

Abstract
Background and purpose:
Recent findings suggest the importance of mast cells in the pathogenesis of rheumatoid arthritis and their potential as a therapeutic target. Tranilast is an anti-allergic compound with a potent membrane-stabilizing effect on mast cells and a wide range of anti-inflammatory effects, thus may be advantageous in the treatment of arthritis. Here, we have evaluated the effects of tranilast on the progression of collagen-induced arthritis in mice.
Experimental approach:
Tranilast (400 mg·kg−1·day−1) was orally administered for 8 weeks to mice with established collagen-induced arthritis. Arthritis was assessed by clinical signs and X-ray scores. In paw tissue, the numbers of mast cells and osteoclasts were measured by histological analysis, and several inflammatory factors were assessed by RT-PCR and Western blot analysis.*
Key results:
TNF-α-positive mast cells were present extensively throughout the inflamed synovium of vehicle-treated arthritic mice, with some mast cells in close proximity to osteoclasts in areas of marked bone and cartilage destruction. Tranilast significantly reduced clinical and X-ray scores of arthritis and decreased numbers of TNF-α-positive mast cells and mRNA levels of TNF-α, chymase (mouse mast cell protease 4), tryptase (mouse mast cell protease 6), stem cell factor, interleukin-6, cathepsin-K, receptor activator of nuclear factor-κB, and of receptor activator of nuclear factor-κB-ligand, but increased interleukin-10 mRNA level in paws of arthritic mice. Osteoclast numbers were decreased by treatment with tranilast.
Conclusions and implications:
Tranilast possesses significant anti-rheumatic efficacy and, probably, this therapeutic effect is partly mediated by inhibition of mast cell activation and osteoclastogenesis.
Keywords: cathepsin-K, chymase, mat cell, osteoclast, RANK, RANKL, SCF, TNF-α, tryptase
Introduction
Rheumatoid arthritis is a chronic autoimmune disorder characterized by inflammation of the joint synovium, resulting in pannus formation and destruction of cartilage and bone (Noss and Brenner, 2008). Although the precise pathogenesis of rheumatoid arthritis remains unknown, synoviocytes, macrophages, lymphocytes and neutrophils are considered to be major contributors to the initiation and development of synovitis and joint destruction. Recent development of new anti-tumour necrosis factor (TNF) biological-response modifiers has led to new therapeutic strategies for rheumatoid arthritis and successful outcomes of clinical trials with these drugs emphasize the importance of TNF-α in the pathogenesis of rheumatoid arthritis (Feldmann and Maini, 2008). The main TNF-α producing cell in synovium has been considered to be the macrophage (Firestein et al., 1990; Chu et al., 1991). However, recent studies have shown that mast cells in human rheumatoid synovial tissue and in the inflamed synovium of mice with collagen-induced arthritis are also important cells producing TNF-α (Juurikivi et al., 2005; Shiota et al., 2006; Eklund, 2007). Notably, the number of mast cells is markedly increased in inflamed synovium of rheumatoid arthritis patients (Bromley et al., 1984; Crisp et al., 1984; Godfrey et al., 1984; Malone et al., 1987). Furthermore, a previous study using mast cell-deficient mice clearly revealed the functional importance of mast cells in the pathogenesis of inflammatory arthritis (Lee et al., 2002). Mast cell-deficient mice were resistant to development of joint inflammation induced by administration of arthritogenic serum from K/BxN mice, and susceptibility to the serum was restored in W/Wv mice by engraftment of mast cells. These results suggest the importance of mast cells in the pathogenesis of rheumatoid arthritis and their potential as a therapeutic target.
Tranilast, N-(3,4-dimethoxycinnamoyl) anthranilic acid, is an effective anti-allergy agent that has been developed in Japan for treatment of diseases such as bronchial asthma, allergic rhinitis and atopic dermatitis (Azuma et al., 1976; Koda et al., 1976). Tranilast is a potent mast cell membrane stabilizer that inhibits the antigen-induced release of mediators from mast cells (Azuma et al., 1976; Komatsu et al., 1988). In addition to its anti-allergy effects, tranilast has been shown to suppress collagen synthesis in human fibroblasts (Suzawa et al., 1992), and its use in the prevention of keloid formation after skin injury has been investigated. Recent studies have further revealed that tranilast inhibits the expression of many cytokines (Platten et al., 2005; Inglis et al., 2007; Pae et al., 2008), chemokines (Chikaraishi et al., 2001; Hida et al., 2008) and proteases (Shimizu et al., 2005; Shimizu et al., 2006) in various cell types including fibroblasts, lymphocytes, macrophages and neutrophils. Interestingly, studies have also revealed that tranilast inhibits the proliferation of B-cells and T-cells, and shifts the cytokine profile of T-cells from Th1 to Th2 (Platten et al., 2005; Inglis et al., 2007). These properties of tranilast may be advantageous in the treatment of arthritis. Recent studies showed that short-term treatment with tranilast reduces the severity of the acute stage of collagen-induced arthritis (Inglis et al., 2007) and inhibits the onset of adjuvant-induced arthritis and streptococcal cell wall-induced arthritis in rats (Nagate et al., 2007). However, it is not known whether tranilast is effective also in the treatment of established arthritis. Furthermore, the precise mechanism underlying the therapeutic effect of tranilast on arthritis is not clear. Therefore, in the present study we administered tranilast for 8 weeks to mice with established arthritis, and evaluated its effects on the progression of chronic collagen-induced arthritis in mice.
Methods
Animal preparation and drug administration
All animal care and experimental procedures were in accordance with the Guiding Principles for the Care and Use of Laboratory Animals and approved by the Committee of Shimane University School of Medicine. Male DBA/1J mice (8 weeks of age; 18–20 g) were obtained from Charles River Japan Inc. (Kanagawa, Japan) and housed under conventional animal laboratory conditions with standard chow and tap water available ad libitum. Collagen-induced arthritis was induced immunologically as described previously (Shiota et al., 2006). Briefly, two milligrams of bovine type II collagen (Sigma, St. Louis, MO, USA) was diluted in 1 mL of 10 mM acetic acid and suspended in 1 mL of Freund's complete adjuvant (Difco Lab., Detroit, MI, USA). The emulsion was injected intradermally (0.1 mL per mouse) into the root of the tail, and a booster injection was given 21 days after the initial immunization. Initial symptoms were observed within 1 week after the booster injection. Three weeks after the booster injection, the mice that showed established arthritis were divided randomly into two groups. One group (n= 12) was given tranilast (provided by Kissei Pharmaceutical Co., Ltd., Nagano, Japan) orally (400 mg·kg−1·day−1, once a day) for 8 weeks. There were no apparent adverse effects. The other group (n= 12) received vehicle during the same period. A previous report showed that adjuvant-induced arthritis and streptococcal cell wall-induced arthritis in rats were inhibited by oral administration of tranilast at a dose of 150 or 300 mg·kg−1 (twice daily) (Nagate et al., 2007). Based on these results, we selected a dose of 400 mg·kg−1·day−1 of tranilast for our study. Mice without immunization with collagen served as a non-immunized normal control group (n= 12).
Collagen-induced arthritis was graded by scoring clinical signs by observers unaware of the treatments, once a week according to criteria described previously (0, normal; 1, swelling in only one toe; 2, swelling in two or more than two toes but not the entire paw; 3, swelling in the entire paw; total score, 0–12 per mouse) (Shiota et al., 2006). At the end of the experiment, all four paws of each mouse were X-rayed using a CMB-2 unit (Softex Co., Ltd., Tokyo, Japan). X-ray scores for joint destruction in each paw were graded by observers unaware of the treatments, as described previously (0, normal; 1, destruction in one joint; 2, destruction in two or more than two joints, but not all joints; 3, destruction in all joints; total score 0–12 per mouse) (Shiota et al., 2006). After animals were killed by administration of an overdose of pentobarbital, forepaws were collected for histological analysis and determinations of mRNA levels of TNF-α, mouse mast cell protease 4 (mMCP4), mouse mast cell protease 6 (mMCP6), stem cell factor (SCF), interleukin (IL)-6, IL-10, cathepsin-K, receptor activator of nuclear factor-κB (RANK) and RANK-ligand (RANKL).
Histological analysis
Longitudinal sections including toe III of the right forepaws were prepared. Tissues were fixed with neutral buffered formalin, paraffin-embedded and cut into 5 µm thick sections. Using a light microscope, morphological characteristics of paw sections from collagen-induced arthritic mice treated with tranilast or vehicle, and from non-immunized normal control mice were analysed. Toluidine blue staining was used to identify mast cells in tissue sections (Shiota et al., 2006). Reacting with toluidine blue, mast cells showed metachromasia, which can be seen as a red-purple colour. Tartrate-resistant acid phosphatase (TRAP) staining was used to identify osteoclasts in tissue sections. Histochemical staining for TRAP was carried out using a commercially available kit (Sigma-Aldrich, Tokyo, Japan). The total numbers of mast cells and osteoclasts around metacarpo-phalangeal joint area of toe III were counted, and the densities of mast cells and osteoclasts were determined as the total number of cells per square millimeter of the total measurement area. For detection of TNF-α and cathepsin-K in paraffin sections of the paws, polyclonal rabbit-antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were used. Paraffin sections were treated overnight at 4°C with primary antibodies against TNF-α (1:250 dilution) or cathepsin-K (1:200 dilution). Normal rabbit IgG in concentrations similar to those used for the primary antibodies was substituted for each primary antibody and produced negative results (control for antibody specificity). The antibodies were then detected with a DAKO ENVISION kit (Dakocytomation, Tokyo, Japan) by standard methods and visualized with diaminobenzidine substrate (Dakocytomation). To distinguish mast cells expressing TNF-α from those that were not expressing TNF-α, tissue sections were first stained with toluidine blue and photographed, then toluidine blue staining was removed by washing with 100% ethanol, and same sections were stained with TNF-α antibody and photographed again. Cells showing both metachromasia and brown stain were identified as TNF-α-positive mast cells.
Quantitative reverse transcriptase polymerase chain reaction (RT-PCR) analysis
Total RNA was isolated from left forepaws, and mRNA levels for mMCP4, TNF-α and SCF were measured by the quantitative RT-PCR method established previously (Kakizoe et al., 2001; Shiota et al., 2006). mRNA levels for mMCP6, IL-6, IL-10, cathepsin-K, RANK and RANKL were measured by the newly established quantitative RT-PCR method. Briefly, to amplify mMCP6 two primers were selected from the 5′ and 3′ regions of the coding sequence (sense primer: 5′-GGGCGACATTGATAATGACGAG-3′; antisense primer: 5′-AGGTCTCAGGAATGCTCAGG-3′). The competitor DNA for mMCP6 was prepared by inserting a 143 bp external DNA fragment at the BamHI site. The PCR products for target mMCP6 and the competitor were 354 and 497 bp respectively. To amplify mouse IL-6 two primers were selected from the 5′ and 3′ regions of the coding sequence (sense primer: 5′-AATTTCCTCTGGTCTTCTGGAG-3′; antisense primer: 5′-CTTAGGCATAACGCACTAGG-3′). The competitor DNA for IL-6 was prepared by inserting a 129 bp external DNA fragment at the BglII site. The PCR products for target IL-6 and the competitor were 316 and 445 bp respectively. To amplify mouse IL-10 two primers were selected from the 5′ and 3′ regions of the coding sequence (sense primer: 5′-TTACCTCTGATACCTCAGTTCC-3′; antisense primer: 5′-CTGAACTCAGGGATGAGAGC-3′). The competitor DNA for IL-10 was prepared by inserting a 129 bp external DNA fragment at the HindIII site. The PCR products for target IL-10 and the competitor were 289 and 418 bp respectively. To amplify mouse cathepsin-K two primers were selected from the 5′ and 3′ regions of the coding sequence (sense primer: 5′-CTGCCTTCCAATACGTGCAGC-3′; antisense primer: 5′-GCATGGTTCACATTATCACGGTCAC-3′). The competitor DNA for cathepsin-K was prepared by inserting a 125 bp external DNA fragment at the HindIII site. The PCR products for target cathepsin-K and the competitor were 280 and 405 bp respectively. To amplify mouse RANK, two primers were selected from the 5′ and 3′ regions of the coding sequence (sense primer: 5′-GACGTTCACACTGGTCAGCG-3′; antisense primer: 5′-CAGTGAAGTCACAGCCCTCAG-3′). The competitor DNA for RANK was prepared by inserting a 129 bp external DNA fragment at the XhoI site. The PCR products for target TNF-α and the competitor were 242 and 371 bp respectively. To amplify mouse RANKL, two primers were selected from the 5′ and 3′ regions of the coding sequence (sense primer: 5′-CATCAATGCTGCCAGCATCC-3′; antisense primer: 5′-CTGAAGATAGTCTGTAGGTACGC-3′). The competitor DNA for RANKL was prepared by inserting a 129 bp external DNA fragment at the KpnI site. The PCR products for target RANKL and the competitor were 205 and 334 bp respectively. Competitive RT-PCR was performed in 100 µL of PCR buffer containing 2 µL of the RT reaction mixture, 25 pmol of the sense and antisense primers, 100 µM each deoxynucleotide, 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, 2.5 units of Taq DNA polymerase and competitor DNA. The integrated density of PCR products was measured as described previously (Kakizoe et al., 2001; Shiota et al., 2006).
Western blot analysis for TNF-α in paw tissues
Right hindpaws were homogenized in phosphate buffer saline supplemented with 5 mM EDTA, 0.05% Tween-20, 1 mM aprotinin and 1 mM phenylmethylsulfonyl fluoride. The homogenates were centrifuged at 12 000×g for 10 min, and supernatants were used for Western blot analysis. Total protein concentration was measured using bicinchoninic acid protein assay reagent (Pierce, Rockford, IL, USA). Supernatants were mixed with equal volume of lysis buffer (500 mM Tris-HCl, 10% SDS, 1% bromophenol blue, 20% glycerol, 1 M DTT) followed by heating at 100°C for 5 min. Then 20 µg of each sample was loaded on 12.5% polyacrylamide gel. The separated proteins were transferred onto Immobilon membranes (Millipore, Billerica, MA, USA). The membranes were treated with 1% skim milk, followed by incubation with rabbit primary antibody against TNF-α (1:1000 dilution) (Pierce) overnight at 4°C. Then the membranes were incubated for 1 h at room temperature with horseradish peroxidase-conjugated secondary antibody (KPL, Gaithersburg, MD, USA). TNF-α proteins were visualized after incubation of the membranes in SuperSignal Chemiluminescent substrate solution (Pierce), and the intensity of the positive signals was quantified with imaging analysis software (Gel-Pro Analyzer, Media Cybernetics, Silver Spring, MD, USA).
Statistical analysis
All data are expressed as means ± SEM. Numerical data were statistically examined by analysis of variance followed by post hoc Bonferroni/Dunn's test. Clinical and X-ray scores were analysed by non-parametric statistics (Kruskal-Wallis test). Differences were considered significant at a level of P < 0.05.
Results
Effect of tranilast on clinical scores and X-ray scores of collagen-induced arthritis
Initial symptoms such as erythema and swelling were observed 1 week after the booster injection. Three weeks after the booster injection, mice with established collagen-induced arthritis were selected and divided randomly into two groups and treated for 8 weeks, one with tranilast and the other with vehicle. Clinical scores of vehicle-treated arthritic mice increased in a time-dependent manner and reached a maximum 7–8 weeks after beginning vehicle treatment (Figure 1A). Treatment with tranilast blunted the increase in clinical scores as soon as 1 week after the start of tranilast administration. Clinical scores by 3–8 weeks after the start of tranilast treatment were significantly lower than in the vehicle-treated group (P < 0.05, Figure 1A). At the end of the experiment, X-ray pictures of all four paws showed severe destruction of the phalangeal joints in the vehicle-treated group (Figure 2B), and less destruction in the tranilast-treated group (Figure 2C). X-ray scores were significantly lower in the tranilast-treated group (P < 0.05, Figure 1B).
Figure 1.
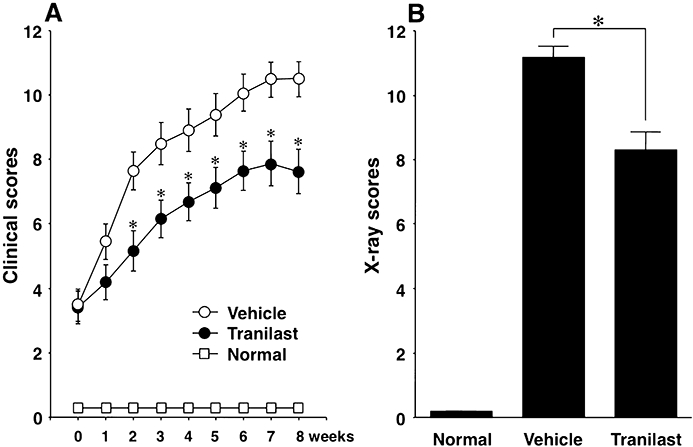
Effect of tranilast on the clinical scores and X-ray scores of mice with collagen-induced arthritis. Arthritic mice were treated with tranilast or vehicle for 8 weeks, starting 3 weeks after booster injection of collagen, and their clinical scores were measured once a week during the same period (A). At the end of the experiment, all four paws of each mouse were X-rayed, and X-ray scores for the destruction of joints of four paws were assessed (B). Results are shown as the mean ± SEM for 12 animals in each group. *P < 0.05 tranilast versus vehicle treatment. Normal, non-immunized normal control mice; Tranilast, tranilast-treated mice with collagen-induced arthritis; Vehicle, vehicle-treated mice with collagen-induced arthritis.
Figure 2.
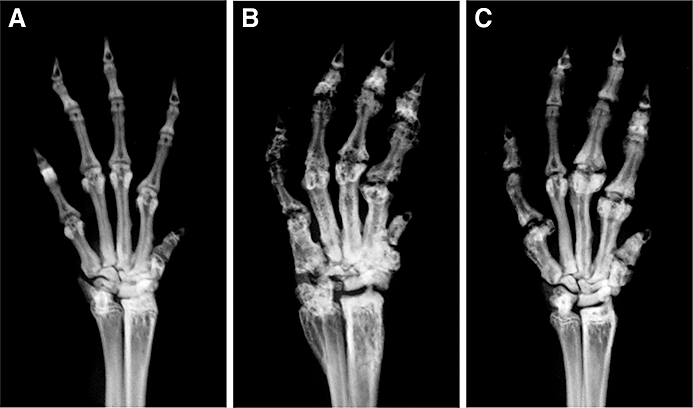
Representative X-ray photographs of left forepaws. At the end of the experiment, mice were X-rayed. Representative X-ray photographs of the left forepaw from a non-immunized normal control mouse (A), vehicle-treated collagen-induced arthritic mouse (B) and tranilast-treated collagen-induced arthritic mouse (C) are depicted. All joints were severely damaged in the vehicle-treated collagen-induced arthritic mouse (B). However, parts of joints remained intact in tranilast-treated collagen-induced arthritic mouse (C).
Localization of mast cells and osteoclasts and immunohistochemical analysis of TNF-α and cathepsin-K
In normal paws, mast cells congregated mainly in the dermis and subcutaneous loose connective tissue layers. Only a few mast cells were found in normal synovium. In the inflamed paws of vehicle-treated arthritic mice, large numbers of mast cells were present throughout the inflamed synovium (Figure 3). Immunohistochemical analysis of the inflamed synovium revealed strong staining of TNF-α in mast cells (Figure 3B), which were also found close to the bone destruction-pannus junction (Figure 3B). Weak staining of TNF-α was also detected in the lining cells, mononuclear cells and vascular endothelial cells of the inflamed synovium. In normal paws, a few osteoclasts were found along the bone and cartilage resorption lacunae, but no osteoclasts were detected on the surface of bones. In inflamed paws of vehicle-treated arthritic mice, large numbers of TRAP-positive osteoclasts were present in the region of bone destruction (Figure 3D). Osteoclasts and mononuclear cells in the inflamed synovium were immunopositive for cathepsin-K (Figure 3F). Interestingly some of the mast cells were in the vicinity of osteoclasts (Figure 3C and E).
Figure 3.
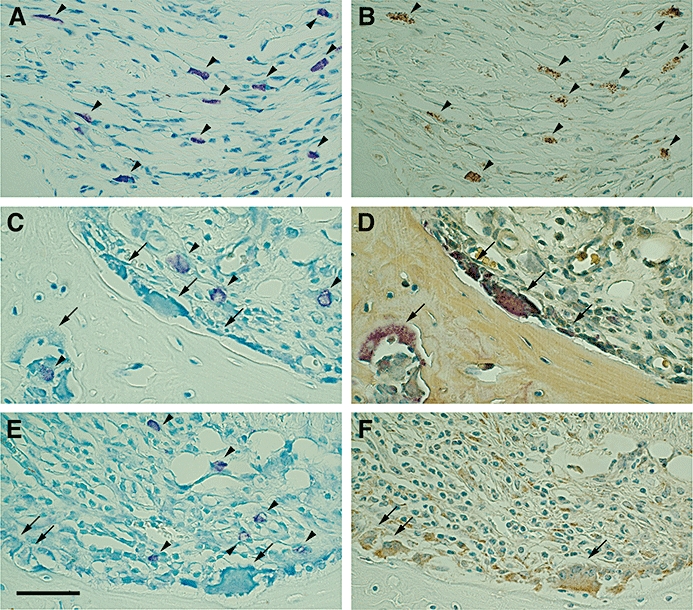
Localization of mast cells and osteoclasts in the inflamed paws of collagen-induced arthritic mice, and immunohistochemical analysis of tumour necrosis factor-α (TNF-α) and cathepsin-K. Representative photographs of toluidine blue staining (A, C and E), tartrate-resistant acid phosphatase (TRAP) staining (D), and immunostaining for TNF-α (B) or cathepsin-K (F) in paw sections from vehicle-treated arthritic mice. Reacting with toluidine blue, mast cells were metachromatically stained red-purple. Mast cells were present extensively throughout the inflamed synovium (A, C and E). After de-staining the toluidine blue-stained sections by washing with 100% ethanol, the same sections were used for staining for TNF-α and TRAP. TNF-α immunopositivity was indicated by the brown colour (B). Mast cells in the inflamed synovium were strongly stained with antibody against TNF-α (B). In TRAP-stained tissue sections, osteoclasts were identified by the red colour (D). To identify cathepsin-K localization, adjacent sections were used. Cathepsin-K immunopositivity was indicated by a brown colour and detected in osteoclasts and mononuclear cells in inflamed synovium (F). Some mast cells were closely localized to TRAP-positive osteoclasts (C,D) or cathepsin-K-positive osteoclasts (E,F). Arrowheads indicate mast cells. Arrows indicate osteoclasts. Scale bar = 50 µm.
Effect of tranilast on the number of mast cells and osteoclasts
The total number of mast cells (TNF-α-positive mast cells plus TNF-α-negative mast cells) and number of TNF-α-positive mast cells in the inflamed paws of vehicle-treated arthritic mice were 2.0 and 2.8 times higher, respectively, than the number found in paws of non-immunized normal control mice (Figure 4A). Tranilast treatment (compared with vehicle treatment) decreased these numbers by 25% and 41% respectively (P < 0.05; Figure 4A). The number of osteoclasts in the inflamed paws of vehicle-treated arthritic mice was 18 times higher than the number of osteoclasts in paws of normal control mice (Figure 4B). Tranilast treatment (relative to vehicle treatment) decreased this number by 63% (P < 0.05; Figure 4B).
Figure 4.
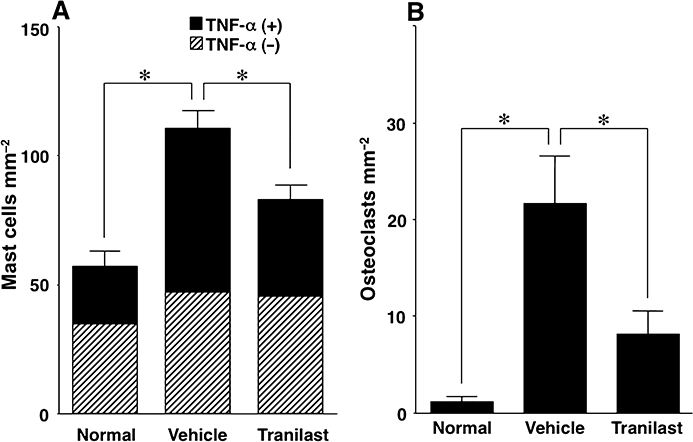
Effect of tranilast on the number of mast cells and osteoclasts in the inflamed paws of collagen-induced arthritic mice. Mast cells (A) and osteoclasts (B) of vehicle-treated (Vehicle), tranilast-treated (Tranilast) collagen-induced arthritic mice and non-immunized normal control mice (Normal) were counted. The tumour necrosis factor-α (TNF-α) positive (+) mast cells can be distinguished from TNF-α negative (−) mast cells. The total number of mast cells, the number of TNF-α (+) mast cells and the number of osteoclasts in inflamed paws were higher in the paws of vehicle-treated arthritic mice than in the paws of normal controls, and were decreased by treatment with tranilast. Results are shown as the mean ± SEM for 12 animals in each group. *P < 0.05.
Effect of tranilast on mMCP4, mMCP6, SCF, TNF-α, IL-6, IL-10, RANK, RANKL and cathepsin-K mRNA levels
Tranilast treatment significantly decreased the mRNA levels of mMCP4, mMCP6, SCF, TNF-α, IL-6, RANK, RANKL, and of cathepsin-K, all of which were elevated in inflamed paws of vehicle-treated arthritic mice compared with normal control mice (P < 0.05; Figures 5 and 6). IL-10 mRNA level was also elevated in inflamed paws compared with normal paws, but tranilast treatment further increased IL-10 mRNA level (P < 0.05; Figure 5).
Figure 5.
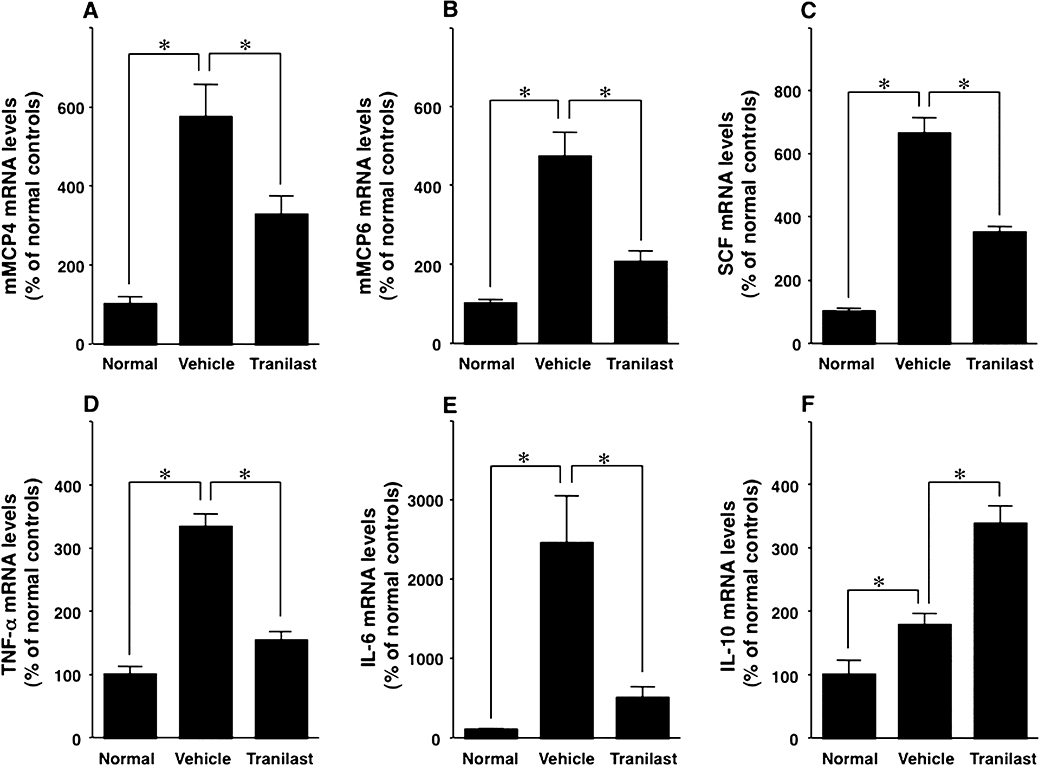
Effect of tranilast on mouse mast cell protease (mMCP)4, mMCP6, stem cell factor (SCF), tumour necrosis factor (TNF)-α, interleukin (IL)-6 and IL-10 mRNA levels in the inflamed paws of collagen-induced arthritic mice. Competitive RT-PCR was used to determine mRNA levels of mMCP4 (A), mMCP6 (B), SCF (C), TNF-α (D), IL-6 (E) and IL-10 (F) in non-immunized normal control mice (Normal), vehicle-treated collagen-induced arthritic mice (Vehicle) and tranilast-treated (Tranilast) collagen-induced arthritic mice. These mRNA levels were higher in inflamed paws than in normal paws. The increase of these mRNA levels except for IL-10 was inhibited by tranilast treatment. IL-10 mRNA level was increased by tranilast treatment. Results are shown as the mean ± SEM for 12 animals in each group. *P < 0.05.
Figure 6.
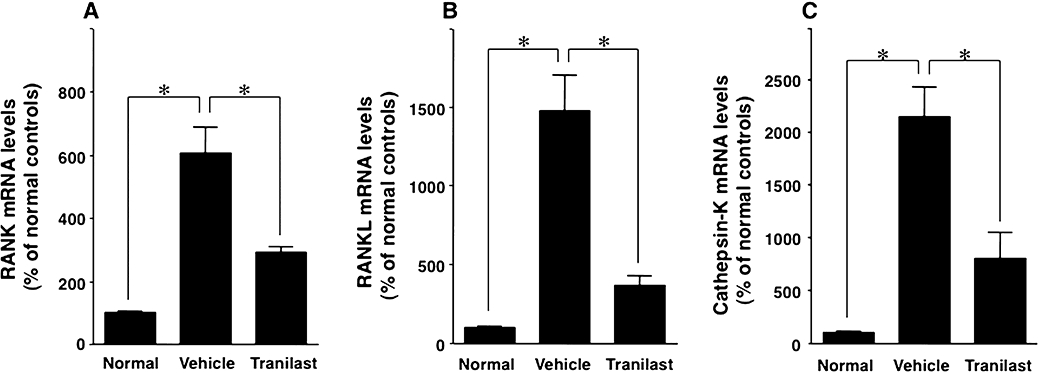
Effect of tranilast on receptor activator of nuclear factor-κB (RANK), RANK-ligand (RANKL) and cathepsin-K mRNA levels in the inflamed paws of collagen-induced arthritic mice. RT-PCR was used to determine mRNA levels of RANK (A), RANKL (B) and cathepsin-K (C) in non-immunized normal control mice (Normal), vehicle-treated collagen-induced arthritic mice (Vehicle) and tranilast-treated (Tranilast) collagen-induced arthritic mice. These levels were higher in the inflamed paws than in normal paws, and this increase was inhibited by treatment with tranilast. Results are shown as the mean ± SEM for 12 animals in each group. *P < 0.05.
Western blot analysis for TNF-α in paw tissues
Tumour necrosis factor-α protein levels in inflamed paws of vehicle-treated collagen-induced arthritic mice were 2.0 times higher (P < 0.05) than those in non-immunized normal control mice (Figure 7). Tranilast treatment blunted the increase in TNF-α protein levels in the inflamed paws of collagen-induced arthritic mice to 67% (P < 0.05) of that observed in vehicle-treated collagen-induced arthritic mice (Figure 7).
Figure 7.
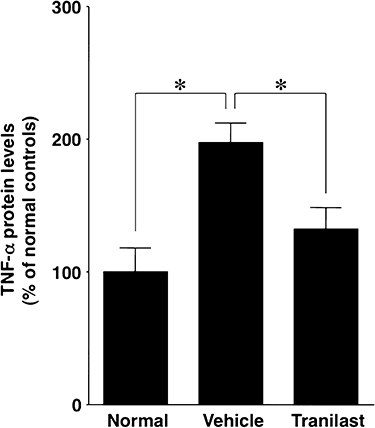
Western blot analysis for tumour necrosis factor-α (TNF-α) in paw tissues. TNF-α protein levels of non-immunized normal control mice (Normal), vehicle-treated collagen-induced arthritic mice (Vehicle) and tranilast-treated (tranilast) collagen-induced arthritic mice were measured by Western blot analysis. TNF-α protein levels in inflamed paws were increased in vehicle-treated mice compared with normal controls, and that increase was inhibited by tranilast treatment. Results are shown as the mean ± SEM for 12 animals in each group. *P < 0.05.
Discussion
The present study demonstrates that long-term (8 weeks) treatment with tranilast effectively prevented the progression of chronic collagen-induced arthritis in mice. Tranilast inhibited gene expression of mast cell-specific proteases, chymase and tryptase, and decreased the number of TNF-α-positive mast cells in the inflamed paws of mice. Furthermore, tranilast decreased the number of osteoclasts, and suppressed the mRNA levels of TNF-α, RANK, RANKL and cathepsin-K. Thus, tranilast may have various beneficial therapeutic effects beyond a simple membrane-stabilizing effect on mast cells.
Tumour necrosis factor-α is a key regulator of immune-inflammatory homeostasis in rheumatoid arthritis (Feldmann and Maini, 2008), and mast cells are one of the important cell types producing TNF-α (Juurikivi et al., 2005; Shiota et al., 2006; Eklund, 2007). Our present study shows that the number of TNF-α-positive mast cells was markedly increased in the inflamed paws of collagen-induced arthritic mice and long-term treatment with tranilast decreased the number of mast cells. Although developed as a membrane stabilizer for mast cells, tranilast may also inhibit proliferation and/or migration of mast cells. Accordingly, tranilast treatment has been shown to prevent the increase of mast cell counts in the adventitia of injured arteries (Shiota et al., 1999) and to reduce mast cell counts in urticaria pigmentosa lesions (Katoh et al., 1996). Cell-to-cell interaction between fibroblasts and mast cells via regulation of the SCF/c-kit signalling system is one of the most important determinants of the mast cell number in local tissues. Synovial membranes in human arthritis express SCF (Ceponis et al., 1998) and a recent report showed that TNF-α promotes SCF mRNA expression in cultured synovial fibroblasts from rheumatoid arthritis patients. Exposure of synovial fibroblasts to TNF-α also increased the migration of HMC-1 cells (Kiener et al., 2000). Our present study showed that TNF-α and SCF mRNA levels in inflamed paws of vehicle-treated arthritic mice were significantly higher than in normal control mice. Importantly, the marked tranilast-induced decrease in SCF mRNA levels in these inflamed paws was concurrent with decreases in the number of mast cells and TNF-α mRNA levels. Thus, tranilast may decrease the number of mast cells by preventing the production of mast cell derived-TNF-α (an up-regulator of SCF expression in the synovial layer). However, tranilast has direct inhibitory effects on fibroblasts (Suzawa et al., 1992; Hida et al., 2008). Therefore we could not exclude the possibility that tranilast acts directly on synovial fibroblasts to inhibit SCF expression. In addition to mast cells, macrophages and T-cells produce TNF-α in the synovium of patients with arthritis (Chu et al., 1991). Recently, it was reported that tranilast diminished TNF-α production from cultured macrophages (Pae et al., 2008) and T-cells (Platten et al., 2005), and inhibited NF-κB-dependent transcriptional activation in endothelial cells (Spiecker et al., 2002). Therefore, the anti-inflammatory effect of tranilast may be mediated by inhibition of gene expression of TNF-α and other pro-inflammatory cytokines regulated by NF-κB-dependent transcriptional activation. An earlier study revealed that tranilast shifted the cytokine profile of T-cells from Th1 to Th2 (Platten et al., 2005). Our present study showed that tranilast treatment significantly decreased the mRNA for the Th1 cytokine IL-6 level but increased that for the anti-inflammatory Th2 cytokine IL-10, in the inflamed paws of arthritic mice. These results suggest that tranilast treatment also shifts the cytokine profile from Th1 to Th2 in the inflamed paws of arthritic mice. Further detailed analysis of several key cytokine levels in tissue and also in serum is necessary to clarify this anti-inflammatory effect of tranilast.
Mast cells also produce specific neutral proteases such as chymase and tryptase, which most likely participate in the inflammatory response by hydrolysing a variety of substrates including extracellular matrix, cytokines and metalloproteases (Pejler et al., 2007). Furthermore, chymase and tryptase have been shown to induce the recruitment of leukocytes (He and Walls, 1998;Shin et al., 2009). Therefore recent studies have showed the importance of these proteases in the pathogenesis of arthritis. Mast cells are classified into two subsets based on their neutral protease content. Human mast cells containing tryptase-β and chymase (MCTC) are connective-tissue-type mast cells, and mast cells containing only tryptase-β (MCT) are the predominant type in mucosal and epithelial surfaces. In human rheumatoid synovium, both types of mast cells are increased (Gotis-Graham and McNeil, 1997). The MCT subset is associated with areas of inflammatory cell infiltration in early rheumatoid arthritis, while the MCTC subset is associated with areas of highly cellular fibrous tissue in more severe chronic rheumatoid arthritis (Gotis-Graham and McNeil, 1997; Gotis-Graham et al., 1998). Humans have a single chymase gene and express a simple chymase isoform in connective-tissue-type mast cells, but mice have more than one chymase isoform. The mMCP4 is specifically expressed by mature connective-tissue-type mast cells, and can be regarded as functionally equivalent to human chymase (Kakizoe et al., 2001; Andersson et al., 2008). Tryptase-β is the most abundant protease in human mast cell granules, and the mouse orthologue of human tryptase-β is mMCP6, which is present in the synovium of arthritic mice (Shin et al., 2009). A recent study showed that collagen-induced arthritis was less severe in mMCP4-deficient mice than in wild-type control mice (Magnusson et al., 2009). Furthermore, mMCP6-deficient mice also developed less severe arthritis than wild-type control mice. In addition, neutrophil infiltration into the arthritic synovium was significantly decreased in mMCP6-deficient mice compared with controls (Shin et al., 2009). These studies showed that chymase and tryptase play important roles in the development of arthritis. Our present study showed that mRNA levels of mMCP4 and mMCP6 were markedly higher in the inflamed paws of collagen-induced arthritic mice than in the paws of non-immunized normal controls. The rate of increase in the mRNA level was clearly higher than the rate of increase in the total number of mast cells. Interestingly, the total number of mast cells was only 25% less in tranilast-treated than vehicle-treated mice, but the levels of mMCP4 and mMCP6 mRNAs were decreased 43% and 57%, respectively, by tranilast treatment. Thus, tranilast not only suppressed proliferation and/or migration of mast cells, but also inhibited gene expression of mast cell-derived factors. The ability of tranilast to regulate several key functions of mast cells makes it a unique mast cell-controlling agent. The limitation of the present study is that we did not study the effect of tranilast on mast cell density in normal control mice. Therefore we could not exclude the possibility that tranilast treatment also affects the mast cell density in normal control mice. However, several previous studies have shown that chronic treatment with tranilast does not induce significant effect on various factors including mast cell density of normal control animals (Shiota et al., 1999; Hara et al., 2002; Bonnet et al., 2003). Tranilast effectively inhibits the function of activated mast cells, whereas tranilast may not induce major effects on mast cells in normal control mice.
Other important findings of the present study are that mast cells are present in large numbers throughout the inflamed synovium, and that some mast cells gather near osteoclasts in regions of bone destruction. Mast cells are also often closely associated with osteoclasts in human rheumatoid joints (Crisp et al., 1984). Significant amounts of presynthesized mediators such as histamine, TNF-α and proteases are stored in the intracellular granules of mast cells. Therefore, once mast cells are activated and degranulated, mast cell-derived mediators may have considerable effects on the surrounding cells. Patients with systemic mastocytosis, which is characterized by abnormal proliferation of mast cells especially in the skin and bone marrow, have increased bone turnover in the regions of mast cell accumulation (Fallon et al., 1981). Furthermore, in mast cell-deficient mice the recruitment of osteoclasts and osteoblast progenitors is delayed and normal bone remodelling is impaired (Silberstein et al., 1991). The RANK/RANKL signalling pathway is essential for osteoclast development (Leibbrandt and Penninger, 2008). TNF-α is one of the most important activators of the RANK/RANKL signalling pathway, but other mast cell-derived factors are also involved in osteoclastogenesis. Histamine (the most abundant mediator in mast cells) activates osteoclastogenesis through increasing RANKL expression in osteoblasts (Deyama et al., 2002; Biosse-Duplan et al., 2009) and promotes osteoclast differentiation (Biosse-Duplan et al., 2009). Moreover, osteoblast attachment to extracellular matrix is impaired by treatment with chymase (Banovac et al., 1993). Interestingly, a recent study showed that mast cells also express RANKL (Ali et al., 2006). Taken together, these observations suggest that mast cell-derived factors may be involved in bone metabolism. Our present study showed that tranilast decreased the number of osteoclasts and mRNA levels of RANK, RANKL and cathepsin-K. Although the precise mechanism is not clear, it seems that tranilast inhibits osteoclastogenesis. Tranilast may indirectly regulate bone metabolism through inhibiting the production of various mast cell-derived factors, but the results of the present study cannot exclude the possibility that tranilast also acts directly on synovial fibroblasts, osteoblasts and osteoclasts.
In conclusion, long-term treatment of collagen-induced arthritic mice with tranilast effectively attenuated the progression of arthritis. Our study showed that the therapeutic effects of tranilast may be due partly to suppression of mast cell functions, but possibly also to direct effects on osteoclasts and stromal cells thus inhibiting pannus formation and osteoclastogenesis. Further detailed in vitro experiments are warranted to uncover the precise mechanism of tranilast in arthritis therapy.
Glossary
Abbreviations:
- mMCP
mouse mast cell protease
- RANK
receptor activator of nuclear factor-κB
- RANKL
RANK-ligand
- SCF
stem cell factor
- TNF
tumour necrosis factor
- TRAP
tartrate-resistant acid phosphatase
Conflicts of interest
N Shibata is an employee of Kissei Pharmaceutical Co., Ltd.
References
- Ali AS, Lax AS, Liljeström M, Paakkari I, Ashammakhi N, Kovanen PT, et al. Mast cells in atherosclerosis as a source of the cytokine RANKL. Clin Chem Lab Med. 2006;44:672–674. doi: 10.1515/CCLM.2006.117. [DOI] [PubMed] [Google Scholar]
- Andersson MK, Karlson U, Hellman L. The extended cleavage specificity of the rodent beta-chymases rMCP-1 and mMCP-4 reveal major functional similarities to the human mast cell chymase. Mol Immunol. 2008;45:766–775. doi: 10.1016/j.molimm.2007.06.360. [DOI] [PubMed] [Google Scholar]
- Azuma H, Banno K, Yoshimura T. Pharmacological properties of N-(3′,4′-dimethoxycinnamoyl) anthranilic acid (N-5′), a new anti-atopic agent. Br J Pharmacol. 1976;58:483–488. doi: 10.1111/j.1476-5381.1976.tb08614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banovac K, Banovac F, Yang J, Koren E. Interaction of osteoblasts with extracellular matrix: effect of mast cell chymase. Proc Soc Exp Biol Med. 1993;203:221–235. doi: 10.3181/00379727-203-43595. [DOI] [PubMed] [Google Scholar]
- Biosse-Duplan M, Baroukh B, Dy M, de Vernejoul MC, Saffar JL. Histamine promotes osteoclastogenesis through the differential expression of histamine receptors on osteoclasts and osteoblasts. Am J Pathol. 2009;174:1426–1434. doi: 10.2353/ajpath.2009.080871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet F, Cao Z, Cooper ME, Cox AJ, Kelly DJ, Gilbert RE. Tranilast attenuates vascular hypertrophy, matrix accumulation and growth factor overexpression in experimental diabetes. Diabetes Metab. 2003;29:386–392. doi: 10.1016/s1262-3636(07)70049-6. [DOI] [PubMed] [Google Scholar]
- Bromley M, Fisher WD, Woolley DE. Mast cells at sites of cartilage erosion in the rheumatoid joint. Ann Rheum Dis. 1984;43:76–79. doi: 10.1136/ard.43.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceponis A, Konttinen YT, Takagi M, Xu JW, Sorsa T, Matucci-Cerinic M, et al. Expression of stem cell factor (SCF) and SCF receptor (c-kit) in synovial membrane in arthritis: correlation with synovial mast cell hyperplasia and inflammation. J Rheumatol. 1998;25:2304–2314. [PubMed] [Google Scholar]
- Chikaraishi A, Hirahashi J, Takase O, Marumo T, Hishikawa K, Hayashi M, et al. Tranilast inhibits interleukin-1beta-induced monocyte chemoattractant protein-1 expression in rat mesangial cells. Eur J Pharmacol. 2001;427:151–158. doi: 10.1016/s0014-2999(01)01215-8. [DOI] [PubMed] [Google Scholar]
- Chu CQ, Field M, Feldmann M, Maini RN. Localization of tumor necrosis factor alpha in synovial tissues and at the cartilage-pannus junction in patients with rheumatoid arthritis. Arthritis Rheum. 1991;34:1125–1132. doi: 10.1002/art.1780340908. [DOI] [PubMed] [Google Scholar]
- Crisp AJ, Chapman CM, Kirkham SE, Schiller AL, Krane SM. Articular mastocytosis in rheumatoid arthritis. Arthritis Rheum. 1984;27:845–851. doi: 10.1002/art.1780270802. [DOI] [PubMed] [Google Scholar]
- Deyama Y, Kikuiri T, Ohnishi G, Feng YG, Takeyama S, Hatta M, et al. Histamine stimulates production of osteoclast differentiation factor/receptor activator of nuclear factor-kappaB ligand by osteoblasts. Biochem Biophys Res Commun. 2002;298:240–246. doi: 10.1016/s0006-291x(02)02440-3. [DOI] [PubMed] [Google Scholar]
- Eklund KK. Mast cells in the pathogenesis of rheumatic diseases and as potential targets for anti-rheumatic therapy. Immunol Rev. 2007;217:38–52. doi: 10.1111/j.1600-065X.2007.00504.x. [DOI] [PubMed] [Google Scholar]
- Fallon MD, Whyte MP, Teitelbaum SL. Systemic mastocytosis associated with generalized osteopenia. Histopathological characterization of the skeletal lesion using undecalcified bone from two patients. Hum Pathol. 1981;12:813–820. doi: 10.1016/s0046-8177(81)80084-6. [DOI] [PubMed] [Google Scholar]
- Feldmann M, Maini SR. Role of cytokines in rheumatoid arthritis: an education in pathophysiology and therapeutics. Immunol Rev. 2008;223:7–19. doi: 10.1111/j.1600-065X.2008.00626.x. [DOI] [PubMed] [Google Scholar]
- Firestein GS, Alvaro-Gracia JM, Maki R. Quantitative analysis of cytokine gene expression in rheumatoid arthritis. J Immunol. 1990;144:3347–3353. [PubMed] [Google Scholar]
- Godfrey HP, Ilardi C, Engber W, Graziano FM. Quantitation of human synovial mast cells in rheumatoid arthritis and other rheumatic diseases. Arthritis Rheum. 1984;27:852–856. doi: 10.1002/art.1780270803. [DOI] [PubMed] [Google Scholar]
- Gotis-Graham I, McNeil HP. Mast cell responses in rheumatoid synovium. Association of the MCTC subset with matrix turnover and clinical progression. Arthritis Rheum. 1997;40:479–489. doi: 10.1002/art.1780400314. [DOI] [PubMed] [Google Scholar]
- Gotis-Graham I, Smith MD, Parker A, McNeil HP. Synovial mast cell responses during clinical improvement in early rheumatoid arthritis. Ann Rheum Dis. 1998;57:664–671. doi: 10.1136/ard.57.11.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara M, Ono K, Hwang MW, Iwasaki A, Okada M, Nakatani K, et al. Evidence for a role of mast cells in the evolution to congestive heart failure. J Exp Med. 2002;195:375–381. doi: 10.1084/jem.20002036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Walls AF. Human mast cell chymase induces the accumulation of neutrophils, eosinophils and other inflammatory cells in vivo. Br J Pharmacol. 1998;125:1491–1500. doi: 10.1038/sj.bjp.0702223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hida RY, Takano Y, Okada N, Dogru M, Satake Y, Fukagawa K, et al. Suppressive effects of tranilast on eotaxin-1 production from cultured conjunctival fibroblasts. Curr Eye Res. 2008;33:19–22. doi: 10.1080/02713680701817366. [DOI] [PubMed] [Google Scholar]
- Inglis JJ, Criado G, Andrews M, Feldmann M, Williams RO, Selley ML. The anti-allergic drug, N-(3′,4′-dimethoxycinnamonyl) anthranilic acid, exhibits potent anti-inflammatory and analgesic properties in arthritis. Rheumatology. 2007;46:1428–1432. doi: 10.1093/rheumatology/kem160. [DOI] [PubMed] [Google Scholar]
- Juurikivi A, Sandler C, Lindstedt KA, Kovanen PT, Juutilainen T, Leskinen MJ, et al. Inhibition of c-kit tyrosine kinase by imatinib mesylate induces apoptosis in mast cells in rheumatoid synovia: a potential approach to the treatment of arthritis. Ann Rheum Dis. 2005;64:1126–1131. doi: 10.1136/ard.2004.029835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakizoe E, Shiota N, Tanabe Y, Shimoura K, Kobayashi Y, Okunishi H. Isoform-selective upregulation of mast cell chymase in the development of skin fibrosis in scleroderma model mice. J Invest Dermatol. 2001;116:118–123. doi: 10.1046/j.1523-1747.2001.00165.x. [DOI] [PubMed] [Google Scholar]
- Katoh N, Hirano S, Yasuno H. Solitary mastocytoma treated with tranilast. J Dermatol. 1996;23:335–339. doi: 10.1111/j.1346-8138.1996.tb04026.x. [DOI] [PubMed] [Google Scholar]
- Kiener HP, Hofbauer R, Tohidast-Akrad M, Walchshofer S, Redlich K, Bitzan P, et al. Tumor necrosis factor alpha promotes the expression of stem cell factor in synovial fibroblasts and their capacity to induce mast cell chemotaxis. Arthritis Rheum. 2000;43:164–174. doi: 10.1002/1529-0131(200001)43:1<164::AID-ANR21>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Koda A, Nagai H, Watanabe S, Yanagihara Y, Sakamoto K. Inhibition of hypersensitivity reactions by a new drug, N(3′,4′-dimethoxycinnamoyl) anthranilic acid (N-5′) J Allergy Clin Immunol. 1976;57:396–407. doi: 10.1016/0091-6749(76)90054-3. [DOI] [PubMed] [Google Scholar]
- Komatsu H, Kojima M, Tsutsumi N, Hamano S, Kusama H, Ujiie A, et al. Study of the mechanism of inhibitory action of tranilast on chemical mediator release. Jpn J Pharmacol. 1988;46:43–51. doi: 10.1254/jjp.46.43. [DOI] [PubMed] [Google Scholar]
- Lee DM, Friend DS, Gurish MF, Benoist C, Mathis D, Brenner MB. Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science. 2002;297:1689–1692. doi: 10.1126/science.1073176. [DOI] [PubMed] [Google Scholar]
- Leibbrandt A, Penninger JM. RANK/RANKL: regulators of immune responses and bone physiology. Ann N Y Acad Sci. 2008;1143:123–150. doi: 10.1196/annals.1443.016. [DOI] [PubMed] [Google Scholar]
- Magnusson SE, Pejler G, Kleinau S, Abrink M. Mast cell chymase contributes to the antibody response and the severity of autoimmune arthritis. FASEB J. 2009;23:875–882. doi: 10.1096/fj.08-120394. [DOI] [PubMed] [Google Scholar]
- Malone DG, Wilder RL, Saavedra-Delgado AM, Metcalfe DD. Mast cell numbers in rheumatoid synovial tissues. Correlations with quantitative measures of lymphocytic infiltration and modulation by antiinflammatory therapy. Arthritis Rheum. 1987;30:130–137. doi: 10.1002/art.1780300202. [DOI] [PubMed] [Google Scholar]
- Nagate T, Tamura T, Sato F, Kuroda J, Nakayama J, Shibata N. Tranilast suppresses the disease development of the adjuvant- and streptococcal cell wall-induced arthritis in rats. J Pharmacol Sci. 2007;105:48–56. doi: 10.1254/jphs.fp0070534. [DOI] [PubMed] [Google Scholar]
- Noss EH, Brenner MB. The role and therapeutic implications of fibroblast-like synoviocytes in inflammation and cartilage erosion in rheumatoid arthritis. Immunol Rev. 2008;223:252–270. doi: 10.1111/j.1600-065X.2008.00648.x. [DOI] [PubMed] [Google Scholar]
- Pae HO, Jeong SO, Koo BS, Ha HY, Lee KM, Chung HT. Tranilast, an orally active anti-allergic drug, up-regulates the anti-inflammatory heme oxygenase-1 expression but down-regulates the pro-inflammatory cyclooxygenase-2 and inducible nitric oxide synthase expression in RAW264.7 macrophages. Biochem Biophys Res Commun. 2008;371:361–365. doi: 10.1016/j.bbrc.2008.04.054. [DOI] [PubMed] [Google Scholar]
- Pejler G, Abrink M, Ringvall M, Wernersson S. Mast cell proteases. Adv Immunol. 2007;95:167–255. doi: 10.1016/S0065-2776(07)95006-3. [DOI] [PubMed] [Google Scholar]
- Platten M, Ho PP, Youssef S, Fontoura P, Garren H, Hur EM, et al. Treatment of autoimmune neuroinflammation with a synthetic tryptophan metabolite. Science. 2005;310:850–855. doi: 10.1126/science.1117634. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Kanai K, Asano K, Hisamitsu T, Suzaki H. Suppression of matrix metalloproteinase production in nasal fibroblasts by tranilast, an antiallergic agent, in vitro. Mediators Inflamm. 2005;2005:150–159. doi: 10.1155/MI.2005.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T, Kanai K, Kyo Y, Asano K, Hisamitsu T, Suzaki H. Effect of tranilast on matrix metalloproteinase production from neutrophils in-vitro. J Pharm Pharmacol. 2006;58:91–99. doi: 10.1211/jpp.58.1.0011. [DOI] [PubMed] [Google Scholar]
- Shin K, Nigrovic PA, Crish J, Boilard E, McNeil HP, Larabee KS, et al. Mast cells contribute to autoimmune inflammatory arthritis via their tryptase/heparin complexes. J Immunol. 2009;182:647–656. doi: 10.4049/jimmunol.182.1.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiota N, Okunishi H, Takai S, Mikoshiba I, Sakonjo H, Shibata N, et al. Tranilast suppresses vascular chymase expression and neointima formation in balloon-injured dog carotid artery. Circulation. 1999;99:1084–1090. doi: 10.1161/01.cir.99.8.1084. [DOI] [PubMed] [Google Scholar]
- Shiota N, Shimoura K, Okunishi H. Pathophysiological role of mast cells in collagen-induced arthritis: study with a cysteinyl leukotriene receptor antagonist, montelukast. Eur J Pharmacol. 2006;548:158–166. doi: 10.1016/j.ejphar.2006.07.046. [DOI] [PubMed] [Google Scholar]
- Silberstein R, Melnick M, Greenberg G, Minkin C. Bone remodeling in W/Wv mast cell deficient mice. Bone. 1991;12:227–236. doi: 10.1016/8756-3282(91)90068-t. [DOI] [PubMed] [Google Scholar]
- Spiecker M, Lorenz I, Marx N, Darius H. Tranilast inhibits cytokine-induced nuclear factor kappaB activation in vascular endothelial cells. Mol Pharmacol. 2002;62:856–863. doi: 10.1124/mol.62.4.856. [DOI] [PubMed] [Google Scholar]
- Suzawa H, Kikuchi S, Arai N, Koda A. The mechanism involved in the inhibitory action of tranilast on collagen biosynthesis of keloid fibroblasts. Jpn J Pharmacol. 1992;60:91–96. doi: 10.1254/jjp.60.91. [DOI] [PubMed] [Google Scholar]