

Abstract
Background and purpose:
Peroxisome proliferator-activated receptor γ (PPARγ) agonists, such as rosiglitazone and pioglitazone, sensitize cells to insulin, and are therefore used to treat type 2 diabetes. However, in some patients, these drugs induce oedema, and the present study tests the hypothesis that this side effect reflects serum and glucocorticoid-inducible kinase 1 (SGK1)-dependent enhancement of epithelia Na+ absorption.
Experimental approach:
Na+ absorbing epithelial cells (H441 cells, mpkCCD cells) on permeable membranes were mounted in Ussing chambers, and the effects of rosiglitazone (2 µM) and pioglitazone (10 µM) on transepithelial Na+ absorption were quantified electrometrically. Changes in SGK1 activity were assessed by monitoring phosphorylation of residues within an endogenous protein.
Key results:
Both cell types absorbed Na+ via an electrogenic process that was enhanced by insulin. In mpkCCD cells, this stimulation of Na+ transport was associated with increased activity of SGK1, whereas insulin regulated Na+ transport in H441 cells through a mechanism that did not involve activation of this kinase. Rosiglitazone and pioglitazone had no discernible effect on transepithelial Na+ absorption in unstimulated or insulin-stimulated cells and failed to alter cellular SGK1 activity.
Conclusions and implications:
Our results do not support the view that PPARγ agonists stimulate epithelial Na+ absorption or alter the control of cellular SGK1 activity. It is therefore likely that other mechanisms are involved in PPARγ-mediated fluid retention, and a better understanding of these mechanisms may help with the identification of patients likely to develop oedema or heart failure when treated with these drugs.
Keywords: rosiglitazone, pioglitazone, SGK1, epithelial Na+ channel, SGK1, type 2 diabetes
Introduction
In addition to its well-documented role in carbohydrate metabolism, it is now clear that insulin also contributes to the control of epithelial Na+ transport (Blazer-Yost et al., 1998, 2003; Alvarez De La Rosa and Canessa, 2003). Early evidence of this came from studies in human subjects, which established that infusion of insulin reduced urinary Na+ excretion (Atchley et al., 1936; Miller and Bogdonoff, 1954), and this Na+ retaining action, which has been confirmed in several other studies (see for example Gupta et al., 1992; Huang et al., 2006), seems to reflect increased Na+ absorption in the collecting duct (reviewed by Tiwari et al., 2007; Loffing and Korbmacher, 2009). Insulin also stimulates Na+ absorption in the Na+ absorbing epithelia of the lungs and airways (Hagiwara et al., 1992), and clinical studies have suggested that this hormone might improve lung function in diabetic patients by stimulating pulmonary Na+ transport (Guazzi et al., 2002a,b;). Moreover, insulin also enhances the glucocorticoid-induced Na+ currents recorded from H441 cells (Thomas et al., 2004; Brown et al., 2008; Inglis et al., 2009), and a role for insulin in the control of Na+ transport in renal and pulmonary Na+ epithelia is therefore well documented.
Peroxisome proliferator-activated receptor γ (PPARγ) agonists are used in the treatment of type 2 diabetes and lower circulating glucose levels by sensitizing cells to insulin, thus facilitating glucose uptake (Henke et al., 1998). However, in some patients, these drugs increase the risk of congestive heart failure by inducing oedema, and this side effect leads to the withdrawal of PPARγ therapy in 10–15% of cases (see Buckingham and Hanna, 2007). While the physiological mechanism underlying this oedema is not clear, it has been suggested that it may reflect an inappropriate stimulation of Na+ transport in the collecting duct (Hong et al., 2003; Chen et al., 2005; Guan et al., 2005). Such an action would be highly significant, since Na+ transport in this region of the nephron is critical to the control of whole body Na+ and water balance, and increased Na+ transport in this tissue would therefore expand body fluid volume (reviewed by Tiwari et al., 2007; Loffing and Korbmacher, 2009). Moreover, since insulin contributes to the control of Na+ absorption in this region of the nephron (see above), this effect of PPARγ agonist stimulation may be related to the insulin-sensitizing actions of these drugs (Henke et al., 1998). It is therefore interesting that studies of human cortical collecting duct (CCD) cells have indicated that PPARγ agonists might activate serum and glucocorticoid-inducible kinase 1 (SGK1), a regulatory kinase that seems to control the abundance of epithelial Na+ channels (ENaC) in the apical membrane (Snyder, 2002; Snyder et al., 2002, 2004; Lang et al., 2006; Loffing et al., 2006). SGK1 activity is therefore thought to be an important determinant of Na+ transport in insulin-stimulated cells (Blazer-Yost et al., 1998, 2003; Alvarez De La Rosa and Canessa, 2003), and this observation provides a possible mechanism that might underlie PPARγ agonist-induced oedema. Moreover, since SGK1 is involved in the control of epithelial Na+ absorption in many different absorptive cell types (Lang et al., 2006; Loffing et al., 2006), this hypothesis also raises the possibility that PPARγ agonists might influence the hormonal control of Na+ transport in epithelial cells derived from non-renal tissues. The present study has therefore tested this hypothesis by exploring the effects of PPARγ agonists and insulin stimulation upon Na+ transport and SGK1 activity in Na+-absorbing epithelial cell lines derived from the human airway (H441 cells) and the mouse renal collecting duct (mpkCCD cells).
Methods
Quantification of transepithelial ion transport
H441 human distal airway epithelial cells were maintained in culture as described in detail elsewhere (Ramminger et al., 2004; Inglis et al., 2009). Experiments were undertaken using cells grown to confluence on Snapwell membranes (5–6 days) in fully defined medium that, unless otherwise stated, contained 0.2 µM dexamethasone (Sigma, Poole, UK) and 20 nM insulin (Sigma). Earlier studies undertaken in this laboratory (Ramminger et al., 2004) have shown that H441 cells maintained in this fully defined medium seldom form electrically resistive epithelial sheets unless exposed to dexamethasone, and the inclusion of this synthetic glucocorticoid is therefore essential. Culture membranes bearing confluent cells were mounted in Ussing chambers and bathed with bicarbonate-buffered physiological salt solution. The cultured epithelia were initially maintained under open circuit conditions while the transepithelial potential difference (Vt) stabilized (40–50 min); the current required to maintain Vt at 0 mV (short circuit current, ISC) was then monitored and recorded as a measure of active ion transport (Ramminger et al., 2004; Inglis et al., 2009). Transepithelial resistance (Rt) was determined from Ohm's law (i.e. Rt= Vt/ISC).
The renally derived mpkCCD cells were routinely cultured in phenol red-free Dulbecco's modified Eagle's Medium (DMEM)/Ham's F12 medium supplemented with glutamine (1 mM), foetal bovine serum (FBS, 2%, vol./vol), penicillin/streptomycin mixture (Sigma, 1 %), sodium selenate (60 nM), transferin (5 µg·mL−1), dexamethasone (50 nM), triiodothyronine (1 nM), epidermal growth factor (10 ng·mL−1), and insulin (5 µg·mL−1). For experiments, cells were plated onto Snapwell membranes (5 × 105 cells cm−2) and cultured for 6 days in complete medium that was replaced every 48 h. This medium was then replaced by hormone-free medium (phenol red-free DMEM/Ham's F12 medium containing antibiotics and 2% charcoal-stripped FBS). After 24 h, serum was withdrawn and the cells used in experiments after a further 18–24 h (Bens et al., 1999).
Assay of SGK1 activity
Aliquots of protein extracted from control and insulin-stimulated cells by scraping and sonication in the presence of phosphatase and protease inhibitors (1% Triton; 50 mM Tris – HCl, pH 7.5; 1 mM EGTA; 1 mM EDTA; 1 mM Na orthovanadate; 10 mM glycerol phosphate; 50 mM NaF; 5 mM Na pyrophosphate; 270 mM sucrose; 0.1% β-mercaptoethanol; 1 Roche Mini protease inhibitor tablet per 10 mL) were fractionated by SDS-polyacrylamide gel electrophoresis and blotted onto Hybond-P membranes (Amersham, Buckinghamshire, UK). Changes in cellular SGK1 activity were monitored using a method based upon the identification of residues within an endogenous protein (n-myc downstream-regulated protein 1 NDRG1) that are phosphorylated by SGK1, but not by other kinases, including the closely related protein kinase B (Murray et al., 2005a,b;). Changes to the phosphorylation status of these residues (NDRG1-Thr346/356/366) thus provide a biomarker of altered cellular SGK1 activity (Murray et al., 2005a; García-Martínez and Alessi, 2008; Inglis et al., 2009). Blots were therefore probed using antibodies provided by Professor Sir P. Cohen, (MRC Protein Phosphorylation Unit, College of Life Sciences, University of Dundee) against the Thr346/356/366-phosphorylated form of NDRG1 and the full length NDRG1 protein (Murray et al., 2005a).
Experimental design
Experiments were undertaken using strictly paired protocols in which control and PPARγ agonist-treated cells were age-matched and at identical passage, and care was taken to ensure that the control and experimental cells were handled identically. The effects of pioglitazone (10 µM) were assessed at 4 h, and effects of rosiglitazone (2 µM) were assessed at 4 and 24 h. Both of these PPARγ agonists were synthesized at Wyeth Research (Collegeville, PA, USA) for use in this project and were prepared as stock solutions in dimethyl sulphoxide, and all control cells were therefore exposed to the appropriate concentration of this solvent (0.1%). The ISC generated by the control and experimental cells was measured concurrently using parallel Ussing chamber systems.
Data analysis
All data are presented as mean ± SEM, and values of n refer to the number of times a protocol was repeated using cells at different passage number. The statistical significance of any differences between mean values was determined using Student's t-test.
Results
Effects of PPARγ agonists on electrogenic Na+ transport in H441 cells
H441 cells grown to confluence on Snapwell membranes became integrated into confluent epithelial sheets (Rt= 200–400 Ωcm2) that generated ISC. This current (∼20 µA cm−2, Table 1) normally remained stable over the time course of the present experiments (∼30 min), but was rapidly (10–20 s) and substantially (∼95%) inhibited by apical amiloride (10 µM). Since amiloride blocks ENaC, this observation confirms that this current is due to electrogenic Na+ absorption (Ramminger et al., 2004; Thomas et al., 2004). Adding insulin (20 nM) to the basolateral solution evoked an initial fall in ISC that became apparent after 2–3 min, but was followed by a slowly developing rise to a plateau value that was maintained throughout the remainder of the experiment. The ISC measured after 15–20 min exposure to insulin was ∼25% greater than the initial value (Figure 1), and, since this response was abolished by apical amiloride (10 µM, n= 4), these data confirm that insulin stimulates Na+ transport in these cells (Thomas et al., 2004; Inglis et al., 2009). Figure 1 also shows responses to insulin measured in age-matched cells at identical passage that had been preincubated (4 h) with pioglitazone (10 µM) or rosiglitazone (2 µM). Analysis of the ISC measured at the onset of these experiments showed that neither compound had a discernible effect upon the basal ISC (Table 1), while Figure 1 shows that these compounds also failed to modify the response to insulin. Further experiments showed that prolonged (24 h) exposure to rosiglitazone (2 µM) also had no effect upon basal ISC (Table 1) or the response to insulin (Figure 2).
Table 1.
Effects of rosiglitazone and pioglitazone on basal ISC in H441 cells
| n |
Basal ISC (µA cm−2) |
||
|---|---|---|---|
| Control | PPARγ agonist treated | ||
| Rosiglitazone (2 µM, 4 h) | 12 | 19.9 ± 2.8 | 20.3 ± 2.8 |
| Pioglitazone (10 µM, 4 h) | 12 | 21.0 ± 3.1 | 19.1 ± 1.8 |
| Rosiglitazone (2 µM, 24 h) | 5 | 22.9 ± 5.3 | 27.3 ± 6.1 |
All experiments were undertaken using a strictly paired experimental protocol in which control and PPARγ agonist treated cells were age matched and at identical passage. Control and PPARγ-agonist-treated cells were both exposed to the same concentration of solvent vehicle throughout the entire experiment. Data are presented as mean ± SEM, and values of n denote the number of times each experiment was repeated using cells at different passage number.
PPARγ, peroxisome proliferators-activated receptor gamma.
Figure 1.
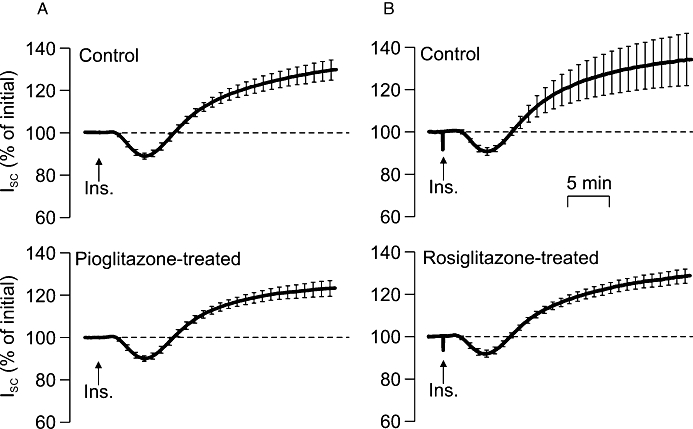
Effects of acute (4 h) exposure to PPARγ agonists upon the response to insulin in H441 cells. Experimental cells were pre-incubated in medium containing pioglitazone (A: 10 µM, n= 12) or rosiglitazone, (B: 2 µM, n= 12) for 4 h, while control cells were exposed to solvent vehicle alone. All cells were stimulated with 20 nM insulin, which was added to the basolateral bath indicated by the arrows (Ins.), and the short circuit current (ISC) generated by each preparation is presented (mean ± SEM) as a percentage of the current measured under unstimulated conditions at the onset of the experiment.
Figure 2.
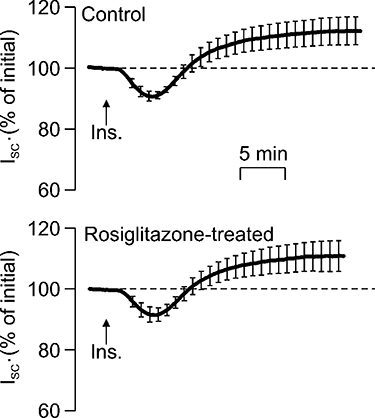
Effects of prolonged (24 h) exposure to rosiglitazone upon the response to insulin in H441 cells. Responses to insulin (20 nM) measured in control cells and in cells that had been treated with 2 µM rosiglitazone for 24 h. The ISC generated by each preparation is presented (mean ± SEM) as a percentage of the current measured under unstimulated conditions at the onset of the experiment (n= 5 paired experiments).
NDRG1-Thr346/356/366 phosphorylation in H441 cells
Pioglitazone (10 µM) and rosiglitazone (2 µM) had no perceptible effect upon the cellular abundance of Thr346/356/366-phosphorylated or total NDRG in unstimulated or insulin-stimulated (20 nM, 30 min) cells (Figure 3), and, since the phosphorylation status of these residues provides a biomarker of cellular SGK1 activity (Murray et al., 2005a; García-Martínez and Alessi, 2008; Inglis et al., 2009), these results indicated that these PPARγ agonists do not alter cellular SGK1 activity in these cells. However, this series of experiments indicate that insulin also failed to activate SGK1 (Figure 3), and this was surprising, since this hormone is thought to activate SGK1 in many different cell types (Cohen, 2006), including H441 cells (Thomas et al., 2004; Inglis et al., 2009). Further experiments therefore re-examined the hormonal control of NDRG1-Thr346/356/366 phosphorylation in H441 cells. The cells used in these studies (n= 5) were age-matched and were maintained in either hormone-free medium or in the standard, dexamethasone-containing medium for 5–6 days before being used in experiments. These studies confirmed (Inglis et al., 2009) that acutely exposing hormone-deprived H441 cells to dexamethasone (0.2 µM, 2 h) evoked NDRG1-Thr346/356/366 phosphorylation, and also showed that insulin (20 nM, 30 min) did not alter the phosphorylation of these residues under these conditions (Figure 4). These experiments also showed that the Thr346/356/366-phosphorylated form of NDRG1 was less abundant in cells that had been maintained in dexamethasone-containing medium for 5–6 days, and, since this effect could not be attributed to a reduction in overall abundance of NDRG1 (Figure 4), this result confirms (Inglis et al., 2009) that prolonged exposure to dexamethasone leads to an apparent loss of cellular SGK1 activity. While insulin did appear to evoke a slight increase in NDRG1-Thr346/356/366 phosphorylation under these conditions (Figure 5), this effect was not statistically significant. Further studies confirmed that dexamethasone evokes NDRG1-Thr346/356/366 phosphorylation in hormone-deprived cells with no effect on overall protein abundance of the NDRG1 protein (Figure 5). This response peaked after ∼2 h, and, thereafter, the phosphorylation of NDRG1-Thr346/356/366 normally declined towards the basal level (Inglis et al., 2009). Figure 5 also includes data derived from age-matched cells that had been exposed to a combination of dexamethasone (0.2 µM) and insulin (20 nM). Inclusion of insulin had no effect upon the initial components of this response to dexamethasone, but abolished the later declining phase so that the dexamethasone-induced NDRG1-Thr346/356/366 phosphorylation was maintained for the remainder of the experiment (Figure 5). Analysis of the data collected after 4–6 h exposure to dexamethasone therefore showed that insulin enhanced (P < 0.02, Student's paired t-test) the dexamethasone-induced activation of SGK1.
Figure 3.
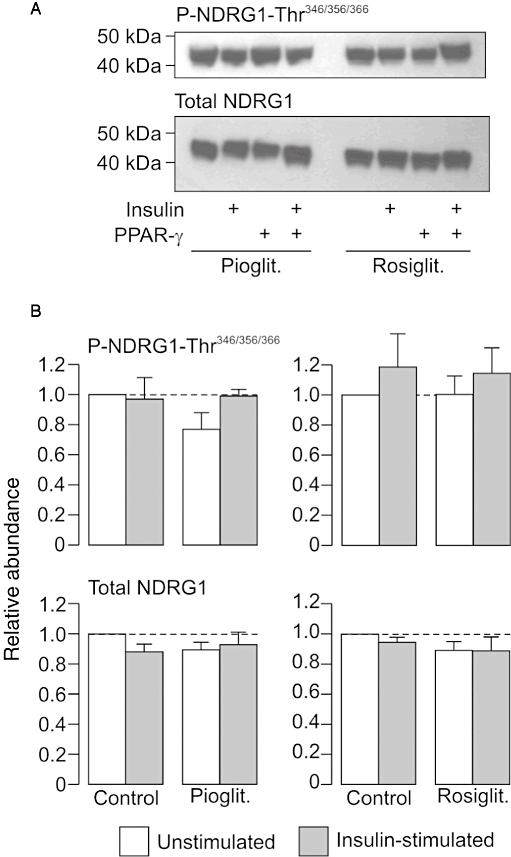
Effects of peroxisome proliferator-activated receptor gamma (PPARγ) agonists on the phosphorylation of n-myc downstream regulated protein 1 (NDRG1)-Thr346/356/366 in H441 cells. (A) Typical Western blot illustrating the effects of pioglitazone (10 µM, 4 h preincubation) and rosiglitazone (2 µM, 4 h pre-incubation) upon the cellular abundance of Thr346/356/366-phosphorylated NDRG1 and total NDRG1. All cells were age matched and at identical passage, and control cells were exposed to solvent vehicle alone. Protein was extracted from cells that had either been maintained in devoid of insulin (unstimulated) or from cells that had been exposed to insulin (20 nM) for 30 min (insulin stimulated). (B) Pooled data (n= 4) showing the effects of insulin upon the cellular abundance of Thr346/356/366-phosphorylated and total NDRG1 in control, pioglitazone- and rosiglitazone-treated cells.
Figure 4.
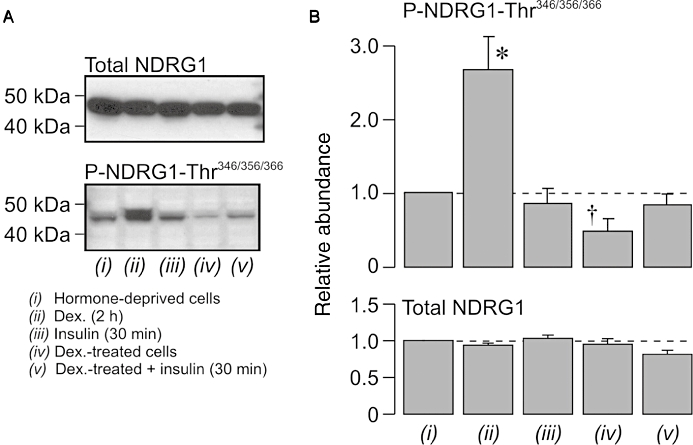
Hormonal control of n-myc downstream regulated protein 1 (NDRG1)-Thr346/356/366 phosphorylation in H441 cells. Experiments were undertaken using a strictly paired experimental design in which cells on 6 well plates were cultured for 5–6 days in either nominally hormone-free medium (hormone-deprived cells) or in medium supplemented with 0.2 µM dexamethasone (Dex.-treated cells). Before the cells were lysed to allow cellular protein to be extracted, the hormone-deprived cells were acutely stimulated with 0.2 µM dexamethasone (2 h) or 20 nM insulin (30 min), while the dexamethasone-treated cells were either untreated or acutely exposed to 20 nM insulin (30 min). (A) Typical Western blot illustrating the effects of these manoeuvres upon the cellular abundance of total and Thr346/356/366-phosphorylated NDRG1. (B) Pooled data (n= 5) showing changes in the abundance of Thr346/356/366-phosphorylated and total NDRG1 evoked by these experimental manoeuvres. *P < 0.05, value greater than that measured in hormone-deprived cells; †P < 0.05, value lower than that measured in hormone-deprived cells.
Figure 5.
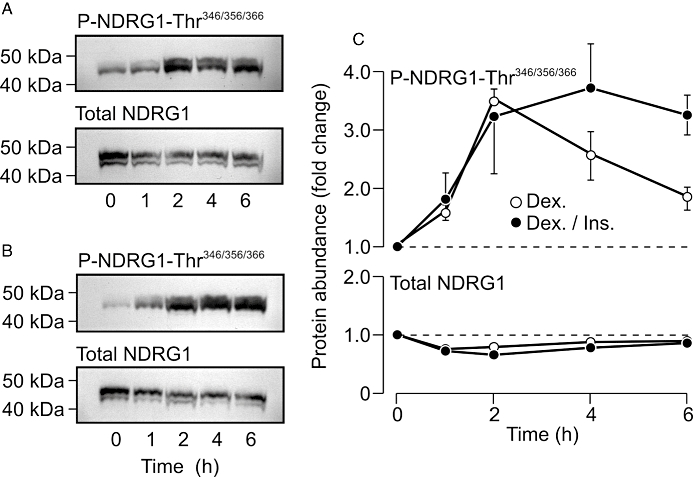
Effects of insulin on the dexamethasone-induced phosphorylation of n-myc downstream regulated protein 1 (NDRG1)-Thr346/356/366 in H441 cells. All cells were cultured in nominally hormone-free medium for 6 days before being used in experiments. (A) Typical Western blots showing the changes to the cellular abundance of total and Thr346/356/366-phosphorylated NDRG1 evoked by acute (0–6 h) exposure to 0.2 µM dexamethasone. (B) Directly analogous blots showing the changes in total and Thr346/356/366-phosphorylated NDRG1 abundance evoked by stimulation (0–6 h) with a combination of 0.2 µM dexamethasone and 20 nM insulin. (C) Pooled data (n= 5) showing the changes in the abundance of Thr346/356/366-phosphorylated (upper panel) and total (lower panel) NDRG1 measured in cells that had been stimulated with 0.2 µM dexamethasone, either alone (Dex.) or in combination with 20 nM insulin (Dex./Ins.).
Bioelectrical properties of mpkCCD cells
Initial studies of mpkCCD cells confirmed (Bens et al., 1999) that these cells became incorporated into coherent epithelial sheets, although the values of Rt measured in these cells (1–2 kΩ cm2) were ∼10-fold greater than the values measured in H441 cells. Even though the mpkCCD cells were maintained in hormone-free medium before being used in experiments (see Methods), these cells consistently generated ISC (10–20 µA·cm−2), and this current, in common with the current recorded from H441 cells, was essentially abolished (∼95% inhibition) by apical amiloride. Experiments (n= 4) in which the concentration of amiloride in the apical bath was increased progressively showed that the concentration required for half maximal inhibition of ISC (IC50) was 0.78 ± 0.08 µM. Although benzamil inhibited ISC as effectively as amiloride, this substance is ∼30-fold more potent (IC50= at 22.6 ± 1.1 nM). While ethylisopropyly amiloride (EIPA) also caused some inhibition of ISC, the highest concentration tested (0.1 mM) reduced this current by only ∼50%, and this substance therefore has a low potency. The rank order of potency amongst these compounds is therefore benzamil > amiloride > EIPA, and this pharmacological profile confirms (Bens et al., 1999) that the ISC generated by these cells is attributed to ENaC-mediated Na+ absorption.
Effects of PPARγ agonists on electrogenic Na+ transport in mpkCCD cells
The mpkCCD cells consistently responded to insulin with a clear increase in ISC, although this response was simpler than that seen in H441 cells, since it consisted of a monotonic increase in ISC that became evident 4–5 min (compare Figure 6 and Figure 1). This response was abolished by apical amiloride (10 µM), and it is therefore clear that this hormone stimulates Na+ absorption in this renally derived cell line (Nofziger et al., 2005). Analysis of current recorded from control and PPARγ agonist-treated (4 h) mpkCCD cells showed that pioglitazone (10 µM) or rosiglitazone (2 µM) both had no effect upon either the basal ISC measured prior to stimulation with insulin (Table 2) and also failed to modify the electrometric response to insulin (Figure 6).
Figure 6.
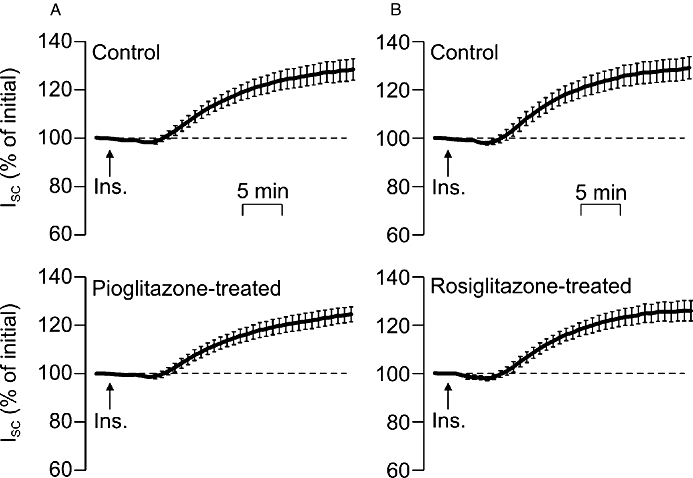
Effects of acute (4 h) exposure to peroxisome proliferator-activated receptor gamma (PPARγ) agonists upon the response to insulin in mpkCCD cells. PPARγ agonist-treated cells were pre-incubated in medium containing pioglitazone (A: 10 µM, n= 12) or rosiglitazone (B: 2 µM, n= 12) for 4 h, while control cells were exposed to solvent vehicle alone. Cells were stimulated with 20 nM insulin added to the basolateral bath as indicated by the arrows (Ins.), and the short circuit current (ISC) generated by each preparation is shown (mean ± SEM) as a percentage of the current measured under unstimulated conditions at the onset of the experiment.
Table 2.
Effects of rosiglitazone and pioglitazone on basal ISC in mpkCCD cells
| n |
Basal ISC (µA cm−2) |
||
|---|---|---|---|
| Control | PPARγ agonist treated | ||
| Rosiglitazone (2 µM, 4 h) | 9 | 10.3 ± 1.3 | 10.6 ± 1.3 |
| Pioglitazone (10 µM, 4 h) | 9 | 9.8 ± 1.3 | 10.0 ± 1.2 |
Experiments were undertaken using the paired protocol described in Table 1, and data shown are mean ± SEM. Values of n denote the number of times each experiment was repeated using cells at different passage number.
PPARγ, peroxisome proliferator-activated receptor gamma.
NDRG1-Thr346/356/366 phosphorylation in mpkCCD cells
Insulin (20 nM) consistently evoked NDRG1-Thr346/356/366-phosphorylation in mpkCCD cells, indicating that this hormone normally causes a robust activation of SGK1 in these cells. This response developed over the first 15 min of stimulation, and, although there was some decline from this initial peak, the abundance of Thr346/356/366-phosphorylated NDRG1 remained elevated for the remainder of the experimental period (Figure 7). Further experiments therefore used a strictly paired experimental design to test the hypothesis (see Introduction) that PPARγ agonists might modify this response. Analysis of data derived from control cells (i.e. cells exposed to solvent vehicle) confirmed that insulin (20 nM, 30 min) normally evokes NDRG1-Thr346/356/366 phosphorylation (Figure 8). Preincubation (4 h) with pioglitazone caused some dephosphorylation of NDRG1-Thr346/356/366 in unstimulated cells, but rosiglitazone had no such effect (Figure 8). However, insulin consistently increased the phosphorylation of NDRG1-Thr346/356/366in PPARγ agonist-treated cells, and analysis of these data showed that neither of the tested PPARγ agonists had a discernible effect upon the phosphorylation of these residues in insulin-stimulated cells. These data therefore indicate that PPARγ-agonists do not alter SGK1 activity in insulin-stimulated cells.
Figure 7.
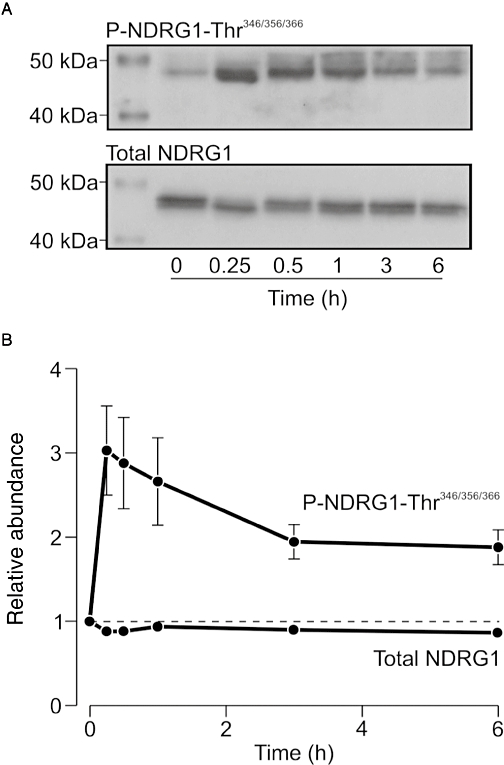
Insulin-induced n-myc downstream regulated protein 1 (NDRG1)-Thr346/356/366 phosphorylation in mpkCCD cells. Cells were stimulated with insulin (20 nM) for 0–6 h before cellular protein was extracted for Western analysis. (A) Typical blot showing the changes in the cellular abundance of Thr346/356/366-phosphorylated and total NDRG1 that occur during exposure to insulin. (B) Pooled data (n= 5) showing the insulin evoked changes to the cellular abundance of Thr346/356/366-phosphorylated and total NDRG1. CCD, cortical collecting duct.
Figure 8.
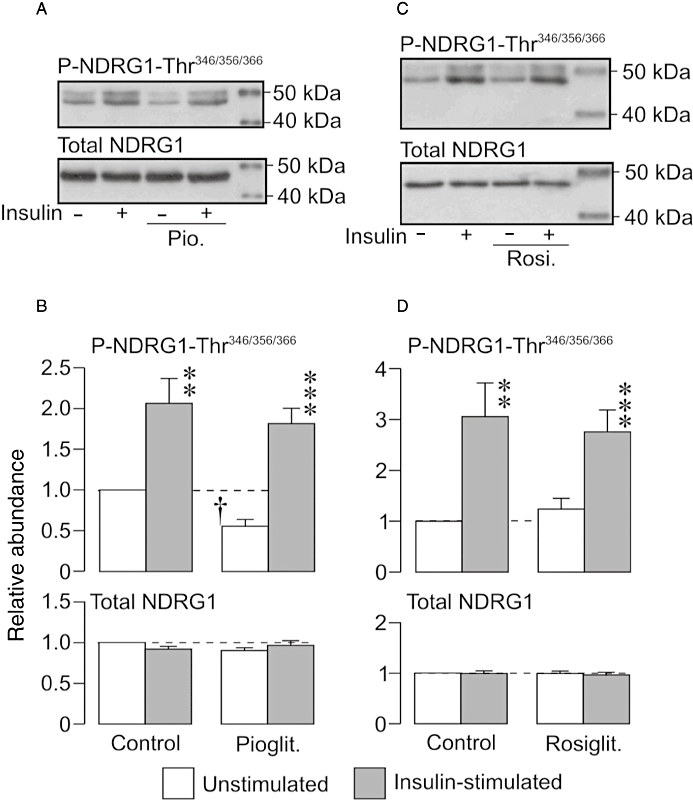
Effects of peroxisome proliferator-activated receptor gamma (PPARγ) agonists on serum and glucocorticoid-inducible kinase 1 (SGK1) activity in mpkCCD cells. (A) Typical blots showing the effects of insulin stimulation upon the cellular abundance of Thr346/356/366-phosphorylated and total n-myc downstream regulated protein 1 (NDRG1) in control and pioglitazone-treated (10 µM, 4 h) cells. (B) Pooled data showing effects of insulin upon the phosphorylation of NDRG1-Thr346/356/366 in control and pioglitazone-treated cells (n= 6). (C) Typical blots showing insulin-induced phosphorylation of NDRG1-Thr346/356/366 in control and rosiglitazone-treated (2 µM, 4 h) cells. (D) Pooled data showing insulin-evoked phosphorylation of NDRG1- Thr346/356/366 in control and rosiglitazone-treated cells (n= 6). **P < 0.002; ***P < 0.001, significant effects of insulin; †P < 0.05, significant effect of pioglitazone; Student's paired t-test. CCD, cortical collecting duct.
Expression of PPARγ receptor protein
Western analysis of protein extracted from H441 cells (n= 3, not shown) using an anti-PPARγ receptor antibody revealed a single band with a molecular weight of ∼44 kDa. Although this is lower than the molecular weight predicted for this protein (Swiss Protein database), this result does accord very well with data presented by Nofziger et al. (2005), who identified a ∼47 kDa band in A6 cells, M1 cells and mpkCCD cells. In our hands, however, mpkCCD cells (n= 3) appeared to express a slightly heavier form of this protein, with a molecular weight (∼54 kDa) close to that predicted for the full length PPARγ receptor protein.
Discussion
Insulin-evoked Na+ transport
Studies in mice have suggested that PPARγ agonist-induced fluid retention can be abolished by co-administration of amiloride or by deleting the PPARγ receptor from the collecting duct, while parallel studies of cultured mouse collecting duct cells indicated that PPARγ agonists can directly enhance amiloride-sensitive Na+ absorption (Guan et al., 2005). It was therefore suggested that the PPARγ agonist-induced expansion of body fluid volume might be due to a stimulation of ENaC-mediated Na+ transport in the distal nephron. However, not all available data support this hypothesis, since Chen et al. (2005) showed that the administration of amiloride, which blocks ENaC, did not prevent the expansion of body fluid induced by GI262570, a PPARγ agonist. Moreover, the present study shows that rosiglitazone and pioglitazone have no effect upon ENaC-mediated Na+ transport in two Na+ absorbing cells lines, both under basal conditions and after stimulation with insulin. Western blot analysis of extracted proteins showed clearly that both cell types used in the present study did express PPARγ receptors, and the concentrations of rosiglitazone and pioglitazone used were sufficient to cause maximal activation of these receptors (Henke et al., 1998). The present data therefore suggest that PPARγ agonists have no direct effect upon ENaC-mediated Na+ transport, and this conclusion accords well with data presented by Nofziger and colleagues (2005), who explored the effects of PPARγ agonists upon basal and insulin-stimulated Na+ transport in three renally derived cell lines. Their data, in common with those presented here, therefore failed to provide any indication of enhanced Na+ transport in unstimulated or insulin-stimulated cells. Moreover, recently published data show that conditional inactivation of the α-ENaC gene in the collecting duct does not prevent the PPARγ agonist-induced expansion of body fluid volume, while direct measurements of ENaC activity in the isolated collecting duct showed that PPARγ agonists have no effect upon the behaviour of these channels (Vallon et al., 2009). At present, almost all available data therefore fail to support the idea that PPARγ agonists enhance ENaC-mediated epithelial Na+ transport.
Although Guan et al. (2005) did make electrometric measurements, their suggestion that PPARγ agonists might evoke increased Na+ transport was based upon data from experiments in which Na+ absorption was assessed by measuring amiloride-sensitive 22Na+ fluxes. While these experiments did reveal an apparent stimulation of Na+ transport, it is important to remember that ENaC-mediated Na+ absorption is electrogenic, and that a substantial stimulation of this ion transport process would therefore hyperpolarize Vt. It is therefore surprising that the electrometric data (Supporting Table S2 in Guan et al., 2005) show that pioglitazone depolarized Vt from ∼−15 to ∼−8 mV, and, since this depolarization was accompanied by a fall in Rt (control: ∼400 Ωcm2; pioglitazone: to ∼240 Ωcm2), these data show clearly that the transepithelial current (i.e. Vt/Rt) is approximately −35 µA cm−2 in both control and the pioglitazone-treated cells. Since electrogenic Na+ transport is the dominant epithelial transport process in such cells, these data, in contrast to the measurements of 22Na+ flux, indicate that pioglitazone does not augment ENaC-mediated Na+ transport. Interestingly, recently published data show that pioglitazone can activate a non-selective cation conductance in cultured collecting duct cells (Vallon et al., 2009). Although these channels might provide a route for absorptive Na+ transport, they would not allow this process to be regulated independently of K+ secretion, and Na+ transport via such non-selective channels would therefore have little effect upon Vt. Such pioglitazone-induced cation channels (Vallon et al., 2009) may therefore provide a route for increased diffusion of 22Na+ and this in turn could explain how this drug can evoke increased 22Na+ flux without altering electrogenic Na+ transport (Guan et al., 2005).
Control of SGK1 activity by insulin
Insulin is thought to control epithelial Na+ transport via SGK1 (Blazer-Yost et al., 1998, 2003; Alvarez De La Rosa and Canessa, 2003), a regulatory kinase that controls the apical abundance of ENaC (Snyder, 2002; Snyder et al., 2002, 2004; Lang et al., 2006; Loffing et al., 2006). The present study therefore explored the effects of insulin upon cellular SGK1 activity by monitoring the phosphorylation of residues within an endogenous protein (NDRG1-Thr346/356/366) that have recently been identified as physiological substrates for SGK1, but not for other, closely related kinases (Murray et al., 2005a,b;). While these assays identified basal SGK1 activity in both cell types, they indicated that insulin had no effect upon SGK1 activity in H441 cells, and this observation was highly surprising, since our recently published data suggest that insulin does activate SGK1 in these cells (Inglis et al., 2009). Moreover, studies of mpkCCD cells undertaken using the same reagents, revealed a clear and consistent increase in SGK1 activity in response to insulin, and the cell types used in the present study therefore appear to display different patterns of SGK1 activation in response to insulin. This was also surprising, since the factors underlying the activation of SGK1 are thought to be well conserved between different cell types (Lang et al., 2006; Loffing et al., 2006), and this discrepancy prompted us to re-examine the hormonal control of SGK1 activity in H441 cells. The first such studies confirmed (Inglis et al., 2009) that dexamethasone acutely activated SGK1 when applied to hormone-deprived cells, but provided no indication that insulin could mimic this action. A subsequent series of experiments confirmed this effect of dexamethasone, and also showed that the acute application of insulin did not directly activate SGK1 in cells that had been maintained in dexamethasone-supplemented medium. However, when acutely administered in combination with dexamethasone, insulin prolonged the glucocorticoid-induced activation SGK1, and so, although this kinase is not entirely insensitive to insulin, glucocorticoids seem to be the principal determinants of SGK1 activity in H441 cells. It thus appears that our previously published data (Inglis et al., 2009) overestimated the importance of insulin to the control of SGK1 activity in H441 cells, although, even in this study, the response to insulin was smaller (∼20%) than the response to dexamethasone (Inglis et al., 2009).
It thus appears insulin stimulates Na+ transport in H441 cells while having no major effect upon SGK1, and it is therefore interesting that the activation of SGK1 is mediated by phosphoinosidide-3 kinase (PI3K) (Kobayashi and Cohen, 1999; Park et al., 1999), and that insulin-evoked transport of Na+ in H441 cells is clearly dependent upon this kinase (Thomas et al., 2004; Inglis et al., 2009). PI3K catalyses the formation of phosphatidylinositol 3,5-bisphosphate (PIP2) and phosphatidylinositol 3,4,5-trisphosphate (PIP3), biologically active phospholipids that control the phosphorylation of SGK1 at Ser422 and Thr256 (Kobayashi and Cohen, 1999; Park et al., 1999). However, PIP2 and PIP3 also control ENaC activity by directly binding to the channel complex, and these biologically active lipids can thus allow PI3K to control ENaC activity independently of SGK1 (Pochynyuk et al., 2005, 2007). Moreover, PIP2 and PIP3 also control the activity of protein kinase B (PKB), a kinase that is closely related to SGK1 (Bayascas and Alessi, 2005), and recent studies of Fisher rat thyroid cells transiently expressing α-, β- and γ-ENaC have indicated that PKB may allow insulin to control Na+ transport (Lee et al., 2007).
Effects of PPARγ agonists on SGK1 activity
Studies of human CCD cells indicated that PPARγ agonists increase sgk1 gene expression (Hong et al., 2003; Chen et al., 2005), and this seems to lead to an increase in cellular SGK1 activity (Hong et al., 2003). Moreover, PPARγ agonists have also been shown to increase the expression of SGK1 mRNA (Chen et al., 2005; Artunc et al., 2008) and protein (Artunc et al., 2008) in the epithelia of the distal nephron, and since SGK1 contributes to the control of ENaC activity (Snyder, 2002; Snyder et al., 2002, 2004; Lang et al., 2006; Loffing et al., 2006; Loffing and Korbmacher, 2009), PPARγ agonist-induced activation of SGK1 may therefore lead to activation of ENaC in the distal nephron, which would provide a physiological basis for the expansion of body fluid volume (Hong et al., 2003; Guan et al., 2005; Artunc et al., 2008). However, although the two cell types used in the present study displayed different patterns of SGK1 activation in response to insulin, the present data show clearly that rosiglitazone and pioglitazone did not increase SGK1 activity in either unstimulated or insulin-stimulated cells. These data do not, therefore, support the view that PPARγ agonists can activate SGK1.
While the reason for this discrepancy with earlier work is unknown, it is important to stress that the approach used in the present study provides a read out of cellular SGK1 activity, whereas most previous studies have monitored changes to the cellular abundance of SGK1 mRNA and/or protein. Such observations do not necessarily indicate alterations to SGK1 activity. Indeed, since the catalytic activity of SGK1 is critically dependent upon the PI3K-regulated phosphorlyation of SGK1-Thr256 and SGK1-Ser422 (Kobayashi and Cohen, 1999; Park et al., 1999), it is perfectly possible for cellular SGK1 activity to increase with no change in mRNA/protein abundance. Although Hong et al. (2003) did assay SGK1 activity, the method used was based upon a generic substrate that would be phosphorylated by several other threonine/serine kinases. The selectivity of this assay system is therefore dependent upon the ability to immunoprecipitate SGK1 selectively from cellular lysates. It is, however, interesting that Nofziger et al. (2005) found that PPARγ agonists had no effect upon the SGK1 protein expression in renally derived epithelia, while physiological studies of sgk1 knock-out mice show that deletion of the sgk1 gene has only a modest effect upon the PPARγ agonist-induced expansion of body fluid volume (Artunc et al., 2008). These findings, in common with the present data, therefore suggest that SGK1 does not play a major role in this response. Interestingly, deletion of the sgk1 gene does prevent the insulin-induced reduction in urinary Na+ excretion (Huang et al., 2006), and this differential requirement for SGK1 makes it extremely unlikely that PPARγ agonists will expand body fluid volume by facilitating insulin-induced Na+ retention (Huang et al., 2006; Artunc et al., 2008). In addition, rosiglitazone has recently been shown to increase fluid retention and body weight in mice with inactivated αENaC in the collecting duct (Vallon et al., 2009), and these observations all provide strong evidence against a central role for SGK1 / ENaC in PPARγ-mediated fluid retention.
Significance of present findings
Although we cannot exclude the possibility that PPARγ agonists might be able to evoke ENaC-mediated Na+ absorption under some experimental conditions, it now appears very unlikely that SGK1-mediated activation of ENaC can account for PPARγ-agonist-induced oedema (present study, Nofziger et al., 2005; Vallon et al., 2009). It is therefore interesting that recent studies have shown that PPARγ agonists can increase the expression of mRNA encoding SGK1, water channel proteins (aquaporin 1/7) and the Na+– H+ exchanger (NHE1) in human proximal tubule cells (Saad et al., 2009). Moreover, the effects on aquaporin 1/7 and NHE1 mRNA were blocked by a small molecule inhibitor of SGK1 (GSK650394A) and by exposing cells to small interfering RNAs designed to disrupt SGK1 expression (Saad et al., 2009). Since Na+ absorption in this nephron segment occurs via NHE1 rather than via ENaC, these data raise the possibility that PPARγ agonist-induced oedema might be due to SGK1-dependent Na+ and water retention in the proximal tubule, rather than the distal tubule (Panchapakesan et al., 2009; Saad et al., 2009). It has, however, also been suggested that PPARγ agonists might evoke Na+ and water retention by activating the renin-angiotensin-aldosterone cascade, by altering blood pressure and by evoking increased vascular permeability. Indeed, there is much evidence that such effects are important to the genesis of PPARγ agonist-induced oedema (Buckingham and Hanna, 2007). Clearly, further studies are needed to identify the precise mechanisms involved in PPARγ-related fluid retention, and this knowledge may help identify patients likely to develop oedema or heart failure when treated with a PPARγ agonist, and may improve the ability to treat fluid retention and oedema in patients treated with PPARγ agonists.
Acknowledgments
This work was supported by an award (Ref: CVMD-DU-086) from the Translational Medicine Research Collaboration – a consortium made up of the Universities of Aberdeen, Dundee, Edinburgh and Glasgow, the four associated NHS Health Boards (Grampian, Tayside, Lothian and Greater Glasgow and Clyde), Scottish Enterprise and Wyeth Pharmaceuticals.
Glossary
Abbreviations:
- CCD
cortical collecting duct
- DMEM
Dulbecco's modified Eagle's Medium
- EIPA
5-(N-ethyl-N-isopropyl)amiloride
- ENaC
epithelial sodium channel
- FBS
fetal bovine serum
- ISC
short circuit current
- NDRG1
n-myc downstream regulated protein 1
- NHE1
Na+– H+ exchanger 1
- PI3K
phosphoinositide-3 kinase
- PIP2
phosphatidylinostiol 3,5-bisphosphate
- PIP3
phosphatidylinositol 3,4,5-trisphosphate
- PKB
protein kinase B
- PPARγ
peroxisome proliferator-activated receptor gamma
- Rt
transepithelial resistance
- SDS
sodium dodecyl sulphate
- SGK1
serum and glucocoritcoid-inducible kinase 1
- Vt
transepithelial potential difference
References
- Alvarez De La Rosa D, Canessa CM. Role of SGK in hormonal regulation of epithelial sodium channel in A6 cells. Am J Physiol Cell Physiol. 2003;284:C404–C414. doi: 10.1152/ajpcell.00398.2002. [DOI] [PubMed] [Google Scholar]
- Artunc F, Sandulache D, Nasir O, Boini KM, Friedrich B, Beier N, et al. Lack of the serum and glucocorticoid-inducible kinase SGK1 attenuates the volume retention after treatment with the PPAR gamma agonist pioglitazone. Pflügers Arch. 2008;456:425–436. doi: 10.1007/s00424-007-0401-5. [DOI] [PubMed] [Google Scholar]
- Atchley D, Loeb RF, Richards DW, Benedict EM, Driscoll ME. On diabetic acicosis. J Clin Invest. 1936;12:297–326. doi: 10.1172/JCI100504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayascas JR, Alessi DR. Regulation of Akt/PKB Ser473 phosphorylation. Mol Cell. 2005;18:143–145. doi: 10.1016/j.molcel.2005.03.020. [DOI] [PubMed] [Google Scholar]
- Bens M, Vallet V, Cluzeaud F, Padcula-Letallec L, Kahn A, Rafestin-Oblin ME, et al. Corticosteroid-dependent sodium transport in a novel immortalized mouse collecting duct principal cell line. J Am Soc Nephrol. 1999;10:923–934. doi: 10.1681/ASN.V105923. [DOI] [PubMed] [Google Scholar]
- Blazer-Yost BL, Liu XH, Helman SI. Hormonal regulation of ENaCs: insulin and aldosterone. Am J Physiol Cell Physiol. 1998;274:1373–1379. doi: 10.1152/ajpcell.1998.274.5.C1373. [DOI] [PubMed] [Google Scholar]
- Blazer-Yost BL, Esterman MA, Vlahos CJ. Insulin-stimulated trafficking of ENaC in renal cells requires PI3-kinase activity. Am J Physiol Cell Physiol. 2003;284:C1645–C1653. doi: 10.1152/ajpcell.00372.2002. [DOI] [PubMed] [Google Scholar]
- Brown SG, Gallacher M, Olver RE, Wilson SM. The regulation of selective and non-selective Na+ conductances in H441 human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2008;294:L942–L954. doi: 10.1152/ajplung.00240.2007. [DOI] [PubMed] [Google Scholar]
- Buckingham RE, Hanna A. Thiazolidinedione insulin sensitizers and the heart: a tale of two organs. Diabetes Obes Metab. 2007;10:312–328. doi: 10.1111/j.1463-1326.2006.00700.x. [DOI] [PubMed] [Google Scholar]
- Chen L, Yang B, McNulty JA, Clifton LG, Binz JG, Grimes AM, et al. GI262570, a peroxisome proliferator-activated receptor γ agonist, changes electrolyte and water reabsorption from the distal nephron on rats. J Pharm Exp Ther. 2005;312:718–725. doi: 10.1124/jpet.104.074088. [DOI] [PubMed] [Google Scholar]
- Cohen P. Timeline – the twentieth century struggle to decipher insulin signalling. Nat Rev Mol Cell Biol. 2006;7:867–873. doi: 10.1038/nrm2043. [DOI] [PubMed] [Google Scholar]
- García-Martínez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-inducible protein kinase 1 (SGK1) Biochem J. 2008;416:375–385. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- Guan Y, Hao C, Cha DR, Rao R, Lu W, Kohan DE, et al. Thiazolidinediones expand body fluid volume through PPARγ stimulation of ENaC-mediated renal salt absorption. Nat Med. 2005;11:861–866. doi: 10.1038/nm1278. [DOI] [PubMed] [Google Scholar]
- Guazzi M, Bramilla R, De Vita S, Guazzi MD. Diabetes worsens pulmonary diffusion in heart failure, and insulin counteracts this effect. Am J Resp Crit Care Med. 2002a;166:978–982. doi: 10.1164/rccm.200203-234OC. [DOI] [PubMed] [Google Scholar]
- Guazzi MD, Oreglia I, Guazzi MD. Insulin improves alveolar-capillary membrane gas conductance in type 2 diabetes. Diabetes Care. 2002b;25:1802–1806. doi: 10.2337/diacare.25.10.1802. [DOI] [PubMed] [Google Scholar]
- Gupta AK, Clark R, Kirchner KA. Verapamil prevents insulin antinaturesis in euglycemic rats. Am J Physiol Regul Integr Comp Physiol. 1992;262:R1145–R1148. doi: 10.1152/ajpregu.1992.262.6.R1145. [DOI] [PubMed] [Google Scholar]
- Hagiwara N, Tohda H, Doi Y, O'Brodovich H, Marunaka Y. Effects of insulin and tyrosine kinase inhibitor on ion transport in the alveolar cell of the fetal lung. Biochem Biophys Res Commun. 1992;187:302–308. doi: 10.1016/0006-291x(92)91267-t. [DOI] [PubMed] [Google Scholar]
- Henke BR, Blanchard SG, Brackeen MF, Brown KK, Cobb JE, Collins JL, et al. N-(2-Benzoylphenyl)-L-tyrosine PPARgamma agonists. 1. Discovery of a novel series of potent antihyperglycemic and antihyperlipidemic agents. J Med Chem. 1998;41:5020–5036. doi: 10.1021/jm9804127. [DOI] [PubMed] [Google Scholar]
- Hong GH, Lockhart A, Davis B, Rahmoune H, Baker S, Ye L, et al. PPARγ activation enhances cell surface ENaCa via upregulation of SGK1 in human collecting duct cells. FASEB J. 2003;17 doi: 10.1096/fj.03-0181fje. [DOI] [PubMed] [Google Scholar]
- Huang DY, Boini KM, Freidrich B, Metzger M, Just L, Osswald H, et al. Blunted hypertensive effect of combined fructose and high salt intake in gene targeted mice lacking serum and gluccocorticoid-inducible kinase SGK1. Am J Physiol Regul Integr Comp Physiol. 2006;290:R935–R944. doi: 10.1152/ajpregu.00382.2005. [DOI] [PubMed] [Google Scholar]
- Inglis SK, Gallacher M, Brown SG, McTavish N, Getty J, Husband EM, et al. SGK1 activity in Na+ asorbing human airway epithelial cells monitored by assaying NDRG1-Thr346/356/366 phosphorylation. Pflügers Arch. 2009;457:1287–1301. doi: 10.1007/s00424-008-0587-1. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Cohen P. Activation of serum- and glucocorticoid-regulated protein kinases by agonists that activate phosphatidylinositol 3-kinase is mediated by 3-phosphoinositide-dependent protein kinase 1 (PDK1) and PDK2. Biochem J. 1999;339:319–328. [PMC free article] [PubMed] [Google Scholar]
- Lang F, Böhmer C, Palmada M, Seebohm G, Strutz-Seebohm N, Vallon V. (Patho)physiological significance of the serum and glucocorticoid-inducible kinase isoforms. Physiol Rev. 2006;86:1151–1178. doi: 10.1152/physrev.00050.2005. [DOI] [PubMed] [Google Scholar]
- Lee I-H, Dinudom A, Sanchez-Perez A, Kumar S, Cook DI. Akt mediates the effects of insulin on epithelial sodium channels by inhibiting Nedd-4-2. J Biol Chem. 2007;282:29866–29873. doi: 10.1074/jbc.M701923200. [DOI] [PubMed] [Google Scholar]
- Loffing J, Flores SY, Staub O. SGK kinases and their role in epithelial transport. Annu Rev Physiol. 2006;68:461–430. doi: 10.1146/annurev.physiol.68.040104.131654. [DOI] [PubMed] [Google Scholar]
- Loffing J, Korbmacher C. Regulated sodium transport in the renal collecting tubule (CNT) via the epithelial sodium channel (ENaC) Pflügers Arch. 2009;458:111–135. doi: 10.1007/s00424-009-0656-0. [DOI] [PubMed] [Google Scholar]
- Miller JH, Bogdonoff MD. Antidiuresis associated with administration of insulin. J Appl Physiol. 1954;6:509–512. doi: 10.1152/jappl.1954.6.8.509. [DOI] [PubMed] [Google Scholar]
- Murray JT, Cambell DG, Morrice N, Auld G, Shpiro N, Marquez R, et al. Exploitation of KESTREL to identify NDRG family members as physiological substrates of SGK1 and GSK3. Biochem J. 2005a;385:1–12. doi: 10.1042/BJ20041057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray JT, Cummings LA, Bloomberg GB, Cohen P. Identification of different specificity requirements between SGK1 and PKBα. FEBS Lett. 2005b;579:991–994. doi: 10.1016/j.febslet.2004.12.069. [DOI] [PubMed] [Google Scholar]
- Nofziger C, Chen L, Shane MA, Smith CD, Brown KK, Blazer-Yost BL. PPARγ agonists do not directly enhance basal or insulin-stimulated Na+ transport via the epithelial Na+ channel. Pflügers Arch. 2005;451:445–453. doi: 10.1007/s00424-005-1477-4. [DOI] [PubMed] [Google Scholar]
- Panchapakesan U, Pollock C, Saad S. Review article: importance of the kidney proximal tubular cells in thizolidinedione-mediated sodium and water uptake. Nephrology. 2009;14:298–301. doi: 10.1111/j.1440-1797.2009.01089.x. [DOI] [PubMed] [Google Scholar]
- Park J, Leong MLL, Buse P, Maiyar AC, Firestone GL, Hemmings BA. Serum and glucocorticoid-inducible kinase (SGK) is a target of the PI 3-kinase-stimulated signaling pathway. EMBO J. 1999;18:3024–3033. doi: 10.1093/emboj/18.11.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pochynyuk O, Staruschenko A, Tong Q, Medina J, Stockand JD. Identification of a functional phosphatidylinositol 3,4,5-trisphosphate binding site in the epithelial Na+ channel. J Biol Chem. 2005;280:37565–37571. doi: 10.1074/jbc.M509071200. [DOI] [PubMed] [Google Scholar]
- Pochynyuk O, Tong Q, Staruschenko A, Stockand JD. Binding and direct activation of the epithelial Na+ channel by phosphoinositides. J Physiol Lond. 2007;580:365–372. doi: 10.1113/jphysiol.2006.127449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramminger SJ, Richard K, Inglis SK, Land SC, Olver RE, Wilson SM. A regulated apical Na+ conductance in dexamethasone-treated H441 airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2004;287:L411–L419. doi: 10.1152/ajplung.00407.2003. [DOI] [PubMed] [Google Scholar]
- Saad S, Agapiou DJ, Chen X-M, Stevens V, Pollack CA. The role of SGK1 in the upregulation of transport proteins by PPAR-γ agonists in human proximal tubule cells. Nephrol Dial Transplant. 2009;24:1130–1141. doi: 10.1093/ndt/gfn614. [DOI] [PubMed] [Google Scholar]
- Snyder PM. The epithelial Na+ channel: cell surface insertion and retrieval in Na+ homeostasis and hypertension. Endocrine Rev. 2002;23:258–275. doi: 10.1210/edrv.23.2.0458. [DOI] [PubMed] [Google Scholar]
- Snyder PM, Olsen DR, Thomas BC. Serum and glucocorticoid-regulated kinase modulates Nedd4-2-mediated inhibition of the epithelial Na+ channel. J Biol Chem. 2002;277:5–8. doi: 10.1074/jbc.C100623200. [DOI] [PubMed] [Google Scholar]
- Snyder PM, Olson DR, Kabra R, Zhou R, Steines JC. cAMP and serum and glucocorticoid-inducible kinase (SGK) regulate the epithelial Na+ channel through convergent phosphorylation of Nedd4-2. J Biol Chem. 2004;279:45753–45758. doi: 10.1074/jbc.M407858200. [DOI] [PubMed] [Google Scholar]
- Thomas CP, Campbell JR, Wright PJ, Husted RF. cAMP-stimulated Na+ transport in H441 distal lung epithelial cells: role of PKA, phosphatidylinositol 3-kinase, and sgk1. Am J Physiol Lung Cell Mol Physiol. 2004;287:L843–L851. doi: 10.1152/ajplung.00340.2003. [DOI] [PubMed] [Google Scholar]
- Tiwari S, Riazi S, Ecelbarger CA. Insulin's impact on renal sodium transport and blood pressure in health, obesity and diabetes. Am J Physiol Renal Physiol. 2007;293:F974–F984. doi: 10.1152/ajprenal.00149.2007. [DOI] [PubMed] [Google Scholar]
- Vallon V, Hummler E, Rieg T, Pochynyuk O, Bugaj V, Schroth J, et al. Thiazolidine-induced fluid retention is independent of collecting duct αENaC activity. J Am Soc Nephrol. 2009;20:721–729. doi: 10.1681/ASN.2008040415. [DOI] [PMC free article] [PubMed] [Google Scholar]