

Abstract
Background and purpose:
Gangliosides, sialic acid-containing glycosphingolipids, abundant in brain, are involved in neuronal function and disease, but the precise molecular mechanisms underlying their physiological or pathological activities are poorly understood. In this study, the pathological role of gangliosides in the extracellular milieu with respect to glial cell death and lipid raft/membrane disruption was investigated.
Experimental approach:
We determined the effect of gangliosides on astrocyte death or survival using primary astrocyte cultures and astrocytoma/glioma cell lines as a model. Signalling pathways of ganglioside-induced autophagic cell death of astrocytes were examined using pharmacological inhibitors and biochemical and genetic assays.
Key results:
Gangliosides induced autophagic cell death in based on the following observations. Incubation of the cells with a mixture of gangliosides increased a punctate distribution of fluorescently labelled microtubule-associated protein 1 light chain 3 (GFP-LC3), the ratio of LC3-II/LC3-I and LC3 flux. Gangliosides also increased the formation of autophagic vacuoles as revealed by monodansylcadaverine staining. Ganglioside-induced cell death was inhibited by either a knockdown of beclin-1/Atg-6 or Atg-7 gene expression or by 3-methyladenine, an inhibitor of autophagy. Reactive oxygen species (ROS) were involved in ganglioside-induced autophagic cell death of astrocytes, because gangliosides induced ROS production and ROS scavengers decreased autophagic cell death. In addition, lipid rafts played an important role in ganglioside-induced astrocyte death.
Conclusions and implications:
Gangliosides released under pathological conditions may induce autophagic cell death of astrocytes, identifying a neuropathological role for gangliosides.
Keywords: ganglioside, autophagy, astrocytes, reactive oxygen species, mTOR
Introduction
Astrocytes, the major glial cell type in brain, provide metabolic and trophic support to neurons and also modulate synaptic activity (Barres and Barde, 2000). Astrocytes play an essential role in regulating neurotransmission and blood flow as well as maintaining a normal brain physiology. In addition to these physiological roles, astrocytes have an important role in the processes of injury and disease in the CNS. Astrocytes are the main responder to CNS insults under various pathological conditions such as ischaemia, infection, autoimmunity and neurodegeneration (Giffard and Swanson, 2005; Maragakis and Rothstein, 2006; Farina et al., 2007). Moreover, astrocytes play multiple roles ranging from passive support to the regulation of inflammation during brain injuries. Multiple signals have been shown to induce the cell death of astrocytes in vitro and in vivo including Ca2+ overload, oxidative stress, mitochondrial dysfunction, endoplasmic reticulum stress and protease activation (Takuma et al., 2004). The survival or death of astrocytes has important implications for neuronal function and survival, because astrocytes are in close contact with neurons providing metabolic and mechanical support.
Gangliosides are sialic acid-containing glycosphingolipids that exist in mammalian cell membranes. Gangliosides are particularly abundant in neuronal cell membranes, and participate in various cellular events of the nervous system (Derry and Wolfe, 1967; Kracun et al., 1984; Rodden et al., 1991; Kotani et al., 1993). GM1, GD1a, GD1b, GT1b and GQ1b are major types of gangliosides found in the brain (Dreyfus et al., 1997). Several lines of evidence point to the importance of brain-derived gangliosides in immune responses and the pathogenesis of brain disease. It has been reported that brain injury can cause the release of gangliosides from damaged neuronal cells into extracellular space, which may lead to pathophysiological conditions (Michikawa et al., 2001). Gangliosides have been reported to play a pivotal role in amyloid β toxicity associated with Alzheimer's disease, as well as in the deposition of amyloid β into senile plaques (McLaurin et al., 1998; Ledesma et al., 2000). Gangliosides activate cultured rat brain microglia (Pyo et al., 1999) and regulate the production of various inflammatory mediators, such as pro-inflammatory cytokines and inducible nitric oxide synthase (Kanda and Watanabe, 2001; Ryu et al., 2002). Individual gangliosides such as GM3 induced inducible nitric oxide synthase expression in murine peritoneal macrophages (Ding et al., 1998), and GM1 enhanced the production of interleukin-1β from reactive astrocytes (Oderfeld-Nowak and Zaremba, 1998). The Toll-like receptors TLR2 and TLR4 have been implicated in glial responses to gangliosides (Jou et al., 2006). On the other hand, gangliosides also induced cell death. For example, GM3 was involved in the apoptotic death of human carcinoma cells and actively dividing astrocytes precursors (Nakatsuji and Miller, 2001). In addition, GD3 induced mitochondrial damage and apoptosis in human hematopoietic cells (Malisan and Testi, 2002), and GT1b increased the apoptotic cell death in thymocytes (Zhou et al., 1998). However, the role of gangliosides in autophagic cell death in astrocytes has not been investigated.
Autophagy is considered to be an evolutionarily conserved process, in which intracellular membrane structures sequester proteins and organelles for lysosomal degradation (Klionsky and Emr, 2000). This process involves the formation of double-membrane structures, termed autophagosomes or autophagic vacuoles, which fuse with the lysosomal membrane to deliver the contents into the autolysosome, where they are degraded (Klionsky and Emr, 2000). The conversion of a microtubule-associated protein light chain LC3-I (cytosolic) into LC3-II (lipidated) is considered to be a general marker for the initiation of autophagy (Mizushima, 2004). The amount of the membrane-bound form of LC3-II correlates with the extent of autophagosome formation (Kabeya et al., 2000). The autofluorescent drug monodansylcadaverine (MDC) is another selective marker for autophagic vacuoles (Biederbick et al., 1995). Cytoplasmic vacuoles can be labelled by MDC in vivo and in vitro in various cell types. Autophagy is a type of programmed cell death (PCD) (Kroemer et al., 2009). As cell injury can occur across an apoptotic-necrotic continuum, autophagy is considered to be the type II PCD (Clarke, 1990). Autophagy plays an important role in many biological processes, such as cellular responses to starvation, cell survival and death (Kroemer and Levine, 2008), cancer and the clearance of inclusion bodies in neurodegenerative disorders (Levine and Klionsky, 2004; Todde et al., 2009). For example, the accumulation of autophagosomes was found in neurites in a transgenic mouse model of Alzheimer's disease (Nixon et al., 2005; Nixon, 2007), in substantia nigra neurons from patients with Parkinson's disease (Zhu et al., 2003) and in cell and animal models for Huntington's disease (Petersen et al., 2001; Qin et al., 2003; Ravikumar et al., 2004).
Oxidative stress has been shown to induce autophagy under starvation and ischaemia/reperfusion conditions (Kabeya et al., 2000). Under oxidative stress, reactive oxygen species (ROS) such as free radicals and H2O2 are generated at high levels, inducing cellular damage and death (Scherz-Shouval et al., 2007). Under starvation conditions, ROS production increases and is required for the induction of autophagy (Pelicano et al., 2004; Scherz-Shouval and Elazar, 2007). ROS also play an important role in inflammatory signalling pathways (Hsu and Wen, 2002). ROS function as second messengers and activate many downstream signalling molecules, including mitogen-activated protein kinases (MAPKs) (Guyton et al., 1996) and the transcription factor NF-κB (Schreck et al., 1991). The membrane-bound NADPH oxidase system is one of the major sources of ROS. It is established that, besides ROS, the Akt/mTOR/p70S6K pathway and the Raf-1/MEK/ERK pathway regulate autophagy. Phosphatidylinositol 3-kinase (PI3K) activates the downstream target Akt, leading to activation of the mammalian target of rapamycin (mTOR), which in turn inhibits autophagy. The p70S6 kinase (p70S6K) is thought to control autophagy downstream of mTOR. In contrast, the class III PI3K complex that includes beclin-1, a homologue of yeast Atg-6, plays a stimulatory role in autophagy (Baehrecke, 2005). The interactions of gangliosides with these autophagic signalling pathways are not understood.
In the present study, we demonstrated that treatment with gangliosides induced ROS-mediated autophagic cell death in astrocytes. Further examination of the signalling pathways indicated that this ganglioside-induced autophagic cell death of astrocytes was subject to either negative or positive regulation by the Akt/mTOR pathway or the ERK1/2 pathway respectively. Finally, lipid rafts were involved in the signalling leading to ganglioside-induced astrocyte death. Our results suggest that gangliosides in the extracellular milieu of the CNS could cause a pathological degeneration of astrocytes through molecular mechanisms that involve ROS and lipid rafts in the plasma membrane.
Methods
Cell cultures
U87MG cells (human glioblastoma cell line) were grown and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Gibco-BRL), penicillin (100 U·mL−1) and streptomycin (100 µg·mL−1; Gibco-BRL, Gaithersburg, MD, USA) at 37°C, 5% CO2. C6 rat glioma cells were maintained in DMEM supplemented with 5% heat-inactivated FBS, gentamicin (50 µg·mL−1). C6 is an astrocytoma cell line that is commonly used as a model of astrocytes.
Primary astrocyte cultures were prepared from the brains of 1–3-day-old ICR mice (Samtako Co., Osan, Korea) by the method of McCarthy and de Vellis (1980). Briefly, whole brains were dissected and dissociated in DMEM, supplemented with 10% FBS, 100 U·mL−1 penicillin and 100 µg·mL−1 streptomycin. Cells were seeded in 75 cm2 tissue culture flasks (Falcon, Becton Dickinson and Company, Franklin Lakes, NJ, USA). Cells were grown at 37°C in a 5% CO2 humidified atmosphere. The culture medium was changed after 5 days in vitro and then every 3 days thereafter. Pure secondary astrocyte cultures were obtained by shaking mixed glial cultures at 100× g overnight, and then culture media were discarded. Astrocytes were dissociated using trypsin-EDTA (Gibco-BRL) and then collected by centrifugation at 184×g for 10 min. The cells were resuspended in DMEM with 10% FBS, 100 U·mL−1 penicillin and 100 µg·mL−1 streptomycin, seeded at 1 × 105 cells·mL−1 onto six-well plates, and cultured for 4 days. The purity of astrocyte cultures was greater than 95%, as determined by glial fibrillary acidic protein immunocytochemical staining (data not shown). Animals used in the current research were acquired and cared for in accordance with guidelines published in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The study was approved by the Institutional Review Board of the Kyungpook National University School of Medicine.
Cell viability assays
Cell viability was assessed by 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT), 2, 3-bis (2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide inner salt (XTT), Trypan blue dye exclusion (Kim et al., 2008) or lactate dehydrogenase (LDH; Kim and Sharma, 2003) assays. For MTT assay, astrocytes, C6 or U87MG cells were seeded in triplicate at a density of 8 × 104 cells per well on 96-well plates. The cells were treated with the ganglioside mixture for 24 h. MTT was added to each well and incubated for 4 h at 37°C. After culture media were discarded, dimethyl sulphoxide was added in order to dissolve the formazan dye. The optical density was measured at 540 nm. Similar results were obtained with lower cell densities (4 × 104 or 6 × 104 cells per well) (data not shown). Cell viability was also evaluated by XTT assay (Wel-Gene, Seoul, Korea). Absorbance (A) was detected with an enzyme calibrator at 450 nm. Cell viability = (A of study group/A of control group) × 100%. For the Trypan blue dye exclusion assay, dead cells were stained with Trypan blue and counted using a haemocytometer.
Both released and total LDH concentrations were determined as described previously for LDH assay (Kim and Sharma, 2003). For the total LDH determination, the cells were lysed by adding 1% of Triton X-100 (final concentration) and incubated for 30 min in the incubator at 37°C. Samples were transferred to plate containing 100 µL of 4.6 mM pyruvic acid in 0.1 M potassium phosphate buffer (pH 7.5). β-NADH (reduced nicotinamide adenine dinucleotide) in 0.1 M potassium phosphate buffer (pH 7.5) was added, mixed, and the absorbance was read kinetically using a Power-Wave x Microplate Scanning spectrophotometer (Bio-Tek Instrument, Winooski, VT, USA). The activity of LDH was normalized to the volume, and the released LDH activity was expressed as a percentage of total cellular LDH. For the cell viability tests and similar assays, either distilled water or dimethyl sulphoxide was used as a vehicle control, which was without effects (data not shown). Viability of the vehicle-treated cells was set to 100%, and the relative viability of the experimental group was calculated accordingly. The 100% injury condition was not used in cell viability assays.
Stable transfection of cDNA for LC3 tagged with green fluorescent protein (GFP-LC3) and fluorescence-detected autophagy
C6 cells in six-well plates were transfected with 4 µg of LC3 cDNA using LipofectAMINE reagent (Invitrogen, Calsbad, CA, USA); all studies of transfection with GFP-LC3 were in C6 cells. The mammalian expression construct of human LC3 cloned into pEGFP was a gift from Dr N Mizushima (Tokyo Medical and Dental University, Tokyo, Japan) (Alexander et al., 2007). An empty pEGFP vector was used as a control for the stable expression of LC3. Stable transfectants were selected in the presence of G418 (500 µg·mL−1) at 2 days after the transfection. The expression of the GFP-LC3 protein in the stable transfectants was confirmed by Western blot and fluorescence microscopy analysis (Olympus BX50, Tokyo, Japan; original magnification, ×400 or ×600). C6 cells were treated with gangliosides either with or without 3-methyladenine (3-MA). The fluorescence of GFP-LC3-labelled vacuoles (autophagosomes) was observed by using a fluorescence microscope. For the quantitative evaluation of LC3 translocation, a minimum of 200 cells were counted for each treatment condition. Fluorescence images were assessed without knowledge of the treatments. The 3-MA was included as a pretreatment for 30 min at 2 mM (Chen et al., 2007).
Visualization of MDC-labelled vacuoles
Autophagic vacuoles were labelled with MDC by incubating astrocytes grown on coverslips with 0.05 mM MDC in phosphate-buffered saline (PBS) at 37°C for 10 min. After incubation, cells were washed four times with PBS and immediately analysed by fluorescence microscopy using an inverted microscope (Olympus BX50) equipped with a filter system (excitation, 380–420 nm; emission, 450 nm; original magnification, ×600).
Quantitative measurement of autophagy by MDC staining
Following the induction of autophagy by gangliosides and amino acid starvation [incubation in Earle's balanced salt solution (EBSS) for 2 h], the astrocytes (1 × 105 cells per well in 24-well plates) were incubated with 0.05 mM MDC in PBS at 37°C for 10 min (Biederbick et al., 1995). After incubation, cells were washed four times with PBS and collected in 10 mM Tris-HCl, pH 8 containing 0.1% Triton X-100. Intracellular MDC was measured by a fluorescent plate reader (Fluostar OPTIMA, BMG LABTECH, Offenburg, Germany) at an excitation of 380 nm and emission of 525 nm and digitized. The fluorescent readings were digitized by using a Soft Max Pro software programme (Molecular Devices, Sunnyvale, CA, USA).
Western blot analysis
Cells were lysed in a triple-detergent lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.02% sodium azide, 0.1% sodium dodecyl sulphate, 1% Nonidet P-40, 0.5% sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride). Protein concentration in cell lysates was determined by using a protein assay kit (Bio-Rad, Hercules, CA, USA). An equal amount of protein (40 µg) from each sample was separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (12% gel) and transferred to Hybond ECL nitrocellulose membranes (Amersham Biosciences, Piscataway, NJ, USA). The membranes were blocked with 5% skim milk and sequentially incubated with primary antibodies [anti-Atg-6, anti-Atg-7, anti-poly (ADP-ribose) polymerase (PARP) and anti-HSC70 antibodies, Santa Cruz Biotechnology (Santa Cruz, CA, USA); anti-phospho-Akt at Ser473, anti-total Akt, anti-phospho-ERK1/2 at Thr202/Tyr204 and anti-total ERK1/2 antibodies, Cell signalling Technology (Beverly, MA, USA); rabbit anti-LC3 polyclonal antibody, MBL International (Woborn, MA, USA); monoclonal anti-α-tubulin clone B-5-1-2 mouse ascites fluid, Sigma] and horseradish peroxidase-conjugated secondary antibodies (anti-rabbit and anti-mouse; Amersham Biosciences) followed by enhanced chemiluminescence detection (Amersham Biosciences).
Small interfering RNAs
The 25-nucleotide small interfering RNA (siRNA) duplexes used in this study were purchased from Invitrogen and have the following sequences: Atg-6, CAG UUU GGC ACA AUC AAU AAC UUC A; Atg-7, CAG AAG GAG UCA CAG CUC UUC CUU A; and GFP, AAG ACC CGC GCC GAG GUG AAG. The siRNA against GFP was used as a control. Another set of siRNAs against Atg-6 or Atg-7 were purchased from Santa Cruz Biotechnology [Atg-6 (sc-29797) and Atg-7 (sc-41447)]. Cells were transfected with siRNA oligonucleotides using LipofectAMINE 2000 (Invitrogen) according to the manufacturer's recommendations.
ROS measurement
For intracellular ROS measurement, either astrocytes or C6 cells were detached with trypsin-EDTA, and incubated with 100 µM of 2′,7′-dichlorofluorescein diacetate (H2DCFDA; Molecular Probes) in a serum-free medium at 37°C for 20 min and then washed with PBS. The cells were then treated with stimulating agents in PBS at 37°C for 12 h and analysed by flow cytometry (excitation, 485 nm; emission, 530 nm).
Flow cytometric analysis of apoptosis
Astrocytes were detached with trypsin-EDTA and washed twice with cold PBS. The cells were then resuspended in 250 µL of binding buffer [10 mM HEPES, 140 mM NaCl and 2.5 mM CaCl2 (pH 7.4)] and incubated with 3 µL of fluorescein isocyanate (FITC)-conjugated annexin V (Molecular Probes) according to the manufacturer's specifications. Afterward, cells were gently vortexed and incubated for 15 min at room temperature in the dark conditions. Propidium iodide (PI; 20 µg·mL−1) was then added, and flow cytometry was performed within 1 h by using FACSAria (BD Biosciences).
Statistical analysis
All results were expressed as mean ± SD. The data were analysed by one-way anova and the Student-Newman-Keul's post hoc analysis by using a spss programme (version 12.0; SPSS Inc., Chicago, IL, USA). A value of P < 0.05 was considered to be statistically significant.
Materials
H2O2, 3-MA, MDC, EBSS, diphenyleneiodonium (DPI), α-tocopherol, trolox, N-acetyl cysteine (NAC), methyl β-cyclodextrin (MβCD) and NH4Cl were purchased from Sigma-Aldrich (St. Louis, MO, USA). Ganglioside mixture (Gmix; 5–500 µg·mL−1; 18% GM1, 55% GD1a, 15% GD1b, 10% GT1b and 2% others), MEK1 inhibitor (PD98059), Akt inhibitor (1L-6-hydroxymethyl-chiro-inositol 2(R)-2-O-methyl-3-O-octadecylcarbonate), rapamycin, benzyloxycarbonyl-Val-Ala-Asp (zVAD-fmk, zVAD) were purchased from Calbiochem (La Jolla, CA, USA). GM1, GD1a and GT1b were purchased from Matreya (Pleasant Gap, PA, USA). Recombinant mouse IFN-γ and soluble recombinant TRAIL were purchased from R&D Systems (Minneapolis, MN, USA). Rottlerin was purchased from Biomol (Plymouth Meeting, PA, USA) and dissolved in dimethyl sulphoxide and freshly diluted in culture media for the experiments. U87MG human glioblastoma cell line and C6 rat glioma cell line were obtained from American Type Culture Collection (ATCC; Manassas, VA, USA).
Results
Gangliosides induced cell death in astrocytes
In order to examine the effect of gangliosides on astrocytes viability, we treated mouse primary astrocyte cultures and C6 rat glioma cell lines with different concentrations of the ganglioside mixture (Gmix; 5–500 µg·mL−1) over a 72 h time period and then measured cell viability by using the MTT assay. The ganglioside mixture (50 µg·mL−1) induced a 28% cell death in astrocytes after 24 h, and cell viability was not greatly reduced by increasing either the time or concentration of the gangliosides (Figure 1A). The ganglioside mixture (50 µg·mL−1) induced a 23% cell death in C6 cells after 72 h (Figure 1B) and in these cells, viability decreased in a concentration- and time-dependent manner. Ganglioside-induced astrocyte cell death was also shown by Trypan blue dye exclusion (Figure 1C) and LDH assays (Figure 1D). As observed with the MTT assay, cell death was increased by gangliosides in astrocytes (24 h) and C6 cells (72 h).
Figure 1.

Gangliosides induced cell death in astrocytes. Primary astrocytes (A) or C6 glioma cells (B) were treated with a ganglioside mixture (Gmix; 5–500 µg·mL−1) for 24–72 h, and then cell viability was assessed by 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay. Also, primary astrocytes (24 h) or C6 cells (72 h) were treated with the Gmix (50 µg·mL−1), and then viability assayed by Trypan blue dye exclusion (C) or lactate dehydrogenase (LDH) release (D). A representative microscopic image of Trypan blue stained cells is shown (C; upper). The LDH release was expressed as a percentage of total cellular LDH. The data were expressed as the mean ± SD (n= 3). The results represent more than three independent experiments (*P < 0.05, **P < 0.001, different from untreated cells at each time point).
Gangliosides induced autophagic cell death in astrocytes
Autophagy is characterized by the formation of double-membraned autophagosomes that fuse with lysosomes in order to form autolysosomes. Autophagosome formation also involves the induction of beclin-1/Atg-6 expression, as well as the localization of the protein LC3 in autophagosomes (Gozuacik and Kimchi, 2004; Codogno and Meijer, 2005). In this study, the autophagy was monitored by measuring: (i) the formation of GFP-LC3-labelled vacuoles; (ii) the conversion of the cytoplasmic form of LC3 (LC3-I, 18 kDa) to the pre-autophagosomal and autophagosomal membrane-bound form of LC3 (LC3-II, 16 kDa); (iii) LC3 flux using the lysosome inhibitor NH4Cl; and (iv) the formation of MDC-labelled vacuoles. GFP-fused LC3, a specific marker for autophagosome formation, was used in order to detect autophagy. GFP-LC3 cDNA was transfected into C6 cells, and cells with GFP-LC3-labelled vacuoles (dots) were observed by fluorescence microscopy. The formation of GFP-LC3-labelled vacuoles was observed after C6 cells were treated with the ganglioside mixture (50 µg·mL−1) for 24 h; the formation of these vacuoles was attenuated by treatment with 3-MA, a specific inhibitor of the early stages of the autophagic process (Figure 2A) (Seglen and Gordon, 1982). As a positive control, C6 cells were placed under starvation conditions (incubation in EBSS for 2 h) known to induce autophagy. Amino acid starvation also increased the amount of GFP-LC3-labelled vacuoles, and this increase was also blocked by 3-MA (data not shown).
Figure 2.
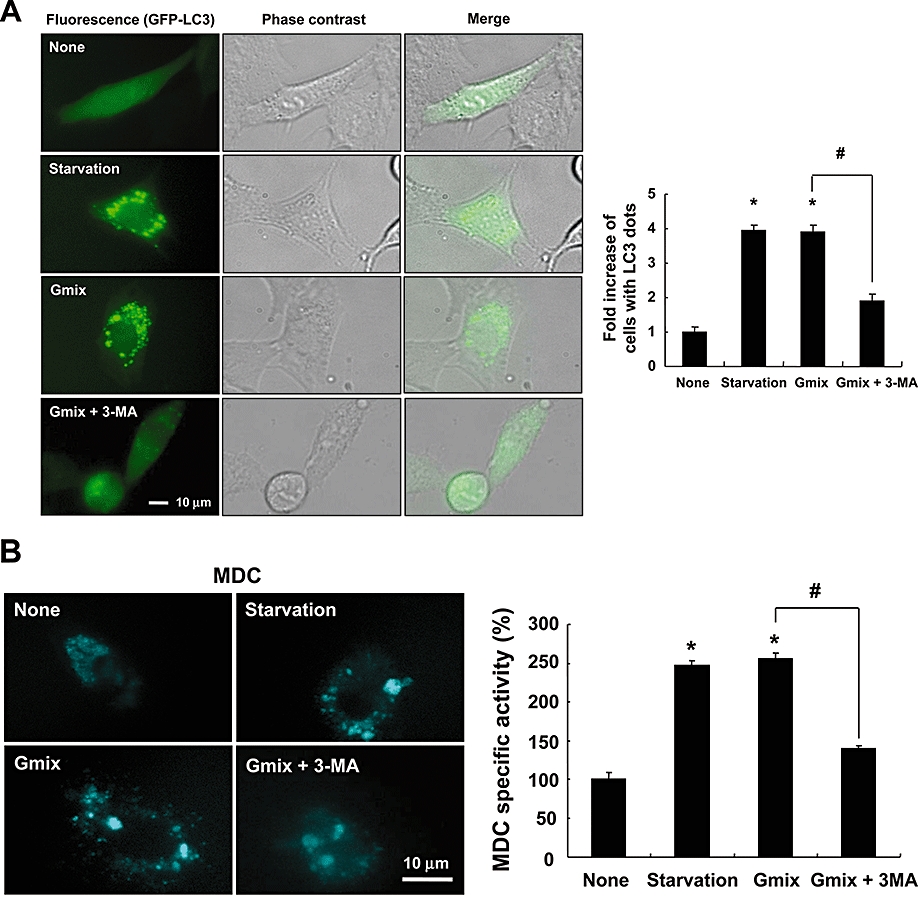

Gangliosides induced autophagy in astrocytes. C6 glioma cells transfected with GFP-LC3 (microtubule-associated protein 1 light chain 3, LC3 tagged with green fluorescent protein) cDNA were either treated with the ganglioside mixture (Gmix; 50 µg·mL−1) in the absence or presence of 3-methyladenine (3-MA) (2 mM) for 24 h or placed under amino acid starvation conditions (Earle's balanced salt solution at 37°C for 2 h). The formation of vacuoles containing GFP-LC3 (dots) was examined by fluorescence or phase contrast microscopy, as shown in the representative microscopic images (original magnification, ×600) (left); a minimum of 200 cells were counted per sample to obtain fold increase of cells with LC3 dots (right) (A). In another set of experiments, C6 cells were similarly treated with the ganglioside mixture or placed under starvation conditions and incubated with 0.05 mM monodansylcadaverine (MDC) for 10 min. Cells were then analysed by fluorescence microscopy (original magnification, ×600), or the intracellular MDC was measured by fluorescence plate reader after cells were lysed (B). Conversion of LC3-I to LC3-II was determined by Western blotting after astrocytes or C6 cells were treated with the Gmix (50 µg·mL−1) or starvation conditions in the presence or absence of the lysosomal inhibitor NH4Cl (30 mM) for 24 h. α-Tubulin was used as a loading control. Quantification of LC3-II protein levels was performed by densitometric analysis, which was then normalized to α-tubulin level (lower). The densitometric data are mean ± SD from more than three experiments (C,D). The formation of GFP-LC3 vacuoles (dots) was examined under by fluorescence microscopy, as shown in the representative microscopic images (original magnification, ×400); a minimum of 200 cells were counted per sample (E). The data were expressed as the mean ± SD (n= 3). Results shown are representative of more than three independent experiments (*P < 0.001 compared with no treatment; #P < 0.001 between the treatments indicated).
Monodansylcadaverine is another specific marker for autolysosomes (Biederbick et al., 1995), and we examined the incorporation of MDC into cells after treatment with gangliosides or starvation. Cells treated with the ganglioside mixture or starved showed an increase in the number and size of MDC-positive vesicles, indicating that these conditions induced the formation of the MDC-labelled vacuoles (Figure 2B). MDC was concentrated in spherical structures distributed within the cytoplasm and incubation with gangliosides or starvation increased MDC uptake, in comparison with untreated cells. As expected, MDC incorporation was attenuated by 3-MA (Figure 2B).
The conversion of LC3-I to LC3-II is another specific marker for autophagy. In astrocytes and C6 cells, both gangliosides and starvation significantly increased the amount of LC3-II protein in comparison with the control after 24 h of treatment (Figure 2C). In the presence of a lysosomal inhibitor NH4Cl, which prevents the degradation of LC3 in autophagosomes (Terman et al., 2006; Chen et al., 2007; 2008;), the amount of LC3-II in astrocytes increased following treatment with the ganglioside mixture (Figure 2D). However, NH4Cl treatment failed to increase the formation of GFP-LC3-labelled vacuoles following ganglioside treatment (Figure 2E) (Terman et al., 2006; Chen et al., 2007; 2008;).
In astrocytes, ganglioside- or starvation-induced cell death was attenuated by the addition of 3-MA (Figure 3A,B), suggesting that autophagy is related with cell death under these conditions. Although starvation-induced autophagy can be a protective mechanism in general, it induced cell death in neurons (Sadasivan et al., 2006; Du et al., 2009) and in brain glial cells. Because the induction of autophagy requires the expression of autophagy-related genes such as beclin-1/Atg-6, Atg-5 and Atg-7 in order to form autophagosomes (Baehrecke, 2005), we hypothesized that the suppression of beclin-1/Atg-6 and Atg-7 expression may reduce the incidence of ganglioside-induced autophagic cell death. In U87MG human glioma cell line, a knockdown of beclin-1/Atg-6 or Atg-7 expression using siRNA against beclin-1/Atg-6 or Atg-7 attenuated ganglioside-induced cell death (Figure 3C) as well as MDC activity (Figure 3D), further supporting that gangliosides induced autophagic cell death in astrocytes. Two different siRNA sequences were used for each Atg gene in order to rule out off-target effects of siRNA. The siRNA-mediated knockdown of Atg-6 or Atg-7 gene expression was confirmed by Western blot analysis (Figure 3E). The effect of Atg7-siRNAs was proportional to the degree of Atg7 gene knockdown: Atg7-siRNA-2 showed greater effects than Atg7-siRNA-1. We also analysed PARP cleavage, which is a hallmark of an unrelated form of PCD, to determine whether the knockdown of Atg-6 or Atg-7 gene expression affects apoptotic cell death. Gangliosides mixtures did not induce a significant cleavage of PARP (Figure 3E). Combination of rottlerin and TRAIL treatment was used as a positive control that caused an increase of protein level of PARP cleavage fragment (Kim et al., 2005; Lim et al., 2009). Taken collectively, these results conclusively indicated that gangliosides induced autophagic cell death in astrocytes (Figure 3).
Figure 3.

Gangliosides induced autophagic cell death in astrocytes. Primary astrocytes were pretreated with 3-methyladenine (3-MA) (2 mM) for 30 min prior to treatment with the ganglioside mixture (Gmix; 50 µg·mL−1) for 24 h or starvation conditions. Cell viability was assessed by 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay (A) or lactate dehydrogenase (LDH) assay (B). U87MG human glioma cells were transfected with 40 nM of control green fluorescent protein siRNA, beclin-1/Atg-6 siRNA or Atg-7 siRNA. At 24 h post transfection, cells were treated with the Gmix (50 µg·mL−1) for 24 h. Then, cellular viability was determined by 2, 3-bis (2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide inner salt (XTT) assay (C), or intracellular monodansylcadaverine (MDC) was measured by fluorescence, after cell lysis (D). Two different siRNA sequences (siRNA-1 and siRNA-2) for each Atg gene were used to rule out off-target effects. U87MG cells were treated with the Gmix (50 µg·mL−1), rottlerin (2.5 µM) and TRAIL (100 ng·mL−1) for 24 h as indicated. Knockdown of Atg-6/Atg-7 expression and poly (ADP-ribose) polymerase (PARP) cleavage was evaluated by Western blot analysis (E). Anti-HSC70 antibody served as control for the loading of protein levels. Quantification of Atg-6 and Atg-7 protein levels was performed by densitometric analysis (lower). All data were expressed as the mean ± SD (n= 3). Results shown are representative of more than three independent experiments (*P < 0.05, **P < 0.001 compared with no treatment; #P < 0.05 between the treatments indicated).
ROS mediated autophagic cell death induced by gangliosides
Because ROS have been previously implicated in autophagy (Xu et al., 2006), we have attempted to determine whether ROS mediate autophagic cell death induced by gangliosides. In astrocytes (Figure 4A) and C6 cells (Figure 4B), ROS scavengers such as α-tocopherol, NAC and trolox attenuated ganglioside-induced cell death. The formation of GFP-LC3-labelled vacuoles and MDC-labelled vacuoles was also induced after C6 cells were treated with H2O2 (500 µM) for 24 h (Figure 4C,D). Ganglioside-induced formation of GFP-LC3-labelled vacuoles was also attenuated by treatment with α-tocopherol (Figure 4E). H2O2 as a ROS donor increased MDC uptake, as observed with the gangliosides. Ganglioside-induced MDC incorporation was attenuated by ROS scavengers (Figure 4F). We next determined whether gangliosides induce ROS production in astrocytes and C6 cells by directly measuring ROS levels as a function of DCF fluorescence (Figure 4G). DCFDA-loaded astrocytes and C6 cells were exposed to gangliosides for 12 h and then subjected to flow cytometric analysis. The DCF fluorescence intensity increased after treatment with the ganglioside mixture. The expression of the NADPH oxidase subunit p47PHOX was detected in both astrocytes and C6 cells in our previous study, indicating that NADPH oxidase is expressed in astrocytes (Kim et al., 2008). We utilized the NADPH oxidase inhibitor DPI (Kim et al., 2008) to determine the role of the NADPH oxidase in the ganglioside-induced autophagic cell death of astrocytes. DPI significantly attenuated the ganglioside-induced astrocyte autophagy, as determined by LC3 translocation and MDC uptake (Figure 4H,I), suggesting a critical role for NADPH oxidase in the ROS generation and autophagic cell death in astrocytes following ganglioside exposure.
Figure 4.


Role of reactive oxygen species (ROS) in the autophagic cell death of astrocytes induced by gangliosides. Primary astrocytes or C6 glioma cells were pretreated with ROS scavengers such as α-tocopherol (α-toco; 10 µM), N-acetyl cysteine (NAC) (1 mM) and trolox (500 µM) for 30 min before treatment with the ganglioside mixture (Gmix; 50 µg·mL−1). The cell viability was assessed by 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay (A,B). Formation of GFP-LC3 (microtubule-associated protein 1 light chain 3, LC3 tagged with green fluorescent protein) vacuoles (dots) was examined, after C6 cells transfected with GFP-LC3 cDNA were treated with H2O2 (500 µM) for 24 h. Representative microscopic images are shown (original magnification, ×600) (C). Primary astrocytes were treated with H2O2 (500 µM) for 24 h, and then incubated with 0.05 mM monodansylcadaverine (MDC) for 10 min. Cells were then analysed by fluorescence microscopy (original magnification, ×600) (D). The formation of GFP-LC3 vacuoles (dots) was examined by fluorescence microscopy, as shown in the representative microscopic images (original magnification, ×400); at least 200 cells were counted per sample (E,H). After primary astrocytes were treated with H2O2 (500 µM) or Gmix (50 µg·mL−1) in the presence or absence of antioxidants or diphenyleneiodonium (DPI) (1 nM or 1 µM), intracellular MDC was similarly measured by fluorescence, after cell lysis (F,I). Astrocytes or C6 cells were treated with the Gmix (50 or 500 µg·mL−1) for 12 h. ROS was measured by H2DCFDA fluorescence by flow cytometric analysis (G). The data were expressed as the mean ± SD (n= 3). Results shown are representative of more than three independent experiments (*P < 0.01, **P < 0.001 compared with Gmix treatment alone; #P < 0.001 compared with no treatment).
Role of Akt/mTOR and ERK pathway in the ganglioside-induced autophagic cell death of astrocytes
The Akt/mTOR/p70S6K pathway is the main regulatory pathway that negatively controls autophagy (Shigemitsu et al., 1999; Arico et al., 2001; Coward et al., 2009), and we therefore examined the effect of gangliosides on this signalling pathway. The mTOR inhibitor rapamycin or the Akt inhibitor augmented ganglioside-induced cell death in astrocytes and C6 cells, indicating that both mTOR and Akt attenuated autophagic death (Figure 5A,B). Because the ERK pathway has been shown to positively regulate autophagy in cancer cells upon starvation (Ogier-Denis et al., 2000; Pattingre et al., 2003), we also examined this pathway. Gangliosides-induced MDC incorporation was reduced by an MEK1 inhibitor PD98059, and increased by the mTOR inhibitor rapamycin and the Akt inhibitor in astrocytes (Figure 5C). These results indicate that gangliosides inhibited the Akt/mTOR pathway while activating the ERK pathway; these two signalling pathways appeared to reciprocally regulate the autophagic cell death of astrocytes induced by gangliosides.
Figure 5.

Gangliosides inhibited the Akt/mTOR (mammalian target of rapamycin) pathway and activated the ERK pathway in astrocytes and C6 glioma cells. Astrocytes and C6 cells were pretreated with the mTOR inhibitor (rapamycin; 1–1000 nM), or the Akt inhibitor (1–4 µM) for 30 min before treatment with gangliosides mixture (Gmix; 50 µg·mL−1) for 24 h (primary astrocytes) or 72 h (C6 glioma cells). Afterwards, either cell viability was assessed by 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay (A,B) or intracellular monodansylcadaverine (MDC) was measured by fluorescence plate reader after cell lysis (C). The data were expressed as the mean ± SD (n= 3). Results represent more than three independent experiments (*P < 0.05, **P < 0.001 compared with Gmix treatment alone; #P < 0.001 compared with no treatment).
Role of lipid rafts in ganglioside-induced cell death
Lipid raft formation has an important role in the dynamic association of multi-protein receptor complexes involved in immune and other cellular responses (Rajendran and Simons, 2005). In astrocytes, the lipid raft-disrupting drug (MβCD) (Jou et al., 2006; Yoon et al., 2008) inhibited ganglioside-induced cell death (Figure 6A). Moreover, the quantification of autophagic cell death indicated that the percentage of MDC-positive cells in ganglioside treatment was significantly reduced by the addition of MβCD (Figure 6B), suggesting that lipid raft formation was important for the autophagic cell death observed. DPI and MβCD also reduced the gangliosides-induced conversion of LC3-I to LC3-II in C6 glia cells, further supporting the involvement of ROS and lipid rafts in astrocyte autophagy (Figure 7). The gangliosides mixture is composed of various types of gangliosides. Thus, we next tested the individual effects of three major types of gangliosides in the brain, GM1, GD1a and GT1b, on astrocyte cell death. GT1b exhibited the greatest inhibitory effect on the viability of astrocytes among the single ganglioside components tested, as determined by MTT or Trypan blue assays (Figure 8A,B). The formation of GFP-LC3-labelled vacuoles was also most strongly increased by GT1b (50 µg·mL−1) after 24 h (Figure 8C). Thus, GT1b may be the major active component of the ganglioside mixture that induced autophagic cell death in astrocytes.
Figure 6.

Lipid rafts were involved in ganglioside-induced autophagic cell death of astrocytes. Astrocytes were pretreated with methyl β-cyclodextrin (MβCD) (100–500 µM) for 30 min prior to treatment with the ganglioside mixture (Gmix; 50 µg·mL−1) for 24–48 h. The cell viability was assessed by 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay (A). Intracellular monodansylcadaverine (MDC) was measured by fluorescence plate reader after cell lysis (B). The data were expressed as the mean ± SD (n= 3). Results shown are representative of more than three independent experiments (*P < 0.001 compared with Gmix treatment alone; #P < 0.001 compared with no treatment).
Figure 7.

Role of reactive oxygen species and lipid rafts in ganglioside-induced conversion of microtubule-associated protein 1 light chain 3 (LC3)-I to LC3-II. C6 cells were treated with the ganglioside mixture (Gmix; 50 µg·mL−1) in the presence of either diphenyleneiodonium (DPI) (1 µM) or methyl β-cyclodextrin (MβCD) (250 µM) for 24 h. LC3 or α-tubulin was detected by Western blot analysis. Quantification of LC3-II protein levels was performed by densitometric analysis, which was then normalized to α-tubulin level (lower). The densitometric data are mean ± SD (n= 3); *P < 0.05 compared with Gmix treatment alone.
Figure 8.

The ganglioside GT1b induced autophagic cell death of astrocytes. Primary astrocytes were treated with indicated concentrations of GM1, GD1a, GT1b or the ganglioside mixture (Gmix; 50 µg·mL−1) for 72 h. Cell viability was measured by 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) (A) or by Trypan blue dye exclusion assays (B) respectively. A representative phase contrast image is shown (B, left). The formation of GFP-LC3 (microtubule-associated protein 1 light chain 3, LC3 tagged with green fluorescent protein) vacuoles (dots) was examined by fluorescence microscopy, as shown in the representative microscopic images (original magnification, ×400); at least 200 cells were counted per sample (C). The data were expressed as the mean ± SD (n= 3). Results shown are representative of more than three independent experiments (*P < 0.001 compared with no treatment).
Discussion
The purpose of this study was to examine whether gangliosides in the extracellular milieu of the CNS induced autophagic death in astrocytes, and if this occurred, to identify the signalling pathway(s) involved. Based on studies using primary astrocytes and glioma cell lines in conjunction with various autophagic markers, we concluded that gangliosides could indeed induce autophagy in astrocytes through molecular mechanisms involving several signalling components (Figure 9). One important component of the ganglioside action in astrocytes was the formation of lipid rafts. Lipid rafts are detergent-resistant and liquid-ordered membrane domains and are enriched for cholesterol, glycosphingolipids and phospholipids with relatively long and saturated acyl chains, and are reported to serve as platforms for several cellular functions, including vesicular trafficking, signal transduction and viral entry and infection (Harder, 2004). In glial cells, gangliosides are thought to be incorporated into the plasma membrane, forming microdomains within lipid membranes (Vyas et al., 2001), and they modulate growth factor receptors and other signalling events (Meuillet et al., 1996). Several lipid signalling molecules are associated with these lipid rafts. And it was possible that lipid raft formation was associated with ganglioside-induced cell death, and influenced by raft-disrupting agents. Indeed, we found that lipid raft formation appeared to be crucial to ganglioside-induced autophagic cell death. Recent studies have suggested that lipid rafts may be associated with several signalling molecules, such as the Src family of tyrosine kinases, Rho A and MAPKs (Iwabuchi et al., 2000). The disruption of lipid rafts down-regulated Kaposi's sarcoma-associated herpes virus (KSHV)-induced PI3K, NF-κB and RhoA-GTPase activation in human microvascular dermal endothelial (HMVEC-d) cells (Raghu et al., 2007) and down-regulated PI3K (Peres et al., 2003). These studies indicate a critical role of lipid rafts in cellular signalling. However, further studies are necessary to gain a better understanding on how lipid rafts regulate the signal transduction pathways of ganglioside-induced cell death in astrocytes. These studies will provide an insight into whether lipid rafts could be targeted in order to regulate the autophagic cell death of astrocytes.
Figure 9.

Possible mechanisms for ganglioside-induced autophagic cell death in astrocytes. Gangliosides, via lipid rafts, initiate autophagic cell death signalling cascades in astrocytes. The ERK pathway and class III PI3K/beclin-1 complex may promote autophagic cell death in astrocytes. In contrast, Akt/mTOR pathway inhibits autophagic cell death. ROS appear to be important mediators of ganglioside-induced astrocyte cell death. The data obtained with various pharmacological inhibitors and siRNAs support the signalling pathways shown here. 3-MA, 3-methyladenine; DPI, diphenyleneiodonium; LC3, microtubule-associated protein 1 light chain 3; MβCD, methyl β-cyclodextrin; mTOR, mammalian target of rapamycin; PI3K, phosphatidylinositol 3-kinase; ROS, reactive oxygen species.
Autophagy is a eukaryotic process, in which long-lived proteins and organelles are turned over throughout the lifecycle of an organism (Seglen and Bohley, 1992). This process may be induced during development, periods of environmental stress or senescence and cell death (Doelling et al., 2002). Most experimental evidence supporting the concept of ‘autophagic cell death’ is based on the presence of autophagic hallmarks (most commonly autophagosomes, an accumulation of LC3-II and LC3 flux) in dying cells (Levine and Yuan, 2005; Gozuacik and Kimchi, 2007; Klionsky et al., 2008), and rescue from cell death via suppression of autophagy (usually achieved via chemical inhibitors or knockdown of Atg genes). A recent study showed that knockdown of beclin-1/Atg-6 gene expression markedly inhibited cell death, suggesting that beclin-1/Atg-6 may be essential for autophagic cell death (Yu et al., 2004). In the present study, the typical morphological characteristics of autophagy were observed in the ganglioside-treated astrocytes. The phenotypic markers of autophagy, including an increase of MDC staining, punctate distribution of GFP-LC3, an increased LC3-II/LC3-I proportion and LC3 turnover, were also noted. Experiments using a lysosomal inhibitor (NH4Cl) revealed that the increase of LC3-II level or the formation of LC3-positive vacuoles was due to the induction of autophagy rather than inhibition of the later stages of the lysosome degradation pathway (Terman et al., 2006; Chen et al., 2007; 2008;). Additionally, ganglioside-induced cell death was inhibited by 3-MA, an inhibitor of autophagy. The knockdown of beclin-1/Atg-6 or Atg-7 expression using siRNA also attenuated the ganglioside-induced cell death. Collectively, these results conclusively indicate that gangliosides induce autophagic cell death of astrocytes. However, sphingolipid-containing gangliosides, sphingosine and ceramide are known to induce apoptotic cell death in various cell types (Kagedal et al., 2001; Nakatsuji and Miller, 2001; Kim et al., 2007b). To define further the nature of ganglioside-induced cell death, we used staining with PI and annexin-V conjugated with FITC, which was followed by flow cytometric analysis. Treatment of astrocytes with the ganglioside mixture (50 and 500 µg·mL−1) resulted in the appearance of some of the characteristics of apoptotic cell death (annexin V+ and PI−, early apoptosis; annexin V+ and PI+, late apoptosis) to a certain extent (data not shown). Also, a caspase inhibitor zVAD-fmk (zVAD) partly reduced ganglioside-induced cell death. When 3-MA and zVAD were combined, cell death was further reduced, suggesting that both autophagic and apoptotic cell death may occur in astrocytes following exposure to gangliosides (data not shown). These results are in accordance with the concept of parallel death pathways in PCD (Degterev and Yuan, 2008). It should be noted that annexin V and PI staining is not absolutely specific in terms of defining apoptosis and necrosis: for instance, annexin V staining can be observed in programmed necrosis with a unique permeabilization of the plasma membrane.
Oxidative stress is involved in signalling pathways that lead to cell death under various conditions (Takeda et al., 1999; Le Bras et al., 2005; Scherz-Shouval and Elazar, 2007). For example, in Parkinson's disease, oxidation of dopamine induced oxidative stress, autophagy and cell death, indicating that autophagic cell death may also occur in the nervous system in response to oxidative stress (Gomez-Santos et al., 2003). Additionally, oxidative stress induced autophagic cell death in transformed or cancer cells (Chen et al., 2008). Recent studies have demonstrated that superoxide and ROS mediate autophagic cell death (Chen et al., 2007; Kim et al., 2007a). It has also been shown that ROS could be involved in the caspase-independent cell death of macrophages (Xu et al., 2006). ROS exhibited a variety of cellular effects, including DNA damage, mitochondrial dysfunction, activation of signalling pathways and activation of transcription factors leading to the up-regulation of gene expression (Schumacker, 2006). Here, we found that ROS may be an important mediator of ganglioside-induced astrocytes cell death. Our results are in agreement with the previous reports that indicate a central role of ROS in cell death. We demonstrated that: (i) ROS scavengers blocked autophagic cell death in ganglioside-treated astrocytes; (ii) H2O2 also induced autophagic cell death; and (iii) gangliosides induced ROS production. However, the precise molecular mechanism whereby ROS induces autophagic cell death of astrocytes is not known at this point.
In this study, we also examined the role of Akt/mTOR and ERK pathways in the autophagic cell death of astrocytes by means of individual manipulation of the regulatory pathways. Both Akt/mTOR and ERK pathways regulated the astrocyte autophagy, but with opposing effects: the Akt/mTOR pathway regulated autophagy negatively, whereas the ERK pathway was a positive regulator. Thus inhibition of the ERK pathway using PD98059 attenuated autophagy whereas inhibition of the Akt/mTOR pathway by using rapamycin or the Akt inhibitor enhanced autophagy. These findings not only add a novel concept to ganglioside-induced cell death pathways, but also indicate that Akt/mTOR and the ERK pathways are two major pathways that regulate autophagy induced by gangliosides in astrocytes. We also examined the effect of gangliosides on these signalling pathways by Western blot analysis, which supported our suggestions. The treatment with gangliosides effectively decreased the level of phosphorylated Akt for a period of 12 h to 24 h in astrocytes as well as for 72 h in C6 cells (data not shown). Gangliosides increased the level of phosphorylated ERK1/2 after 24 h in astrocytes and 72 h in C6 cells (data not shown). Additionally, the results in this study showed that gangliosides induced more than one form of cell death (apoptosis and autophagic cell death). This is similar to the effect of arsenic trioxide (As2O3) on cell death of human T-lymphocytic leukaemia and the myelodysplastic syndrome cell line (Qian et al., 2007). In that report, As2O3 treatment led to not only apoptosis but also autophagic cell death via the up-regulation of beclin-1 in leukaemia cells.
In this study, we demonstrated that ganglioside treatment induced autophagic cell death of primary astrocytes in culture. Recent studies reported autophagy of astrocytes under different conditions. For example, tryptamine induced autophagy in mouse HT22 and human SK-N-SH neuroblastoma cell lines and in primary astrocytes (Herrera et al., 2006). However, there was a discrepancy in the results between glioma cells and primary astrocytes in some cases. Sodium selenite induced autophagic cell death in human glioma cells but not in normal human astrocytes (Kim et al., 2007b). Rotenone, thenoyltrifluoroacetone, H2O2 and 2-methoxyestradiol also induced autophagic cell death in transformed and cancer cells, but failed to induce autophagic cell death in non-transformed astrocytes (Perez-Ortiz et al., 2004; Chen et al., 2007; Chen et al., 2008). Transformed glioma cells appear to be more sensitive to autophagic cell death than primary astrocytes. Currently, it is not clear why gangliosides induced greater cell death response in primary astrocytes than in glioma cells. Nevertheless, it should be noted that the primary astrocytes were derived from rat brain cortex in one of the previous reports (Perez-Ortiz et al., 2004), while, in the present study, primary astrocytes were prepared from mouse whole brain. Although the autophagy of glioma and cancer cells has been widely reported, less is known about the autophagic cell death process in normal astrocytes. Accordingly, the mechanism of autophagic cell death of astrocytes has not been thoroughly investigated. Nevertheless, in a recent in vivo study, gangliosides have been shown to induce autophagy in brains under β-galactosidase-deficient (β-gal−/−) conditions (Takamura et al., 2008). It was reported that GM1-gangliosidosis in β-gal−/− mouse brains enhanced autophagy and mitochondrial alterations. In that report, mitochondrial cytochrome c oxidase activity had significantly decreased in cultured astrocytes obtained from β-gal−/− mice (Takamura et al., 2008). However, the autophagic cell death of astrocytes in vivo has not been convincingly demonstrated. Gangliosides have been also regarded as neuroprotective agents. For example, gangliosides at low (nM and µM) concentrations inhibited glutamate-induced free radical reactions (Avrova et al., 1998). Gangliosides enhanced survival of serum-deprived dopaminergic neurons in culture (Leon et al., 1988) and protected neuroblastoma cells against calcium ionophore cytotoxicity (Nakamura et al., 1992). These previous reports on neurons and neuroblastoma systems appear to contradict what has been observed for astrocytes in this study. The molecular mechanism underlying this discrepancy remains to be determined.
Gangliosides are abundantly found in neuronal cell membranes. Gangliosides could be released from damaged neuronal cells to the extracellular space in injured brain. Several studies support this possibility; the amount of gangliosides in cerebrospinal fluid increases in patients with neurodegenerative diseases and in HIV-infected brain (Blennow et al., 1991; Gisslen et al., 1997). Under pathological states, the composition and volume of the extracellular space change, thereby slowing down the movement of various molecules and increasing their local concentrations at injured sites (Sykova, 2005). Thus, the concentration of gangliosides in the extracellular space at injured sites can be greatly increased, up to µg·mL−1 (Jou et al., 2006). Abnormally released gangliosides under pathological conditions may influence cell survival or death of neurons and astrocytes. Our results have important implications in the role of gangliosides in brain pathologies and may provide a link between astrocyte autophagy and the pathological role of gangliosides in brain. Astrocytes play a key role in the maintenance of normal brain physiology and, in many neuropathologies, and their dysfunction leads to disruption of neuronal function (Barres and Barde, 2000; Maragakis and Rothstein, 2006; Farina et al., 2007). Current findings not only provide insights into ganglioside-induced autophagic cell death pathways in astrocytes, but also suggest the potential of gangliosides-targeted therapy for CNS pathologies, such as neurodegenerative diseases and gliomas. However, further studies are necessary in order to elucidate the precise molecular mechanisms underlying the ganglioside-induced autophagic cell death of astrocytes, as well as to better understand how gangliosides participate in the control of astrocyte death, in relation to neurons and other glia cell types in brain.
Acknowledgments
We are grateful to Dr EJ Park (National Cancer Center, Koyang, Korea) for providing GM1, GD1a and GT1b. This work was supported by the Korea Science and Engineering Foundation (KOSEF) grant funded by the Korea government (MEST) (No. 2009-0078941) and Bio R&D programme through the Korea Science and Engineering Foundation funded by the Ministry of Education, Science and Technology (2008-04090). JH was supported by the Brain Korea 21 Project in 2008–2009. KS is a recipient of the Korea Research Foundation Grant funded by the Korean Government (MOEHRD, Basic Research Promotion Fund) (KRF-2006-005-J04202).
Glossary
Abbreviations:
- 3-MA
3-methyladenine
- EBSS
Earle's balanced salt solution
- GFP
green fluorescent protein
- LC3
microtubule-associated protein 1 light chain 3
- MAPKs
mitogen-activated protein kinases
- MβCD
methyl β-cyclodextrin
- MDC
monodansylcadaverine
- mTOR
mammalian target of rapamycin
- PARP
poly (ADP-ribose) polymerase
- PI3K
phosphatidylinositol 3-kinase
- ROS
reactive oxygen species
Conflict of interest
The authors have no conflict of interest.
References
- Alexander DE, Ward SL, Mizushima N, Levine B, Leib DA. Analysis of the role of autophagy in replication of herpes simplex virus in cell culture. J Virol. 2007;81:12128–12134. doi: 10.1128/JVI.01356-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, et al. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem. 2001;276:35243–35246. doi: 10.1074/jbc.C100319200. [DOI] [PubMed] [Google Scholar]
- Avrova NF, Victorov IV, Tyurin VA, Zakharova IO, Sokolova TV, Andreeva NA, et al. Inhibition of glutamate-induced intensification of free radical reactions by gangliosides: possible role in their protective effect in rat cerebellar granule cells and brain synaptosomes. Neurochem Res. 1998;23:945–952. doi: 10.1023/a:1021076220411. [DOI] [PubMed] [Google Scholar]
- Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6:505–510. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- Barres BA, Barde Y. Neuronal and glial cell biology. Curr Opin Neurobiol. 2000;10:642–648. doi: 10.1016/s0959-4388(00)00134-3. [DOI] [PubMed] [Google Scholar]
- Biederbick A, Kern HF, Elsasser HP. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol. 1995;66:3–14. [PubMed] [Google Scholar]
- Blennow K, Davidsson P, Wallin A, Fredman P, Gottfries CG, Karlsson I, et al. Gangliosides in cerebrospinal fluid in ‘probable Alzheimer's disease. Arch Neurol. 1991;48:1032–1035. doi: 10.1001/archneur.1991.00530220048018. [DOI] [PubMed] [Google Scholar]
- Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J Cell Sci. 2007;120:4155–4166. doi: 10.1242/jcs.011163. [DOI] [PubMed] [Google Scholar]
- Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2008;15:171–182. doi: 10.1038/sj.cdd.4402233. [DOI] [PubMed] [Google Scholar]
- Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 2005;12(Suppl. 2):1509–1518. doi: 10.1038/sj.cdd.4401751. [DOI] [PubMed] [Google Scholar]
- Coward J, Ambrosini G, Musi E, Truman JP, Haimovitz-Friedman A, Allegood JC, et al. Safingol (L-threo-sphinganine) induces autophagy in solid tumor cells through inhibition of PKC and the PI3-kinase pathway. Autophagy. 2009;5:184–193. doi: 10.4161/auto.5.2.7361. [DOI] [PubMed] [Google Scholar]
- Degterev A, Yuan J. Expansion and evolution of cell death programmes. Nat Rev Mol Cell Biol. 2008;9:378–390. doi: 10.1038/nrm2393. [DOI] [PubMed] [Google Scholar]
- Derry DM, Wolfe LS. Gangliosides in isolated neurons and glial cells. Science. 1967;158:1450–1452. doi: 10.1126/science.158.3807.1450. [DOI] [PubMed] [Google Scholar]
- Ding Y, Ma K, Tsui ZC. Induction of nitric oxide production by ganglioside GM3 in murine peritoneal macrophages activated for tumor cytotoxicity. In Vivo. 1998;12:357–361. [PubMed] [Google Scholar]
- Doelling JH, Walker JM, Friedman EM, Thompson AR, Vierstra RD. The APG8/12-activating enzyme APG7 is required for proper nutrient recycling and senescence in Arabidopsis thaliana. J Biol Chem. 2002;277:33105–33114. doi: 10.1074/jbc.M204630200. [DOI] [PubMed] [Google Scholar]
- Dreyfus H, Guerold B, Freysz L, Hicks D. Successive isolation and separation of the major lipid fractions including gangliosides from single biological samples. Anal Biochem. 1997;249:67–78. doi: 10.1006/abio.1997.2143. [DOI] [PubMed] [Google Scholar]
- Du L, Hickey RW, Bayir H, Watkins SC, Tyurin VA, Guo F, et al. Starving neurons show sex difference in autophagy. J Biol Chem. 2009;284:2383–2396. doi: 10.1074/jbc.M804396200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28:138–145. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Giffard RG, Swanson RA. Ischemia-induced programmed cell death in astrocytes. Glia. 2005;50:299–306. doi: 10.1002/glia.20167. [DOI] [PubMed] [Google Scholar]
- Gisslen M, Hagberg L, Norkrans G, Lekman A, Fredman P. Increased cerebrospinal fluid ganglioside GM1 concentrations indicating neuronal involvement in all stages of HIV-1 infection. J Neurovirol. 1997;3:148–152. doi: 10.3109/13550289709015804. [DOI] [PubMed] [Google Scholar]
- Gomez-Santos C, Ferrer I, Santidrian AF, Barrachina M, Gil J, Ambrosio S. Dopamine induces autophagic cell death and alpha-synuclein increase in human neuroblastoma SH-SY5Y cells. J Neurosci Res. 2003;73:341–350. doi: 10.1002/jnr.10663. [DOI] [PubMed] [Google Scholar]
- Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–2906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- Gozuacik D, Kimchi A. Autophagy and cell death. Curr Top Dev Biol. 2007;78:217–245. doi: 10.1016/S0070-2153(06)78006-1. [DOI] [PubMed] [Google Scholar]
- Guyton KZ, Gorospe M, Kensler TW, Holbrook NJ. Mitogen-activated protein kinase (MAPK) activation by butylated hydroxytoluene hydroperoxide: implications for cellular survival and tumor promotion. Cancer Res. 1996;56:3480–3485. [PubMed] [Google Scholar]
- Harder T. Lipid raft domains and protein networks in T-cell receptor signal transduction. Curr Opin Immunol. 2004;16:353–359. doi: 10.1016/j.coi.2004.03.013. [DOI] [PubMed] [Google Scholar]
- Herrera F, Martin V, Carrera P, Garcia-Santos G, Rodriguez-Blanco J, Rodriguez C, et al. Tryptamine induces cell death with ultrastructural features of autophagy in neurons and glia: Possible relevance for neurodegenerative disorders. Anat Rec A Discov Mol Cell Evol Biol. 2006;288:1026–1030. doi: 10.1002/ar.a.20368. [DOI] [PubMed] [Google Scholar]
- Hsu HY, Wen MH. Lipopolysaccharide-mediated reactive oxygen species and signal transduction in the regulation of interleukin-1 gene expression. J Biol Chem. 2002;277:22131–22139. doi: 10.1074/jbc.M111883200. [DOI] [PubMed] [Google Scholar]
- Iwabuchi K, Zhang Y, Handa K, Withers DA, Sinay P, Hakomori S. Reconstitution of membranes simulating ‘glycosignaling domain’ and their susceptibility to lyso-GM3. J Biol Chem. 2000;275:15174–15181. doi: 10.1074/jbc.275.20.15174. [DOI] [PubMed] [Google Scholar]
- Jou I, Lee JH, Park SY, Yoon HJ, Joe EH, Park EJ. Gangliosides trigger inflammatory responses via TLR4 in brain glia. Am J Pathol. 2006;168:1619–1630. doi: 10.2353/ajpath.2006.050924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagedal K, Zhao M, Svensson I, Brunk UT. Sphingosine-induced apoptosis is dependent on lysosomal proteases. Biochem J. 2001;359:335–343. doi: 10.1042/0264-6021:3590335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda N, Watanabe S. Gangliosides GD1b, GT1b, and GQ1b enhance IL-2 and IFN-gamma production and suppress IL-4 and IL-5 production in phytohemagglutinin-stimulated human T cells. J Immunol. 2001;166:72–80. doi: 10.4049/jimmunol.166.1.72. [DOI] [PubMed] [Google Scholar]
- Kim EH, Kim SU, Choi KS. Rottlerin sensitizes glioma cells to TRAIL-induced apoptosis by inhibition of Cdc2 and the subsequent downregulation of survivin and XIAP. Oncogene. 2005;24:838–849. doi: 10.1038/sj.onc.1208241. [DOI] [PubMed] [Google Scholar]
- Kim EH, Sohn S, Kwon HJ, Kim SU, Kim MJ, Lee SJ, et al. Sodium selenite induces superoxide-mediated mitochondrial damage and subsequent autophagic cell death in malignant glioma cells. Cancer Res. 2007a;67:6314–6324. doi: 10.1158/0008-5472.CAN-06-4217. [DOI] [PubMed] [Google Scholar]
- Kim NH, Kim K, Park WS, Son HS, Bae Y. PKB/Akt inhibits ceramide-induced apoptosis in neuroblastoma cells by blocking apoptosis-inducing factor (AIF) translocation. J Cell Biochem. 2007b;102:1160–1170. doi: 10.1002/jcb.21344. [DOI] [PubMed] [Google Scholar]
- Kim S, Hwang J, Lee WH, Hwang DY, Suk K. Role of protein kinase Cdelta in paraquat-induced glial cell death. J Neurosci Res. 2008;86:2062–2070. doi: 10.1002/jnr.21643. [DOI] [PubMed] [Google Scholar]
- Kim SH, Sharma RP. Cytotoxicity of inorganic mercury in murine T and B lymphoma cell lines: involvement of reactive oxygen species, Ca(2+) homeostasis, and cytokine gene expression. Toxicol In Vitro. 2003;17:385–395. doi: 10.1016/s0887-2333(03)00040-7. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotani M, Kawashima I, Ozawa H, Terashima T, Tai T. Differential distribution of major gangliosides in rat central nervous system detected by specific monoclonal antibodies. Glycobiology. 1993;3:137–146. doi: 10.1093/glycob/3.2.137. [DOI] [PubMed] [Google Scholar]
- Kracun I, Rosner H, Cosovic C, Stavljenic A. Topographical atlas of the gangliosides of the adult human brain. J Neurochem. 1984;43:979–989. doi: 10.1111/j.1471-4159.1984.tb12833.x. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004–1010. doi: 10.1038/nrm2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bras M, Clement MV, Pervaiz S, Brenner C. Reactive oxygen species and the mitochondrial signaling pathway of cell death. Histol Histopathol. 2005;20:205–219. doi: 10.14670/HH-20.205. [DOI] [PubMed] [Google Scholar]
- Ledesma MD, Da Silva JS, Crassaerts K, Delacourte A, De Strooper B, Dotti CG. Brain plasmin enhances APP alpha-cleavage and Abeta degradation and is reduced in Alzheimer's disease brains. EMBO Rep. 2000;1:530–535. doi: 10.1093/embo-reports/kvd107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon A, Dal Toso R, Presti D, Benvegnu D, Facci L, Kirschner G, et al. Development and survival of neurons in dissociated fetal mesencephalic serum-free cell cultures: II. Modulatory effects of gangliosides. J Neurosci. 1988;8:746–753. doi: 10.1523/JNEUROSCI.08-03-00746.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115:2679–2688. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JH, Park JW, Choi KS, Park YB, Kwon TK. Rottlerin induces apoptosis via death receptor 5 (DR5) upregulation through CHOP-dependent and PKC delta-independent mechanism in human malignant tumor cells. Carcinogenesis. 2009;30:729–736. doi: 10.1093/carcin/bgn265. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaurin J, Franklin T, Fraser PE, Chakrabartty A. Structural transitions associated with the interaction of Alzheimer beta-amyloid peptides with gangliosides. J Biol Chem. 1998;273:4506–4515. doi: 10.1074/jbc.273.8.4506. [DOI] [PubMed] [Google Scholar]
- Malisan F, Testi R. GD3 ganglioside and apoptosis. Biochim Biophys Acta. 2002;1585:179–187. doi: 10.1016/s1388-1981(02)00339-6. [DOI] [PubMed] [Google Scholar]
- Maragakis NJ, Rothstein JD. Mechanisms of disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol. 2006;2:679–689. doi: 10.1038/ncpneuro0355. [DOI] [PubMed] [Google Scholar]
- Meuillet E, Cremel G, Dreyfus H, Hicks D. Differential modulation of basic fibroblast and epidermal growth factor receptor activation by ganglioside GM3 in cultured retinal Muller glia. Glia. 1996;17:206–216. doi: 10.1002/(SICI)1098-1136(199607)17:3<206::AID-GLIA3>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Michikawa M, Gong JS, Fan QW, Sawamura N, Yanagisawa K. A novel action of alzheimer's amyloid beta-protein (Abeta): oligomeric Abeta promotes lipid release. J Neurosci. 2001;21:7226–7235. doi: 10.1523/JNEUROSCI.21-18-07226.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol. 2004;36:2491–2502. doi: 10.1016/j.biocel.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Wu G, Ledeen RW. Protection of neuro-2a cells against calcium ionophore cytotoxicity by gangliosides. J Neurosci Res. 1992;31:245–253. doi: 10.1002/jnr.490310205. [DOI] [PubMed] [Google Scholar]
- Nakatsuji Y, Miller RH. Selective cell-cycle arrest and induction of apoptosis in proliferating neural cells by ganglioside GM3. Exp Neurol. 2001;168:290–299. doi: 10.1006/exnr.2000.7602. [DOI] [PubMed] [Google Scholar]
- Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120:4081–4091. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- Oderfeld-Nowak B, Zaremba M. GM1 ganglioside potentiates trimethyltin-induced expression of interleukin-1 beta and the nerve growth factor in reactive astrocytes in the rat hippocampus: an immunocytochemical study. Neurochem Res. 1998;23:443–453. doi: 10.1023/a:1022482106152. [DOI] [PubMed] [Google Scholar]
- Ogier-Denis E, Pattingre S, El Benna J, Codogno P. Erk1/2-dependent phosphorylation of Galpha-interacting protein stimulates its GTPase accelerating activity and autophagy in human colon cancer cells. J Biol Chem. 2000;275:39090–39095. doi: 10.1074/jbc.M006198200. [DOI] [PubMed] [Google Scholar]
- Pattingre S, Bauvy C, Codogno P. Amino acids interfere with the ERK1/2-dependent control of macroautophagy by controlling the activation of Raf-1 in human colon cancer HT-29 cells. J Biol Chem. 2003;278:16667–16674. doi: 10.1074/jbc.M210998200. [DOI] [PubMed] [Google Scholar]
- Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat. 2004;7:97–110. doi: 10.1016/j.drup.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Peres C, Yart A, Perret B, Salles JP, Raynal P. Modulation of phosphoinositide 3-kinase activation by cholesterol level suggests a novel positive role for lipid rafts in lysophosphatidic acid signalling. FEBS Lett. 2003;534:164–168. doi: 10.1016/s0014-5793(02)03832-2. [DOI] [PubMed] [Google Scholar]
- Perez-Ortiz JM, Tranque P, Vaquero CF, Domingo B, Molina F, Calvo S, et al. Glitazones differentially regulate primary astrocyte and glioma cell survival. Involvement of reactive oxygen species and peroxisome proliferator-activated receptor-gamma. J Biol Chem. 2004;279:8976–8985. doi: 10.1074/jbc.M308518200. [DOI] [PubMed] [Google Scholar]
- Petersen A, Larsen KE, Behr GG, Romero N, Przedborski S, Brundin P, et al. Expanded CAG repeats in exon 1 of the Huntington's disease gene stimulate dopamine-mediated striatal neuron autophagy and degeneration. Hum Mol Genet. 2001;10:1243–1254. doi: 10.1093/hmg/10.12.1243. [DOI] [PubMed] [Google Scholar]
- Pyo H, Joe E, Jung S, Lee SH, Jou I. Gangliosides activate cultured rat brain microglia. J Biol Chem. 1999;274:34584–34589. doi: 10.1074/jbc.274.49.34584. [DOI] [PubMed] [Google Scholar]
- Qian W, Liu J, Jin J, Ni W, Xu W. Arsenic trioxide induces not only apoptosis but also autophagic cell death in leukemia cell lines via up-regulation of Beclin-1. Leuk Res. 2007;31:329–339. doi: 10.1016/j.leukres.2006.06.021. [DOI] [PubMed] [Google Scholar]
- Qin ZH, Wang Y, Kegel KB, Kazantsev A, Apostol BL, Thompson LM, et al. Autophagy regulates the processing of amino terminal huntingtin fragments. Hum Mol Genet. 2003;12:3231–3244. doi: 10.1093/hmg/ddg346. [DOI] [PubMed] [Google Scholar]
- Raghu H, Sharma-Walia N, Veettil MV, Sadagopan S, Caballero A, Sivakumar R, et al. Lipid rafts of primary endothelial cells are essential for Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8-induced phosphatidylinositol 3-kinase and RhoA-GTPases critical for microtubule dynamics and nuclear delivery of viral DNA but dispensable for binding and entry. J Virol. 2007;81:7941–7959. doi: 10.1128/JVI.02848-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran L, Simons K. Lipid rafts and membrane dynamics. J Cell Sci. 2005;118:1099–1102. doi: 10.1242/jcs.01681. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Rodden FA, Wiegandt H, Bauer BL. Gangliosides: the relevance of current research to neurosurgery. J Neurosurg. 1991;74:606–619. doi: 10.3171/jns.1991.74.4.0606. [DOI] [PubMed] [Google Scholar]
- Ryu JK, Shin WH, Kim J, Joe EH, Lee YB, Cho KG, et al. Trisialoganglioside GT1b induces in vivo degeneration of nigral dopaminergic neurons: role of microglia. Glia. 2002;38:15–23. doi: 10.1002/glia.10047. [DOI] [PubMed] [Google Scholar]
- Sadasivan S, Waghray A, Larner SF, Dunn WA, Jr, Hayes RL, Wang KK. Amino acid starvation induced autophagic cell death in PC-12 cells: evidence for activation of caspase-3 but not calpain-1. Apoptosis. 2006;11:1573–1582. doi: 10.1007/s10495-006-7690-6. [DOI] [PubMed] [Google Scholar]
- Scherz-Shouval R, Elazar Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007;17:422–427. doi: 10.1016/j.tcb.2007.07.009. [DOI] [PubMed] [Google Scholar]
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991;10:2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacker PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell. 2006;10:175–176. doi: 10.1016/j.ccr.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Seglen PO, Bohley P. Autophagy and other vacuolar protein degradation mechanisms. Experientia. 1992;48:158–172. doi: 10.1007/BF01923509. [DOI] [PubMed] [Google Scholar]
- Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci USA. 1982;79:1889–1892. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemitsu K, Tsujishita Y, Hara K, Nanahoshi M, Avruch J, Yonezawa K. Regulation of translational effectors by amino acid and mammalian target of rapamycin signaling pathways. Possible involvement of autophagy in cultured hepatoma cells. J Biol Chem. 1999;274:1058–1065. doi: 10.1074/jbc.274.2.1058. [DOI] [PubMed] [Google Scholar]
- Sykova E. Glia and volume transmission during physiological and pathological states. J Neural Transm. 2005;112:137–147. doi: 10.1007/s00702-004-0120-4. [DOI] [PubMed] [Google Scholar]
- Takamura A, Higaki K, Kajimaki K, Otsuka S, Ninomiya H, Matsuda J, et al. Enhanced autophagy and mitochondrial aberrations in murine G(M1)-gangliosidosis. Biochem Biophys Res Commun. 2008;367:616–622. doi: 10.1016/j.bbrc.2007.12.187. [DOI] [PubMed] [Google Scholar]
- Takeda M, Shirato I, Kobayashi M, Endou H. Hydrogen peroxide induces necrosis, apoptosis, oncosis and apoptotic oncosis of mouse terminal proximal straight tubule cells. Nephron. 1999;81:234–238. doi: 10.1159/000045282. [DOI] [PubMed] [Google Scholar]
- Takuma K, Baba A, Matsuda T. Astrocyte apoptosis: implications for neuroprotection. Prog Neurobiol. 2004;72:111–127. doi: 10.1016/j.pneurobio.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Terman A, Gustafsson B, Brunk UT. The lysosomal-mitochondrial axis theory of postmitotic aging and cell death. Chem Biol Interact. 2006;163:29–37. doi: 10.1016/j.cbi.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Todde V, Veenhuis M, van der Klei IJ. Autophagy: principles and significance in health and disease. Biochim Biophys Acta. 2009;1792:3–13. doi: 10.1016/j.bbadis.2008.10.016. [DOI] [PubMed] [Google Scholar]
- Vyas KA, Patel HV, Vyas AA, Schnaar RL. Segregation of gangliosides GM1 and GD3 on cell membranes, isolated membrane rafts, and defined supported lipid monolayers. Biol Chem. 2001;382:241–250. doi: 10.1515/BC.2001.031. [DOI] [PubMed] [Google Scholar]
- Xu Y, Kim SO, Li Y, Han J. Autophagy contributes to caspase-independent macrophage cell death. J Biol Chem. 2006;281:19179–19187. doi: 10.1074/jbc.M513377200. [DOI] [PubMed] [Google Scholar]
- Yoon HJ, Jeon SB, Suk K, Choi DK, Hong YJ, Park EJ. Contribution of TLR2 to the initiation of ganglioside-triggered inflammatory signaling. Mol Cells. 2008;25:99–104. [PubMed] [Google Scholar]
- Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- Zhou J, Shao H, Cox NR, Baker HJ, Ewald SJ. Gangliosides enhance apoptosis of thymocytes. Cell Immunol. 1998;183:90–98. doi: 10.1006/cimm.1998.1247. [DOI] [PubMed] [Google Scholar]
- Zhu JH, Guo F, Shelburne J, Watkins S, Chu CT. Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol. 2003;13:473–481. doi: 10.1111/j.1750-3639.2003.tb00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]