K. Nicotine reverses GABAergic inhibition of long-term potentiation induction in the hippocampal CA1 region. Brain Res. 2000;863:259–265. doi: 10.1016/s0006-8993(00)02119-3. [DOI] [PubMed] [Google Scholar]
- Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- Hunter BE, de Fiebre CM, Papke R, Kem WR, Meyer EM. A novel nicotinic agonist facilitates induction of long-term potentiation in the rat hippocampus. Neurosci Lett. 1994;168:130–134. doi: 10.1016/0304-3940(94)90433-2. [DOI] [PubMed] [Google Scholar]
- Jeggo RD, Wang Y, Jordan D, Ramage AG. Activation of 5-HT1B and 5-HT1D receptors in the rat nucleus tractus solitarius: opposing action on neurons that receive an excitatory vagal C-fibre afferent input. Br J Pharmacol. 2007;150:987–995. doi: 10.1038/sj.bjp.0707169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji K, Dani JA. Inhibition and disinhibition of pyramidal neurons by activation of nicotinic receptors on hippocampal interneurons. J Neurophysiol. 2000;83:2682–2690. doi: 10.1152/jn.2000.83.5.2682. [DOI] [PubMed] [Google Scholar]
- Ji D, Lape R, Dani JA. Timing and location of nicotinic activity enhances or depresses hippocampal synaptic plasticity. Neuron. 2001;31:131–141. doi: 10.1016/s0896-6273(01)00332-4. [DOI] [PubMed] [Google Scholar]
- Jones S, Yakel JL. Functional nicotinic Ach. receptors on interneurones in the rat hippocampus. J Physiol. 1997;504:603–610. doi: 10.1111/j.1469-7793.1997.603bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasa P. The cholinergic systems in brain and spinal cord. Prog Neurobiol. 1986;26:211–272. doi: 10.1016/0301-0082(86)90016-x. [DOI] [PubMed] [Google Scholar]
- Leonard S, Breese C, Adams C, Benhammou K, Gault J, Stevens K, et al. Smoking and schizophrenia: abnormal nicotinic receptor expression. Eur J Pharmacol. 2000;393:237–242. doi: 10.1016/s0014-2999(00)00035-2. [DOI] [PubMed] [Google Scholar]
- Levin ED. Nicotinic systems and cognitive function. Psychopharmacology. 1992;108:417–431. doi: 10.1007/BF02247415. [DOI] [PubMed] [Google Scholar]
- Levin ED, Bettegowda C, Blosser J, Gordon J. AR-R17779, an α7 nicotinic agonist, improves learning and memory in rats. Behav Pharmacol. 1999;10:675–680. doi: 10.1097/00008877-199911000-00014. [DOI] [PubMed] [Google Scholar]
- Li X, Rainnie DG, McCarley RW, Greene RW. Presynaptic nicotinic receptors facilitate monoaminergic transmission. J Neurosci. 1998;18:1904–1912. doi: 10.1523/JNEUROSCI.18-05-01904.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggi L, Sher E, Cherubini E. Regulation of GABA release by nicotinic acetylcholine receptors in the neonatal rat hippocampus. J Physiol. 2001;536:89–100. doi: 10.1111/j.1469-7793.2001.00089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggi L, Le Magueresse C, Changeux JP, Cherubini E. Nicotine activates immature ‘silent’ connections in the developing hippocampus. Proc Natl Acad Sci U S A. 2003;100:2059–2064. doi: 10.1073/pnas.0437947100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S, Matsumoto A, Enomoto T, Nishizaki T. Activation of nicotinic acetylcholine receptors induces long-term potentiation in vivo in the intact mouse dentate gyrus. Eur J Neurosci. 2000;12:3741–3747. doi: 10.1046/j.1460-9568.2000.00259.x. [DOI] [PubMed] [Google Scholar]
- Moore C, Wang Y, Ramage AG. Cardiovascular effects of activation of central α7 and α4β2 nAChRs: a role for vasopressin in anaesthetized rats. Br J Pharmacol. 2008;153:1728–1738. doi: 10.1038/bjp.2008.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen G, Napier J, Balestra M, DeCory T, Hale G, Macor J, et al. Spiro[1-azabicyclo[2.2.2]octane-3,5′-oxazolidin-2′-one], a conformationally restricted analogue of acetylcholine, is a highly selective full agonist at the alpha 7 nicotinic acetylcholine receptor. J Med Chem. 2000;43:4045–4050. doi: 10.1021/jm000249r. [DOI] [PubMed] [Google Scholar]
- Nakauchi S, Brennan RJ, Boulter J, Sumikawa K. Nicotine gates long-term potentiation in the hippocampal CA1 region via the activation of α2* nicotinic ACh receptors. Eur J Neurosci. 2007;25:2666–2681. doi: 10.1111/j.1460-9568.2007.05513.x. [DOI] [PubMed] [Google Scholar]
- Paterson D, Nordberg A. Neuronal nicotinic receptors in the human brain. Prog Neurobiol. 2000;61:75–111. doi: 10.1016/s0301-0082(99)00045-3. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 2nd. San Diego, CA: Academic Press Inc; 1986. [Google Scholar]
- Prendergast MA, Terry AV, Jr, Jackson WJ, Marsh KC, Decker MW, Arneric SP, et al. Improvement in accuracy of delayed recall in aged and non-aged mature monkeys after intramuscular or transdermal administration of the CNS nicotinic receptor agonist abt-418. Psychopharmacology. 1997;130:276–284. doi: 10.1007/s002130050240. [DOI] [PubMed] [Google Scholar]
- Radcliffe KA, Dani JA. Nicotinic stimulation produces multiple forms of increased glutamatergic synaptic transmission. J Neurosci. 1998;18:7075–7083. doi: 10.1523/JNEUROSCI.18-18-07075.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezvani AH, Levin ED. Cognitive effects of nicotine. Biol Psychiatry. 2001;49:258–267. doi: 10.1016/s0006-3223(00)01094-5. [DOI] [PubMed] [Google Scholar]
- Schilstrom B, Svensson HM, Svensson TH, Nomikos GG. Nicotine and food induced dopamine release in the nucleus accumbens of the rat: putative role of alpha7 nicotinic receptors in the ventral tegmental area. Neuroscience. 1998;85:1005–1009. doi: 10.1016/s0306-4522(98)00114-6. [DOI] [PubMed] [Google Scholar]
- Seguela P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain α7: a nicotinic cation channel highly permeable to calcium. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G, Vijayaraghavan S. Modulation of presynaptic store calcium induces release of glutamate and postsynaptic firing. Neuron. 2003;38:929–939. doi: 10.1016/s0896-6273(03)00322-2. [DOI] [PubMed] [Google Scholar]
- Sharma G, Grybko M, Vijayaraghavan S. Action potential-independent and nicotinic receptor-mediated concerted release of multiple quanta at hippocampal CA3–mossy fiber synapses. J Neurosci. 2008;28:2563–2575. doi: 10.1523/JNEUROSCI.5407-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudweeks SN, Yakel JL. Functional and molecular characterization of neuronal nicotinic ACh receptors in rat CA1 hippocampal neurons. J Physiol. 2000;527:515–528. doi: 10.1111/j.1469-7793.2000.00515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers KL, Kem WR, Giacobini E. Nicotinic agonist modulation of neurotransmitter levels in the rat frontoparietal cortex. Jpn J Pharmacol. 1997;74:139–146. doi: 10.1254/jjp.74.139. [DOI] [PubMed] [Google Scholar]
- Wanaverbecq N, Semyanov A, Pavlov I, Walker MC, Kullmann DM. Cholinergic axons modulate GABAergic signaling among hippocampal interneurons via postsynaptic α7 nicotinic receptors. J Neurosci. 2007;27:5683–5693. doi: 10.1523/JNEUROSCI.1732-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Sherwood JL, Mile CP, Whiffin G, Lodge D. TC-2559 excites dopaminergic neurones in the ventral tegmental area by stimulating alpha4beta2-like nicotinic acetylcholine receptors in anaesthetised rats. Br J Pharmacol. 2006a;147:379–390. doi: 10.1038/sj.bjp.0706621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Sherwood JL, Lodge D. The α4β2 nicotinic acetylcholine receptor agonist TC-2559 impairs long-term potentiation in the dentate gyrus in vivo. Neurosci Lett. 2006b;406:183–188. doi: 10.1016/j.neulet.2006.06.075. [DOI] [PubMed] [Google Scholar]
- White HK, Levin ED. Chronic transdermal nicotine patch treatment effects on cognitive performance in age-associated memory impairment. Psychopharmacology. 2004;171:465–471. doi: 10.1007/s00213-003-1614-8. [DOI] [PubMed] [Google Scholar]
- Wittner L, Henze DA, Záborszky L, Buzsáki G. Three-dimensional reconstruction of the axon arbor of a CA3 pyramidal cell recorded and filled in vivo. Brain Struct Funct. 2007;21:75–83. doi: 10.1007/s00429-007-0148-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y, Jia Y, Hamaue N, Sumikawa K. Nicotine-induced switch in the nicotinic cholinergic mechanisms of facilitation of long-term potentiation induction. Eur J Neurosci. 2005;22:845–860. doi: 10.1111/j.1460-9568.2005.04259.x. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Oka H. Topographical projections from the medial septum-diagonal band complex to the hippocampus: a retrograde tracing study with multiple fluorescent dyes in rats. Neurosci Res. 1995;21:199–209. doi: 10.1016/0168-0102(94)00852-7. [DOI] [PubMed] [Google Scholar]