

Abstract
Elevated muscle sympathetic nerve activity (MSNA) features in many cardiovascular diseases, but how this sympathoexcitation is brought about differs across pathologies. Unitary recordings from post-ganglionic muscle vasoconstrictor neurones in human subjects have shown that the augmented MSNA in the obstructive sleep apnoea syndrome (OSAS) is associated with an increase in firing probability and mean firing rate, and an increase in multiple within-burst firing. Here we characterize the firing properties of muscle vasoconstrictor neurones in patients with chronic obstructive pulmonary disease (COPD), who are chronically asphyxic. We tested the hypothesis that this elevated chemical drive would shift the firing pattern from that seen in healthy subjects to that seen in OSAS. The mean firing probability (52%) and mean firing rate (0.92 Hz) of 17 muscle vasoconstrictor neurones recorded in COPD were comparable to those previously recorded in OSAS (51% and 0.96 Hz), but significantly higher than those recorded in a group of healthy subjects with high levels of resting MSNA (35% and 0.33 Hz). In COPD single neurones fired once in 63% of cardiac intervals, comparable to OSAS (59%), but significantly lower than in the healthy group (78%). Conversely, single neurones fired twice in 25% of cardiac intervals, similar to OSAS (27%), but significantly higher than in the healthy group (18%). We conclude that the chronic asphyxia associated with COPD results in an increase in the firing probability and mean firing frequency of muscle vasoconstrictor neurones and causes a shift towards multiple firing, reflecting an increase in central muscle vasoconstrictor drive.
Introduction
Chronic obstructive pulmonary disease (COPD) is a disease characterized by marked airflow limitation, particularly in expiration, and compromised gas exchange. Clinically, COPD patients have been described classically as one of two types: ‘pink puffers’ exhibit breathlessness and hyperinflation, and typically present with mild hypoxaemia and a normal 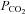 , whereas ‘blue bloaters’ are cyanotic, with marked hypoxaemia, polycythaemia and hypercapnia; the latter also develop pulmonary hypertension and cor pulmonale (Flenley, 1988; Howard, 1990; Ryu & Scanlon, 2001). However, there is significant overlap in these two phenotypes and the predominant limiting symptom of moderate to severe COPD is shortness of breath on activity and chronic hypoxaemia, which tends to be worse in sleep, and results in increasing pulmonary artery pressures (McKenzie et al. 2003). COPD is often associated with co-morbidities such as cardiovascular disease, diabetes and hypertension, which contribute importantly to a higher risk of mortality (Sin & Man, 2003; Sin et al. 2006; Mannino et al. 2008). There are many cardiac manifestations of COPD, with right ventricular dysfunction and pulmonary vascular disease being known to complicate the clinical course of COPD and correlate inversely with survival; coronary artery disease and cardiovascular disease also occurs commonly in people with COPD (Sin & Man, 2003; Sin et al. 2006; Falk et al. 2008). Moreover, COPD can often occur with the obstructive sleep apnoea syndrome (OSAS), the so-called overlap syndrome, in which affected individuals have a greater risk for pulmonary hypertension, right heart failure, and hypercapnia than patients who have either disorder alone (Hiestand & Phillips, 2008). As in patients with OSAS, treatment of overlap patients with continuous positive airway pressure is beneficial, with supplemental oxygen added for selected patients (Zamarrón et al. 2008).
, whereas ‘blue bloaters’ are cyanotic, with marked hypoxaemia, polycythaemia and hypercapnia; the latter also develop pulmonary hypertension and cor pulmonale (Flenley, 1988; Howard, 1990; Ryu & Scanlon, 2001). However, there is significant overlap in these two phenotypes and the predominant limiting symptom of moderate to severe COPD is shortness of breath on activity and chronic hypoxaemia, which tends to be worse in sleep, and results in increasing pulmonary artery pressures (McKenzie et al. 2003). COPD is often associated with co-morbidities such as cardiovascular disease, diabetes and hypertension, which contribute importantly to a higher risk of mortality (Sin & Man, 2003; Sin et al. 2006; Mannino et al. 2008). There are many cardiac manifestations of COPD, with right ventricular dysfunction and pulmonary vascular disease being known to complicate the clinical course of COPD and correlate inversely with survival; coronary artery disease and cardiovascular disease also occurs commonly in people with COPD (Sin & Man, 2003; Sin et al. 2006; Falk et al. 2008). Moreover, COPD can often occur with the obstructive sleep apnoea syndrome (OSAS), the so-called overlap syndrome, in which affected individuals have a greater risk for pulmonary hypertension, right heart failure, and hypercapnia than patients who have either disorder alone (Hiestand & Phillips, 2008). As in patients with OSAS, treatment of overlap patients with continuous positive airway pressure is beneficial, with supplemental oxygen added for selected patients (Zamarrón et al. 2008).
While it is known that sustained hypoxaemia causes a long-lasting increase in muscle sympathetic nerve activity (MSNA) and blood pressure, that largely persists following the return to normoxia (Morgan et al. 1995; Hansen & Sander, 2003; Tamisier et al. 2005), to our knowledge there have been only two studies that have investigated whether muscle vasoconstrictor drive is elevated in COPD. Heindl and colleagues (Heindl et al. 2001) studied a mixed group of patients with COPD or restrictive lung disease (pulmonary fibrosis). Compared to a small group of age- and sex-matched healthy subjects MSNA was significantly increased in the patients: mean burst frequency was elevated from 34 ± 2 (s.e.m.) to 61 ± 5 bursts min−1. In a more recent study of 15 COPD patients, Raupach et al. (2008) reported a comparable elevation of mean burst frequency: 39 ± 3 bursts min−1 in the controls to 61 ± 5 bursts min−1 in the patients. This level of sympathoexcitation is comparable to that seen in the obstructive sleep apnoea syndrome, in which episodes of airway obstruction in sleep cause repeated bouts of hypoxaemia and an increase in MSNA that persists in the awake state (Hedner et al. 1988; Carlsson et al. 1993, 1996; Narkiewicz et al. 1998, 1999; Elam et al. 2002).
The purpose of the present study was to characterize the sympathoexcitation in COPD by recording from single muscle vasoconstrictor neurones, so as to increase our understanding of the physiological mechanisms by which the sympathetic nervous system grades its output in health and disease. Unitary recordings provide a more sensitive measure of central sympathetic drive than standard multi-unit recordings (Macefield et al. 1994, 2002; Macefield, 2009). In multi-unit recordings the absolute strength (intensity) of the recorded activity cannot be determined because of the dependence on the proximity of the electrode tip to the active nerve fibres, but this is not a limitation with single-unit recordings. Differences in the strength of multi-unit sympathetic activity (i.e. the number and the intensity of multi-unit bursts) may be brought about either by differences in (i) the number of active neurones or (ii) the mean firing frequency of the neurones (or a combination of both). A difference in mean firing frequency may, in turn, be due to either (i) a difference in firing probability of active neurones, i.e. the percentage of heart beats in which the neurones are active, or (ii) a difference in the number of spikes the neurones discharge within the bursts (or a combination of both). Using this reductionist approach we can tease out the mechanisms by which the sympathoexcitation is brought about in different pathophysiological states. There is increasing interest in the single-unit approach to identify patterns of central sympathetic drive, as evidenced by a recent paper on the greater increases in firing probability and multiple firing of muscle vasoconstrictor neurones during hand-grip exercise in congestive heart failure (CHF) (Murai et al. 2009; see Macefield, 2009). In the present study we tested the hypothesis that the elevated chemical drive resulting from the chronic hypoxaemia and hypercapnia associated with COPD would increase the firing probability and mean firing rates of individual muscle vasoconstrictor neurones, and cause a shift towards multiple firing, similar to that seen in OSAS. We also characterized sympathetic outflow in bronchiectasis associated with chronic airflow limitation, in which the airways are dilated and inflamed but blood gases are only compromised in severe stages of the disease (Ilowite et al. 2008; Lazarus et al. 2008).
Methods
Subjects
Data were obtained from 11 female and 7 male COPD patients ranging in age from 61 to 84 years (mean ±s.e.m.= 72 ± 2 years), and 4 female and 1 male bronchiectasis patients ranging in age from 63 to 75 years (70 ± 3 years), recruited from the Department of Respiratory Medicine. All patients remained on their scheduled pharmacological and physiotherapeutic treatment. Each patient provided informed written consent to the procedures, which were conducted under the approval of the Human Research Ethics Committee of the University of New South Wales and conformed to the standards set by the Declaration of Helsinki.
General procedures
All studies were performed in the morning. Patients lay semi-recumbent in a chair with their backs at 45 deg and their legs supported horizontally. Continuous blood pressure was recorded using radial arterial tonometry (Colin 7000 NIBP, Colin Corp., Japan). Muscle sympathetic nerve activity was recorded from fascicles of the common peroneal nerve supplying the ankle and toe extensor and foot everter muscles via tungsten microelectrodes (FHC, Bowdoinham, ME, USA) inserted percutaneously at the level of the fibular head. A nearby subdermal electrode with a larger uninsulated tip served as the reference electrode. Neural activity was amplified (gain 20 000, bandpass 0.3–5.0 kHz) using an isolated amplifier (DAM 80i, World Precision Instruments, Sarasota, FL, USA) and stored on a computer (20 kHz sampling) using a computer-based data acquisition and analysis system (PowerLab 16SP hardware and Chart 5 software; ADInstruments, Sydney, Australia). ECG (0.3 Hz to 1.0 kHz) was recorded with Ag–AgCl surface electrodes on the chest and sampled at 2 kHz. Respiration (DC to 100 Hz) was recorded using a strain-gauge transducer (Pneumotrace, UFI, Morro Bay, CA, USA) wrapped around the chest. Resting multi-unit bursts and heart rate were recorded during 5 min of quiet breathing, so as to allow measurement of burst incidence (bursts per 100 heart beats) and burst frequency (bursts per minute). The microelectrode was then manipulated until large unitary discharges appeared out of the multi-unit sympathetic bursts and a stable unitary site was obtained.
Data acquisition and analysis
During off-line analysis a copy of the nerve signal was RMS processed to emulate a leaky integrator (time constant 100 ms). The morphology of every spike of a candidate unit was carefully checked using spike recognition software (Spike Histogram, ADInstruments). The computer measured all interspike intervals and the number of spikes a unit fired in each cardiac interval. For each unit the following parameters were determined: (i) probability of firing (the percentage of heart beats during which one or more spikes occurred), (ii) the numbers of cardiac intervals in which a unit generated 1, 2, 3 or 4 spikes (in %), and (iii) mean firing frequency (the mean of the inverse of all interspike intervals).
Statistics
All values are expressed as means and standard error of the mean (s.e.m.). All statistical evaluation of the data was performed using Prism 5 (GraphPad Software, Inc., USA) using ANOVA, Tukey's HSD test and linear regression analysis. P < 0.05 was considered statistically significant.
Results
Patient details
Typical of the profiles of disease progression, all COPD and bronchiectasis (BE) patients included in the study were elderly. Secondary diagnoses included ischaemic heart disease, peptic ulcer disease, osteoporosis and gastro-oesophageal reflux disease. Two COPD patients had undergone lung volume reduction surgery at least 2 years prior to the study, but nevertheless still suffered from respiratory insufficiency. Respiratory medications included salbutamol, ipatropium, fluticasone propionate, salmeterol, and beclomethasone dipropionate. One COPD patient was also on long-term oxygen therapy (2 l min−1); this was withdrawn 1 h prior to the experimental procedures.
Clinical details of the patients are provided in Table 1. Both sets of patients had clear evidence of severe respiratory insufficiency: forced vital capacity (FVC) ranged from 0.78 to 3.22 l (1.80 ± 0.15 l) and 1.43 to 2.60 l (1.89 ± 0.22 l) for the COPD and BE patients, respectively. On average, the forced expiratory volume in 1 s (FEV1) was 0.82 ± 0.07 l in the COPD patients and 1.03 ± 0.15 l in the BE patients. Expressed as a percentage of FVC, expiratory airflow limitation was significantly lower in COPD than in BE. Laboratory blood gas analysis revealed evidence of significant hypoxaemia and hypercapnia in the patients with COPD, but not in those with bronchiectasis (Table 1). Mean heart rates were normal in both COPD and BE, and neither group showed evidence of systemic hypertension. However, diastolic pressure was significantly higher in COPD than in BE (Table 1).
Table 1.
Clinical details of the patients with obstructive pulmonary disease (COPD) or bronchiectasis (BE)
| COPD (n= 18) | BE (n= 5) | |
|---|---|---|
| Age (years) | 71 ± 2 | 70 ± 3 |
| BMI (kg m−2) | 24.8 ± 0.8 | 28.7 ± 2.3 |
| FVC (l) | 1.80 ± 0.15 | 1.89 ± 0.22 |
| FEV1 (l) | 0.82 ± 0.07 | 1.03 ± 0.15 |
| FEV1 (%) | 46.7 ± 2.6 | 54.7 ± 3.2* |
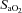 (%) (%) |
88.3 ± 3.2 | 97.6 ± 0.7* |
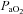 (mmHg) (mmHg) |
69.3 ± 5.3 | 93.5 ± 8.6* |
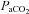 (mmHg) (mmHg) |
59.0 ± 3.3 | 38.5 ± 1.5*** |
| pH | 7.37 ± 0.01 | 7.41 ± 0.02 |
| Systolic BP (mmHg) | 138 ± 4 | 131 ± 4 |
| Diastolic BP (mmHg) | 73 ± 3 | 64 ± 2* |
| Heart rate (beats min−1) | 70 ± 2 | 76 ± 8 |
Values presented are mean ±s.e.m. BMI, body mass index; FVC, forced vital capacity; FEV1, forced expiratory volume in 1 s; FEV1 as a percentage of FVC; 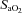 , O2 saturation;
, O2 saturation; 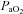 , partial pressure of arterial O2;
, partial pressure of arterial O2; 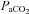 , partial pressure of arterial CO2. Parameters for which COPD and BE significantly differed are indicated by asterisks:
, partial pressure of arterial CO2. Parameters for which COPD and BE significantly differed are indicated by asterisks:
P < 0.05;
P < 0.001.
Muscle sympathetic nerve activity: multi-unit recordings
Multi-unit recordings of muscle sympathetic nerve activity (MSNA) were obtained in all patients. Experimental records are shown for one female COPD patient in Fig. 1. As can be seen in the RMS-processed signal, overall muscle sympathetic nerve activity (MSNA) was high, with sympathetic bursts occurring in 83% of cardiac intervals in this patient (68 bursts min−1). Across the COPD patients, burst incidence ranged from 71 to 97 bursts (100 heart beats)−1 (mean ±s.e.m., 85.1 ± 1.9%). Measured as burst frequency, MSNA ranged from 53 to 72 bursts min−1 (mean ±s.e.m., 61.8 ± 1.3 min−1). The five bronchiectasis patients also exhibited high levels of MSNA that were slightly lower than those observed in COPD: 77.6 ± 4.3 (range 69–91) bursts (100 heart beats)−1 and 58.4 ± 2.0 (55–66) bursts min−1. Correlation analyses revealed no significant relationships between burst incidence (or burst frequency) and FVC, FEV1 or FEV1%; given the consistently severe respiratory insufficiency, and the uniformly high levels of MSNA, this is perhaps not surprising.
Figure 1.

Multi-unit recording from a muscle fascicle of the common peroneal nerve in an awake female patient with COPD, showing a high incidence of muscle sympathetic nerve activity. Sympathetic bursts occurred in almost every cardiac interval in this patient.
Muscle sympathetic nerve activity: single-unit recordings
Stable unitary recordings were made from 17 muscle vasoconstrictor neurones, obtained from nine COPD patients. As shown in Fig. 2, individual neurones generated a low level of spontaneous activity. Average firing rates, calculated as the inverse of the mean of all interspike intervals for each unit, ranged from 0.31 to 2.10 Hz (0.92 ± 0.11 Hz). Moreover, individual units never discharged in every cardiac interval; firing probability (the percentage of cardiac intervals in which a neurone was active) ranged from only 21% to a maximum of 69% (52.2 ± 4.9%). Finally, multiple within-burst firing was rare: measured from those cardiac intervals in which the neurones were active, solitary spikes were generated in 63.4 ± 3.3% of intervals. This prevalence for solitary firing, despite the evidently high level of sympathetic activity, can readily be seen in Fig. 2.
Figure 2.

Unitary recording from a single muscle vasoconstrictor neurone (negative-going spikes) in an awake male patient with COPD. This unit generally fired only one spike (indicated by asterisks) per cardiac interval, but occasionally fired multiple spikes per cardiac interval. A spontaneously active muscle spindle (positive-going spikes) can be seen in the background. Because the sympathetic recording was so focal in this case, the RMS nerve signal does not adequately represent the population discharge.
All units showed overt cardiac rhythmicity, consistent with their identification as muscle vasoconstrictor neurones. Examples of cross-correlation histograms between unit firing and ECG are shown for three units recorded in COPD in Fig. 3A–C; similar histograms are shown for one unit recorded in bronchiectasis in Fig. 3D.
Figure 3.

Cross-correlation histograms between unit firing and ECG, and autocorrelation histograms for ECG. Data from three units recorded in COPD (A–C) and one unit recorded in bronchiectasis (D). n= number of spikes comprising the histogram.
Mean firing properties of these 17 neurones, and of 6 recorded from four BE patients, are shown graphically in Fig. 4. Numerical data, including data previously obtained in healthy subjects with high levels of MSNA, and from patients with obstructive sleep apnoea syndrome (OSAS), are provided in Table 2. It can be seen that the COPD patients had comparable levels of multi-unit MSNA to the OSAS and BE patients, as well as to the healthy subjects, but that unitary firing probabilities in BE were no different to those in healthy subjects. However, patients with COPD, OSAS and BE all shared a significant tendency to generate more double spikes – and fewer solitary spikes – within a burst, and to generate higher mean firing rates, than the healthy subjects.
Figure 4.

Single-unit data from 17 muscle vasoconstrictor neurones recorded in chronic obstructive pulmonary disease (COPD) and from 6 neurones recorded in bronchiectasis (BE). Histograms showing pooled data on the percentage of cardiac intervals (mean ±s.e.m.) in which units were quiescent, fired a single spike or multiple spikes. In the top panels, all cardiac intervals have been included: the open columns represent those cardiac intervals in which the neurones were quiescent. The lower panels show data in which only those cardiac intervals were included in which a unit was active.
Table 2.
Firing properties (mean ±s.e.m.) of muscle vasoconstrictor neurones in healthy subjects with high resting levels of MSNA (Macefield & Wallin, 1999), and patients with bronchiectasis (BE), obstructive sleep apnoea syndrome (OSAS;Elam et al. 2002) or chronic obstructive pulmonary disease (COPD)
| Units (n) | Burst incidence (%) | Firing probability (%) | Mean frequency (Hz) | One spike (%) | Two spikes (%) | Three spikes (%) | Four spikes (%) | |
|---|---|---|---|---|---|---|---|---|
| Healthy | 19 | 74.9 ± 0.5 | 34.9 ± 3.6 | 0.33 ± 0.04 | 77.6 ± 3.8 | 18.1 ± 2.9 | 3.6 ± 1.1 | 0.5 ± 0.3 |
| BE | 6 | 77.6 ± 4.3 | 37.8 ± 6.8 | 0.72 ± 0.17* | 60.0 ± 6.2* | 23.2 ± 2.6* | 10.6 ± 1.9* | 1.5 ± 0.7 |
| OSAS | 12 | 77.2 ± 5.2 | 50.7 ± 4.4* | 0.96 ± 0.11* | 58.7 ± 2.8* | 27.3 ± 1.3* | 9.7 ± 1.5* | 2.9 ± 0.7 |
| COPD | 17 | 85.1 ± 1.9* | 52.2 ± 4.9* | 0.92 ± 0.12* | 63.4 ± 3.3* | 24.8 ± 2.0* | 8.7 ± 1.0* | 2.2 ± 0.6 |
Significantly different (P < 0.05) from healthy subjects.
Discussion
The present study has documented for the first time the firing properties of individual muscle vasoconstrictor neurones in awake patients with chronic obstructive pulmonary disease. We have shown that mean firing rates are high, mean firing probability is high and there is a significant shift away from firing only once per sympathetic burst to firing more often. We believe each of these changes reflects an increase in central muscle vasoconstrictor drive, brought about by the chronic asphyxia of COPD.
Chemoreceptor drive and muscle sympathetic outflow
It is known that both hypoxia and hypercapnia cause an increase in muscle sympathetic nerve activity (MSNA), and that the muscle vasoconstrictor response is greater to asphyxia than to either hypoxia or hypercapnia alone (Somers et al. 1989). However, it is also recognized that chronic hypoxia causes a sustained increase in MSNA that outlasts the stimulus (Morgan et al. 1995; Hansen & Sander, 2003; Tamisier et al. 2005). It is the repeated bouts of nocturnal hypoxaemia that are believed to be responsible for the elevated MSNA seen in the awake state in patients with the obstructive sleep apnoea syndrome (Hedner et al. 1988; Carlsson et al. 1993; Somers et al. 1995), and an increase in sensitivity of the peripheral chemoreceptors has been show to increase MSNA in OSAS (Narkiewicz et al. 1998, 1999). Unlike COPD, OSAS patients are not hypoxaemic in the awake state, but it is highly likely that the ongoing hypoxaemia (and hypercapnia) in COPD is (are) responsible for the augmented MSNA. Indeed, oxygen administration has been shown to lower MSNA in COPD patients, supporting the idea that peripheral chemoreceptors contribute to the elevated MSNA (Heindl et al. 2001), and slow, deep breathing (which may improve gas exchange) has also been shown to be effective in reducing MSNA (Raupach et al. 2008). It is known that the elevated chemical drive causes an increase in central respiratory drive in COPD, as evidenced by the increased firing rates of single motor units recorded from the diaphragm (De Troyer et al. 1997) or from the inspiratory scalene and parasternal muscles (Gandevia et al. 1996). Moreover, lung volume reduction surgery – which is effective in improving lung function, exercise performance and quality of life in patients with COPD – reduces central inspiratory drive to the diaphragm and scalene muscles (Gorman et al. 2005); it is not known whether this surgical treatment also reduces the elevated MSNA.
Increases in MSNA in other pathophysiological states
Elevated levels of muscle sympathetic nerve activity feature in many cardiovascular and cardiorespiratory diseases. As noted above, MSNA is high in patients with congestive heart failure (Leimbach et al. 1986; Grassi et al. 1995; Macefield et al. 1999; Murai et al. 2009) and the obstructive sleep apnoea syndrome (Hedner et al. 1988; Carlsson et al. 1993; Somers et al. 1995; Narkiewicz et al. 1999; Elam et al. 2002). There is also evidence that MSNA is increased in essential hypertension (Grassi et al. 1998; Schlaich et al. 2004), pregnancy-induced hypertension (Schobel et al. 1996; Fischer et al. 2004), renovascular hypertension (Johansson et al. 1999; Miyajima et al. 2001) and the hypertension associated with chronic kidney disease (Tuncel et al. 2002; Hausberg et al. 2002; Schlaich et al. 2009). However, a high level of MSNA does not necessarily mean there is any underlying pathology. Indeed, many healthy young individuals have high MSNA at rest yet normal blood pressures and no evidence of cardiovascular disease (Macefield & Wallin, 1999). The major advantage of single-unit recordings is that they can contribute more information than multi-unit recordings: by looking for increases in firing probability, mean firing rate or multiple firing we can see whether an elevated level of MSNA reflects any underlying pathology (Macefield et al. 1994, 2002; Macefield, 2009). We have already shown that individual muscle vasoconstrictor neurones in healthy subjects with elevated resting levels of MSNA have low firing probabilities, low firing rates and a low incidence of multiple firing (Macefield & Wallin, 1999). We have also shown that these parameters change with pathology, but that the nature of the changes differs according to the type of pathology.
Firing properties in COPD vs. other forms of cardiovascular and respiratory disease
In many respects, the firing properties of the single muscle vasoconstrictor neurones recorded in our COPD patients were similar to those seen in other pathophysiological states of elevated muscle vasoconstrictor drive. Overall burst incidence in COPD (86%) was comparable to that seen in moderate to severe heart failure (88%; Macefield et al. 1999), but higher than that of patients with either bronchiectasis (78%) or obstructive sleep apnoea (77%; Elam et al. 2002), or of healthy subjects with high levels of MSNA (75%; Macefield & Wallin, 1999). The firing probability of muscle vasoconstrictor neurones was also as high in COPD (52%) as in CHF (55%), but firing probability in OSAS was also high (51%). The latter supports our contention that single-unit firing properties can provide a more sensitive measure of central sympathetic drive than multi-unit recordings alone. Indeed, it is interesting that the firing probability of single neurones recorded in the bronchiectasis patients (38%) was no different from that of the healthy subjects (35%); we know that hypoxaemia and hypercapnia are not clinical features of bronchiectasis, so perhaps this accounts for the low firing probabilities seen in this disease. It also leads one to ask: if multi-MSNA is high, but single-unit firing probability is not, can a multi-unit recording really reflect an elevated sympathetic drive? Unless some other unitary firing properties – indicative of an increase in central sympathetic drive – have changed, we think not.
Mean firing rates in our COPD patients (0.92 Hz) were similar to those found in OSAS (0.96 Hz) and CHF patients (0.98 Hz), but higher than those in the bronchiectasis patients (0.72 Hz) and the healthy subjects (0.33 Hz). However, unlike the CHF patients, who showed no decrease in the proportion of solitary spikes generated within a burst, the patients with COPD, OSAS and BE each showed a significant trend towards multiple firing. We had already demonstrated that a shift towards multiple firing can occur in CHF, but only during the prolonged cardiac intervals following ventricular ectopic beats (Elam & Macefield, 2001). While the shift towards multiple firing can be explained by the elevated chemical drive in COPD and OSAS, it is difficult to account for the increase seen in BE; perhaps it is related to the increased work of breathing in BE. Another possible mechanism is nitric oxide (NO), a potent vasodilator. Indeed, it is known that alveolar NO production is reduced in OSAS, and it has been suggested that this may contribute to the systemic hypertension (Foresi et al. 2007), but whether NO production is elevated in COPD is unknown. One piece of evidence against this comes from the fact that stimulated NO production is reduced in lung explants from patients with COPD (Cremona et al. 1999).
Regardless of the underlying mechanisms by which multiple within-burst firing occurs, it is reasonable to posit that a shift towards multiple firing will increase the liberation of noradrenaline (and co-localized neurotransmitters) from the vasoconstrictor nerve terminals, thereby increasing the degree of neurally mediated vasoconstriction. Such a mechanism has also been proposed in hyperhidrosis: compared to sudomotor neurones active in thermally induced sweating, those spontaneously active in hyperhidrotic subjects show a significant shift towards multiple firing which, it is argued, would be expected to increase the release of acetylcholine from the nerve terminals and hence the liberation of sweat from the skin (Macefield et al. 2008).
Limitations
Perhaps the greatest limitation of this study is that we did not withdraw pharmacological treatment from our patients prior to the study, our aim being to avoid rebound cardiovascular responses and associated baroreceptor-mediated effects on sympathetic nerve traffic. Nevertheless, given that all medications were inhaled they should have limited systemic effects. Moreover, optimal pharmacological treatment was maintained in our previous studies of CHF and OSAS patients, and despite this ongoing treatment MSNA remained very high. Another important consideration, referred to in the Introduction, is the presence of co-morbidities, such as obstructive sleep apnoea or ischaemic heart disease, which are very common in this population, as are most of the smoking-related diseases. Nevertheless, the co-morbidities in this population were not primary diagnoses. The patients all presented with primary symptoms of respiratory insufficiency, detailed clinical and laboratory assessments being used to diagnose the research participants as suffering from either chronic obstructive pulmonary disease or bronchiectasis. It should be acknowledged that the number of bronchiectasis patients was small, but despite this limitation we felt it instructive to include their data.
Unlike the high incidence of OSAS with obesity – the OSAS patients we had studied previously had a mean BMI of 32 ± 2 (Elam et al. 2002) – our COPD subjects had, on average, a normal BMI; conversely, on this criterion the BE patients were defined as being overweight, but not obese. It is known that a BMI above 30 (obese) itself increases the resting level of MSNA, but the levels typically encountered (burst incidence 53%) are much lower than those we see in COPD, OSAS, BE or CHF; moreover, individual muscle vasoconstrictor neurones do not show an increased incidence of multiple firing in obesity (Lambert et al. 2007), so we do not believe that the firing characteristics observed in our BE (or COPD) patients can be accounted for by their body weight.
In addition, unlike the healthy subjects with high levels of resting MSNA, whose ages ranged from 24 to 61 years (mean 36 years; Macefield & Wallin, 1999), all of our COPD and BE patients were elderly. It is known that MSNA increases with age, particularly after the age of 60 (Sverrisdóttir et al. 2001; Hart et al. 2009). However, the overall burst incidence we found in these pathophysiological conditions was considerably higher than that found in healthy elderly male subjects (e.g. 64%; Hart et al. 2009). Moreover, that the average firing probability of the units recorded in COPD was higher than that found in BE, despite their similar ages, would argue against an age-related increase in MSNA as being the primary determinant of the firing pattern we report.
Finally, despite the elevated levels of central sympathetic drive evident in COPD and BE, none of our patients were hypertensive, with blood pressures being what one would expect in older individuals (e.g. Hart et al. 2009 found a mean value of 136/71 mmHg). Still, diastolic pressures were significantly higher in COPD than in BE, suggesting a tendency towards hypertension. By comparison, in our study of obstructive sleep apnoea average blood pressure was 145/96; these individuals were clearly hypertensive (Elam et al. 2002). Given that asphyxia causes vasodilatation and hypotension in brain-dead patients (Lahana et al. 2005), who have no sympathetic vasoconstrictor drive, we may be seeing the direct effects of hypoxia and hypercapnia on blood vessels; the increase in muscle vasoconstrictor drive, via the baroreflex, serves to offset this local vasodilatation.
Conclusions
In conclusion, we have shown that the increase in resting levels of muscle sympathetic nerve activity in patients with COPD is characterized by an increase in firing probability, mean firing rates and incidence of multiple firing of individual muscle vasoconstrictor neurones. While the overall level of sympathoexcitation, as measured from multi-unit burst incidence, is comparable to that seen in CHF, the unitary firing properties observed in COPD align with those seen in OSAS. We believe that the chronic hypoxaemia associated with COPD is responsible for the elevated levels of muscle vasoconstrictor drive seen in this disease. Moreover, we believe that the shift towards multiple firing in COPD and OSAS would presumably cause greater neurotransmitter release from the sympathetic nerve terminals and hence a greater vasoconstriction. However, as noted above, our patients were not hypertensive, so other – presumably vascular – factors must be tempering any tendency towards the neurogenic hypertension one sees in OSAS.
Acknowledgments
This work was supported by the National Health and Medical Research Council of Australia, grant no. 963206. Y. B. Sverrisdottir was supported in Australia by the Swedish Medical Research Council. M. Sander was supported in Australia by the Michaelsen Foundation and the Danish Medical Research Council.
Glossary
Abbreviations
- BE
bronchiectasis
- CHF
congestive heart failure
- COPD
chronic obstructive pulmonary disease
- MSNA
muscle sympathetic nerve activity
- OSAS
obstructive sleep apnoea syndrome
Author contributions
V.G.M. and D.K.McK. were responsible for the conception and design of the experiments. C.A., Y.B.S., M.S. and V.G.M. were each involved in data acquisition. C.A., D.B. and V.G.M. were involved in analysis and interpretation of the data. C.A. and V.G.M. drafted the article and all authors contributed to the final version. All experiments were performed at The Prince of Wales Medical Research Institute.
References
- Carlsson JT, Hedner J, Elam M, Ejnell H, Sellgren J, Wallin BG. Augmented resting sympathetic activity in awake patients with obstructive sleep apnea. Chest. 1993;103:1763–1768. doi: 10.1378/chest.103.6.1763. [DOI] [PubMed] [Google Scholar]
- Carlsson JT, Hedner J, Sellgren J, Elam M, Wallin BG. Depressed baroreflex sensitivity in patients with obstructive sleep apnea. Am J Respir Crit Care Med. 1996;154:1490–1496. doi: 10.1164/ajrccm.154.5.8912770. [DOI] [PubMed] [Google Scholar]
- Cremona G, Higenbottam TW, Bower EA, Wood AM, Stewart S. Hemodynamic effects of basal and stimulated release of endogenous nitric oxide in isolated human lungs. Circulation. 1999;100:1316–1321. doi: 10.1161/01.cir.100.12.1316. [DOI] [PubMed] [Google Scholar]
- De Troyer A, Leeper JB, McKenzie DK, Gandevia SC. Neural drive to the diaphragm in patients with severe COPD. Am J Respir Crit Care Med. 1997;155:1335–1340. doi: 10.1164/ajrccm.155.4.9105076. [DOI] [PubMed] [Google Scholar]
- Elam M, Macefield VG. Multiple firing of single muscle vasoconstrictor neurons during cardiac dysrhythmias in human heart failure. J Appl Physiol. 2001;91:717–724. doi: 10.1152/jappl.2001.91.2.717. [DOI] [PubMed] [Google Scholar]
- Elam M, Macefield VG, McKenzie D. The firing pattern of single muscle vasoconstrictor neurons in awake patients with the obstructive sleep apnoea syndrome. J Appl Physiol. 2002;93:297–303. doi: 10.1152/japplphysiol.00899.2001. [DOI] [PubMed] [Google Scholar]
- Falk JA, Kadiev S, Criner GJ, Scharf SM, Minai OA, Diaz P. Cardiac disease in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2008;5:543–548. doi: 10.1513/pats.200708-142ET. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer T, Schobel HP, Frank H, Andreae M, Schneider KY, Heusser K. Pregnancy-induced sympathetic hyperactivity: a precursor of preeclampsia. Eur J Clin Invest. 2004;34:443–448. doi: 10.1111/j.1365-2362.2004.01350.x. [DOI] [PubMed] [Google Scholar]
- Flenley DC. Chronic obstructive pulmonary disease. Dis Mon. 1988;34:537–599. doi: 10.1016/0011-5029(88)90015-6. [DOI] [PubMed] [Google Scholar]
- Foresi A, Leone C, Olivieri D, Cremona G. Alveolar-derived exhaled nitric oxide is reduced in obstructive sleep apnea syndrome. Chest. 2007;132:860–867. doi: 10.1378/chest.06-3124. [DOI] [PubMed] [Google Scholar]
- Gandevia SC, Leeper JB, McKenzie DK, De Troyer A. Discharge frequencies of parasternal intercostal and scalene motor units during breathing in normal and COPD subjects. Am J Respir Crit Care Med. 1996;153:622–628. doi: 10.1164/ajrccm.153.2.8564108. [DOI] [PubMed] [Google Scholar]
- Gorman RB, McKenzie DK, Butler JE, Tolman JF, Gandevia SC. Diaphragm length and neural drive after lung-volume reduction surgery. Am J Respir Crit Care Med. 2005;172:1259–1266. doi: 10.1164/rccm.200412-1695OC. [DOI] [PubMed] [Google Scholar]
- Grassi G, Colombo M, Seravalle G, Spaziani D, Mancia G. Dissociation between muscle and skin sympathetic nerve activity in essential hypertension, obesity, and congestive heart failure. Hypertension. 1998;31:64–67. doi: 10.1161/01.hyp.31.1.64. [DOI] [PubMed] [Google Scholar]
- Grassi G, Seravalle G, Cattaneo BM, Lanfranchi A, Vailati S, Giannattasio C, Del Bo A, Sala C, Bolla GB, Pozzi M. Sympathetic activation and loss of reflex sympathetic control in mild congestive heart failure. Circulation. 1995;92:3206–3211. doi: 10.1161/01.cir.92.11.3206. [DOI] [PubMed] [Google Scholar]
- Hansen J, Sander M. Sympathetic neural overactivity in healthy humans after prolonged exposure to hypobaric hypoxia. J Physiol. 2003;546:921–929. doi: 10.1113/jphysiol.2002.031765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart EC, Joyner MJ, Wallin BG, Johnson CP, Curry TB, Eisenach JH, Charkoudian N. Age-related changes in the sympathetic-hemodynamic balance in men. Hypertension. 2009;54:127–133. doi: 10.1161/HYPERTENSIONAHA.109.131417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausberg M, Kosch M, Harmelink P, Barenbrock M, Hohage H, Kisters K, Dietl KH, Rahn KH. Sympathetic nerve activity in end-stage renal disease. Circulation. 2002;106:1974–1979. doi: 10.1161/01.cir.0000034043.16664.96. [DOI] [PubMed] [Google Scholar]
- Hedner J, Ejnell H, Sellgren J, Hedner T, Wallin G. Is high and fluctuating muscle nerve sympathetic activity in the sleep apnoea syndrome of pathogenetic importance for the development of hypertension? J Hypertens Suppl. 1988;6:S529–S531. doi: 10.1097/00004872-198812040-00166. [DOI] [PubMed] [Google Scholar]
- Heindl S, Lehnert M, Criée CP, Hasenfuss G, Andreas S. Marked sympathetic activation in patients with chronic respiratory failure. Am J Respir Crit Care Med. 2001;164:597–601. doi: 10.1164/ajrccm.164.4.2007085. [DOI] [PubMed] [Google Scholar]
- Hiestand D, Phillips B. The overlap syndrome: chronic obstructive pulmonary disease and obstructive sleep apnea. Crit Care Clin. 2008;24:551–563. doi: 10.1016/j.ccc.2008.02.005. [DOI] [PubMed] [Google Scholar]
- Howard P. Natural history of obstructive airways disease and hypoxia: implications for therapy. Lung. 1990;168(Suppl.):743–750. doi: 10.1007/BF02718203. [DOI] [PubMed] [Google Scholar]
- Ilowite J, Spiegler P, Chawla S. Bronchiectasis: new findings in the pathogenesis and treatment of this disease. Curr Opin Infect Dis. 2008;21:163–167. doi: 10.1097/QCO.0b013e3282f4f237. [DOI] [PubMed] [Google Scholar]
- Johansson M, Elam M, Rundqvist B, Eisenhofer G, Herlitz H, Lambert G, Friberg P. Increased sympathetic nerve activity in renovascular hypertension. Circulation. 1999;18:2537–2542. doi: 10.1161/01.cir.99.19.2537. [DOI] [PubMed] [Google Scholar]
- Lahana A, Constantopulos S, Nakos G. The local component of the acute cardiovascular response to simulated apneas in brain-dead humans. Chest. 2005;128:634–639. doi: 10.1378/chest.128.2.634. [DOI] [PubMed] [Google Scholar]
- Lambert E, Straznicky N, Schlaich M, Esler M, Dawood T, Hotchkin E, Lambert G. Differing pattern of sympathoexcitation in normal-weight and obesity-related hypertension. Hypertension. 2007;50:862–868. doi: 10.1161/HYPERTENSIONAHA.107.094649. [DOI] [PubMed] [Google Scholar]
- Lazarus A, Myers J, Fuhrer G. Bronchiectasis in adults: a review. Postgrad Med. 2008;120:113–121. doi: 10.3810/pgm.2008.09.1912. [DOI] [PubMed] [Google Scholar]
- Leimbach WN, Wallin BG, Victor RG, Aylward PE. Direct evidence from intraneural recordings for increased central sympathetic outflow in patients with heart failure. Circulation. 1986;73:913–919. doi: 10.1161/01.cir.73.5.913. [DOI] [PubMed] [Google Scholar]
- Macefield VG. Single-minded about heart failure. J Physiol. 2009;587:2421. doi: 10.1113/jphysiol.2009.173914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macefield VG, Elam M, Wallin BG. Firing properties of single postganglionic sympathetic neurones recorded in awake human subjects. Autonom Neurosci. 2002;95:146–159. doi: 10.1016/s1566-0702(01)00389-7. [DOI] [PubMed] [Google Scholar]
- Macefield VG, Rundqvist B, Sverrisdottir YB, Wallin BG, Elam M. Firing properties of single muscle vasoconstrictor neurons in the sympathoexcitation associated with congestive heart failure. Circulation. 1999;100:1708–1713. doi: 10.1161/01.cir.100.16.1708. [DOI] [PubMed] [Google Scholar]
- Macefield VG, Sverrisdottir YB, Elam E, Harris J. Firing properties of sudomotor neurones in hyperhidrosis and thermal sweating. Clin Autonom Res. 2008;18:325–330. doi: 10.1007/s10286-008-0507-7. [DOI] [PubMed] [Google Scholar]
- Macefield VG, Wallin BG. Firing properties of single vasoconstrictor motoneurones in subjects with high levels of muscle sympathetic activity. J Physiol. 1999;516:293–301. doi: 10.1111/j.1469-7793.1999.293aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macefield VG, Wallin BG, Vallbo ÅB. The discharge behaviour of single vasoconstrictor motoneurones in human muscle nerves. J Physiol. 1994;481:799–809. doi: 10.1113/jphysiol.1994.sp020482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie DK, Frith PA, Burdon JGW, Town GI. The COPDX Plan: Australian and New Zealand Guidelines for the management of Chronic Obstructive Pulmonary Disease 2003. Med J Aust. 2003;178(Suppl.):S1–S40. doi: 10.5694/j.1326-5377.2003.tb05213.x. [DOI] [PubMed] [Google Scholar]
- Mannino DM, Thorn D, Swensen A, Holguin F. Prevalence and outcomes of diabetes, hypertension and cardiovascular disease in COPD. Eur Respir J. 2008;32:962–969. doi: 10.1183/09031936.00012408. [DOI] [PubMed] [Google Scholar]
- Miyajima E, Yamada Y, Yoshida Y, Matsukawa T, Shionoiri H, Tochikubo O, Ishii M. Muscle sympathetic nerve activity in renovascular hypertension and primary aldosteronism. Hypertension. 2001;17:1057–1062. doi: 10.1161/01.hyp.17.6.1057. [DOI] [PubMed] [Google Scholar]
- Morgan BJ, Crabtree DC, Palta M, Skatrud JB. Combined hypoxia and hypercapnia evokes long-lasting sympathetic activation in humans. J Appl Physiol. 1995;79:205–213. doi: 10.1152/jappl.1995.79.1.205. [DOI] [PubMed] [Google Scholar]
- Murai H, Takamura M, Maruyama M, Nakano M, Ikeda T, Kobayashi D, Otowa K, Ootsuji H, Okajima M, Furusho H, Takata S, Kaneko S. Altered firing pattern of single-unit muscle sympathetic nerve activity during handgrip exercise in chronic heart failure. J Physiol. 2009;587:2613–2622. doi: 10.1113/jphysiol.2009.172627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narkiewicz K, Pesek CA, Kato M, Phillips BG, Davison DE, Somers VK. Baroreflex control of sympathetic nerve activity and heart rate in obstructive sleep apnea. Hypertension. 1998;32:1039–1043. doi: 10.1161/01.hyp.32.6.1039. [DOI] [PubMed] [Google Scholar]
- Narkiewicz K, van de Borne PJH, Pesek CA, Dyken ME, Montano N, Somers VK. Selective potentiation of peripheral chemoreflex sensitivity in obstructive sleep apnea. Circulation. 1999;99:1183–1189. doi: 10.1161/01.cir.99.9.1183. [DOI] [PubMed] [Google Scholar]
- Raupach T, Bahr F, Herrmann P, Luethje L, Heusser K, Hasenfuss G, Bernardi L, Andreas S. Slow breathing reduces sympathoexcitation in COPD. Eur Respir J. 2008;32:387–392. doi: 10.1183/09031936.00109607. [DOI] [PubMed] [Google Scholar]
- Ryu JH, Scanlon PD. Obstructive lung diseases: COPD, asthma, and many imitators. Mayo Clin Proc. 2001;76:1144–1153. doi: 10.4065/76.11.1144. [DOI] [PubMed] [Google Scholar]
- Schlaich MP, Lambert E, Kaye DM, Krozowski Z, Campbell DJ, Lambert G, Hastings J, Aggarwal A, Esler MD. Sympathetic augmentation in hypertension: role of nerve firing, norepinephrine reuptake, and angiotensin neuromodulation. Hypertension. 2004;43:169–175. doi: 10.1161/01.HYP.0000103160.35395.9E. [DOI] [PubMed] [Google Scholar]
- Schlaich MP, Socratous F, Hennebry S, Eikelis N, Lambert EA, Straznicky N, Esler MD, Lambert GW. Sympathetic activation in chronic renal failure. J Am Soc Nephrol. 2009;20:933–939. doi: 10.1681/ASN.2008040402. [DOI] [PubMed] [Google Scholar]
- Schobel HP, Fischer T, Heuszer K, Geier H, Schmeider RE. Preeclampsia – a state of sympathetic hyperactivity. N Engl J Med. 1996;335:1480–1485. doi: 10.1056/NEJM199611143352002. [DOI] [PubMed] [Google Scholar]
- Sin DD, Anthoniesen NR, Soriano JB, Agusti AG. Mortality in COPD: Role of comorbidities. Eur Respir J. 2006;28:1245–1257. doi: 10.1183/09031936.00133805. [DOI] [PubMed] [Google Scholar]
- Sin DD, Man SFP. Why are patients with chronic obstructive pulmonary disease at increased risk of cardiovascular diseases? The potential role of systemic inflammation in chronic obstructive pulmonary disease. Circulation. 2003;107:1514–1519. doi: 10.1161/01.cir.0000056767.69054.b3. [DOI] [PubMed] [Google Scholar]
- Somers VK, Dyken ME, Clary MP, Abboud FM. Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest. 1995;96:1897–1904. doi: 10.1172/JCI118235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somers VK, Mark AL, Zavala DC, Abboud FM. Contrasting effects of hypoxia and hypercapnia on ventilation and sympathetic activity in humans. J Appl Physiol. 1989;67:2101–2106. doi: 10.1152/jappl.1989.67.5.2101. [DOI] [PubMed] [Google Scholar]
- Sverrisdóttir YB, Johannsson G, Jungersten L, Wallin BG, Elam MJ. Is the somatotropic axis related to sympathetic nerve activity in healthy ageing men? Hypertension. 2001;19:2019–2024. doi: 10.1097/00004872-200111000-00012. [DOI] [PubMed] [Google Scholar]
- Tamisier R, Anand A, Nieto LM, Cunnington D, Weiss JW. Arterial pressure and muscle sympathetic nerve activity are increased after two hours of sustained but not cyclic hypoxia in healthy humans. J Appl Physiol. 2005;98:343–349. doi: 10.1152/japplphysiol.00495.2004. [DOI] [PubMed] [Google Scholar]
- Tuncel M, Augustyniak R, Zhang W, Toto RD, Victor RG. Sympathetic nervous system function in renal hypertension. Curr Hypertens Rep. 2002;4:229–236. doi: 10.1007/s11906-002-0012-7. [DOI] [PubMed] [Google Scholar]
- Zamarrón C, García Paz V, Morete E, del Campo Matías F. Association of chronic obstructive pulmonary disease and obstructive sleep apnea consequences. Int J Chron Obstruct Pulmon Dis. 2008;3:671–682. doi: 10.2147/copd.s4950. [DOI] [PMC free article] [PubMed] [Google Scholar]