

Abstract
Oxidative stress results in protein oxidation and is involved in the pathogenesis of lung diseases such as chronic obstructive pulmonary disorder (COPD). Sulfiredoxin-1 (Srx1) catalyzes reduction of cysteine sulfinic acid to sulfenic acid in oxidized proteins and protects them from inactivation. This study examined the mechanism of transcriptional regulation of Srx1 and its possible protective role during oxidative stress associated with COPD. Nrf2, a transcription factor known to influence susceptibility to pulmonary diseases, upregulates Srx1 expression during oxidative stress caused by cigarette smoke exposure in the lungs of mice. Disruption of Nrf2 signaling by genetic knockout in mice or RNAi in cells downregulated the expression of Srx1. In silico analysis of the 5′-promoter flanking region of Srx1 identified multiple antioxidant response elements that are highly conserved. Reporter and chromatin-immunoprecipation assays demonstrated that ARE1 at −228 is critical for the Nrf2-mediated response. Attenuation of Srx1 expression with RNAi potentiated the toxicity of hydrogen peroxide (H2O2), whereas overexpression of Srx1 protected against H2O2 mediated cell death in vitro. Immunoblot analysis revealed dramatic decreases in Srx1 expression in lungs from patients with COPD relative to non-emphysematous lungs together with a decline in Nrf2 protein. Thus, Srx1, a key Nrf2-regulated gene, contributes to protection against oxidative injury in the lung.
Keywords: Srx1, Nrf2, oxidative stress, antioxidant response element, chronic obstructive pulmonary disease, emphysema
INTRODUCTION
The lung represents a unique tissue as it is directly exposed to high oxygen tension and oxidative insults. Many of these insults induce formation of reactive oxygen species such as hydrogen peroxide, oxyradicals, and organic hydroperoxides [1]. Endogenous reactive oxygen species (ROS) are constantly generated in the course of common metabolic process such as in the mitochondrial respiratory chain. Since excess ROS can lead to severe cell damage and malignant transformation, cells in the lungs are equipped with robust antioxidant enzyme defense system, which counteracts oxidative stress and maintains cellular redox status [2]. The major ROS detoxification enzymes are glutathione peroxidase, superoxide dismutase, catalase, and peroxiredoxins (Prxs). Prxs catalyze the reduction of a broad spectrum of peroxides [1, 3, 4].
Chronic exposure to oxidative insult due to cigarette smoke (CS) and air pollutants are the primary factors for pathogenesis of chronic obstructive pulmonary disease (COPD), which is defined by progressive airflow limitation that is caused by advanced obstruction of the smaller conducting airways by mucus and/or emphysematous destruction of the lung [5, 6]. Both cause progressive reduction in the volume of air leaving the lungs following forced expiration [6]. COPD has become a global epidemic and is likely to become the third largest cause of death worldwide by 2020 [7]. Oxidant-antioxidant imbalance in lungs has been strongly implicated in COPD severity [8, 9]. Strong epidemiological and genetic evidence support an individual’s ability to defend against CS-induced oxidative stress by up-regulation of lung antioxidant defenses represents a critical event in the pathogenesis of COPD [10].
Nuclear factor, erythroid derived 2, like 2 (Nrf2), is a central transcription factor that regulates the anti-oxidant defense system and acts as a modifier of several lung diseases that involve oxidative stress and inflammation. Nrf2 has an overarching protective role in several lung inflammatory diseases as it increases sensitivity of Nrf2-disrupted mice to allergen-induced asthma [11, 12], bacterial lipopolysaccharide-induced sepsis [13], hyperoxia-induced acute injury [14], and diesel exhaust-induced DNA damage [15]. Nrf2 gene deletion in mice provided the first evidence of a direct link between the regulation of antioxidant genes and alveolar destruction in the CS-induced emphysema in murine model [16]. More recently, decline in Nrf2 has been linked to an increase in the oxidative stress and pathogenesis in lungs of patients with COPD [17–19].
In response to oxidants including CS, a network of cytoprotective antioxidant genes are induced in lungs in a rapid and highly coordinated fashion through the activation of the Nrf2 pathway [2, 16]. Following CS exposure [20], Nrf2, a basic leucine zipper transcription factor, dissociates from its cytosolic repressor kelch like ECH-associated protein 1 (KEAP1), accumulates in the nucleus, and binds to the antioxidant response element (ARE) [21] in the promoter of its target genes, leading to their transcriptional induction in lungs [22]. Nrf2 regulates genes involved in two major redox systems, the glutathione and thioredoxin systems. γ-glutamyl cysteine synthetase catalytic (Gclc) and regulatory subunits (Gclm), members of the Glutathione-S-transferase (GST) family, Glutathione reductase (GSR), Glutathione peroxidase 2 (GPx2), Glutathione peroxidase 3 (GPx3), Thioredoxin reductase (TxnR), and Peroxiredoxin-1 (Prx1)and were all induced in the lungs of Nrf2+/+ mice in response to CS [16, 23].
Srx1, also known as neoplastic progression 3, is a member of a conserved family of antioxidants present in eukaryotes and contains a C-terminal cysteine residue that is highly conserved and crucial for its antioxidant function [3, 4]. It plays a key role in cellular responses to oxidative insults by restoring the activity of over-oxidized Prxs [3, 4, 24, 25]. Gene deletion studies in yeast revealed that Srx1 gene expression confers resistance against hydrogen peroxide toxicity [25]. A study by Rangasamy et al (2004) showed that Srx1 expression was selectively induced in the lungs of Nrf2+/+ mice in response to CS [16]. Two agents known to affect Nrf2 signaling in the liver, 3H-1,2-dithiole-3-thione [26] and sodium arsenite [27] also upregulated Srx1 expression. However, the molecular mechanism underlying Nrf2 dependent transcriptional upregulation of Srx1 expression and the functional significance of Nrf2 dependent Srx1 induction in response to CS exposure or treatment with Nrf2 activators are not known [53]. A better understanding of the role of Nrf2 in the regulation of Srx1 expression will help elucidate its possible protective role in lungs in response to oxidative stress caused by CS and environmental pollutants that play a role in pathogenesis of COPD and other lung diseases.
MATERIALS AND METHODS
Animals and Care
Nrf2-disrupted CD-1 (ICR) mice were generated as described previously [28]. Mice were genotyped for Nrf2 status by PCR amplification of genomic DNA extracted from blood as described [16]. All experimental protocols conducted in mice were performed in accordance with the standards established by the US Animal Welfare Acts, set forth in the National Institutes of Health guidelines and the Policy and Procedures Manual of the Johns Hopkins University Animal Care and Use Committee.
Cell Culture
A549 cells, human alveolar type II epithelial cells, and BEAS2B cells were obtained from American Type Culture Collection (Rockville, MD, USA) and were cultured in recommended medium supplemented with 10% fetal bovine serum and 1% penicillin –streptomycin.
Exposure to Cigarette Smoke
Eight wk old mice were divided into 4 groups (n = 3 per group): I, control Nrf2+/+ mice; II, experimental Nrf2+/+ mice; III, control Nrf2−/− mice; and IV, experimental Nrf2−/− mice. Control groups I and III were kept in a filtered air environment, and treatment groups II and IV were subjected to CS according as described previously [16]. Mice were exposed to acute CS for 5 h, using a TE-10 smoking machine (Teague Enterprises, Davis, CA, USA) and 2R4F reference cigarettes (University of Kentucky, Tobacco Research Institute, Lexington, KY, USA). Chamber atmosphere was monitored for total suspended particles and carbon monoxide, with concentrations of 90 mg/m3 and 150 ppm, respectively. Immediately after 5h exposure, mice were sacrificed and lungs were collected. Cigarette smoke extract (CSE dissolved in DMSO, 40mg/ml total particulate matter) was purchased from Murthy Pharmaceuticals (Lexington, KY, USA).
Quantitation of Srx1 mRNA Expression after CS Treatment
To measure the expression of Srx1, mice were exposed to CS for 5 h. Total RNA was extracted from lung tissues and or cells with TRIzol reagent (Invitrogen, CA, USA) and reverse transcribed using a high capacity cDNA synthesis kit (Applied Biosystems, Foster city, CA, USA) as per the manufacturer’s instructions. Quantitative real-time reverse transcriptase-polymerase chain reaction (RT-PCR) analyses of murine Srx1, was performed using assay on demand primers and probe sets from Applied Biosystems. Assays were performed by using the ABI 7000 Taqman system (Applied Biosystems, Foster City, CA, USA). GAPDH was used for normalization.
Western Blot
Western blot analyses of Srx1 and Nrf2 were performed as described previously [29]. Briefly, 50 μg of total protein were separated by 15% sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride (PVDF) transfer membrane by semi-dry blotting. The PVDF membrane was blocked in 2% non-fat milk and incubated with polyclonal goat anti-Srx1 antibody (1:200), polyclonal rabbit anti-Nrf2 antibody (1:500) or GAPDH (1:1000) followed by incubation with Horseradish-peroxidase conjugated secondary antibody, and developed using an ECL chemiluminescence detection system (Amersham Biosciences, Piscataway, NJ, USA). All the antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Generation of A549 cells Stably Expressing Nrf2 shRNA
To inhibit the expression of Nrf2, a short hairpin RNA targeting the 3′ end of the Nrf2 transcript was developed as described previously [29, 30]. The Nrf2 shRNA duplex contained the following sense and antisense sequences: 5′-GATCCGTAAGAAGCCAGATGTTAATTCAAGAGACATTCTTCGGTCTACAATTTTTTTTGGAAA-3′ (sense) and 5′-AGCTTTTCCAAAAAAAATTGTAGACCGAAGAATG TCTCTTGAATTAACATCTGGCTTCTTACG-3′ (antisense) [29]. The short hairpin RNA cassette was subcloned into pSilencer vector and transfected into A549 cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). A short hairpin RNA targeting luciferase gene was used as control. Stable cell clones with reduced Nrf2 expression were generated. All the clones were screened by real-time RT-PCR and immunoblotting.
Identification of ARE(s) in the Promoter of Srx1
To identify the presence and location of antioxidant response elements (AREs) in the Srx1 promoter, the 4 kb upstream region from the translation start site was downloaded from the NCBI database (Human Genome resources). The 5 kb sequence was used to search for AREs with the help of Genamics Expression 1.1 software (Hamilton, New Zealand) using the primary core sequence of ARE (RTGABNNNGCR) as the probe [31]. The promoter sequence of Rattus norvegicus, Mus musculus, Pongo pygmaeus, Pan troglodytes, Macaca mulatta, Bos taurus, Canis familiaris, Fugu rubripes, Gasterosteus aculeatus, Tetraodon nigroviridis orthologues of Srx1 were downloaded from the NCBI database and scanned for the presence of AREs.
Plasmids and Mutagenesis
The 5′ flanking region of human Srx1 promoter region (−3664 to +19) was PCR amplified from human Bacterial Artificial Chromosome (BAC) [32] clone corresponding to Srx1 gene using high-fidelity Taq polymerase (Applied Biosystems, Foster city, CA, USA). The isolated PCR product was ligated into pCR2.1 (Invitrogen, Carlsbad, CA, USA), and a KpnI-XhoI fragment from this construct was cloned into pGL3 basic vector (Promega, WI, USA). Three deletion constructs (−1518 to +19 and −319 to +19 and −233 to +19) were generated from the full-length promoter construct. The primers used for amplification were forward primer (−3664), CGCTACGGATGAAGACAACA; and reverse primer 3 (+19), TCCGGCCGGGCCATACCATT. The full length 3683 bp construct consisting of all 3 AREs (All 3ARE-pGL3b) was digested with KpnI and EcoRV to remove 2164 bp fragment including ARE 3 and religated. The resulting second ~1.5 kb deletion construct comprising of ARE2 and ARE1 (ARE1&2-pGL3b) was further digested with BglI to remove ARE2 and generate ~336 bp 3rd deletion construct (ARE1-pGL3b). Finally, 3rd deletion construct was further cleaved with SmaI to generate the smallest deletion construct (~252 bp) lacking all AREs (No-ARE-pGL3b). Oligonucleotides of all the 3 ARE core sequences plus the flanking sequence (81 bp in length) were annealed and cloned at KpnI-XhoI site in the pTAL luciferase reporter vector (BD Biosciences, San Jose, CA, USA) (ARE1-pTAL, ARE2-pTAL and ARE3-pTAL). The sequence of ARE oligo’s cloned in pTAL vector is as follows; ARE1 oligo, CGGTGCGTCACCCGCGCCCCTCCCGCCCCGCGACCTGCAAACTCACCCTGAGTCAGCCCGCCCGGGCGTTTCCACTCCCCG; ARE2 oligo, ACCCCAGCAGCGGGGCAGCAGCGCACCGCGGGCTTTGCCGACTCACCCTGGCTGGGCAGGTCCGCCGCGCTCTCCCCCGCC; ARE3 oligo, GGAGGTGACCCTGAGAATGAAAACTCTGCTGGAAAATGATGGAGCGGCAAGATAGAAAGGGCCTATGCCACTAGTGACCAT. Mutation in ARE1-enhancer sequence cloned in pTAL vector (Mut-ARE1-pTAL) was generated by using a site-directed mutagenesis kit from Stratagene (La Jolla, CA, USA). Primers containing the mutated ARE sequences CCGCGACCTGCAAACGCCCCCTGAGTCAGC (ARE1-Mu) were used for PCR amplification of the mutated Srx1 promoter, and PCR products were digested with DpnI for 1 h to cleave the wild-type promoter. The mutation of each promoter was verified by sequencing.
Validation of Srx1 siRNA Duplex
A small interfering RNA duplex target human Srx1 mRNA has been reported by Chang et al 2004 [33]. The sequence of siRNA targeting human Srx1 is 5′-GGAGGUGACUACUUCUACU-3′. The sequence of control non-targeting NS siRNA is 5′-UAGCGACUAAACACAUCAAUU-3′. A549 cells in the exponential growth phase were plated in 6-well plates at a density of 5 × 105 cells/well, grown for 12 h, and then transfected with 20 pmol of siRNA duplexes by using Lipofectamine 2000 and OPTI-MEMI reduced serum medium (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. Concentrations of siRNAs were chosen on the basis of dose-response studies. Knockdown of the target gene was quantified by real-time RT-PCR at 72 h after transfection using assay on demand primers and by using probe sets from Applied Biosystems. Assays were performed using the ABI 7000 Taqman system (Applied Biosystems, Foster city, CA, USA). GAPDH was used for normalization.
DNA Transfection and Luciferase Activity
Cells were transfected at 85% confluency using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Briefly, cells were seeded in 24-well plates at a density of 2 × 105 cells/mL and grown overnight. After ~ 12 h, the medium was removed and transfection complex containing 200 ng of plasmid DNA, 1 ng of pRL-TK plasmid (Promega, Madison, WI, USA), and transfection reagent were added to each well in the presence of fetal bovine serum. Cells were incubated for another 36 h. After 48 h, cells were lysed; Renilla and firefly luciferase activities were measured using the dual luciferase assay kit (Promega, Madison, WI, USA) with a luminometer (EG&G Wallac, MD, USA). Luciferase activity was normalized relative to Renilla luciferase activity using it as an internal control.
Chromatin Immunoprecipiation (CHIP) Assay
CHIP assay was performed by using a commercially available kit (Millipore, Billerica, MA, USA). Immunoprecipitates and total chromatin input were reverse cross-linked, DNA was isolated, and 1 μl of DNA was used for PCR (35 cycles) with primers specific for the Srx1 promoter. The Srx1 promoter primer sequence is as follows: Forward, AATGTTCGGAACGACTCCAG; Reverse, CAGGGTGAGTCGGCAAAG.
Measurement of ROS levels
Cells were incubated with 10 μM 2′, 7′-dichlorodihydrofluorescein diacetate (c-H2DCFDA) (Invitrogen, CA, USA) for 30 mins at 37oC to assess the ROS mediated oxidation to the fluorescent compound c-H2DCF. Fluorescence of oxidized c-H2DCF was measured at an excitation wavelength of 480 nM and an emission wavelength of 525 nM using a FAC Scan flow cytometer (Becton Dickinson, San Jose, USA).
MTT Cell Viability Assay
Cells were transfected with 20 nM siRNAs two times with a 48 h interval and, 24 h after the second transfection, cells were plated at a density of 7,500 cells/well in 180 μl of growth media in 96-well plates. After 12 h, cells were treated with various concentrations of H2O2 for 24 h. Cell viability was evaluated by using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT; Sigma, St. Louis, MO, USA) reduction conversion assay [34]. Cell survival was expressed as absorbance relative to that of untreated controls. Results are presented as means ± SD.
Human Tissues
Frozen lung tissue samples were obtained from cardiopulmonary pathology tissue bank, The Johns Hopkins University, Baltimore, MD, USA and the Interstitial Lung Disease Tissue Bank at National Jewish Medical Health, Denver, CO, USA. The non-COPD lungs (3 non-smokers and 2 ex-smokers) had normal spirometric lung function and no evidence of emphysema. Study protocols were approved by the institutional review boards for human studies.
Statistical Analysis
Results are presented as the means ± S.E. Statistical comparisons were performed by paired Student’s t-tests. A value of p<0.05 was considered statistically significant.
RESULTS
Transcriptional induction of Srx1 gene expression in lungs, in response to CS exposure, is dependent on Nrf2
Nrf2+/+ and Nrf2−/− mice were exposed to CS for 5 h and then examined for expression of Srx1 by real-time RT-PCR. Srx1 mRNA level was upregulated by 8-fold in response to CS in Nrf2+/+ mice. No induction was observed in the lungs of CS-exposed Nrf2−/− mice (Fig. 1A). Western blot analysis of lung proteins showed that Srx1 protein was present constitutively, and its level increased significantly on exposure to CS in Nrf2+/+ mice. By contrast, expression of Srx1 protein was low in Nrf2−/− mice and did not change significantly with CS exposure (Fig. 1B and C). The data indicate that pulmonary Srx1 expression is induced by CS, and the induction is abrogated in Nrf2−/− mice, implicating a critical role of Nrf2 in induction of Srx1 gene expression.
Fig. 1. CS exposure induces expression of Srx1 in lungs in Nrf2+/+ mice.
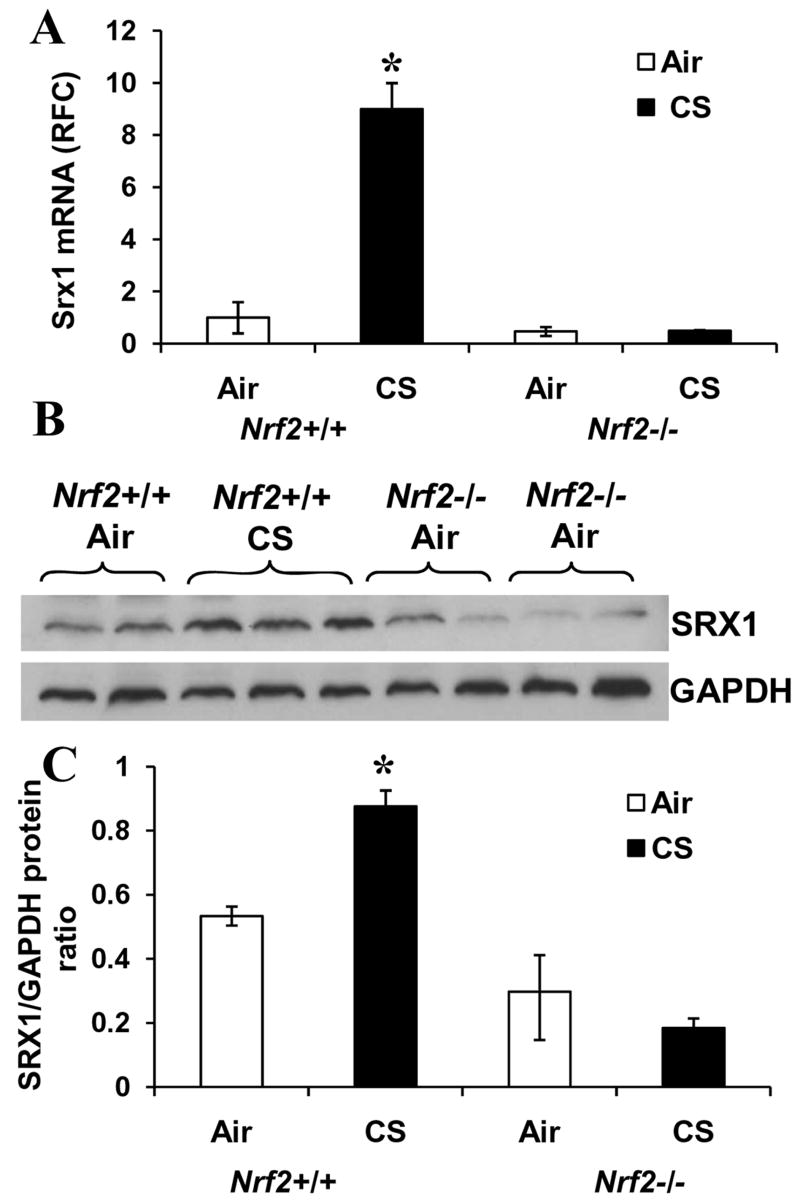
(A) Nrf2+/+ and Nrf2−/− mice were exposed to CS for 5 h and total RNA was isolated. Expression level of Srx1 was analyzed by real-time RT-PCR. GAPDH was used for normalization. Data are presented as fold change over air exposed Nrf2+/+ group. (B) SRX1 protein levels in the lungs of Nrf2+/+ and Nrf2−/− mice. Mice were exposed to CS for 5 h and lung protein was isolated after 24 h and used for western blot analysis. C) Quantification of Srx1 protein expression in the lungs of air and CS-exposed Nrf2+/+ and Nrf2−/− mice. (Mean ± SD of the Srx1/GAPDH protein ratio in three independent samples). ‘*’, significant when compared to air exposed mice (P ≤0.05).
Nrf2 activation increases Srx1 mRNA and protein level in lung epithelial cells
To further elucidate the role of Nrf2 in regulating transcription of Srx1, siRNA targeting Nrf2 was used to inhibit Nrf2 activity. Conversely, KEAP1 siRNA was used to activate the expression of Nrf2 dependent genes. The siRNA duplexes were transfected into BEAS2B cells, 72 h later cells were harvested, and mRNA expression for Nrf2, KEAP1, and Srx1 were quantified by real-time RT-PCR. The siRNA targeting Nrf2 and KEAP1 led to >85% decrease in mRNA of these genes (Fig. 2A–B). To further confirm the specificity of the siRNA effect on Nrf2, we measured the transcript level of NQO1, another well-characterized target of Nrf2. The siRNA mediated decrease in Nrf2 lead to a 65% decrease in NQO1 transcript, whereas siRNA targeting KEAP1 upregulated the expression of NQO1 by 600% (Fig. 2A–B). To further confirm that Nrf2 is essential for NQO1 induction in response to oxidative stress, the NS siRNA and Nrf2 siRNA transfected BEAS2B cells were retransfected NQO1- ARE reporter vector. Twenty four hours after the second transfection, cells were treated with H2O2 (200 μM), 2,5-t-butylhydroquinone (TBHQ) (50 μM), and CS extract (100 μg/ml) for 12h and luciferase activity was measured. As shown in Fig. 2C., BEAS2B cells transfected with NS siRNA displayed a significant increase in the amount of ARE luciferase activity in response to treatment with oxidative stress inducing agents where as Nrf2siRNA transfected cells showed significantly reduced NQO1 ARE-luciferase induction. Thus, this non-tumorigenic lung cell line model was further used for determining the regulation of Srx1 by Nrf2. Depletion of Nrf2 by siRNA resulted in a decline in mRNA expression of Srx1 and conversely, inhibition of expression of KEAP1 upregulated the transcript of Srx1 in BEAS2B cells (Fig. 3A). Thus, the siRNA data obtained in non-tumorigenic lung epithelial cells confirm the results obtained from mice indicating that increase in Srx1 mRNA level is Nrf2 dependent. Next, we examined whether oxidative stress inducing agents, which activate Nrf2, also induce Srx1 expression in BEAS2B cells. The cells were exposed to various oxidative stressors such as H2O2 (200 μM), 2,5-t-butylhydroquinone (TBHQ) (50 μM), and CS extract (100 μg/ml) for 6 h to 24 h. The NQO1 and Srx1 induction in response to these treatments was quantified by real-time RT-PCR. Srx1 expression was upregulated in response to treatment with all three oxidative stress inducing agents (Fig. 3B–D). To determine whether increase in Srx1 transcript correlates with increase in Srx1 protein, cells were treated with TBHQ and levels of Srx1 protein was measured. Treatment with TBHQ led to a parallel increase in Srx1 protein levels (Fig. 3E–F). These results clearly indicate that oxidative stress mediated increase in Srx1 levels is dependent on activation of Nrf2 signaling.
Fig. 2. Modulation of Nrf2 activity in BEAS2B cells by RNAi approach and oxidative stress inducing agents.
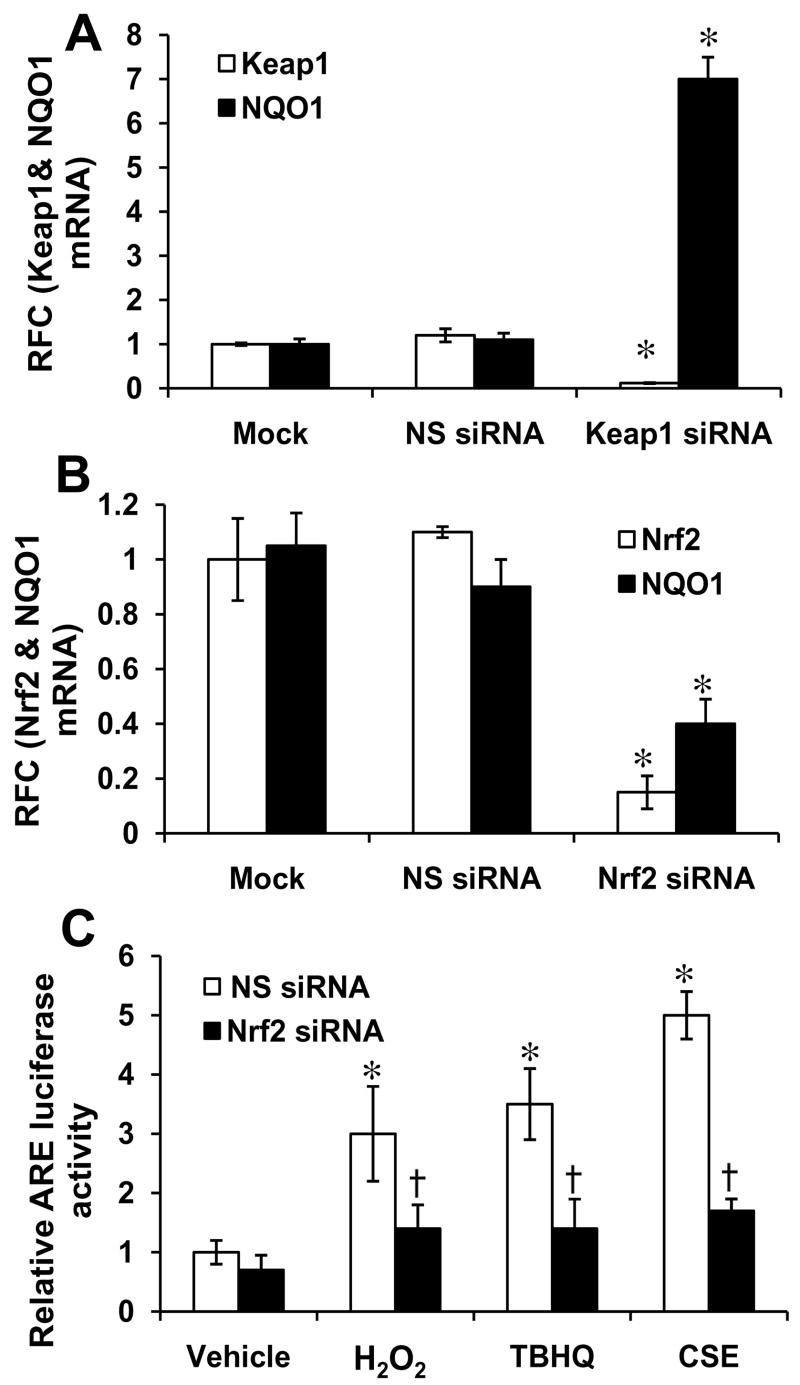
(A) siRNA targeting KEAP1 decreased KEAP1 mRNA level and upregulated the expression of Nrf2 dependent gene NQO1. (B) siRNA targeting Nrf2 lowered Nrf2 transcript level and inhibited the expression of downstream target gene NQO1. (C) Treatment with oxidative stress inducing agents like CS extract (100 μg/ml), H2O2 (200 μM) and tert-butyl hydroquinone (50 μM) upregulated the NQO1-ARE-luciferase activity in Nrf2 dependent manner. ‘*’ Significant when compared with vehicle (DMSO) treated cells; ‘†’ significant when compared with non-targeting NS siRNA (non-specific) -transfected BEAS2B cells (P ≤0.05).
Fig. 3. Nrf2 activation increases Srx1 transcript and protein level in normal human lung epithelial cells.

(A) Transient transfection of BEAS2B cells with KEAP1 siRNA unregulated the expression of Srx1 mRNA, whereas transfection with Nrf2 siRNA lowered the expression of Srx1 transcript in BEAS2B cells. ‘*’, significant when compared to non-targeting, non-specific (NS) siRNA transfected cells. (B–D) Induction of Srx1 expression in response to treatment with multiple oxidative stress inducing agents. BEAS2B cells were exposed to CS extract (100 μg/ml), H2O2 (200 μM) and 2,5-t-butylhydroquinone (TBHQ, 50 μM), and expression of Srx1 was measured by real-time RT-PCR. ’*’, significant when compared to vehicle (DMSO) treated sample. (E) Western blot analysis of BEAS2B cells treated with TBHQ. Total protein was isolated and SRX1 protein levels were determined. GAPDH was used as the loading control. (F) Quantification of SRX1 protein in the Western blots of BEAS2B cells treated with TBHQ’*’, significant when compared to vehicle (DMSO) treated cells (P ≤0.05).
Decreased Srx1 RNA levels in lung cancer cells expressing Nrf2shRNA
A loss of function mutation in KEAP1 results in upregulation of the Nrf2 pathway in A549 lung cancer cells [29]. To attenuate the expression of Nrf2, we designed a short hairpin RNA targeting the 3′ end of the Nrf2 mRNA as described earlier [29, 30]. A short hairpin RNA targeting luciferase gene was used as a control. Stable cell clones with decreased Nrf2 expression were generated. All clones were screened by real-time RT-PCR and immunoblotting. After initial screening, we selected one independent clone of A549 cells expressing Nrf2 shRNA, which demonstrated a stable 85% downregulation of Nrf2 mRNA and a corresponding 70% decrease in NQO1 expression (Fig. 4A). Measurement of Nrf2 protein by immunoblotting showed similar decrease in Nrf2 protein levels (Fig. 4B). The expression of Nrf2 in the control cells transfected with luciferase shRNA and the untransfected cancer cells was comparable (Fig. 4B). Expression of Srx1 was drastically lower (>85%) in A549 cells expressing Nrf2 shRNA (Fig. 4C–D).
Fig. 4. Increased Srx1 mRNA levels in A549 lung cancer cells with constitutively activated Nrf2.

(A) Generation of cell lines stably expressing Nrf2 shRNA. Real-time RT-PCR analysis of Nrf2 and NQO1 expression in A549 cells stably expressing Nrf2 shRNA and luciferase shRNA. Total RNA from stable clones harboring Nrf2 shRNA or non-targeting luciferase shRNA were analyzed for expression of Nrf2, and NQO1. GAPDH was used as normalization control. ’*’, significant when compared to A549 parent cells and A549 cells expressing Luciferase shRNA (P ≤0.05) (B). Immunoblot detection of Nrf2 in A549 cells stably transfected with shRNAs targeting Nrf2 and luciferase. (C) Comparison of Srx1 expression between cells expressing Nrf2 shRNA and control cells expressing luciferase shRNA. D) Western blot analysis of Srx1 expression in A549 cells stably transfected with the Nrf2 shRNA and control cells expressing luciferase shRNA.
ARE mediates the transcriptional regulation of Srx1
To identify putative AREs in Srx1 promoter, we examined the human Srx1 genomic locus in detail. Functional AREs, such as the one in HO-1, GPx2 or NQO1 gene, are often represented by a extended ARE consensus sequence, 5′-TMAnnRTGAYnnnGCR-wwww-3′ and detailed in silico analysis of the human Srx1 genomic locus revealed 3 potential ARE sequences within the ~3.4 kb of the 5′ promoter region (Fig. 5) [31]. Two of these AREs are located in the proximal ~−340 bp region. ARE1 and ARE2 are located at −228 and −338 bp, respectively. In silico analysis of Srx1 promoter revealed putative AREs in: Rattus norvegicus, Mus musculus, Pongo pygmaeus, Pan troglodytes, Macaca mulatta, Bos taurus, Canis familiaris, Fugu rubripes, Gasterosteus aculeatus and Tetraodon nigroviridis. (Table 1). The ARE1 core sequence is remarkably well conserved from humans to fish. The location and orientation of ARE enhancer sequence from transcription start site is similar in Homo sapiens, Rattus norvegicus, Mus musculus, Pongo pygmaeus, Pan troglodytes, Macaca mulatta, Bos taurus, Canis familiaris (Table 1).
Fig 5. Deletion analysis of Srx1 promoter by transient transfection in A549 cells.

(A)Three putative AREs were identified in the proximal promoter region of the human Srx1gene. They are located at −3.4 kb, −338 bp, and −228 bp relative to the transcription start site. Four luciferase reporter deletion constructs were made that contained all three ARE (All 3ARE-pGL3b; 3683 bp), two ARE (ARE1&2-pGL3b; 1.537 kb), one ARE (ARE1-pGL3b; 336 bp) or no AREs (No-ARE-pGL3b; 252 bp). (B) Reporter constructs were transfected into A549 cells or A549-Nrf2shRNA cells constitutively expressing Nrf2shRNA. A 3683 bp portion of the Srx1 promoter was cloned into pGL3 basic luciferase reporter vector (−3664 to +19) to monitor the activity of this promoter. Three deletion constructs ARE1&2-pGL3b (−1518 to +19), ARE1-pGL3b (−319 to +19) and no AREs-pGL3b (−233 to +19) were prepared by restriction digestion of the full length 3683bp promoter construct in pGL3basic vector. All 4 constructs (All 3ARE-pGL3b, ARE1&2-pGL3b, ARE1-pGL3b, No ARE-pGL3b) were transfected into A549 control cells and A549-Nrf2shRNA cells and luciferase activities were measured after 48 h. Luciferase activity was normalized to co-transfected pRL-TK activities and expressed in arbitrary units. The three constructs having ARE had similar reporter activities. The −233 to +19 bp deletion construct lacking an ARE sequence displayed the minimum reporter activity. ‘*’, significant when compared to the smallest construct lacking all 3 ARE; ‘†’, significant when compared to A549 control cells (P ≤0.05). (C) ARE enhancer sequence oligonucleotides were cloned in pTAL vector (ARE1-pTAL, ARE2-pTAL and ARE3-pTAL) and sequence verified. Oligonucleotide containing mutations in ARE1 core sequence was also cloned in pTAL vector (Mut-ARE1-pTAL). These constructs were transfected into A549 cells and A549-Nrf2shRNA cells and the luciferase activities were measured. ‘*’, significant when compared to ARE2-pTAL, ARE3-pTAL and mutant ARE1 (Mut-ARE1-pTAL). (D) ARE1 enhancer sequence cloned in pTAL (ARE1-pTAL) vector was transfected into BEAS2B cells. After 36 h, cells were treated with TBHQ for 12 h and luciferase activities were measured. Luciferase activities were normalized by measuring the Renilla luciferase activity from a cotransfected reporter vector. ‘*’, significant when compared to vector transfected or ARE2, ARE3 transfected cells; ‘†’ significant when compared to vehicle treated BEAS2B cells (P ≤0.05).
Table 1. ARE mediates the transcriptional regulation of the Srx1 gene.
(A) Comparison of ARE sequences from Homo sapiens, Rattus norvegicus, Mus musculus, Pongo pygmaeus, Pan troglodytes, Macaca mulatta, Bos taurus, Canis familiaris, Fugu rubripes, Gasterosteus aculeatus, Tetraodon nigroviridis orthologues of Srx1. Promoter sequences were downloaded from the NCBI database and scanned for the presence of ARE. The sequence RTGAY/GNNNGCR was used as the probe.
| Species | Location | Orientation of ARE | ARE Sequence |
|---|---|---|---|
| Homo sapiens (human) | −228 | 3′-5′ | CCGCGACCTGCAAACTCACCCTGAGTCAGC |
| −338 | 3′-5′ | GCGGGCTTTGCCGACTCACCCTGGCTGGGC | |
| Rattus norvegicus (Rat) | −252 | 3′-5′ | CCGCGACCTGCAAACTCACCCTGAGTCAGC |
| −613 | 5′-3′ | GAGCCTAGATGAGCCTGCGCTGGGATCCAG | |
| Mus musculus (mouse) | +27 | 3′-5′ | CAGTGGGCAGCTAGGTCAGAGTGAGACACG |
| −2218 | 5′-3′ | TAAAATGTTTGACATAGCTCAGCTTATTTC | |
| Pongo pygmaeus (Chimpanzee) | −394 | 3′-5′ | GCTAGCTTTGCCGACTCATCCTGGCTGGGC |
| −1958 | 3′-5′ | CTTGCTCTTGCTCTGTCACCCAGGCTGTAG | |
| Pan troglodytes (Orangutan) | −824 | 3′-5′ | CCGCGACCTGCAAACTCACCCTGAGTCAGC |
| −931 | 3′-5′ | GCGGGCTTTGCCGACTCATCCCGGCTGGGC | |
| Macaca mulatta (Monkey) | −308 | 3′-5′ | CCGCGACCTGCAAACTCACCCTGAGTCAGC |
| −401 | 3′-5′ | GCAGGATTTGCCGACTCATCCCGGCTGGGC | |
| Bos Taurus (Cow) | −305 | 3′-5′ | CCACGACCTGCAGACTCACCCTGAGTCAGC |
| −415 | 3′-5′ | GCGAGTTTTGCCGACTCACCCTCGCTGGGC | |
| Canis familiaris (Dog) | +89 | 3′-5′ | GCCGCTTCTGCCGACTCATCCTGGCTCCGC |
| −1415 | 5′-3′ | CTAAGAGGGTGACTATGCATTCCCCATGCA | |
| Fugu rubripes (Fugu fish) | −3681 | 5′-3′ | GCAGAATAATGAGACAGCAGACAGAGCGGG |
| −4873 | 5′-3′ | GCAATAATATGACAATGCATGTAGACATTT | |
| Gasterosteus aculeatus (three-spined stickleback fish) | −34 | 5′-3′ | TCCAGGAAGTGAGGAAGCGCAAACACGTCT |
| Tetraodon nigroviridis (green spotted puffer fish) | −3716 | 5′-3′ | GTGGAATAATGAGACGGCAGACAGAGCGGG |
| −4846 | 5′-3′ | GCAATAATATGACAATGCATGTAGACATTT |
A 3683 bp human Srx1 promoter region was cloned, and deletion reporter constructs containing all three putative AREs (All 3 ARE-pGL3b), two AREs (ARE1&2-pGL3b), one ARE (ARE1-pGL3b), or no ARE (No-ARE-pGL3b) were generated (Fig. 5A; Table 2). Nrf2-depdendent activities of these constructs were first investigated in human lung non-small-cell lung cancer cell (A549) in which Nrf2 is constitutively activated due to KEAP1 mutation. All 3 deletion constructs containing the ARE1 sequence exhibited over 10- fold higher activity compared with the smallest reporter construct (−233 to +19 bp) lacking all ARE sequences (Fig. 5B). All 3 constructs containing the ARE1 sequence exhibited over 4-fold higher activities in A549 control cells compared with A549-Nrf2shRNA cells (Fig. 5B). We used A549 untransfected cells as a control, because the control cells expressing luciferase shRNA were not compatible with the luciferase reporter assay. Of these 3 deletion constructs containing ARE sequences, the 3rd deletion construct (−319 to +19 bp) exhibited highest reporter activity. These data strongly suggest that −319 +19 region of the Srx1 promoter contains an active ARE consistent with the localization of a putative ARE (−228bp) in this region. To further confirm this finding and to dissect functionalities of individual ARE sequences to act as enhancers, we cloned individual AREs with sufficient flanking sequences into a pTAL-Luc reporter vector containing a minimal promoter. The reporter assay using pTAL-Luc derived constructs showed that only ARE1 (pTAL-ARE1) demonstrates substantially higher reporter activity in control A549 cells as compared with A549-Nrf2shRNA cells (Fig. 5C). ARE2 (pTAL-ARE2) and ARE3 (pTAL-ARE3) showed minimal reporter activity. Similar to the data obtained with A549 cells, ARE1 facilitated TBHQ inducer activity in BEAS-2B cells, while ARE2 and ARE3 showed no TBHQ inducer activity (Fig. 5D). These observations indicate that only ARE1 is functional, and it is involved in Nrf2-dependent regulation of Srx1 promoter.
Table 2.
Details of the Srx1 promoter deletion constructs.
| Construct name | Details of the Srx1 promoter region cloned in the reporter vector |
|---|---|
| All 3ARE-pGL3b | A 3683 bp Srx1 promoter region (−3664 to +19) containing all 3 ARE sequences, cloned in pGL3basic promoterless vector |
| ARE1&2-pGL3b | A 1537 bp Srx1 promoter region (−1518 to +19) containing ARE1 and ARE2, cloned in pGL3basic promoterless vector |
| ARE1-pGL3b | A 336 bp Srx1 promoter region (−319 to +19) containing only ARE1, cloned in pGL3basic promoterless vector |
| No ARE-pGL3b | A 252bp Srx1 promoter region (−233 to +19) lacking all the 3 ARE, cloned in pGL3basic promoterless vector |
| ARE1-pTAL | ARE1 core sequence (−274 to−193) cloned in pTAL luciferase minimal promoter vector |
| ARE2-pTAL | ARE2 core sequence (−384 to −303) cloned in pTAL luciferase minimal promoter vector |
| ARE3-pTAL | ARE3 core sequence (−3444 to −3363) cloned in pTAL luciferase minimal promoter vector |
| Mut-ARE1-pTAL | Mutant ARE1 core sequence (−274 to −193; 2 single base substitutions at −240 and −242) cloned in pTAL luciferase minimal promoter vector |
Nrf2 binds to the Srx1 promoter in vivo
We used CHIP assay to determine whether Nrf2 binds to the endogenous Srx1 promoter constitutively. Furthermore, we determined whether increase in the oxidative stress mediated activation of Nrf2 signaling leads to an increase in its recruitment to the promoter of Srx1. In A549 control cells, constitutive recruitment of Nrf2 to the promoter of the Srx1 was observed (Fig. 6A). As expected, in A549-Nrf2shRNA cells, Nrf2 was present at undetectable levels and low amplification was detected using Srx1 promoter primers. As shown in (Fig. 6B), Nrf2 was present in the basal transcription complex formed with Srx1 promoter in BEAS2B cells. Activation of Nrf2 by TBHQ treatment enhanced the recruitment of Nrf2 to the Srx1 promoter as demonstrated by increased amplification of the Srx1 promoter region (Fig. 6B). Immunoprecipitations with control IgG failed to select the Srx1 promoter (Fig. 6B), suggesting that the sites were not enriched in nonspecific fashion. Collectively, the results from both A549 and BEAS2B cells conclusively show that Nrf2 regulates transcription of Srx1 by binding to ARE1 enhancer sequence in Srx1 promoter.
Fig. 6. Nrf2 binds to Srx1 promoter in vivo.

(A) CHIP assay was performed with A549 cells stably expressing Nrf2shRNA and the control cells expressing luciferase shRNA. PCR reactions were performed to amplify the Srx1 regulatory region encompassing ARE1. (B) Increased recruitment of Nrf2 to ARE1 consensus sequence in the Srx1 promoter in response to TBHQ treatment.
Human Srx1 protects cells from oxidative stress by H2O2
To assess the importance of human Srx1 in response to oxidative stress, we used A549 cells, a lung cancer cell line with KEAP1 mutation leading to constitutive activation of Nrf2. Increased Nrf2 activity in A549 leads to increased Srx1 expression thus making A549 cells as an ideal Srx1 overexpression model system and siRNA targeting Srx1 transcript downregulated Srx1 expression in A549 cells (Fig. 7A). Similarly, ecotopic expression of Srx1 cDNA in BEAS2B cells led to substantial increase in endogenous SRX1 protein levels (Fig. 7B). We speculate that the higher levels of Srx1 may enhance the recycling of cysteine sulfinic acid forms of Prxs with reactivation of hyperoxidized Prxs resulting in augmentation of the antioxidant capacity of the cell. To determine whether attenuating Srx1 expression in A549 cells leads to an overall increase in oxidative stress due to decreased Prx activity, we downregulated Srx1 expression by RNAi followed by H2O2 treatment. Intracellular ROS levels were monitored using c-H2DCFDA and flow cytometry. The results demonstrated a 2-fold increase in fluorescence in vehicle treated A549 Srx1 siRNA transfectedcells compared to vehicle treated non-specific (NS) siRNA transfected cells (Fig. 7C). Treatment of Srx1 siRNA transfected A549 cells with H2O2 for 30 mins led to a dramatic 10-fold increase in ROS level, whereas H2O2 treatment induced only 2-fold increase in ROS level in non-targeting, non-specific (NS) siRNA transfected A549 cells suggesting that peroxide detoxification is altered in Srx1 siRNA transfected cells (Fig. 7C). Attenuation of Srx1 expression in A549 cells potentiated the toxicity of H2O2, whereas overexpression of Srx1 cDNA in BEAS2B cells protected the cells against H2O2 mediated cell death (Fig. 7A–B, D–E). Thus, Srx1 protects against oxidative stress in the lung cells, and it may contribute to the overall Nrf2 mediated cytoprotection against environmental pollutants such as CS.
Fig. 7. Srx1 confers protection against oxidative damage in lung epithelial cells.
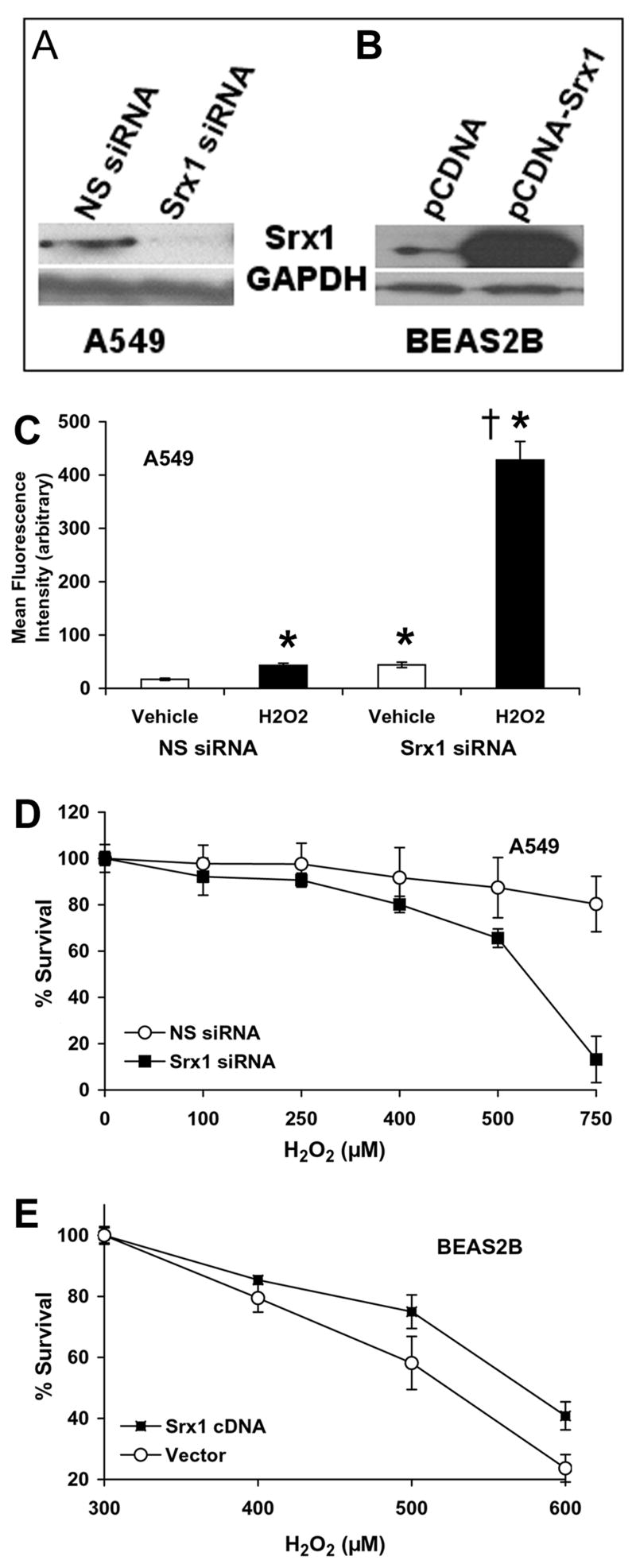
(A) A549 cells were transfected with Srx1 siRNA and non targeting NS siRNA. Cells were harvested after 72 h and Srx1 protein level was determined by immuloblotting (B) BEAS2B cells were transfected with Srx1 cDNA expression plasmid and Srx1 protein levels was measured 48 h after transfection. (C) Increased ROS levels in A549 cells transfected with Srx1 siRNA and treated with H2O2. A549 cells transfected with Srx1 siRNA and NS siRNA were treated with 500 μM H2O2 for 30 mins. The cells were washed and stained with H2DCF-DA, and the mean fluorescence was measured using flow cytometry. Data are presented as mean ± SD. ‘*’, significant when compared to NS siRNA transfected and vehicle treated cells; ‘†’, significant when compared to NS siRNA transfected and H2O2 treated cells. (D) Attenuation of Srx1 expression in A549 cells by RNAi approach enhanced the cytotoxicity of H2O2. Cells were exposed to H2O2 for 24 h and viable cells were determined by MTT assay. Data are represented as percentage of viable cells relative to the vehicle (culture medium) treated control. Data are mean of 6 independent replicates, combined to generate the mean ± SD for each concentration. ‘*’, P<0.01 relative to NS siRNA. (E) Overexpression of Srx1 in BEAS2B cells protected against H2O2 cytotoxicity. ‘*’, P<0.05 relative to vector control.
Srx1 expression declines in lungs of advanced COPD patients
We have recently reported that there is a decline in Nrf2 activity and the expression of Nrf2-dependent antioxidant enzymes like HO-1, NQO1, GCLM, and GPx2 in the lung tissues of patients with progressive COPD, which is associated with increased oxidative stress [27]. To determine whether expression of Srx1 is affected by diminished Nrf2 expression, we isolated total RNA from 18 advanced stage COPD lung tissues and 5 normal healthy smokers lung tissue and measured the expression of Srx1. Real-time RT-PCR revealed marked decrease in the expression of Srx1 in COPD tissues. The average value for relative Srx1 expression in normal healthy smokers was 1.07 (SD±0.32). As compared with normal healthy lungs, the relative Srx1 transcript level was significantly lower in lung tissues from patients with COPD. (Average- 0.47, SD±0.18, p=0.003) (Fig. 8A). Immunoblot analysis of randomly selected 11 representative COPD lungs confirmed the decrease in Srx1 mRNA and protein expression and a decline in Nrf2 protein level in the COPD lungs as compared with normal lungs (Fig. 8B–C). The overall correlation coefficient (r) is 0.75 (p-value <0.001) (Fig. 8D).
Fig. 8. Srx1 expression declines in patients with Advanced COPD.
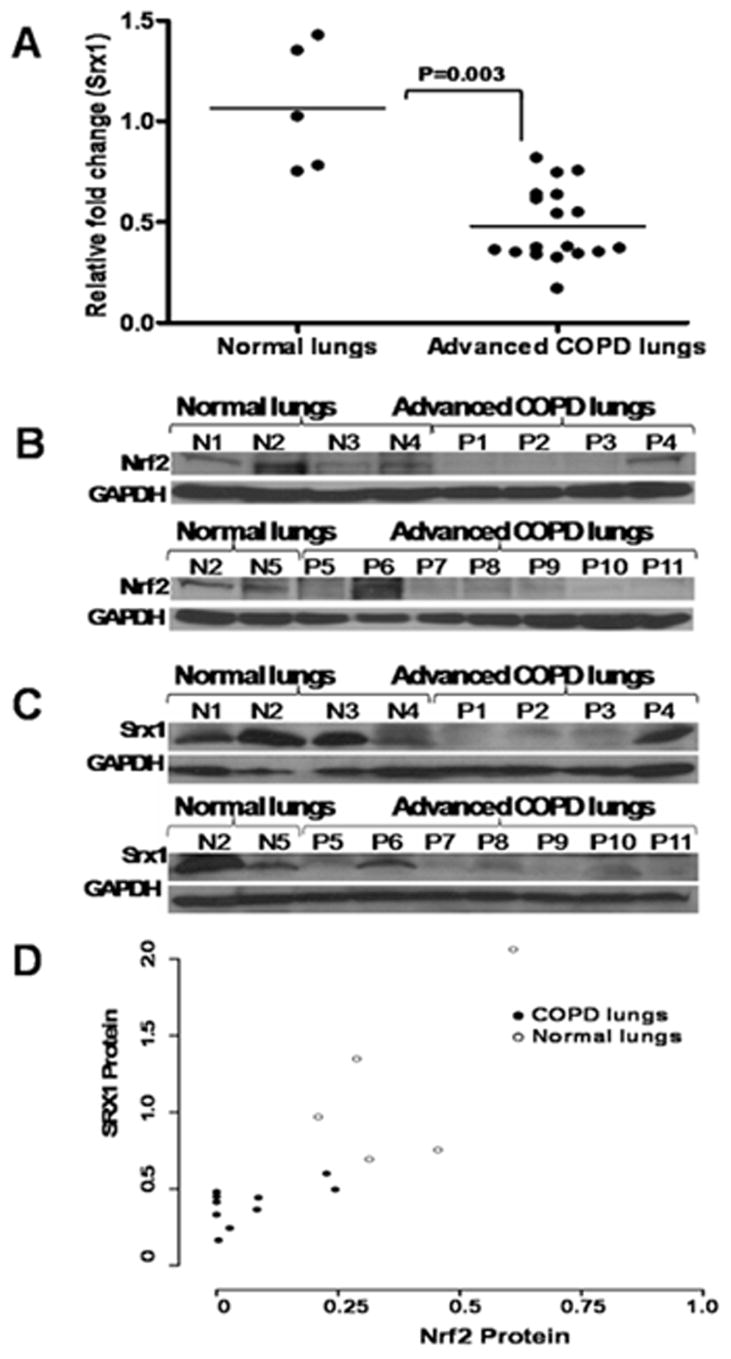
(A) Dot plot showing relative mRNA levels of Srx1 in normal healthy individuals (n=5) and advanced COPD lungs n=18). Srx1 expression in normal healthy individuals was significantly higher than patients with COPD (P=0.003). Data were analyzed using two-sample Wilcoxon rank-sum (Mann-Whitney) test. Immunoblot analysis of Nrf2 (B) and Srx1 (C) protein levels in normal healthy lungs (n=5) and advanced COPD lungs (n=11). Blots were normalized with GAPDH as loading control. ‘P’, lungs from patients with COPD; ‘N’, Normal healthy lungs. (D) Spearman correlation analysis showed significant correlation between Nrf2 and Srx1 protein levels. The overall correlation coefficient (r) is 0.75 (P <0.001).
DISCUSSION
Cigarette smoking is the most important etiological factor for the development of COPD. There is a dose dependent relationship between the cumulative amount of smoking and incidence of COPD. Nrf2, a redox-sensitive basic leucine zipper transcription factor, is a critical determinant of susceptibility to lung inflammation and oxidative stress and the development of emphysema caused by chronic exposure to CS [16]. Consistent with the central role of Nrf2 in protection against CS-induced oxidative stress, we have previously observed the enhanced formation of 4HNE, a marker of lipid peroxidation, and 8-Oxo-dG, a marker of oxidative DNA damage, in the lungs of Nrf2−/− mice exposed to CS relative to wild-type mice [16, 30]. There is also a strong correlation between decline in Nrf2 protein levels and severity of COPD along with increase in oxidative stress [17–19].
The potent oxidants in CS include superoxide anion (O2•−), nitric oxide (NO•), and reactive peroxynitrite (ONOO−), which is formed due to the interaction of O2•− and NO• (25). Semiquinones and benzoquinones present in the cigarette tar along with superoxide radicals lead to production of reactive hydroxyl radicals (•OH) and hydrogen peroxide (H2O2) [35]. Some of these reactive oxygen species cause lipid peroxidation, leading to destruction of membrane lipids [36, 37]. Lipid peroxidation, a well-established mechanism of cellular injury is used as an indicator of oxidative stress. Glutathione peroxidases, peroxiredoxins, and catalases decompose H2O2 and organic hydroperoxides produced during oxidative insults and prevent peroxide-induced DNA damage, lipid peroxidation, and protein degradation [3, 38–40]. Peroxiredoxins are a family of enzymes that reduce H2O2 and alkyl hydroperoxides to water and alcohol, with the use of reducing equivalents provided by a thiol like thioredoxin [3, 39, 40]. The Prx enzymes containing 2 cysteine residues are intrinsically susceptible to reversible hyperoxidation to cysteine sulfinic acid. Cysteine hyperxoidation of Prx results in loss of peroxidase activity. Sulfiredoxin catalyzes reduction of sulfinic Prx enzymes to sulfenic acid thus restoring the functional cysteine residues and activity of Prx enzymes [3, 4, 24, 39, 40]. Srx1 is the first protein identified that has been implicated in removal of glutathione moiety from proteins, a process also known as deglutathionylation [3, 41]. The results of the present study show that Srx1 expression is selectively upregulated in the lungs of Nrf2+/+ mice in response to CS exposure. Both basal and CS-inducible expressions of Srx1 are Nrf2 dependent in murine lungs. Consistent with the Nrf2-dependent gene expression patterns, Srx1 mRNA and protein were significantly higher in the lungs of Nrf2+/+ mice in response to acute CS exposure. Induction of Srx1 expression in CS exposed Nrf2+/+ mice lungs correlated with the increase in total Prx activity in the lungs of wild type CS exposed mice [16].
Exposures of lung epithelial cells (BEAS2B) to various oxidative stressors like H2O2, TBHQ, and CSE induced the expression of Srx1, suggesting that Srx1 is an oxidative stress responsive gene. Down-regulation of Nrf2 by RNAi in human lung epithelial cells like A549 cells and BEAS2B attenuated the expression of Srx1. Targeting the cytosolic repressor of Nrf2, KEAP1, in BEAS2B cells resulted in upregulation of Srx1 expression. A549 cells, which harbor KEAP1 mutation leading to constitutive activation of Nrf2 and its target genes, demonstrated increased expression of Srx1 further suggesting that transcription of Srx1 is tightly controlled by Nrf2 status. These results clearly show that basal and inducible expression of Srx1 mRNA is dependent upon cellular levels of Nrf2.
We further explored the molecular basis underlying the Nrf2-dependent Srx1 induction in lungs. A common feature of the promoter regions of genes induced by oxidative and electrophilic insult in an Nrf2-dependent fashion is the presence of a cis-acting enhancer sequence, the antioxidant response element (ARE) [22]. Enhancers are DNA sequences that increase transcription independent of their orientation and distance relative to the RNA start site [42]. AREs have been identified in several antioxidant and xenobiotic genes, including mouse Gsta1 [43], Ho-1 [44], and Ferritin H [45] and rat Gsta2 [46], Gsta3 [47], Nqo1 [48], Gclc [49], Gpx2 [30], Prx1 [31, 50]. We searched for AREs in the human Srx1 and identified 3 putative AREs in the proximal promoter region. The Srx1 AREs are similar to the AREs found in the HO-1 or NQO1 regulatory regions. Detailed sequence analysis revealed that the sequences of the Srx1 AREs are highly conserved across different species. Identification of putative ARE sequences in Srx1 promoter in mammals, rodents, and fish suggests that the regulation of Srx1 expression is evolutionarily conserved [51].
To identify functional ARE binding sequences in human Srx1 promoter, we generated several Srx1 promoter deletion constructs. The activity of the luciferase reporter construct (−3683 to +19) harboring the all the 3 Srx1-ARE in pGL3 basic vector was 4-fold higher in A549 control cells as compared to A549-Nrf2shRNA cells. Studies with nested deletion constructs differing in their 5′ ends indicated that only the ARE sequence located between −300 and +19 (ARE1) is active. The reporter construct containing the putative ARE1 ligated to pTAL luciferase vector (containing TATA-like promoter region) was significantly activated in response to TBHQ treatment in BEAS2B cells. The pTAL-ARE1 reporter activity was reduced substantially in A549-Nrf2shRNA cells. Mutation of the core sequence of ARE1 in the pGL3 reporter vector or pTAL vector completely abolished Srx1 promoter or luciferase reporter activity. Recruitment of Nrf2 to the ARE1 sequence in the Srx1 promoter was further confirmed by CHIP assay with Nrf2 antibody. Collectively, these results suggest that Nrf2 binds to the Srx1-ARE1 and regulates its expression. Studies focused on understanding the functional significance of Nrf2 dependent Srx1 induction in lungs revealed that attenuation of Srx1 expression by RNAi potentiated the toxicity of H2O2 where as overexpression of Srx1 protected against H2O2 mediated cell death.
Increased H2O2 levels in breath condensates from patients with COPD have been reported [52, 53] and it may result from impaired detoxification or decomposition of H2O2. We have recently reported that decline in NRF2-regulated antioxidant defense in COPD patient lungs accounts for the greater oxidant–antioxidant imbalance. Lung tissues exhibiting severe emphysema demonstrated reduced mRNA and protein levels of HO-1, GCLM, NQO1, GPx2 [17–19]. To determine whether suboptimal Nrf2 activity leads to a parallel decrease in Srx1 activity as well, we measured Srx1 expression in lung tissues from patients with COPD. Interestingly, expression of Srx1 was downregulated at the transcriptional level as well as protein level in the lung tissues form patients with COPD. We speculate that low levels of Srx1 in COPD tissues may lead to accumulation of oxidized sulfinic acid forms of Prx enzymes, thereby attenuating the total Prx activity in lungs. Decline in the activity of Srx1, which modulates Prx activity, total glutathione peroxidase activity may lead to impaired H2O2 detoxification thereby enhancing the oxidative burden in the lungs of COPD.
In conclusion, we found that expression of Srx1, an important oxidative stress-inducible gene is regulated by Nrf2. Acute exposure to oxidative stress induces Srx1 expression through Nrf2 dependent pathway, which has a protective role in the anti-oxidative defense. However, extremely low level of Srx1 protein level in some severe COPD lung tissues indicates that chronic exposure of cigarette smoke can lead to a decline in Srx1 expression, which in turn, confers increased susceptibility to oxidative stress related diseases. Failure to maintain Srx1 transcript and protein levels in lung tissues from patients with COPD correlates with the decreased Nrf2 activity in these samples, suggesting an important functional relationship between decline in Nrf2 activity and Srx1 levels. Taken together, we present strong evidence that the Nrf2-dependent transcriptional regulation of Srx1 may play an important role in counteracting oxidative stress. Preservation of basal and inducible expression of Srx1, along with other Nrf2 regulated antioxidative enzymes, may help in maintaining the pulmonary antioxidant defense and protect against COPD.
Acknowledgments
This work was supported by NIH grants R01 HL081205 (SB), SCCOR P50 HL084945, P50 CA058184, P30ES03819, and young clinical innovator award (SB, AN) and young clinical scientist award to Anju Singh from Flight Attendant Medical Research Institute. We thank James Watkins III for providing frozen COPD tissues.
ABBREVIATIONS
- Srx1
Sulfiredoxin
- COPD
chronic obstructive pulmonary disorder
- CS
cigarette smoke
- TSS
transcription start site
- ARE
antioxidant response element
- RNAi
RNA interference
- shRNA
short hairpin RNA
- siRNA
short interfering RNA
- GSH
glutathione
- GCLc
γ-glutamyl cysteine synthetase catalytic subunit
- GCLm
γ-glutamyl cysteine synthetase modifier subunit
- GSR
glutathione reductase
- GST
glutathione-S-transferase
- ROS
reactive oxygen species
- CSE
Cigarette smoke extract
- TBHQ
tert-butyl hydroquinone
- KEAP1
Kelch-like ECH-associated protein 1
- Nrf2
nuclear factor erythroid-2 related factor 2
- GSH
glutathione
- GSR
glutathione reductase
- GST
glutathione-S-transferase
- PRDX
peroxiredoxin
- H2O2
hydrogen peroxide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Peltoniemi MJ, Rytila PH, Harju TH, Soini YM, Salmenkivi KM, Ruddock LW, Kinnula VL. Modulation of glutaredoxin in the lung and sputum of cigarette smokers and chronic obstructive pulmonary disease. Respir Res. 2006;7:133. doi: 10.1186/1465-9921-7-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 3.Findlay VJ, Tapiero H, Townsend DM. Sulfiredoxin: a potential therapeutic agent? Biomed Pharmacother. 2005;59:374–379. doi: 10.1016/j.biopha.2005.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rhee SG, Jeong W, Chang TS, Woo HA. Sulfiredoxin, the cysteine sulfinic acid reductase specific to 2-Cys peroxiredoxin: its discovery, mechanism of action, and biological significance. Kidney Int Suppl. 2007:S3–8. doi: 10.1038/sj.ki.5002380. [DOI] [PubMed] [Google Scholar]
- 5.Barnes PJ. Mediators of chronic obstructive pulmonary disease. Pharmacol Rev. 2004;56:515–548. doi: 10.1124/pr.56.4.2. [DOI] [PubMed] [Google Scholar]
- 6.Yoshida T, Tuder RM. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev. 2007;87:1047–1082. doi: 10.1152/physrev.00048.2006. [DOI] [PubMed] [Google Scholar]
- 7.Lopez AD, Shibuya K, Rao C, Mathers CD, Hansell AL, Held LS, Schmid V, Buist S. Chronic obstructive pulmonary disease: current burden and future projections. Eur Respir J. 2006;27:397–412. doi: 10.1183/09031936.06.00025805. [DOI] [PubMed] [Google Scholar]
- 8.Barnes PJ. Chronic obstructive pulmonary disease. N Engl J Med. 2000;343:269–280. doi: 10.1056/NEJM200007273430407. [DOI] [PubMed] [Google Scholar]
- 9.Rahman I, MacNee W. Role of oxidants/antioxidants in smoking-induced lung diseases. Free Radic Biol Med. 1996;21:669–681. doi: 10.1016/0891-5849(96)00155-4. [DOI] [PubMed] [Google Scholar]
- 10.Sandford AJ, Silverman EK. Chronic obstructive pulmonary disease. 1: Susceptibility factors for COPD the genotype-environment interaction. Thorax. 2002;57:736–741. doi: 10.1136/thorax.57.8.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li N, Alam J, Venkatesan MI, Eiguren-Fernandez A, Schmitz D, Di Stefano E, Slaughter N, Killeen E, Wang X, Huang A, Wang M, Miguel AH, Cho A, Sioutas C, Nel AE. Nrf2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals. J Immunol. 2004;173:3467–3481. doi: 10.4049/jimmunol.173.5.3467. [DOI] [PubMed] [Google Scholar]
- 12.Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, Yamamoto M, Kensler TW, Tuder RM, Georas SN, Biswal S. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med. 2005;202:47–59. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thimmulappa RK, Scollick C, Traore K, Yates M, Trush MA, Liby KT, Sporn MB, Yamamoto M, Kensler TW, Biswal S. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem Biophys Res Commun. 2006;351:883–889. doi: 10.1016/j.bbrc.2006.10.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cho HY, Jedlicka AE, Reddy SP, Kensler TW, Yamamoto M, Zhang LY, Kleeberger SR. Role of NRF2 in protection against hyperoxic lung injury in mice. Am J Respir Cell Mol Biol. 2002;26:175–182. doi: 10.1165/ajrcmb.26.2.4501. [DOI] [PubMed] [Google Scholar]
- 15.Aoki Y, Sato H, Nishimura N, Takahashi S, Itoh K, Yamamoto M. Accelerated DNA adduct formation in the lung of the Nrf2 knockout mouse exposed to diesel exhaust. Toxicol Appl Pharmacol. 2001;173:154–160. doi: 10.1006/taap.2001.9176. [DOI] [PubMed] [Google Scholar]
- 16.Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, Yamamoto M, Petrache I, Tuder RM, Biswal S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 2004;114:1248–1259. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goven D, Boutten A, Lecon-Malas V, Marchal-Somme J, Amara N, Crestani B, Fournier M, Leseche G, Soler P, Boczkowski J, Bonay M. Altered Nrf2/Keap1-Bach1 equilibrium in pulmonary emphysema. Thorax. 2008 doi: 10.1136/thx.2007.091181. [DOI] [PubMed] [Google Scholar]
- 18.Malhotra D, Thimmulappa R, Navas-Acien A, Sandford A, Elliott M, Singh A, Chen L, Zhuang X, Hogg J, Pare P, Tuder RM, Biswal S. Decline in NRF2 Regulated Antioxidants in COPD Lungs due to Loss of its Positive Regulator DJ-1. Am J Respir Crit Care Med. 2008 doi: 10.1164/rccm.200803-380OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Suzuki M, Betsuyaku T, Ito Y, Nagai K, Nasuhara Y, Kaga K, Kondo S, Nishimura M. Downregulated NF-E2-related Factor 2 in Pulmonary Macrophages of Aged Smokers and COPD Patients. Am J Respir Cell Mol Biol. 2008 doi: 10.1165/rcmb.2007-0424OC. [DOI] [PubMed] [Google Scholar]
- 20.Gao AC, Lou W, Ichikawa T, Denmeade SR, Barrett JC, Isaacs JT. Suppression of the tumorigenicity of prostatic cancer cells by gene(s) located on human chromosome 19p13.1–13.2. Prostate. 1999;38:46–54. doi: 10.1002/(sici)1097-0045(19990101)38:1<46::aid-pros6>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 21.Bondareva AA, Capecchi MR, Iverson SV, Li Y, Lopez NI, Lucas O, Merrill GF, Prigge JR, Siders AM, Wakamiya M, Wallin SL, Schmidt EE. Effects of thioredoxin reductase-1 deletion on embryogenesis and transcriptome. Free Radic Biol Med. 2007;43:911–923. doi: 10.1016/j.freeradbiomed.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003;43:233–260. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- 23.Adair-Kirk TL, Atkinson JJ, Griffin GL, Watson MA, Kelley DG, DeMello D, Senior RM, Betsuyaku T. Distal airways in mice exposed to cigarette smoke: Nrf2-regulated genes are increased in Clara cells. Am J Respir Cell Mol Biol. 2008;39:400–411. doi: 10.1165/rcmb.2007-0295OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425:980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 25.Vivancos AP, Castillo EA, Biteau B, Nicot C, Ayte J, Toledano MB, Hidalgo E. A cysteine-sulfinic acid in peroxiredoxin regulates H2O2-sensing by the antioxidant Pap1 pathway. Proc Natl Acad Sci U S A. 2005;102:8875–8880. doi: 10.1073/pnas.0503251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwak MK, Wakabayashi N, Itoh K, Motohashi H, Yamamoto M, Kensler TW. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J Biol Chem. 2003;278:8135–8145. doi: 10.1074/jbc.M211898200. [DOI] [PubMed] [Google Scholar]
- 27.Liu J, Xie Y, Ducharme DM, Shen J, Diwan BA, Merrick BA, Grissom SF, Tucker CJ, Paules RS, Tennant R, Waalkes MP. Global gene expression associated with hepatocarcinogenesis in adult male mice induced by in utero arsenic exposure. Environ Health Perspect. 2006;114:404–411. doi: 10.1289/ehp.8534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 29.Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, Herman JG, Baylin SB, Sidransky D, Gabrielson E, Brock MV, Biswal S. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3:e420. doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh A, Rangasamy T, Thimmulappa RK, Lee H, Osburn WO, Brigelius-Flohe R, Kensler TW, Yamamoto M, Biswal S. Glutathione peroxidase 2, the major cigarette smoke-inducible isoform of GPX in lungs, is regulated by Nrf2. Am J Respir Cell Mol Biol. 2006;35:639–650. doi: 10.1165/rcmb.2005-0325OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baker AF, Landowski T, Dorr R, Tate WR, Gard JM, Tavenner BE, Dragovich T, Coon A, Powis G. The antitumor agent imexon activates antioxidant gene expression: evidence for an oxidative stress response. Clin Cancer Res. 2007;13:3388–3394. doi: 10.1158/1078-0432.CCR-06-0873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JK, Gazdar AF, Hartigan J, Wu L, Liu C, Parmigiani G, Park BH, Bachman KE, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 33.Chang TS, Jeong W, Woo HA, Lee SM, Park S, Rhee SG. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J Biol Chem. 2004;279:50994–51001. doi: 10.1074/jbc.M409482200. [DOI] [PubMed] [Google Scholar]
- 34.Alley MC, Scudiero DA, Monks A, Hursey ML, Czerwinski MJ, Fine DL, Abbott BJ, Mayo JG, Shoemaker RH, Boyd MR. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1988;48:589–601. [PubMed] [Google Scholar]
- 35.Zang LY, Stone K, Pryor WA. Detection of free radicals in aqueous extracts of cigarette tar by electron spin resonance. Free Radic Biol Med. 1995;19:161–167. doi: 10.1016/0891-5849(94)00236-d. [DOI] [PubMed] [Google Scholar]
- 36.Rahman I, van Schadewijk AA, Crowther AJ, Hiemstra PS, Stolk J, MacNee W, De Boer WI. 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002;166:490–495. doi: 10.1164/rccm.2110101. [DOI] [PubMed] [Google Scholar]
- 37.Rahman I, Morrison D, Donaldson K, MacNee W. Systemic oxidative stress in asthma, COPD, and smokers. Am J Respir Crit Care Med. 1996;154:1055–1060. doi: 10.1164/ajrccm.154.4.8887607. [DOI] [PubMed] [Google Scholar]
- 38.Sun Y. Free radicals, antioxidant enzymes, and carcinogenesis. Free Radical Biology and Medicine. 1990;8:583–599. doi: 10.1016/0891-5849(90)90156-d. [DOI] [PubMed] [Google Scholar]
- 39.Rhee SG, Kang SW, Jeong W, Chang TS, Yang KS, Woo HA. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol. 2005;17:183–189. doi: 10.1016/j.ceb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 40.Rhee SG, Yang KS, Kang SW, Woo HA, Chang TS. Controlled elimination of intracellular H(2)O(2): regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxid Redox Signal. 2005;7:619–626. doi: 10.1089/ars.2005.7.619. [DOI] [PubMed] [Google Scholar]
- 41.Findlay VJ, Townsend DM, Morris TE, Fraser JP, He L, Tew KD. A novel role for human sulfiredoxin in the reversal of glutathionylation. Cancer Res. 2006;66:6800–6806. doi: 10.1158/0008-5472.CAN-06-0484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blackwood EM, Kadonaga JT. Going the distance: a current view of enhancer action. Science. 1998;281:61–63. doi: 10.1126/science.281.5373.60. [DOI] [PubMed] [Google Scholar]
- 43.Friling RS, Bergelson S, Daniel V. Two adjacent AP-1-like binding sites form the electrophile-responsive element of the murine glutathione S-transferase Ya subunit gene. Proc Natl Acad Sci U S A. 1992;89:668–672. doi: 10.1073/pnas.89.2.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Inamdar NM, Ahn YI, Alam J. The heme-responsive element of the mouse heme oxygenase-1 gene is an extended AP-1 binding site that resembles the recognition sequences for MAF and NF-E2 transcription factors. Biochem Biophys Res Commun. 1996;221:570–576. doi: 10.1006/bbrc.1996.0637. [DOI] [PubMed] [Google Scholar]
- 45.Tsuji Y, Ayaki H, Whitman SP, Morrow CS, Torti SV, Torti FM. Coordinate transcriptional and translational regulation of ferritin in response to oxidative stress. Mol Cell Biol. 2000;20:5818–5827. doi: 10.1128/mcb.20.16.5818-5827.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rushmore TH, King RG, Paulson KE, Pickett CB. Regulation of glutathione S-transferase Ya subunit gene expression: identification of a unique xenobiotic-responsive element controlling inducible expression by planar aromatic compounds. Proc Natl Acad Sci U S A. 1990;87:3826–3830. doi: 10.1073/pnas.87.10.3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jowsey IR, Jiang Q, Itoh K, Yamamoto M, Hayes JD. Expression of the aflatoxin B1-8,9-epoxide-metabolizing murine glutathione S-transferase A3 subunit is regulated by the Nrf2 transcription factor through an antioxidant response element. Mol Pharmacol. 2003;64:1018–1028. doi: 10.1124/mol.64.5.1018. [DOI] [PubMed] [Google Scholar]
- 48.Favreau LV, Pickett CB. Transcriptional regulation of the rat NAD(P)H:quinone reductase gene. Identification of regulatory elements controlling basal level expression and inducible expression by planar aromatic compounds and phenolic antioxidants. J Biol Chem. 1991;266:4556–4561. [PubMed] [Google Scholar]
- 49.Mulcahy RT, Wartman MA, Bailey HH, Gipp JJ. Constitutive and beta-naphthoflavone-induced expression of the human gamma-glutamylcysteine synthetase heavy subunit gene is regulated by a distal antioxidant response element/TRE sequence. J Biol Chem. 1997;272:7445–7454. doi: 10.1074/jbc.272.11.7445. [DOI] [PubMed] [Google Scholar]
- 50.Kim YJ, Ahn JY, Liang P, Ip C, Zhang Y, Park YM. Human prx1 gene is a target of Nrf2 and is up-regulated by hypoxia/reoxygenation: implication to tumor biology. Cancer Res. 2007;67:546–554. doi: 10.1158/0008-5472.CAN-06-2401. [DOI] [PubMed] [Google Scholar]
- 51.Nioi P, Hayes JD. Contribution of NAD(P)H:quinone oxidoreductase 1 to protection against carcinogenesis, and regulation of its gene by the Nrf2 basic-region leucine zipper and the arylhydrocarbon receptor basic helix-loop-helix transcription factors. Mutat Res. 2004;555:149–171. doi: 10.1016/j.mrfmmm.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 52.Kirkham P, Rahman I. Oxidative stress in asthma and COPD: antioxidants as a therapeutic strategy. Pharmacol Ther. 2006;111:476–494. doi: 10.1016/j.pharmthera.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 53.MacNee W. Oxidants/antioxidants and COPD. Chest. 2000;117:303S–317S. doi: 10.1378/chest.117.5_suppl_1.303s-a. [DOI] [PubMed] [Google Scholar]