

Abstract
Background and purpose:
K+ channels play a role in the proliferation of cancer cells. We have investigated the effects of specific K+ channel inhibitors on basal and oestrogen-stimulated proliferation of breast cancer cells.
Experimental approach:
Using the mammary adenocarcinoma cell line MCF-7 we assayed cell proliferation by radiolabelled thymidine incorporation in the absence or presence of various K+ channel inhibitors with or without 17β-oestradiol.
Key results:
Inhibitors of Kv10.1 and KCa3.1 K+ channels suppressed basal proliferation of MCF-7 cells, but not oestrogen-stimulated proliferation. TRAM-34, a specific inhibitor of KCa3.1 channels increased or decreased cell proliferation depending on the concentration. At intermediate concentrations (3–10 µM) TRAM-34 increased cell proliferation, whereas at higher concentrations (20–100 µM) TRAM-34 decreased cell proliferation. The enhancement of cell proliferation caused by TRAM-34 was blocked by the oestrogen receptor antagonists ICI182,780 and tamoxifen. TRAM-34 also increased progesterone receptor mRNA expression, decreased oestrogen receptor-α mRNA expression and reduced the binding of radiolabelled oestrogen to MCF-7 oestrogen receptor, in each case mimicking the effects of 17β-oestradiol.
Conclusions and implications:
Our results demonstrate that K+ channels Kv10.1 and KCa3.1 play a role in basal, but not oestrogen-stimulated MCF-7 cell proliferation. TRAM-34, as well as inhibiting KCa3.1, directly interacts with the oestrogen receptor and mimics the effects of 17β-oestradiol on MCF-7 cell proliferation and gene modulation. Our finding that TRAM-34 is able to activate the oestrogen receptor suggests a novel action of this supposedly specific K+ channel inhibitor and raises concerns of interpretation in its use.
Keywords: TRAM-34, oestrogen, potassium channel, proliferation, MCF-7, cancer
Introduction
Potassium channels play an important role in a wide range of biological functions. In addition to their traditional roles in electrical excitability and cell signalling, it has been demonstrated that specific K+ channels are important for the proliferation of normal (Panyi, 2005) and cancerous cells (Wang, 2004; Kunzelmann, 2005; Pardo et al., 2005). Aberrant expression of K+ channels has been demonstrated in cancer cells compared with normal cells from the same tissue (Meyer et al., 1999; Wang, 2004). It has been suggested that aberrantly expressed K+ channels may be useful as biomarkers for the detection of cancer cells (Farias et al., 2004; Stuhmer et al., 2006) and as novel targets for cancer therapy (Schonherr, 2005; Felipe et al., 2006). Specific K+ channel inhibition reduces cell proliferation of numerous cancer cell types, including breast cancer cells (Pardo et al., 2005; Ouadid-Ahidouch and Ahidouch, 2008). How K+ channel activity is linked to cell proliferation is not completely understood, but it has been suggested that such activity is important for cell cycle progression because it controls the resting membrane potential. According to this model, K+ channel inhibition inhibits proliferation because it leads to membrane potential depolarization (Wonderlin and Strobl, 1996;Ouadid-Ahidouch and Ahidouch, 2008).
MCF-7 cells are a mammary adenocarcinoma cell line commonly used to study the pathophysiology of breast cancer. MCF-7 cells express the mRNAs for numerous K+ channels (channel nomenclature follows Alexander et al., 2008), including voltage-gated [KCNQ1 (vanTol et al., 2007), KCNH1 (Ouadid-Ahidouch et al., 2001) and KCNH2 (Roy et al., 2008)] and calcium-activated K+ channels [KCNN4 (Ouadid-Ahidouch et al., 2004b) and KCNMA1 (Ouadid-Ahidouch et al., 2004a)]. Channel activity has also been observed for most of these K+ channels detected in MCF-7 cells (Ouadid-Ahidouch et al., 2001; Ouadid-Ahidouch et al., 2004a,b; vanTol et al., 2007). In addition, cell cycle variations in expression and channel activity have been demonstrated for certain K+ channels (Ouadid-Ahidouch et al., 2001; Ouadid-Ahidouch et al., 2004a,b;). Both pharmacological tools (channel inhibitors) and molecular tools (agents for mRNA knockdown) have been used to demonstrate the role of specific K+ channels in MCF-7 cell proliferation. For example, clotrimazole has been used to implicate KCa3.1 (KCNN4) in cell proliferation (Ouadid-Ahidouch et al., 2004b), whereas astemizole or imipramine have been used to implicate Kv10.1 (KCNH1) in that process (Roy et al., 2008). Furthermore, small interfering RNA targeting Kv10.1 has recently confirmed the contribution of this channel to MCF-7 cell proliferation (Weber et al., 2006).
The oestrogens play a key role in the development and progression of breast cancer (Doisneau-Sixou et al., 2003). Oestrogens increase the proliferation of both normal and cancerous mammary gland cells that express the appropriate receptor [oestrogen receptor-(ER)α or β]. Not surprisingly, MCF-7 cells, which express both ER-α (Brooks et al., 1973) and ER-β (Kuiper et al., 1996), proliferate much faster when treated with oestrogens (Brooks and Skafar, 2004).
Recently, it has been demonstrated that specific K+ channels contribute to the proliferative effects of certain growth factors on MCF-7 cells (Coiret et al., 2005; Borowiec et al., 2007). We therefore sought to determine the relative contribution of different specific K+ channels to the proliferative effect of oestrogen on MCF-7 cells. Firstly, using RT-PCR we confirmed the mRNA expression of specific K+ channels, including KCNH1, KCNH2, KCNJ8, KCNMA1, KCNN4 and KCNQ1. Then using specific channel inhibitors we determined the contribution of each K+ channel type to basal and oestrogen-stimulated proliferation. One specific channel inhibitor, TRAM-34, which blocks KCa3.1 (KCNN4), produced an unusual biphasic response, either stimulating or inhibiting proliferation depending on its concentration. Therefore, we also sought to understand the additional possible mechanisms by which TRAM-34 can modulate the proliferation of MCF-7 cells.
Methods
Cell culture
MCF-7 cells were obtained from the American Tissue Culture Collection (Rockville, MD, USA). Stock cultures were maintained in minimal essential medium (MEM) containing 5% fetal bovine serum (FBS) and supplemented with 10 µM non-essential amino acids, 100 U·mL−1 penicillin, 100 µg·mL−1 streptomycin (all from Invitrogen, Burlington, ON, Canada) and 100 µg·mL−1 human insulin (Sigma-Aldrich, Oakville, ON, Canada) in a humidified atmosphere at 37°C with 5% CO2. For mRNA expression (detection and quantitative PCR) and cell counting, 5 × 104 cells were seeded in each well of a 6-well plate. For [3H]-thymidine incorporation, 2.5 × 104 cells were seeded in each well of a 24-well plate.
RNA extraction, reverse transcription (RT) and PCR
MCF-7 total RNA was extracted using TRIzol™ reagent (Invitrogen) following the manufacturer's protocol. Turbo DNA-free™ Kit (Ambion, Austin, TX, USA) was then used to remove any contaminating genomic DNA. RNA (2 µg) was reverse-transcribed using 5 mM deoxynucleotide triphosphate (dNTP) mix (Invitrogen), 1 µM oligo(dT) (GE Healthcare Life Sciences, Baie d'Urfé, PQ, Canada) and M-MLV reverse transcriptase enzyme (200 U; Invitrogen) in a total volume of 100 µL. Primers (Invitrogen) were each designed to amplify exon–exon junctions and tested to confirm the absence of genomic DNA amplification by performing a negative RT reaction, omitting the M-MLV reverse transcriptase enzyme. Primer sequences, annealing temperatures and predicted amplicon sizes are given in Table S1. PCR conditions were as follows: an initial melting step at 95°C for 1 min followed by 40 cycles of denaturation at 95°C for 45 s, an appropriate annealing temperature (Table S1) for 45 s and extension at 72°C for 1 min. The PCR was then given a final extension step of 72°C for 10 min and held at 4°C. Each PCR reaction contained the following reagents in a total of 25 µL: 100 pM of corresponding forward and reverse primers, 200 µM dNTP mix (Invitrogen), 1× PCR buffer (Fermentas Canada, Burlington, ON, Canada), 1.5 mM MgCl2 (Fermentas), 2.5 U Taq DNA polymerase (Fermentas) and 2 µL cDNA. PCR amplicons were visualized by gel electrophoresis, extracted and sequenced by a commercial sequencing facility to confirm identity (DalGEN Microbial Genomics Centre, Dalhousie University, Halifax, NS, Canada).
Quantitative RT-PCR (qRT-PCR) using the Roche LightCycler® system (Roche Applied Science, Laval, PQ, Canada) was performed to detect and quantify changes in gene expression, as described previously (Roy et al., 2006). The levels of ER-α and progesterone receptor (PR) transcript were normalized relative to the level of the housekeeping gene, hypoxanthine phosphoribosyltransferase (HPRT), and expressed as a percentage of control for each trial.
Materials
Glibenclamide, chromanol 293B, astemizole, E-4031, clotrimazole, iberiotoxin, TRAM-34 and 17β-oestradiol (E2) were obtained from Sigma-Aldrich. ICI182,780 (ICI) was obtained from Tocris Bioscience (Ellisville, MO, USA). All drugs were dissolved and diluted in appropriate vehicles according to the manufacturer's instructions. For proliferation assays, the concentration of dimethyl sulphoxide (vehicle) did not exceed 0.7% v/v. Channel blockers were used at a single high concentration that was confirmed to be non-toxic as determined by a Trypan blue exclusion assay (data not shown).
[3H]-Thymidine incorporation
The quantification of MCF-7 cell proliferation was carried out as described previously (Roy et al., 2008). Briefly, MCF-7 cells were initially seeded in regular MEM containing 5% FBS and supplements. After 24 h the media was changed to 5% dextran-coated charcoal-FBS (DCC-FBS, Hyclone, Logan, UT, USA) in phenol red-free MEM (Invitrogen) containing supplements. After 72 h, cells were partially synchronized with respect to cell cycle in 1% DCC-FBS phenol red-free MEM containing supplements for 48 h. After synchronization, cells were treated with indicated drug in 5% DCC-FBS phenol red-free MEM containing supplements in the presence of [methyl-3H]-thymidine (TRK-300, GE Healthcare Bio-Sciences Inc.; 1 µCi·mL−1) and 1 µM non-radioactive thymidine for 36–48 h. DNA was extracted as previously described (Roy et al., 2008), and radioactivity was measured on a Beckman LS 5000TA scintillation counter (Beckman Coulter Canada, Mississauga, ON, Canada) as counts per minute (cpm). Each treatment was performed on six separate wells, and cpm values were converted to a percentage of control values on the same plate.
Cell counting
MCF-7 cells were cultured as for [3H]-thymidine incorporation (see above) except that following synchronization the cells were treated with the indicated drug in 5% DCC-FBS phenol red-free MEM containing supplements for 72 h. Cells were then dissociated with 1 mL of 0.25% trypsin/EDTA (Invitrogen) and the diluted cell suspension counted using a Coulter Counter® model ZM30383 (Beckman Coulter Canada).
ER competitive binding assay
The collection of MCF-7 ER protein was carried out as reported previously (Kramer et al., 1997). Briefly, MCF-7 cells were plated onto 10 cm dishes and grown in 5% FBS MEM containing supplements for 4 days, after which the medium was changed to 5% DCC-FBS phenol red-free MEM containing supplements and cells were grown to 85% confluence. The cells were then suspended by incubation in 1 mM EDTA Ca2+-Mg2+-free phosphate-buffered saline for 30 min at 37°C. Cells from multiple dishes were combined and collected by centrifugation at 1000×g for 5 min, following which they were disrupted with ice-cold 10 mM TRIS 1.5 mM EDTA, 1 mM dithiothreitol and 10% glycerol, pH 7.4 (TEDG buffer) containing HALT™ Protease-inhibitor Cocktail (Fisher Scientific, Ottawa, ON, Canada) and disrupted by ultrasonification. Cytosolic protein was obtained by centrifuging the cell lysate at 100 000×g at 4°C for 30 min and collecting the supernatant. Protein concentrations were determined using the Bradford method (Bio-Rad, Hercules, CA, USA) and stored in 500 µL aliquots at −80°C.
Competitive binding assays were performed as follows. MCF-7 cell protein (250 µg) was incubated at room temperature for 2 h in TEDG buffer in the presence of 0.1 nM [2,4,6,7,16,17-3H(N)]-oestradiol ([3H]-E2) (110 Ci·mmol−1; Perkin-Elmer, Waltham, MA, USA) in a total final volume of 500 µL. Non-specific binding was assessed in the presence of a 100-fold excess of non-radioactive E2. TRAM-34 and E2 standards were diluted in phenol red-free 5% DCC-FBS MEM containing supplements before being added to the cytosolic protein. A vehicle control comprised of 5% DCC-FBS MEM containing supplements with 0.7% DMSO. To separate ER-bound [3H]-E2 from unbound [3H]-E2, 250 µL of hydroxylapatite (HAP, 60% in TEDG buffer, Sigma-Aldrich) was added, the mixture was vortexed every 5 min over 15 min and centrifuged at 1000×g for 10 min. The HAP-[3H]-E2-ER complex was washed with TEDG buffer, centrifuged and the wash step repeated. To elute [3H]-E2 from the HAP-[3H]-E2-ER complex, 500 µL of 100% ethanol was added and the mixture then incubated for 15 min and centrifuged at 1034×g for 10 min. The separated [3H]-E2 was removed and added to 2 mL of scintillation fluid. Radioactivity was quantified using a Beckman LS 5000TA scintillation counter (Beckman Coulter Canada). Competition of [3H]-E2 with TRAM-34 was assayed in quadruplicate on four independent protein extractions. An apparent dissociation constant of 0.135 ± 0.034 nM (n= 3) and a maximum binding capacity of 48.3 ± 5.4 fmol·mg−1 (n= 3) were determined by Scatchard analysis.
Statistical analysis
For [3H]-thymidine incorporation experiments data were analysed using Student's t-test for unpaired values. For cell counting, qRT-PCR and competitive binding data were analysed using anova with Tukey's post hoc test. Data were analysed prior to normalization for [3H]-thymidine incorporation and qRT-PCR. A P-value of <0.05 was considered significant.
Results
Involvement of K+ channels in MCF-7 proliferation
In order to identify the mRNA expression of specific K+ channels in MCF-7 cells, RT-PCR was performed on MCF-7 total RNA (Figure 1A). Primers designed for KCNJ8 (RefSeq: NM-004982), KCNQ1 (NM-000218.2, NM-181798 and NM-181797), KCNH1 (NM-172362 and NM-002238), KCNH2 (NM-000238, NM-172056 and NM-172057), KCNN4 (NM-002250) and KCNMA1 (NM-001014797 and NM-002247) amplified PCR products 336, 154, 177, 172, 158 and 153 bp in length respectively. All amplicons were excised from the agarose gel and sequenced, confirming the correct predicted sequence (data not shown). Figure 1A therefore confirms that, as consistent with previous findings (Ouadid-Ahidouch et al., 2001; Ouadid-Ahidouch et al., 2004a,b; vanTol et al., 2007; Roy et al., 2008), our MCF-7 cells express mRNA transcripts for KCNQ1, KCNH1, KCNH2, KCNN4 and KCNMA1. However we have further demonstrated that MCF-7 cells also express mRNA transcripts for the ATP-sensitive K+ channel KCNJ8.
Figure 1.
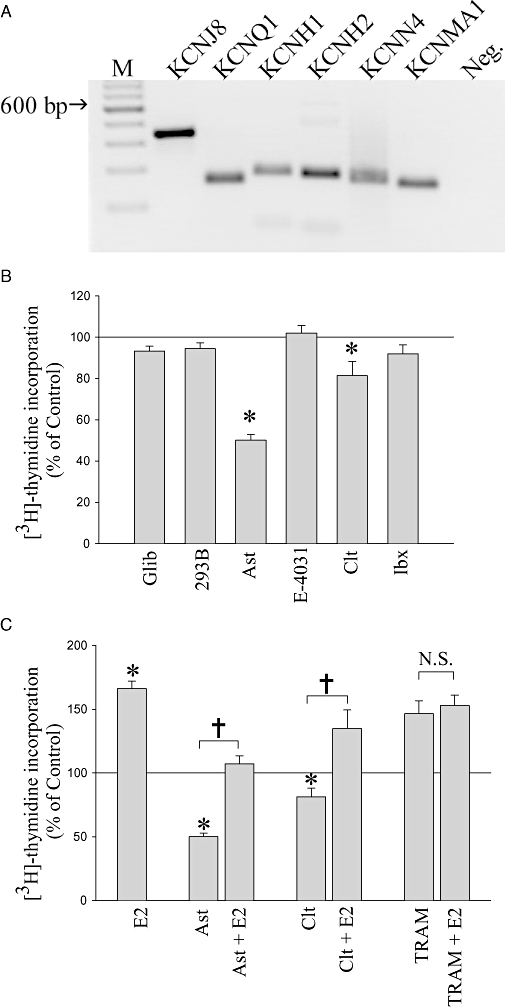
Detection of specific K+ channels expressed by MCF-7 cells and their contribution to basal and 17β-oestradiol (E2)-stimulated proliferation. (A) Agarose gel depicting PCR amplicons for the mRNA transcripts for KCNJ8 (336 bp), KCNQ1 (154 bp), KCNH1 (177 bp), KCNH2 (172 bp), KCNN4 (158 bp) and KCNMA1 (158 bp). Lane M is a DNA ladder, and Neg. is a negative control containing H2O instead of cDNA. (B) [3H]-thydimine incorporation by MCF-7 cells in the presence of K+ channel blockers specifically targeting these K+ channel types – glibenclamide (Glib, 30 µM), chromanol 293B (293B, 100 µM), astemizole (Ast, 3 µM), E-4031 (3 µM), clotrimazole (Clt, 10 µM) and iberiotoxin (Ibx, 100 nM). Mean normalized data from 3–11 experiments ± standard error of the mean. (C) [3H]-thymidine incorporation of MCF-7 cells in the presence of specific K+ channel blockers with or without co-application of E2. Concentrations of drugs were: E2 (1 nM), astemizole (Ast, 3 µM), clotrimazole (Clt, 10 µM) and TRAM-34 (TRAM, 10 µM). Mean normalized data from 4–28 experiments ± standard error of the mean. *P < 0.05 when compared with control, †P < 0.05, and N.S., not statistically significant between two indicated treatments.
The [3H]-thymidine incorporation approach to measure DNA synthesis was used together with selective channel blockers to determine the contribution of each of these K+ channel types to the proliferation of MCF-7 cells. Glibenclamide was used to block Kir6.1 (KCNJ8), chromanol 293B (293B) for Kv7.1(KCNQ1), astemizole for Kv10.1 (KCNH1), E-4031 for Kv11.1 (KCNH2), clotrimazole for KCa3.1 (KCNN4) and iberiotoxin for KCa1.1 (KCNMA1). Figure 1B shows the normalized data pooled from 3–11 experiments. Only astemizole and clotrimazole had a significant effect on proliferation, both causing a decrease. Astemizole (3 µM, n= 11) decreased [3H]-thymidine incorporation by ∼50% compared with controls (P < 0.05), as described previously (Roy et al., 2008), whereas clotrimazole (10 µM, n= 6) decreased [3H]-thymidine incorporation by ∼20% compared with controls (P < 0.05). Glibenclamide (30 µM, n= 4), chromanol 293B (100 µM, n= 3), E-4031 (3 µM, n= 5) and iberiotoxin (100 nM, n= 3) were without effect on [3H]-thymidine incorporation. These data are therefore consistent with previous suggestions (Ouadid-Ahidouch et al., 2001; Ouadid-Ahidouch et al., 2004b; Roy et al., 2008) that the activities of Kv10.1 and KCa3.1 channels are involved in the proliferation of MCF-7 cells. In contrast, we find no evidence that the activities of K+ channels Kir6.1, Kv7.1, Kv11.1 or KCa1.1 contribute to cell proliferation.
We next sought to determine whether Kv10.1 and KCa3.1 channel activity contribute to oestrogen-induced proliferation. Figure 1C shows pooled data for K+ channel blockers applied with or without E2 (1 nM). Across all experiments E2 alone increased [3H]-thymidine incorporation by ∼60% compared with controls (n= 28, P < 0.05). Astemizole (Kv10.1 inhibitor, 3 µM) itself decreased [3H]-thymidine incorporation by ∼50%; however, the decrease caused by astemizole was reversed by the presence of E2, so that E2 was able to stimulate substantially [3H]-thymidine incorporation in the presence of astemizole (P < 0.05). The same result was observed with clotrimazole (KCa3.1 inhibitor); at 10 µM this drug itself significantly decreased [3H]-thymidine incorporation by ∼20%; however, [3H]-thymidine incorporation was still increased by the addition of E2 (P < 0.05). The inhibition of basal proliferation by astemizole and clotrimazole implicates Kv10.1 and KCa3.1 channels in constitutive pathways of cell growth regulation. However, in the presence of astemizole or clotrimazole there was a consistent increase due to E2 of ∼50%, showing an intact response to E2. The similar increases in [3H]-thymidine incorporation (∼50–60%) observed with E2 in the absence or presence of both inhibitors provide evidence that the pharmacological blockade of Kv10.1 or KCa3.1 activity did not affect the ability of oestrogen to stimulate the proliferation of MCF-7 cells. Thus Kv10.1 and KCa3.1 channels may contribute to basal MCF-7 proliferation but are not essential for oestrogen-stimulated proliferation.
Another KCa3.1 channel blocker, TRAM-34, which is regarded as more specific than clotrimazole, gave quite different results. Thus, 10 µM TRAM-34 unexpectedly increased [3H]-thymidine incorporation ∼50% compared with controls (P < 0.05) and prevented any further increase due to E2 (Figure 1C). The stimulatory effects of E2 and TRAM-34 did not appear to be additive, and there was no difference in [3H]-thymidine incorporation between TRAM-34 alone and TRAM-34 plus E2 (P > 0.6). This was an unexpected result because clotrimazole, which is a structurally similar KCa3.1 channel blocker, had inhibited [3H]-thymidine incorporation at similar concentrations and not affected the E2 response. Given the lack of additivity with the E2 response, one possible explanation of this phenomenon was that TRAM-34 might be acting in a similar way to E2 to increase MCF-7 cell proliferation.
Effects of ER antagonist on responses to TRAM-34
In order to investigate further the effects of TRAM-34 on MCF-7 cell proliferation a wider concentration range was studied. Figure 2A shows raw data from one representative [3H]-thymidine incorporation experiment, and Figure 2B shows the normalized data pooled from 3–11 experiments. At low doses (0.1–1 µM) TRAM-34 had no significant effect on [3H]-thymidine incorporation (Figure 2B). At intermediate concentrations (3–10 µM) TRAM-34 increased [3H]-thymidine incorporation (P < 0.05), but at the highest concentrations tested (20–100 µM) TRAM-34 inhibited [3H]-thymidine incorporation (P < 0.05, Figure 2B). In contrast, clotrimazole was always inhibitory at all doses that produced an effect (Figure 2C). Thus, TRAM-34, but not clotrimazole, had biphasic and divergent effects on MCF-7 [3H]-thymidine incorporation depending on the concentration used.
Figure 2.
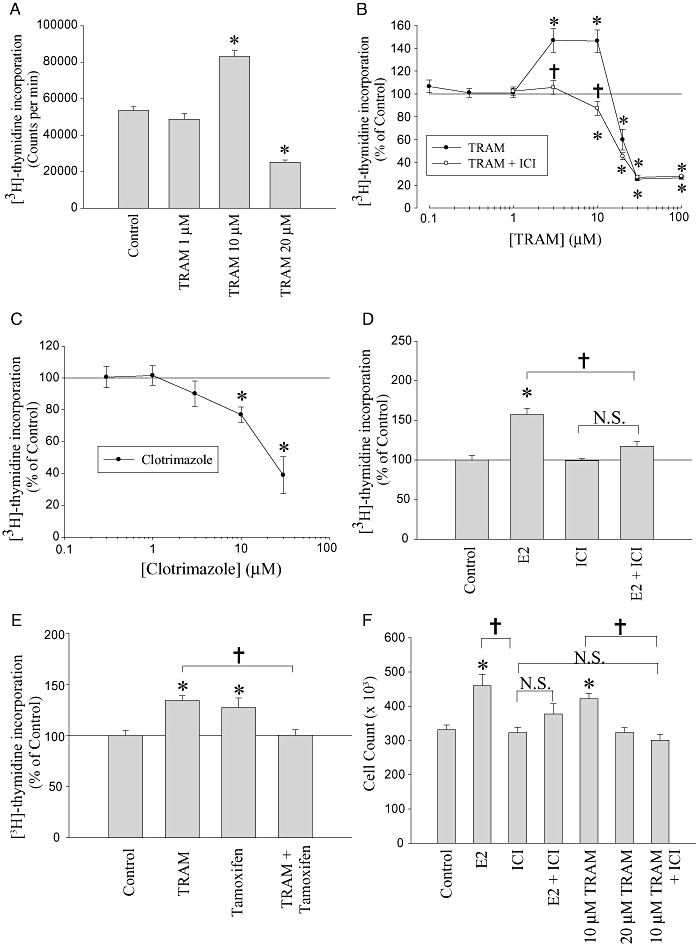
Stimulation and inhibition of proliferation by TRAM-34. (A) Raw data from a single representative [3H]-thymidine incorporation experiment. Data are mean counts per minute ± SEM from six wells. (B) The mean data from multiple trials converted to percentage of control. Each data point is the mean of 3–10 trials ± standard error of the mean. Results are shown for TRAM-34 treatment alone (TRAM) and for TRAM-34 treatment in the presence of ICI182,780 (1 nM, TRAM + ICI). (C) Mean [3H]-thymidine incorporation data in the presence of clotrimazole. Each data point represents data from 3–4 trials ± standard error of the mean expressed as percentage of control. (D) Mean [3H]-thymidine incorporation data in the presence of 17β-oestradiol (E2) (0.3 nM), ICI182,780 (ICI, 1 nM) or both (E2 + ICI). (E) Mean [3H]-thymidine incorporation in the presence of TRAM-34 alone (10 µM, TRAM), tamoxifen alone (1 µM) or both (TRAM + tamoxifen). For (D) and (E) data are expressed as percentage of control ± standard error of the mean (n= 3). (F) Mean cell counting data from three separate trials ± standard error of the mean. Concentration of E2 and ICI182,780 used was 0.3 and 1 nM respectively. *P < 0.05 when compared with control and †P < 0.05 between two indicated treatments. N.S., not signicicant.
To examine whether TRAM-34 might be increasing cell proliferation at intermediate concentrations by acting via the ER, we used the potent ER antagonist ICI in an attempt to block the stimulation. At 1 nM, ICI itself had no significant effect on [3H]-thymidine incorporation (P > 0.5) and was able to inhibit the stimulatory effect of 0.3 nM E2 (P < 0.05, Figure 2D). ICI (1 nM) also blocked the mitogenic effect of 3 and 10 µM TRAM-34 (P < 0.05; Figure 2B) and was also able to unmask an anti-proliferative effect of TRAM-34 at 10 µM (P < 0.05; Figure 2B). As a result, the concentration–response relationship for TRAM-34 applied in the presence of ICI appeared monophasic, reflecting a simple dose-dependent inhibition of [3H]-thymidine incorporation (Figure 2B) and revealing a previously masked inhibitory effect at lower concentrations. The increase in [3H]-thymidine incorporation caused by TRAM-34 (10 µM) was also inhibited by the selective ER modulator tamoxifen (Figure 2E). Tamoxifen (1 µM) itself increased [3H]-thymidine incorporation (P < 0.05), probably due to its activity as a partial agonist at the ER, but inhibited the increase caused by TRAM-34 at 10 µM (P < 0.05), again unmasking an inhibitory effect of this concentration of TRAM-34 when compared with tamoxifen alone (Figure 2E).
In confirmation of our key results from [3H]-thymidine incorporation, cell counting experiments were performed, and the combined data from three separate experiments are shown in Figure 2F. Oestrogen (0.3 nM) significantly increased the number of cells (P < 0.05), as expected. TRAM-34 (10 µM) also significantly increased the number of MCF-7 cells compared with controls (P < 0.05), whereas at 20 µM TRAM-34 had no effect (P > 0.05), which is consistent with the concentration–response curve for the effect of TRAM-34 on DNA synthesis ([3H]-thymidine incorporation, Figure 2B). ICI (1 nM) alone had no effect on cell number (P > 0.05) but was able to block the stimulatory effects both of E2 (P < 0.05) and of 10 µM TRAM-34 (P < 0.05). This confirms our findings in the [3H]-thymidine incorporation assay and provides further evidence for an oestrogenic activity of TRAM-34.
In addition to stimulating growth, activation of ERs also causes well-described changes in the mRNA expression profile of MCF-7 cells. Two widely studied mRNA expression changes caused by ER activation in MCF-7 cells are the up-regulation of PR and the down-regulation of ER-α mRNA (Ree et al., 1989). To assess whether TRAM-34 can mimic functional activation of the ER, qRT-PCR was used to analyse the expression profile of PR and ER-α mRNA in response to 24 h TRAM-34 treatment. Figure 3 shows the combined, normalized PR/HPRT (Figure 3A) and ER-α/HPRT (Figure 3B) ratios from four separate qRT-PCR experiments (24 h treatments). As expected, E2 (1 nM) significantly increased PR mRNA expression ∼250% (P < 0.05) and decreased ER-α mRNA expression ∼60% (P < 0.05). Similarly and consistent with an oestrogen-like action, TRAM-34 (10 µM) also significantly increased PR mRNA expression ∼100% (P < 0.05, n= 4) and decreased ER-α expression ∼50% (P < 0.05, n =4). The ER antagonist ICI (1 nM) by itself had no significant effects on either PR or ER-α expression (P > 0.05). Nevertheless, ICI was able to completely (Figure 3A) or partially (Figure 3B) block the increase in PR expression or the decrease in ER-α expression caused by TRAM-34 respectively. Similar results were also obtained from semi-quantitative RT-PCR (see Figure S1). We also examined whether the structurally similar compound clotrimazole had any effect on PR or ER-α expression. Clotrimazole (10 µM) was able to slightly but significantly increase PR expression ∼23% (P < 0.05, n= 3, Figure 3A), as did TRAM-34, but had no effect at all on ER-α expression (P > 0.05, n= 3, Figure 3B), providing further evidence for a unique property of TRAM-34 at these concentrations. These results demonstrate that TRAM-34 can mimic the effects of E2 by increasing the expression of PR and decreasing the expression of ER-α and may be activating the ERs.
Figure 3.
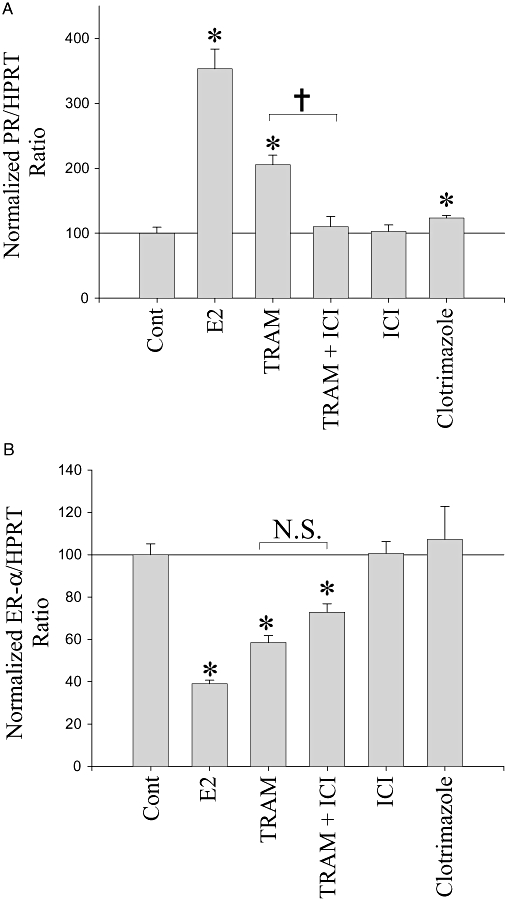
TRAM-34 mimics the effects of 17β-oestradiol (E2) on progesterone receptor (PR) and oestrogen receptor (ER)-α mRNA expression. (A) and (B) Normalized mean PR/hypoxanthine phosphoribosyltransferase (HPRT) or ER-α/HPRT ratios from quantitative RT-PCR ± standard error of the mean, from four separate trials. MCF-7 cells were treated for 24 h with E2 (1 nM), TRAM-34 (TRAM, 10 µM), ICI182,780 (ICI, 1 nM) and clotrimazole (10 µM). *P < 0.05 when compared with controls, †P < 0.05 and N.S., not statistically significant between two indicated treatments.
Direct interaction of TRAM-34 with the ER
In order to study the binding of TRAM-34 to the MCF-7 ERs, competitive ligand binding studies were performed. Figure 4 shows a competition curve depicting the percentage of [3H]-E2 bound to MCF-7 ER protein in the presence of competitors. Unlabelled E2 (0.01 µM) was used as a positive control and reduced the specific binding of [3H]-E2 to MCF-7 ER protein at that concentration by ∼50% (P < 0.05, n= 4). TRAM-34 (1–100 µM) also decreased the amount of specific [3H]-E2 bound to ER protein from MCF-7 cells (P < 0.05, n= 3). Lower concentrations of TRAM-34 (0.01–0.1 µM) did not reduce [3H]-E2 binding, and values were near vehicle control (n= 1). Fitting the data to a one-site competition curve yielded an IC50 of 1.51 µM. This result demonstrates that, at concentrations similar to those used in proliferation assays (3–10 µM), TRAM-34 is able to interact directly with the ER protein of MCF-7 cells. As TRAM-34 is able to compete with [3H]-E2 for binding to ER protein this suggests that TRAM-34 binds close to the oestrogen-binding site of ERs. We also tested the ability of clotrimazole to compete with [3H]-E2. Surprisingly, at 100 µM clotrimazole reduced the binding of [3H]-E2 to ER protein by 35 ± 3.5% compared with control (P < 0.05, n= 3). Thus, clotrimazole also binds to ERs, but unlike TRAM-34 appears only very weakly able to activate the receptor and cause downstream effects such as changes in gene expression and increases in proliferation.
Figure 4.
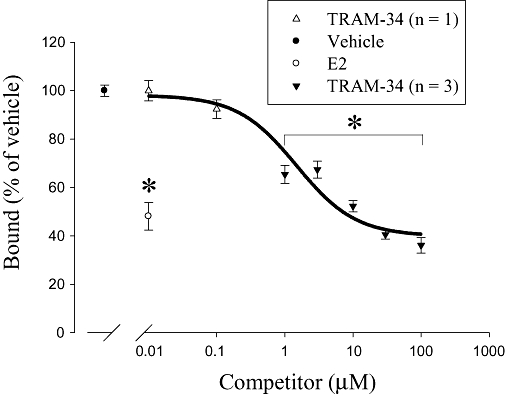
TRAM-34 competes with [3H]-17β-oestradiol (E2) for binding to the oestrogen receptor (ER). Data are expressed as a percentage of vehicle control ± standard error of the mean. Unlabelled E2 (0.01 µM) reduced the binding of [3H]-E2 by ∼50% (P < 0.05, n= 4). TRAM-34 (at low concentrations, 0.01–0.1 µM) did not reduce the binding of [3H]-E2. These data were determined from four sample replicates from one protein extraction, therefore only n= 1. Statistical analysis (Student's t-test) performed on this one protein extraction comparing the mean of four control samples versus four TRAM-34 samples yielded a P > 0.05, and was determined to be not statistically significant. TRAM-34 at higher concentrations (1–100 µM) significantly reduced the specific binding of [3H]-E2 (P < 0.05, n= 3 independent protein extractions). *P < 0.05 compared with vehicle control.
Furthermore, we also tested the effect of TRAM-34 on displacement of radiolabelled oestrogen in cytosolic protein preparations from two other breast cancer cell lines. MDA-MB-231 cells, a cell line with a markedly invasive phenotype, were found to be ER-negative. However, we found that protein from T47D cells showed specific [3H]-E2 binding at a level ∼40% of that for MCF-7 cell protein. A high dose of TRAM-34 (100 µM) caused a highly variable displacement of [3H]-E2 in different trials, which we attribute to the lower apparent yield of ER protein we were able to obtain from these cells. Overall, this concentration of TRAM-34 reduced [3H]-E2 binding to 76.0 ± 14.6% of control levels (n= 12), suggesting that TRAM-34 has the ability to bind to ER protein isolated from at least two different human breast carcinoma cell lines.
Discussion
K+ channels and MCF-7 cell proliferation
The activity of specific K+ channel types has been shown to play a role in MCF-7 cell proliferation (Pardo et al., 2005; Ouadid-Ahidouch and Ahidouch, 2008). Pharmacological agents that block Kv10.1 and KCa3.1 inhibit the proliferation of MCF-7 cells (Ouadid-Ahidouch et al., 2004b; Roy et al., 2008). Our results, using astemizole, clotrimazole and TRAM-34, are consistent with those previous reports that both Kv10.1 and KCa3.1 play a role in basal MCF-7 cell proliferation, while indicating that a number of other K+ channels expressed in these cells (Kir6.1, Kv7.1, Kv11.1 and KCa1.1) are unlikely to play a crucial role in basal cell proliferation. We also suggest that Kv10.1 and KCa3.1 channels are not necessary for oestrogen-stimulated cell proliferation. This result contrasts with the mitogenic effect of insulin-like growth factor-1 in MCF-7 cells, which has been demonstrated to be dependent on the function of Kv10.1 channels and modulated by the Akt signalling pathway (Borowiec et al., 2007).
TRAM-34 is a novel ER agonist
TRAM-34 was originally developed as a derivative of the commonly used KCa3.1 channel blocker clotrimazole (Wulff et al., 2000) and was selected from a group of prepared clotrimazole derivatives because of its high affinity for KCa3.1 channels (Kd= 20 ± 3 nM) and lack of cytochrome P450 inhibition (Wulff et al., 2000). TRAM-34 differs structurally from clotrimazole by a substituted pyrazole ring in place of the imidazole ring. TRAM-34 has frequently been used to demonstrate a role for KCa3.1 in cell proliferation, for example in human vascular smooth muscle cells (Toyama et al., 2008), B lymphocytes (Wang et al., 2007a), T lymphocytes (Ghanshani et al., 2000; Wulff et al., 2000), endothelial cells (Grgic et al., 2005), endometrial cancer cells (Wang et al., 2007b), pancreatic cancer cells (Jager et al., 2004) and prostate cancer cells (Lallet-Daher et al., 2009).
Our present results show that TRAM-34 has novel biphasic effects on MCF-7 cell proliferation. At moderate concentrations TRAM-34 increased cell proliferation (Figure 2B), but at higher concentrations TRAM-34 inhibited proliferation (Figure 2B), mimicking the effects of clotrimazole (Figure 2C). In the presence of the ER antagonist ICI TRAM-34 failed to stimulate proliferation and instead inhibited cell proliferation at lower concentrations, such that ICI was able to unmask this inhibitory effect of TRAM-34 (Figure 2B). Consistent with observations in many other cell types (Ghanshani et al., 2000; Wulff et al., 2000; Jager et al., 2004; Grgic et al., 2005; Wang et al., 2007a,b; Toyama et al., 2008; Lallet-Daher et al., 2009) we suggest the anti-proliferative effect of TRAM-34 (and also clotrimazole) is due to inhibition of KCa3.1 channels. In contrast, the mitogenic effect of TRAM-34 observed at moderate concentrations does not appear to be the result of K+ channel inhibition. This effect of TRAM-34 on cell proliferation appeared to be non-additive with that of E2 and was inhibited by the ER-selective modulators ICI and tamoxifen, suggesting that TRAM-34 may be mimicking the effects of E2 to increase cell proliferation. TRAM-34 was also capable of increasing PR and decreasing ER-α mRNA expression, two characteristic effects of ER activation in MCF-7 cells, and these effects were also sensitive to the ER antagonist ICI. Interestingly, clotrimazole had a small, but statistically significant effect on PR mRNA expression, suggesting that clotrimazole may also be a very weak ER agonist. Unlike TRAM-34, clotrimazole had no stimulatory effect on proliferation, consistent with clotrimazole being a much weaker ER agonist. Competitive ligand binding assays pointed to a direct effect of TRAM-34 mediated through ERs, as both TRAM-34 and clotrimazole were able to specifically reduce the binding of [3H]-E2 to ER protein. This confirms that TRAM-34 and clotrimazole bind directly to ERs close to the E2 binding site, suggesting that interactions with the ER may be a common effect of triarylmethanes. However, in our studies only TRAM-34 can activate pathways leading to modulation of gene expression and increases in cell proliferation, whereas clotrimazole has no, or only very weak, agonist activity in spite of its ability to bind to the ER.
In conclusion, TRAM-34 is a high-affinity KCa3.1 channel blocker that is routinely used to study the role of KCa3.1 in cell physiology. Caution however should be taken when interpreting results using TRAM-34 as our results demonstrate complex effects not previously described. In MCF-7 cells TRAM-34 mimics the effects of E2 most likely by direct interaction with the ER. We therefore suggest that TRAM-34 is a novel non-steroidal ER agonist that would have K+ channel-independent effects in cells that are ER-positive. Our research raises caveats to the use of TRAM-34 or related molecules in vivo to target KCa3.1 channels for therapeutic purposes (Wulff et al., 2007; Chou et al., 2008).
Acknowledgments
We would like to thank Eileen Denovan-Wright for help with qRT-PCR. This work was supported by grants from Nova Scotia Health Research Foundation-Student Research Award (JW Roy), Nova Scotia Health Research Foundation (EA Cowley), the Natural Sciences and Engineering Research Council of Canada (J Blay) and Canadian Breast Cancer Foundation-Atlantic Chapter (P Linsdell).
Glossary
Abbreviations:
- cpm
counts per minute
- DCC
dextran-coated charcoal
- dNTP
deoxynucleotide triphosphate
- E2
17β-oestradiol
- ER
oestrogen receptor
- FBS
fetal bovine serum
- HAP
hydroxylapatite
- HPRT
hypoxanthine phosphoribosyltransferase
- ICI
ICI182,780
- MEM
minimal essential media
- PR
progesterone receptor
- qRT-PCR
quantitative RT-PCR
- RT
reverse transcription
- TEDG
TRIS-EDTA-dithiothreitol-glycerol
- TRAM
TRAM-34
Statement of conflicts of interest
None.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1 TRAM-34 modulates the expression of oestrogen receptor-α (ER-α) and progesterone receptor (PR) mRNA. Semi-quantitative RT-PCR was performed, as previously described (Roy et al., 2006), to detect changes in ER-α and progesterone receptor mRNA caused by TRAM-34. The optimal numbers of PCR cycles were 24, 35 and 29 for ER-α, progesterone receptor PR and hypoxanthine-guanine phosphoribosyltransferase (HPRT) respectively. The concentrations used were: 17β-oestradiol (E2, 1 nM), TRAM-34 (TRAM, 10 μM), ICI182,780 (ICI, 1 nM) and TRAM-34 (10 μM) + ICI182,780 (1 nM). (A) Agarose gel demonstrating the results from semi-quantitative RT-PCR of ER-α (top panel) and HPRT (bottom panel) mRNA expression. (B) Agarose gel demonstrating the results from semi-quantitative RT-PCR of PR (top panel) and HPRT (bottom panel). M is a 100 base pair DNA ladder, and Neg. is an H2O control. The amplicon density was quantified using NIH Image J software (developed at the US National Institutes of Health and available on the internet at http://rsb.info.nih.gov/nih-image/) and the density of each amplicon converted to a ratio of ER-α or PR to HPRT mRNA expression and normalized as a percentage of control. (C) and (D) Average ER-α/HPRT or PR/HPRT ratios ± standard error of the mean, from four separate trials. *P < 0.05 when compared with controls, †P < 0.05 and N.S., not statistically significant between two indicated treatments.
Table S1 Primer sequences, annealing temperature and predicted amplicon sizes for mRNA expression studies
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edn. Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowiec AS, Hague F, Harir N, Guenin S, Guerineau F, Gouilleux F, et al. IGF-1 activates hEAG K(+) channels through an Akt-dependent signaling pathway in breast cancer cells: role in cell proliferation. J Cell Physiol. 2007;212:690–701. doi: 10.1002/jcp.21065. [DOI] [PubMed] [Google Scholar]
- Brooks SC, Skafar DF. From ligand structure to biological activity: modified estratrienes and their estrogenic and antiestrogenic effects in MCF-7 cells. Steroids. 2004;69:401–418. doi: 10.1016/j.steroids.2004.03.014. [DOI] [PubMed] [Google Scholar]
- Brooks SC, Locke ER, Soule HD. Estrogen receptor in a human cell line (MCF-7) from breast carcinoma. J Biol Chem. 1973;248:6251–6253. [PubMed] [Google Scholar]
- Chou CC, Lunn CA, Murgolo NJ. KCa3.1: target and marker for cancer, autoimmune disorder and vascular inflammation? Expert Rev Mol Diagn. 2008;8:179–187. doi: 10.1586/14737159.8.2.179. [DOI] [PubMed] [Google Scholar]
- Coiret G, Matifat F, Hague F, Ouadid-Ahidouch H. 17-beta-estradiol activates maxi-K channels through a non-genomic pathway in human breast cancer cells. FEBS Lett. 2005;579:2995–3000. doi: 10.1016/j.febslet.2005.02.085. [DOI] [PubMed] [Google Scholar]
- Doisneau-Sixou SF, Sergio CM, Carroll JS, Hui R, Musgrove EA, Sutherland RL. Estrogen and antiestrogen regulation of cell cycle progression in breast cancer cells. Endocr Relat Cancer. 2003;10:179–186. doi: 10.1677/erc.0.0100179. [DOI] [PubMed] [Google Scholar]
- Farias LM, Ocana DB, Diaz L, Larrea F, Avila-Chavez E, Cadena A, et al. Ether a go-go potassium channels as human cervical cancer markers. Cancer Res. 2004;64:6996–7001. doi: 10.1158/0008-5472.CAN-04-1204. [DOI] [PubMed] [Google Scholar]
- Felipe A, Vicente R, Villalonga N, Roura-Ferrer M, Martinez-Marmol R, Sole L, et al. Potassium channels: new targets in cancer therapy. Cancer Detect Prev. 2006;30:375–385. doi: 10.1016/j.cdp.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Ghanshani S, Wulff H, Miller MJ, Rohm H, Neben A, Gutman GA, et al. Up-regulation of the IKCa1 potassium channel during T-cell activation. Molecular mechanism and functional consequences. J Biol Chem. 2000;275:37137–37149. doi: 10.1074/jbc.M003941200. [DOI] [PubMed] [Google Scholar]
- Grgic I, Eichler I, Heinau P, Si H, Brakemeier S, Hoyer J, et al. Selective blockade of the intermediate-conductance Ca2+-activated K+ channel suppresses proliferation of microvascular and macrovascular endothelial cells and angiogenesis in vivo. Arterioscler Thromb Vasc Biol. 2005;25:704–709. doi: 10.1161/01.ATV.0000156399.12787.5c. [DOI] [PubMed] [Google Scholar]
- Jager H, Dreker T, Buck A, Giehl K, Gress T, Grissmer S. Blockage of intermediate-conductance Ca2+-activated K+ channels inhibit human pancreatic cancer cell growth in vitro. Mol Pharmacol. 2004;65:630–638. doi: 10.1124/mol.65.3.630. [DOI] [PubMed] [Google Scholar]
- Kramer VJ, Helferich WG, Bergman A, Klasson-Wehler E, Giesy JP. Hydroxylated polychlorinated biphenyl metabolites are anti-estrogenic in a stably transfected human breast adenocarcinoma (MCF7) cell line. Toxicol Appl Pharmacol. 1997;144:363–376. doi: 10.1006/taap.1997.8163. [DOI] [PubMed] [Google Scholar]
- Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci USA. 1996;93:5925–5930. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunzelmann K. Ion channels and cancer. J Membr Biol. 2005;205:159–173. doi: 10.1007/s00232-005-0781-4. [DOI] [PubMed] [Google Scholar]
- Lallet-Daher H, Roudbaraki M, Bavencoffe A, Mariot P, Gackiere F, Bidaux G, et al. Intermediate-conductance Ca2+-activated K+ channels (IKCa1) regulate human prostate cancer cell proliferation through a close control of calcium entry. Oncogene. 2009;28:1792–1806. doi: 10.1038/onc.2009.25. [DOI] [PubMed] [Google Scholar]
- Meyer R, Schonherr R, Gavrilova-Ruch O, Wohlrab W, Heinemann SH. Identification of ether a go-go and calcium-activated potassium channels in human melanoma cells. J Membr Biol. 1999;171:107–115. doi: 10.1007/s002329900563. [DOI] [PubMed] [Google Scholar]
- Ouadid-Ahidouch H, Ahidouch A. K+ channel expression in human breast cancer cells: involvement in cell cycle regulation and carcinogenesis. J Membr Biol. 2008;221:1–6. doi: 10.1007/s00232-007-9080-6. [DOI] [PubMed] [Google Scholar]
- Ouadid-Ahidouch H, Le Bourhis X, Roudbaraki M, Toillon RA, Delcourt P, Prevarskaya N. Changes in the K+ current-density of MCF-7 cells during progression through the cell cycle: possible involvement of a h-ether.a-gogo K+ channel. Receptors channels. 2001;7:345–356. [PubMed] [Google Scholar]
- Ouadid-Ahidouch H, Roudbaraki M, Ahidouch A, Delcourt P, Prevarskaya N. Cell-cycle-dependent expression of the large Ca2+-activated K+ channels in breast cancer cells. Biochem Biophys Res Commun. 2004a;316:244–251. doi: 10.1016/j.bbrc.2004.02.041. [DOI] [PubMed] [Google Scholar]
- Ouadid-Ahidouch H, Roudbaraki M, Delcourt P, Ahidouch A, Joury N, Prevarskaya N. Functional and molecular identification of intermediate-conductance Ca(2+)-activated K(+) channels in breast cancer cells: association with cell cycle progression. Am J Physiol Cell Physiol. 2004b;287:C125–C134. doi: 10.1152/ajpcell.00488.2003. [DOI] [PubMed] [Google Scholar]
- Panyi G. Biophysical and pharmacological aspects of K+ channels in T lymphocytes. Eur Biophys J. 2005;34:515–529. doi: 10.1007/s00249-005-0499-3. [DOI] [PubMed] [Google Scholar]
- Pardo LA, Contreras-Jurado C, Zientkowska M, Alves F, Stuhmer W. Role of voltage-gated potassium channels in cancer. J Membr Biol. 2005;205:115–124. doi: 10.1007/s00232-005-0776-1. [DOI] [PubMed] [Google Scholar]
- Ree AH, Landmark BF, Eskild W, Levy FO, Lahooti H, Jahnsen T, et al. Autologous down-regulation of messenger ribonucleic acid and protein levels for estrogen receptors in MCF-7 cells: an inverse correlation to progesterone receptor levels. Endocrinology. 1989;124:2577–2583. doi: 10.1210/endo-124-5-2577. [DOI] [PubMed] [Google Scholar]
- Roy J, Denovan-Wright EM, Linsdell P, Cowley EA. Exposure to sodium butyrate leads to functional downregulation of calcium-activated potassium channels in human airway epithelial cells. Pflugers Arch. 2006;453:167–176. doi: 10.1007/s00424-006-0128-8. [DOI] [PubMed] [Google Scholar]
- Roy J, vanTol B, Cowley EA, Blay J, Linsdell P. Pharmacological separation of hEAG and hERG K+ channel function in the human mammary carcinoma cell line MCF-7. Oncol Rep. 2008;19:1511–1516. [PubMed] [Google Scholar]
- Schonherr R. Clinical relevance of ion channels for diagnosis and therapy of cancer. J Membr Biol. 2005;205:175–184. doi: 10.1007/s00232-005-0782-3. [DOI] [PubMed] [Google Scholar]
- Stuhmer W, Alves F, Hartung F, Zientkowska M, Pardo LA. Potassium channels as tumour markers. FEBS Lett. 2006;580:2850–2852. doi: 10.1016/j.febslet.2006.03.062. [DOI] [PubMed] [Google Scholar]
- vanTol BL, Missan S, Crack J, Moser S, Baldridge WH, Linsdell P, et al. Contribution of KCNQ1 to the regulatory volume decrease in the human mammary epithelial cell line MCF-7. Am J Physiol Cell Physiol. 2007;293:C1010–C1019. doi: 10.1152/ajpcell.00071.2007. [DOI] [PubMed] [Google Scholar]
- Toyama K, Wulff H, Chandy KG, Azam P, Raman G, Saito T, et al. The intermediate-conductance calcium-activated potassium channel KCa3.1 contributes to atherogenesis in mice and humans. J Clin Invest. 2008;118:3025–3037. doi: 10.1172/JCI30836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Xu YQ, Liang YY, Gongora R, Warnock DG, Ma HP. An intermediate-conductance Ca(2+)-activated K (+) channel mediates B lymphoma cell cycle progression induced by serum. Pflugers Arch. 2007a;454:945–956. doi: 10.1007/s00424-007-0258-7. [DOI] [PubMed] [Google Scholar]
- Wang Z. Roles of K+ channels in regulating tumour cell proliferation and apoptosis. Pflugers Arch. 2004;448:274–286. doi: 10.1007/s00424-004-1258-5. [DOI] [PubMed] [Google Scholar]
- Wang ZH, Shen B, Yao HL, Jia YC, Ren J, Feng YJ, et al. Blockage of intermediate-conductance-Ca(2+) -activated K(+) channels inhibits progression of human endometrial cancer. Oncogene. 2007b;26:5107–5114. doi: 10.1038/sj.onc.1210308. [DOI] [PubMed] [Google Scholar]
- Weber C, Mello de Queiroz F, Downie BR, Suckow A, Stuhmer W, Pardo LA. Silencing the activity and proliferative properties of the human EagI Potassium Channel by RNA Interference. J Biol Chem. 2006;281:13030–13037. doi: 10.1074/jbc.M600883200. [DOI] [PubMed] [Google Scholar]
- Wonderlin WF, Strobl JS. Potassium channels, proliferation and G1 progression. J Membr Biol. 1996;154:91–107. doi: 10.1007/s002329900135. [DOI] [PubMed] [Google Scholar]
- Wulff H, Miller MJ, Hansel W, Grissmer S, Cahalan MD, Chandy KG. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: a potential immunosuppressant. Proc Natl Acad Sci USA. 2000;97:8151–8156. doi: 10.1073/pnas.97.14.8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff H, Kolski-Andreaco A, Sankaranarayanan A, Sabatier JM, Shakkottai V. Modulators of small- and intermediate-conductance calcium-activated potassium channels and their therapeutic indications. Curr Med Chem. 2007;14:1437–1457. doi: 10.2174/092986707780831186. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.