

Abstract
Monoacylglycerol lipase (MAGL) and fatty acid amide hydrolase (FAAH) are two enzymes from the serine hydrolase superfamily that degrade the endocannabinoids 2-arachidonoylglycerol and anandamide, respectively. We have recently discovered that MAGL and FAAH are both inhibited by carbamates bearing an N-piperidine/piperazine group. Piperidine/piperazine carbamates show excellent in vivo activity, raising brain endocannabinoid levels and producing CB1-dependent behavioral effects in mice, suggesting that they represent a promising class of inhibitors for studying the endogenous functions of MAGL and FAAH. Herein, we disclose a full account of the syntheses, structure-activity relationships, and inhibitory activities of piperidine/piperazine carbamates against members of the serine hydrolase family. These scaffolds can be tuned for MAGL-selective or dual MAGL-FAAH inhibition by the attachment of an appropriately substituted bisarylcarbinol or aryloxybenzyl moiety, respectively, on the piperidine/piperazine ring. Modifications to the piperidine/piperazine ring ablated inhibitory activity, suggesting a strict requirement for a six-member ring to maintain potency.
The endogenous cannabinoid system is composed of the G-protein coupled receptors CB1 and CB2, their endogenous ligands anandamide (AEA) and 2-arachidonoylglycerol (2-AG) (the “endocannabinoids”), and the enzymes that biosynthesize and degrade endocannabinoids.1 The magnitude and duration of brain endocannabinoid signaling is tightly regulated by enzymatic hydrolysis, a process that involves distinct enzymes for each endocannabinoid. Fatty acid amide hydrolase (FAAH) is the principal hydrolytic enzyme for AEA.2 Genetic or pharmacological disruption of FAAH in rodents causes dramatic elevations (> 10-fold) in brain AEA levels and CB1-dependent analgesic responses in several acute and chronic pain models.3–5 Although several enzymes can hydrolyze 2-AG in vitro, this activity appears to be principally mediated by monoacylglycerol lipase (MAGL) in the rodent brain.6, 7 Recently, we described a highly selective and efficacious MAGL inhibitor 1 (JZL184, Figure 1), a piperidine carbamate that, upon administration to mice, reduces brain 2-AG hydrolysis activity, elevates total brain 2-AG levels by > 8-fold, and causes CB1-dependent hypomotility and analgesia.7, 8 We have also described 2 (JZL195, Figure 1), a dual FAAH-MAGL inhibitor based on a piperazine carbamate scaffold, that elevates total brain AEA and 2-AG to levels comparable to selective inhibitors of each individual enzyme.9 Interestingly, 2 produces strong antinociceptive and cataleptic effects that provide evidence for crosstalk between AEA and 2-AG signaling pathways in vivo. Concurrently with these studies, highly selective inhibitors of FAAH have been introduced that use a piperazine or piperidine urea to inactivate this enzyme. Examples include 3 (PF-622) and 4 (PF-3845) (Figure 1).5, 10 Collectively, these data indicate that piperazine/piperidine carbamates and ureas may represent privileged chemical scaffolds11, 12 for generating covalent, selective, and efficacious inhibitors of endocannabinoid hydrolases. Herein, we provide a full account of the discovery, synthesis, structure-activity relationships, and inhibitory activities of compounds that lead to the identification of 1 and 2, and show that, by attaching an appropriately substituted 4-bisarylcarbinol or 4-aryloxybenzyl moiety, the versatile piperidine/piperazine carbamate scaffold can be tuned to generate MAGL-selective or dual FAAH-MAGL inhibitors.
Figure 1.

Structures of the MAGL-selective inhibitor 1, the dual FAAH-MAGL inhibitor 2, and the FAAH-selective inhibitors 3 and 4.
Results and Discussion
Lead endocannabinoid hydrolase inhibitors discovered by competitive activity-based protein profiling (ABPP)
Both FAAH and MAGL are members of the serine hydrolase superfamily of enzymes that use a conserved serine nucleophile for catalysis.13–15 Our search for selective inhibitors of MAGL or dual inhibitors for FAAH-MAGL has therefore benefited from some unusual features of this family. First, the catalytic serine is susceptible to covalent inactivation by several electrophilic groups, including fluorophosphonates and carbamates, that show little cross-reactivity with other enzyme classes (Figure 2A).16–18 Carbamates are a privileged scaffold in this respect because their selectivity among members of the serine hydrolases can be tuned in two ways, either by modulation of the carbonyl electrophilicity (reactivity) or by modification of substituents distal to the reactive carbonyl (binding). A second attribute of serine hydrolases is that their activity state can be concurrently profiled in complex biological samples using the functional proteomic method activity-based protein profiling (ABPP).16, 19, 20 ABPP of serine hydrolases is typically performed using fluorophore- or biotin-conjugated fluorophosphonate probes [e.g., FP-rhodamine (FP-Rh) or FP-biotin] that covalently label serine hydrolases, which are then resolved by SDS-PAGE and detected by in-gel fluorescence scanning (for FP-Rh) or enriched by avidin chromatography and identified by liquid chromatography-mass spectrometry (for FP-biotin).21, 22 When performed in a competitive mode, where inhibitors are pre-incubated with cell or tissue proteomes prior to addition of FP probes, ABPP provides a highly versatile screen for serine hydrolase inhibitors.7, 23–26 Competitive ABPP has the important advantage of testing inhibitors against numerous serine hydrolases in parallel (i.e., all of the FP-reactive enzymes that are expressed in a given cell or tissue) and, therefore, offers a powerful way to concurrently optimize the potency and selectivity of inhibitors directly in native proteomes (Figure 2B).
Figure 2.

Competitive activity-based protein profiling (ABPP) for discovering lead carbamates that selectively inhibit MAGL. (A) Carbamates irreversibly inactivate serine hydrolases (shown as grey shapes) by carbamoylation of the nucleophilic serine. (B) Schematic depiction of competitive ABPP. Native tissue proteomes containing various serine hydrolases (grey shapes) are first incubated with a carbamate inhibitor (green box) and then with an ABPP probe such as fluorphosphonate conjugated rhodamine (FP-Rh, red circles). Separation and visualization of FP-Rh-labeled proteins by SDS-PAGE and in-gel fluorescence scanning, respectively, reveal the disappearance of bands that represent the serine hydrolase targets of the carbamate inhibitor (fluorescent gel shown in grayscale). (C) Structures of the lead compounds 5 and 6. (D) Competitive ABPP showing the concentration-dependent effects of 5 and 6 on serine hydrolases activities in the mouse brain membrane proteome. 5 and 6 were incubated with proteomes for 30 min at 37 °C before addition of FP-Rh. Note that MAGL migrates as several bands between 33 and 35 kDa, as reported previously.45
In our initial screen of a diverse carbamate library against the mouse brain membrane proteome, we identified two compounds (5 and 6) that showed good potency and promising selectivity for MAGL over the other serine hydrolases (Figure 2C, D and Table 1). At higher concentrations, both compounds also inhibited FAAH and a second brain 2-AG hydrolase, ABHD6.6 Compounds 5 and 6 were structurally similar, each containing a six-membered ring in the form of a piperidine or piperazine, an activated N-4-nitrophenoxy carbamate, and a bisarylcarbinol moiety distal to the electrophilic carbonyl. However, despite their 27- and 68-fold greater activity for MAGL over FAAH in brain proteomes (Table 1), compounds 5 and 6 were not sufficiently selective for in vivo studies, since their administration to mice at doses that reduced MAGL activity also significantly blocked FAAH activity (data not shown). We therefore undertook an effort to improve the selectivity of piperazine/piperidine carbamates for inhibiting MAGL.
Table 1.
In vitro activity of lead compounds. IC50’s are shown in nM unless otherwise indicated.
| Cmpd | FAAH | MAGL | ABHD6 | Selectivity |
|---|---|---|---|---|
| 5 | 5410 | 200 | 900 | 27 |
| 6 | 2660 | 40 | 2110 | 68 |
Structure-activity relationships of MAGL-selective inhibitors
We first asked whether the six-membered piperazine/piperidine ring was required for activity. Carbamates 9a and 9b, which maintained the N,N-dialkyl 4-nitrophenoxy carbamate moiety, but had unbranched alkyl linkers to a bisphenyl substituted terminal carbon, and carbamate 15, in which the piperidine was contracted into a pyrrolidine ring, were synthesized as outlined in Schemes 1 and 2. Carbamoylation of the commercially available secondary amines 7a and 7b using 4-nitrophenylchloroformate (8) afforded 9a and 9b. Addition of 4-methoxyphenylmagnesium bromide (11) to methyl 1-benzyl-5-oxopyrrolidine-3-carboxylate (10) in refluxing THF gave the amide 12, which was reduced to 13 using LiAlH4. Benzyl deprotection using standard Pd/C hydrogenation conditions to afford secondary amine 14, and finally carbamoylation afforded pyrrolidine carbamate 15. When 9a, 9b, and 15 were assessed by competitive ABPP against the mouse brain membrane proteome, they were found to be inactive against FAAH, MAGL, and other serine hydrolases (Table 2). These data therefore suggested that the 6-membered ring in compounds 5 and 6 was required for MAGL inhibition.
Scheme 1.

Synthesis of carbamates 9a and 9b.
Scheme 2.

Synthesis of carbamate 15.
Table 2.
In vitro activity of carbamates lacking a six-membered ring. IC50’s are shown in nM unless otherwise indicated.
| Cmpd | FAAH | MAGL | ABHD6 |
|---|---|---|---|
| 9a | >50 μM | >50 μM | >50 μM |
| 9b | >50 μM | >50 μM | >50 μM |
| 15 | >50 μM | >50 μM | >50 μM |
We next focused on making structural modifications to the portion of the molecules distal to the piperazine or piperidine ring. The synthesis of these compounds is outlined in Schemes 3–5. Carbamoylation under the usual conditions of commercially available diphenyl(piperidin-4-yl)methanol (16) afforded 17 (Scheme 3). Alternatively, the dibenzylic tertiary hydroxyl of 16 could be readily eliminated using TFA to afford 18, which was then converted to the corresponding carbamate 19 using chloroformate 8. Treatment of olefin 19 with mCPBA afforded epoxide 20 (Scheme 3). Compound 23, containing a fully saturated bisaryl to piperidine linker, was synthesized starting from commercially available 1-tert-butyl 4-ethyl piperidine-1,4-dicarboxylate 21 (Scheme 4). Addition of Grignard 11 under refluxing conditions afforded Boc-protected piperidine 22, which was then subjected to a three step procedure involving sequential treatment with TFA/CH2Cl2, which concomitantly removed the Boc group and eliminated the hydroxyl, Pd/C under the usual hydrogenation conditions, which saturated the incipient olefin, and chloroformate 8, to afforded the saturated piperidine carbamate 23. We also rigidified the bisaryl ring system by installing an ortho-oxygen bridge. This compound (29) was synthesized starting from xanthen-9-one 24, which was reduced with NaBH4 and chlorinated with SOCl2 to afford 26. Piperazine 28, which was made using a two step carbamoylation/deprotection sequence from Boc-piperazine, was used to alkylate 26 and afford 29.
Scheme 3.

Synthesis of carbamates 17, 19, and 20.
Scheme 5.

Synthesis of carbamate 29.
Scheme 4.

Synthesis of carbamate 23.
17, 19, 20, 23, and 29 were evaluated against brain membrane proteomes using competitive ABPP. Comparison of 17 and 6 revealed that the 4-methoxy group on the distal aryl rings conferred both additional potency and selectivity for MAGL (Table 3). However, rigidification of any portion of this distal substitutent, either by installation of an olefin (19) or epoxide (20) linker between the phenyl groups and the piperidine ring, or by covalently tethering the aryl rings with an oxygen bridge (29), reduced the activity and/or selectivity for MAGL in comparison to lead carbamates 5 and 6 (IC50 = 200 nM–6 μM for 19, 20 and 29, versus IC50 = 200 and 40 nM for 5 and 6, respectively). 23, in which the methanol linker of compound 17 was replaced with a methane, exhibited excellent selectivity for MAGL over FAAH (> 500-fold), despite being of slightly lower potency compared to lead compound 6 (IC50 = 70 nM versus 40 nM). Compound 23 provided the first evidence that it would be possible to achieve selectivity for MAGL over FAAH on the order of 300- to 1000-fold with the piperidine carbamate scaffold. We therefore continued to search for compounds that displayed this high degree of selectivity while also being more potent for MAGL than the original compound 6.
Table 3.
In vitro activity of carbamates modified with different linkers. IC50’s are shown in nM unless otherwise indicated.
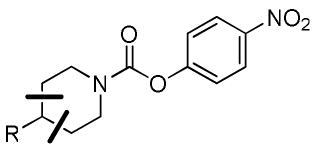 | |||||
|---|---|---|---|---|---|
| Cmpd | R = | FAAH | MAGL | ABHD6 | Selectivity |
| 17 | 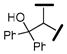 |
4340 | 690 | 1100 | 6 |
| 19 | 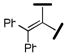 |
3500 | 1390 | 1300 | 3 |
| 20 | 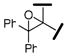 |
5850 | 200 | 380 | 29 |
| 23 | 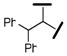 |
40080 | 73 | 550 | 549 |
| 29 | 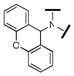 |
9000 | 6120 | 2120 | 2 |
Our next series of carbamates was motivated by the improved potency of 6 compared to 17, which suggested that further gains in MAGL active site binding interactions could be achieved by additional substitution and/or increasing the steric bulk of the arene rings. We reasoned that such modifications might have the further benefit of increasing the selectivity window for MAGL versus FAAH, since we hypothesized that these compounds’ selectivity was in part due to the inability of the sterically encumbering bisaryl groups to fit into the relatively narrow FAAH acyl-chain binding pocket.27 Since 6 remained at this point the most potent compound identified, we chose to maintain the piperidine carbinol scaffold while varying the aryl groups. Compounds 33a–f and 1 were prepared according to Scheme 6. Addition of variously substituted aryl groups to the ester of 1-benzyl 4-ethyl piperidine-1,4-dicarboxylate (30) was accomplished using aryl lithium anions at −78°C, which were generated by treating the appropriate aryl bromide with t-BuLi at the same temperature. The resultant Cbz-protected 4-bisarylcarbinol piperidine compounds 32a–h were deprotected by hydrogenation using Pd/C (32a–d, 32f–h) or aqueous KOH (32e) to give the crude amines, which were used directly in the carbamoylation reaction with 8 to afford the carbamates 33a–f.
Scheme 6.

Synthesis of carbamates 33a–f.
Evaluation of 33a and 33b by competitive ABPP showed that a single methoxy substituent at the 3- or 2-position of the distal aryl ring could be tolerated and that these compounds could still maintained good selectivity for MAGL (Table 4, ~10-fold selective). Remarkably, when we incorporated two oxygen substituents, either in the form of the 3,4-dimethoxybenzene (33c) or 3,4-methylenedioxybenzene (1), we achieved excellent selectivity for MAGL (~40–400-fold selective) without inhibiting any of the other serine hydrolases in mouse brain membranes as judged by our competitive ABPP gels. Compound 1, which was ultimately advanced to in vivo pharmacological and behavioral studies, maintained a comparable MAGL-selectivity window to 23 but gained a 7-fold improvement in potency. An additional advantage was that, unlike many other compounds in the piperidine/piperazine carbamate series, 1 exhibited low activity for ABHD6, another enzyme that has been shown to hydrolyze 2-AG in vitro.6 Further efforts to refine the aryl substitution patterns by expanding the steric bulk, by using 4-N,N-dimethylanilinebenzene (33d) or 2-naphthyl (33f) groups, or by modifying the aryl group electronics, with substituents such as 4-chlorobenzene (33e), did not produce more potent or more selective compounds. Since both larger (33c, 33d and 33f) and smaller (17) substituents reduce the potency of these piperidine carbamates for MAGL, we attribute, in part, the high potency and selectively of 1 to be due to its unique substitution pattern and steric properties, since the methylenedioxy group exploits two points of substitution along the arene ring while maintaining a relatively small Van der Waals volume.
Table 4.
In vitro activity of carbamates modified at the distal arene group. IC50’s are shown in nM unless otherwise indicated.
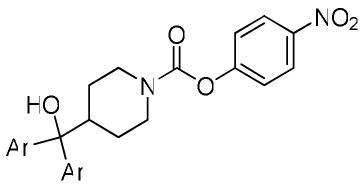 | |||||
|---|---|---|---|---|---|
| Cmpd | Ar = | FAAH | MAGL | ABHD6 | Selectivity |
| 33a | 5830 | 560 | 460 | 10 | |
| 33b | 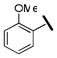 |
15100 | 510 | 1100 | 11 |
| 33c | >50 μM | 1300 | 6960 | >38 | |
| 1 | 4690 | 10 | 3270 | 426 | |
| 33d | 10540 | 2120 | 1020 | 5 | |
| 33e | 3360 | 1100 | 180 | 3 | |
| 33f | 4690 | 810 | 3430 | 6 | |
Tuning the piperazine carbamate scaffold to create dual FAAH-MAGL inhibitors
Our competitive ABPP screens revealed that many compounds of the piperidine/piperazine carbamate class with activity for MAGL also tended to inhibit a select number of additional serine hydrolases in the mouse brain proteome, principally FAAH and ABHD6. This suggested that, despite their low sequence homology, MAGL, FAAH, and ABHD6 share active site similarity, a hypothesis that is reinforced by the structural relatedness of some FAAH-selective (3 and 4) and MAGL-selective (1) inhibitors, which all shared an N-electrophile tethered to a piperidine or piperazine ring motif. Since 3 and 4 derive their selectivity for FAAH by containing a relatively unreactive urea electrophile, and 1 derives its selectivity for MAGL from the bulky piperidine bisarylcarbinol scaffold, we reasoned that an appropriate combination of 1 and 3 could potentially provide a dual FAAH-MAGL inhibitor.28, 29 In such a compound, the FAAH-selective urea electrophile would be replaced with a carbamate to enable inactivation of MAGL (4-nitrophenoxy carbamate), and the MAGL-selective bulky bisarylcarbinol motif would be substituted with a distal group that interacted with both the FAAH and MAGL active sites.
With these general concepts in mind, we targeted the piperazine p-nitrophenoxy carbamate as a scaffold that was both likely to yield a dual inhibitor and also amenable to facile N-modification (36a–q). These compounds were synthesized by one of two routes shown in Scheme 7. Boc-piperazine was alkylated by displacement of substituted benzyl bromides using K2CO3 in refluxing CH2Cl2, and the resultant products were deprotected and carbamoylated in the usual way to afford the products. Alternatively, alkylation could be accomplished by reductive amination of Boc-piperazine with aryl aldehydes using Na(OAc)3BH at room temperature.
Scheme 7.

Synthesis of carbamates 36a–q.
Our initial compound in this series, the 2-napthyl substituted carbamate 36a, showed good activity for FAAH and MAGL, but also inhibited another serine hydrolase, the 150-kDa neurpathy target esterase (NTE) (Figure 3).30, 31 Since the inhibition of NTE is believed to be responsible for the delayed onset mortality induced by organophosphate reagents and also lower limb paralysis in humans,32–34 we sought to reduce the potency of the piperazine carbamates for this target and pursued derivatives of 36a (Table 5). While many compounds of this series fully blocked labeling of NTE at high concentrations and also inhibited NTE when administered to mice (for example, compounds 36h at 20 mg/kg, i.p., data not shown), a small subset of three compounds showed only partially blocked NTE activity at high concentrations (100 μM, Table 6). Of these compounds, 2 was most promising since it showed the least inhibition towards NTE while maintaining excellent activity for MAGL and FAAH. Additional derivatives that also contained the N-3-aryloxybenzyl motif of 2, such as 36m–q, were also unable to fully block NTE activity at high doses (Table 7), suggesting that the poor inhibition of NTE exhibited by these compounds is due to the unique steric and/or electronic properties of the aryloxybenzyl substitution. That 3,5-dichlorobenzene phenyl ether 36q was unable to inhibit any of the hydrolases in our screen indicated that there is an upper limit to the steric bulk that can be appended to the distal arene ring. Thus, while the piperidine bisarylcarbinol scaffold results in MAGL-selective inhibition, a piperazine aryloxybenzyl scaffold can be used as a scaffold for the selective dual blockade of FAAH and MAGL.
Figure 3.

Concentration-dependent effects of 36a on serine hydrolases activities in the mouse brain membrane proteome as determined by competitive ABPP. 36a was incubated with proteomes at the indicated concentrations for 30 min at 37 °C before addition of FP-Rh probe.
Table 5.
In vitro activity of initial dual FAAH-MAGL inhibitors. IC50’s are shown in nM unless otherwise indicated.
 | |||||
|---|---|---|---|---|---|
| Cmpd | Ar = | FAAH | MAGL | ABHD6 | NTE |
| 36a | 380 | 70 | 150 | 1100 | |
| 36b | 1040 | 80 | 490 | 910 | |
| 36c | 930 | 900 | 740 | 610 | |
| 36d |  |
1050 | 1530 | 1270 | 480 |
| 36e | 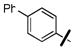 |
4600 | 15 | 1650 | 4270 |
| 36f | 3570 | 670 | 2860 | 330 | |
| 36g | 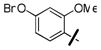 |
930 | 90 | 310 | 420 |
| 36h | 40 | 50 | 220 | 80 | |
| 36i | 280 | 10 | 1000 | 890 | |
| 36j | 20 | 90 | 220 | 250 | |
Table 6.
In vitro activity of dual FAAH-MAGL inhibitors that showed selectivity away from NTE. IC50’s are shown in nM unless otherwise indicated.
 | ||||||
|---|---|---|---|---|---|---|
| Cmpd | Ar = | FAAH | MAGL | ABHD6 | NTE | % inhibition of NTE at 100 μM |
| 36k | 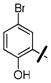 |
720 | 90 | 130 | 3240 | 73 |
| 36l | 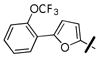 |
5 | 70 | 180 | 570 | 81 |
| 2 | 13 | 19 | 50 | >50 μM | 50 | |
Table 7.
In vitro activity of dual FAAH-MAGL inhibitors modified at the 3-aryloxybenzyl position. IC50’s are shown in nM unless otherwise indicated.
 | ||||||
|---|---|---|---|---|---|---|
| Cmpd | Ar = | FAAH | MAGL | ABHD6 | NTE | % inhibition of NTE at 100 μM |
| 36m | 40 | 500 | 1130 | 3000 | 70 | |
| 36n | 40 | 50 | 60 | 1270 | 73 | |
| 36o | 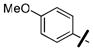 |
90 | 130 | 460 | 2900 | 60 |
| 36p | 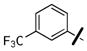 |
5 | 300 | 320 | 14400 | 57 |
| 36q | 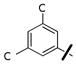 |
>100 μM | >100 μM | >100 μM | >100 μM | 0 |
Though 2 showed weak inhibition of NTE in vitro (~50% blockade at 100 μM), no inhibition of NTE was observed following in vivo administration to mice at doses that completely blocked FAAH and MAGL (20 mg/kg intraperitoneally, or 100 mg/kg orally), and chronic administration of 2 to mice (20 mg/kg i.p. daily for 6 days) did not produce death or overt signs of discomfort.9 That ABHD6 was also inhibited at higher concentrations suggests that 2 has the potential to function as a poly-pharmacology inhibitor for several endocannabinoid hydrolases (FAAH and two of the major brain 2-AG hydrolases, MAGL and ABHD6). The FAAH-selective inhibitor 4, the MAGL-selective inhibitor 1, and the dual FAAH-MAGL inhibitor 2 thus constitute a versatile arsenal of efficacious and selective pharmacological probes that can be used to interrogate the function of endocannabinoid-degrading enzymes in vivo.
Conclusions
Herein, we have described the discovery, synthesis, structure-activity relationships, and inhibitory activities of a series of piperidine/piperazine carbamates that function either as selective inhibitors of MAGL or dual inhibitors of FAAH and MAGL in vivo. The development of compounds such as 1 and 2 underscores the utility of competitive ABPP to concurrently optimize inhibitor potency and selectivity directly in native proteomes. Indeed, considering that NTE shares little to no sequence homology with either FAAH or MAGL,31 this common “off-target” of dual FAAH-MAGL inhibitors would likely have gone undetected without the broad and unbiased screening output provided by competitive ABPP. From a structure-activity perspective, our data suggest that the MAGL-selective inhibitors derive much of their selectivity from the piperidine ring as well as the bulky bisarylcarbinol motif distal to the electrophilic carbonyl. We have also demonstrated that, by rationally changing the substitution at the 4-position of the 6-membered ring, from bisarylcarbinol to 3-aryloxybenzyl, we can selectively tune the inhibitory properties of the piperidine/piperazine carbamate scaffold from being a MAGL-selective inhibitor to a dual inhibitor of MAGL and FAAH.
Since their disclosure, 1 and 2 have proven useful for selectively enhancing 2-AG signaling or concurrently augmenting both 2-AG and anandamide signaling, respectively, in a variety of in vitro and in vivo systems.35–39 Nevertheless, we anticipate that even within this general chemical class there remains much potential for improvements in the potency and selectivity of future MAGL-selective inhibitors or dual FAAH-MAGL inhibitors. For instance, compound 36e, while originally designed to be a dual FAAH-MAGL inhibitor, in fact displayed remarkable selectivity for MAGL over FAAH. We attribute this unique property of 36e to the rigid and linear biphenyl group, and speculate that this compound might serve as a lead for the development of next generation MAGL inhibitors with a scaffold distinct from the bisarylcarbinol of 1. Moreover, we elected to use a 4-nitrophenoxy carbamate since our initial structure-activity relationship efforts indicated that MAGL inhibition requires an activated leaving group. However, we hypothesize that, similar to the urea series of FAAH inhibitors,5 hydroxypyridine leaving groups and derivatives thereof may also be sufficiently activated for MAGL inhibition, and that incorporation of additional substitutions on the pyridine ring might offer a new way to tune the selectivity window of these inhibitors for MAGL versus FAAH. Recently, two crystal structures of human MAGL have been solved, one of which contained the piperazine inhibitor SAR629 bound to the catalytic serine of MAGL.40, 41 These crystal structures may help in the design of future irreversible or reversible MAGL inhibitors. Lastly, piperazine carbamates have been also reported to act as dual FAAH inhibitors/transient receptor potential V1 (TRPV1) antagonists.42 That TRP channels can also be activated by endogenous FAAH substrates such as AEA43 or the N-acyl taurines44 suggests a high degree of homology between the binding pockets of these enzymes, and furthermore indicates that the piperidine/piperazine scaffold could potentially be exploited to generate polypharmacological tools that interact with MAGL, FAAH, and members of the TRP channel family.
Experimental Section
Chemistry
General methods
All reagents were purchased from Sigma-Aldrich, Acros, Fisher, Fluka, or Maybridge and used without further purification, except where noted. Dry solvents were obtained by passing commercially available pre-dried, oxygen-free formulations through activated alumina columns. All reactions were carried out under a nitrogen atmosphere using oven-dried glassware unless otherwise noted. Flash chromatography was performed using 230–400 mesh silica gel. NMR spectra were recorded in CDCl3 on a Varian Inova-400 or a Bruker DMX-600 spectrometer and were referenced to trimethylsilane (TMS) or the residual solvent peak. Chemical shifts are reported in ppm relative to TMS and J values are reported in Hz. High resolution mass spectrometry (HRMS) experiments were performed at The Scripps Research Institute Mass Spectrometry Core on an Agilent mass spectrometer using electrospray ionization-time of flight (ESI-TOF). LC/MS analysis was performed on an Agilent 1100 LC/MS instrument, using a ZORBAX SB-C18, 3.5 mm, 4.6 × 50 column, a flow rate of 0.75 ml/min, detection at 220 and 254 nm, and a 10–98% acetonitrile/water/0.1% formic acid gradient and a 50–98% acetonitrile/water/0.1% formic acid gradient. The purity of compounds (≥ 95%) was confirmed by clean NMR spectra and elution as a single peak by LC/MS. Compounds 1–6 have been described previously.5, 7, 9, 10
4-nitrophenyl 3,3-diphenylpropyl(methyl)carbamate (9a)
General Procedure A
To a stirring solution of N-methyl-3,3-diphenylpropan-1-amine (70 mg, 0.27 mmol) in CH2Cl2 (3 ml) was sequentially added triethylamine (0.1 ml, 0.7 mmol) and 4-nitrochloroformate (143 mg, 0.71 mmol). After 2 h, TLC indicated complete consumption of the starting material. The reaction was diluted with CH2Cl2 and poured onto saturated aqueous Na2CO3. The organic layer was washed thrice with 2N NaOH, once with brine, dried over Na2SO4, and concentrated in vacuo. Purification of the crude oil by flash chromatography (1–2% MeOH in CH2Cl2) gave 9a (52 mg, 49% yield): 1H NMR (CDCl3, 400 MHz) δ 8.09 (d, J = 9.2 Hz, 2H), 7.28-7.16 (m, 10H), 6.90 (d, J = 9.2 Hz, 2H), 3.78 (t, J = 7.8 Hz, 2H), 3.12 (t, J = 7.8 Hz, 2H), 2.81 (s, 3H), 2.31 (m, 2H). MS (ESI+) m/z 391 [M+H]+. HRMS calculated for C23H23N2O4 [M+H]+ 391.1652, found 391.1650.
4-nitrophenyl benzhydryl(methyl)carbamate (9b)
Prepared according to General Procedure A, using N-methyl-1,1-diphenylmethanamine (280 mg, 1.42 mmol), 4-nitrochloroformate (313 mg, 1.56 mmol), triethylamine (1 ml, 7 mmol), and CH2Cl2 (10 ml). Purification of the crude oil by flash chromatography (12:1 Hex:EtOAc) gave 9b (201 mg, 39% yield): 1H NMR (CDCl3, 400 MHz) δ 8.24 (d, J = 8.1 Hz, 2H), 7.42-7.25 (m, 12H), 6.73 (s, 1H), 2.93 (s, 3H). HRMS calculated for C21H19N2O4 [M+H]+ 363.1339, found 363.1364.
1-benzyl-4-(hydroxybis(4-methoxyphenyl)methyl)pyrrolidin-2-one (12)
To a stirring solution of methyl 1-benzyl-5-oxopyrrolidine-3-carboxylate (494 mg, 2.12 mmol) in dry ether (10 ml) was added 4-methoxyphenylmagesium bromide (0.5M in THF, 10 ml, 5 mmol). The reaction was heated to reflux and TLC indicated completion consumption of the starting material after 12 h. The reaction was diluted with CH2Cl2 and poured onto saturated aqueous Na2CO3. The organic layer was washed once with brine, dried over Na2SO4, and concentrated in vacuo. Purification of the crude oil by flash chromatography (1:1 Hex:EtOAc) gave 12 (190 mg, 21% yield): 1H NMR (CDCl3, 400 MHz) δ 7.31-7.19 (m, 7H), 7.15 (d, J = 7.4 Hz, 2H), 6.81-6.64 (m, 4H), 4.43 (d, J = 14.8 Hz, 1H), 4.11 (d, J = 15.3 Hz, 1H), 3.70 (s, 6H), 3.48-3.33 (m, 1H), 3.23 (dd, J = 10.0, 6.3 Hz, 1H), 3.08 (t, J = 9.5 Hz, 1H), 2.45 (ddd, J = 27.2, 17.5, 8.5 Hz, 2H). MS (ESI+) m/z 440 (M+Na)+.
(1-benzylpyrrolidin-3-yl)bis(4-methoxyphenyl)methanol (13)
To a −78°C stirring solution of 12 (185 mg, 0.44 mmol) in dry ether:CH2Cl2 (4:1 v/v, 25 ml total) was added LiAlH4 (4M in ether, 0.5 ml, 2 mmol). The dry ice bath was removed and the reaction was heated to reflux. After 2 h, TLC indicated complete consumption of the starting material. The reaction was diluted with CH2Cl2 and poured onto water. The organic layer was washed once with brine, dried over Na2SO4, and concentrated in vacuo. The crude oil was passed through a pad of silica to afford 13 (160 mg, 90% yield): 1H NMR (CDCl3, 400 MHz) δ 7.30-7.15 (m, 9H), 6.78-6.52 (m, 4H), 3.65 (d, J = 5.2 Hz, 6H), 3.52 (d, J = 12.8 Hz, 1H), 3.41 (d, J = 12.9 Hz, 1H), 3.16-3.08 (m, 1H), 2.95-2.87 (m, 1H), 2.77 (d, J = 9.5 Hz, 1H), 2.16 (dd, J = 9.5, 6.5 Hz, 1H), 2.03 (q, J = 8.9 Hz, 1H), 1.91-1.74 (m, 2H). MS (ESI+) m/z 404 [M+H]+.
4-nitrophenyl 3-(hydroxybis(4-methoxyphenyl)methyl)pyrrolidine-1-carboxylate (15)
General Procedure B
To a stirring solution of the 13 (90 mg, 0.22 mmol) in EtOH (5 ml) was added 10% Pd/C (20 mg) and H2 gas was bubbled through the reaction. After 4 h, TLC indicated complete consumption of the starting material. The reaction was diluted with CH2Cl2, filtered over a pad of Celite, and concentrated in vacuo. The crude was taken up in CH2Cl2 (10 ml) and triethylamine (1 ml, 7 mmol) and 4-nitrochloroformate (80 mg, 0.4 mmol) were sequentially added. After 2 h, TLC indicated complete consumption of the starting material. The reaction was diluted with CH2Cl2 and poured onto saturated aqueous Na2CO3. The organic layer was washed thrice with 2N aqueous NaOH, once with brine, dried over Na2SO4, and concentrated in vacuo. Purification of the crude oil by flash chromatography (6:1 Hex:EtOAc) gave the 15 (30 mg, 28% yield over two steps): 1H NMR (CDCl3, 400 MHz) δ 8.28-8.17 (m, 2H), 7.40-7.22 (m, 6H), 6.85 (dd, J = 12.5, 5.8 Hz, 4H), 3.78 (dd, J = 3.2, 1.3 Hz, 6H), 3.71-3.30 (m, 4H), 2.34 (d, J = 14.1 Hz, 1H), 2.01-1.89 (m, 2H). HRMS calculated for C26H26N2NaO7 [M+Na]+ 501.1632, found 508.1681.
4-nitrophenyl 4-(hydroxydiphenylmethyl)piperidine-1-carboxylate (17)
Prepared according to General Procedure A, using 4-benzhydrylpiperidine (192 mg, 0.72 mmol), 4-nitrochloroformate (230 mg, 1.1 mmol), triethylamine (0.2 ml, 1.4 mmol), and CH2Cl2 (10 ml). Purification of the crude oil by flash chromatography (2–3% MeOH in CH2Cl2) gave 17 (100 mg, 32% yield): 1H NMR (CDCl3, 400 MHz) δ 8.22 (d, J = 9.1 Hz, 2H), 7.47 (d, J = 8.0 Hz, 4H), 7.35-7.22 (m, 8H), 4.30 (bs, 2H), 3.02 (t, J = 12.5 Hz, 1H), 2.89 (t, J = 12.2 Hz, 1H), 2.64 (m, 1H), 1.64-1.60 (m, 2H), 1.47 (m, 2H). HRMS calculated for C25H24N2NaO5 [M+Na]+ 455.1577, found 455.1586.
4-(diphenylmethylene)piperidine (18)
To a stirring solution of 4-benzhydrylpiperidine (1.77 g, 6.7 mmol) in CH2Cl2 (15 ml) was added TFA (5 ml). After 7 h, TLC indicated complete consumption of the starting material. The reaction was concentrated in vacuo and then diluted with CH2Cl2 and saturated aqueous Na2CO3. The aqueous layer was extracted twice with CH2Cl2 and the combined organic layers were dried over Na2SO4 and concentrated in vacuo. The crude was passed through a pad of silica (5% MeOH in CH2Cl2) to give 18 (1.6 g, 96% yield): 1H NMR (CDCl3, 400 MHz) δ 7.34-7.23 (m, 6H), 7.11-7.08 (m, 4H), 3.24 (bs, 4H), 2.64 (m, 4H). MS (ESI+) m/z 250 [M+H]+.
4-nitrophenyl 4-(diphenylmethylene)piperidine-1-carboxylate (19)
Prepared according to General Procedure A, using 18 (182 mg, 0.73 mmol), 4-nitrochloroformate (142 mg, 0.71 mmol), triethylamine (0.3 ml, 2.1 mmol), and CH2Cl2 (10 ml). The crude product was passed through a pad of silica (CH2Cl2) to afford 19 (281 mg, 93% yield): 1H NMR (CDCl3, 400 MHz) δ 8.23 (d, J = 9.3 Hz, 1H), 7.36-7.09 (m, 12H), 3.69 (m, 2H), 3.61 (m, 2H), 2.47 (m, 4H). HRMS calculated for C25H25N2O4 [M+H]+ 417.1808, found 417.1761.
4-nitrophenyl 2,2-diphenyl-1-oxa-6-azaspiro[2.5]octane-6-carboxylate (20)
To a stirring solution of 19 (23 mg, 0.056 mmol) in CH2Cl2 (5 ml) was added mCPBA (< 72%, 49 mg, < 0.2 mmol). After 2 h, TLC indicated complete consumption of the starting material, and a saturated aqueous solution of Na2S2O3 (1 ml) was added to the reaction. After stirring for 30 min, the reaction was diluted with EtOAc and the organic layer was washed twice with water, once with saturated aqueous Na2CO3, once with brine, dried over Na2SO4 and concentrated in vacuo. The crude was passed through a pad of silica to afford 20 (15 mg, 63% yield): 1H NMR (CDCl3, 400 MHz) δ 8.24 (d, J = 9.2 Hz, 2H), 7.47 (d, J = 7.6 Hz, 2H), 7.39-7.22 (m, 8H), 4.03 (m, 2H), 3.51 (t, J = 11 Hz, 1H), 3.39 (t, J = 11 Hz, 1H), 1.87 (m, 2H), 1.38 (d, J = 13.8 Hz, 1H). HRMS calculated for C25H23N2O5 [M+H]+ 431.1602, found 431.1626.
tert-butyl 4-(hydroxybis(4-methoxyphenyl)methyl)piperidine-1-carboxylate (22)
To a 0°C stirring solution of 1-tert-butyl 4-ethyl piperidine-1,4-dicarboxylate (1.16 g, 4.5 mmol) in dry THF (10 ml) was added 4-methoxyphenylmagnesium bromide (26 ml, 13 mmol) dropwise. After the reagent addition was complete, the ice bath was removed and the reaction was heated to 60°C. TLC indicated complete consumption of the starting material after 9 h. The reaction was diluted with EtOAc and poured onto water. The organic layer was washed once with water, once with brine, dried over Na2SO4 and concentrated in vacuo. Purification of the crude oil by flash chromatography (12:1 then 3:1 Hex:EtOAc) gave 22 (1.6 g, 82% yield): 1H NMR (CDCl3, 400 MHz) δ 7.34 (d, J = 8.8 Hz, 2H), 7.34 (d, J = 8.8 Hz, 2H), 4.11 (s, 2H), 3.75 (s, 6H), 2.68 (t, J = 11.7 Hz, 2H), 2.44 (dd, J = 13.2, 10.5 Hz, 1H), 2.16 (s, 1H), 1.60-1.45 (m, 2H), 1.42 (s, 9H), 1.33-1.19 (m, 2H). MS (ESI+) m/z 410 (M-H2O+H)+.
4-nitrophenyl 4-(hydroxybis(4-methoxyphenyl)methyl)piperidine-1-carboxylate (23)
To a stirring solution of 22 (90 mg, 0.21 mmol) in CH2Cl2 (2 ml) was added TFA (2 ml). The reaction turned red and vigorous bubbling ensued. After 1 h, TLC indicated complete consumption of the starting material. Saturated aqueous Na2CO3 was added until the reaction was completely neutralized, and the aqueous layer was extracted thrice with CH2Cl2. The combined organic layers were washed once with brine, dried over Na2SO4 and concentrated in vacuo. The crude was taken up in EtOH (3 ml) and 10% Pd/C (100 mg) was added. After 12 h, the reaction was diluted with CH2Cl2, filtered over a pad of Celite, and concentrated in vacuo. The crude was taken up in CH2Cl2 (5 ml) and triethylamine (0.15 ml, 1.1 mmol) and 4-nitrochloroformate (66 mg, 0.33 mmol) were sequentially added. After 2 h, TLC indicated complete consumption of the starting material. The reaction was diluted with CH2Cl2 and poured onto saturated aqueous Na2CO3. The organic layer was washed once with brine, dried over Na2SO4, and concentrated in vacuo. Purification of the crude oil by flash chromatography (12:1 then 3:1 Hex:EtOAc) gave 23 (16 mg, 15% yield over three steps): 1H NMR (CDCl3, 400 MHz) δ 8.25 (d, J = 9.2 Hz, 2H), 8.25 (d, J = 9.2 Hz, 2H), 7.09-6.99 (m, 4H), 6.90-6.78 (m, 4H), 3.80 (s, 6H), 3.68 (m, 2H), 3.61 (m, 2H), 2.52-2.43 (m, 4H). HRMS calculated for C27H29N2O6 [M+H]+ 477.2020, found 477.2054.
benzyl 4-(hydroxybis(3-methoxyphenyl)methyl)piperidine-1-carboxylate (32a)
General Procedure C
To a −78°C stirring solution of 1-bromo-3-methoxybenzene (980 mg, 5.2 mmol) in dry THF (10 ml) was added t-BuLi (1.7M in pentane, 3 ml, 5.1 mmol) dropwise. After 30 min, a solution of 30 (300 mg, 1.03 mmol) in dry THF (3 ml) was added to the reaction. After 4 h of stirring at the same temperature, TLC indicated complete consumption of the starting material. The reaction was diluted with CH2Cl2 and poured onto water. The organic layer was washed twice with water, once with brine, dried over Na2SO4, and concentrated in vacuo. Purification of the crude oil by flash chromatography (10:1 then 4:1 Hex:EtOAc) gave 32a (290 mg, 61% yield): 1H NMR (CDCl3, 400 MHz) δ 7.37-7.26 (m, 5H), 7.20 (t, J = 8.0 Hz, 2H), 7.09-6.99 (m, 4H), 6.71 (ddd, J = 8.2, 2.5, 0.7 Hz, 2H), 5.06 (s, 2H), 4.20 (bs, 2H), 3.75 (s, 6H), 2.77 (bs, 2H), 2.61-2.43 (m, 1H), 2.31 (s, 1H), 1.51 (s, 1H), 1.34 (qd, J = 12.6, 4.4 Hz, 1H). MS (ESI+) m/z 444 (M-H2O+H)+.
benzyl 4-(hydroxybis(2-methoxyphenyl)methyl)piperidine-1-carboxylate (32b)
Prepared according to General Procedure C, using 1-bromo-2-methoxybenzene (980 mg, 5.2 mmol), 1-benzyl 4-ethyl piperidine-1,4-dicarboxylate (280 mg, 0.96 mmol), t-BuLi (1.7M in pentane, 3 ml, 5.1 mmol), and dry THF (20 ml). Purification of the crude oil by flash chromatography (10:1 then 4:1 Hex:EtOAc) gave 32b (210 mg, 49% yield): 1H NMR (CDCl3, 400 MHz) δ 7.66 (d, J = 7.1 Hz, 2H), 7.41-7.26 (m, 5H), 7.20 (ddd, J = 8.1, 7.4, 1.7 Hz, 2H), 7.02 (td, J = 7.7, 1.1 Hz, 2H), 6.78 (dd, J = 8.2, 1.0 Hz, 2H), 5.14 (s, 2H), 4.24 (bs, 2H), 3.51 (s, 6H), 3.03 (t, J = 10.8 Hz, 1H), 2.85 (bs, 2H), 1.74-1.40 (m, 4H). MS (ESI+) m/z 444 (M-H2O+H)+.
benzyl 4-(bis(3,4-dimethoxyphenyl)(hydroxy)methyl)piperidine-1-carboxylate (32c)
Prepared according to General Procedure C, using 4-bromo-1,2-dimethoxybenzene (485 mg, 2.2 mmol), 1-benzyl 4-ethyl piperidine-1,4-dicarboxylate (215 mg, 0.74 mmol), t-BuLi (1.7M in pentane, 1.2 ml, 2.0 mmol), and dry THF (10 ml). Purification of the crude oil by flash chromatography (6:1 Hex:EtOAc) gave 32c (50 mg, 13% yield): 1H NMR (CDCl3, 400 MHz) δ 7.40-7.28 (m, 5H), 7.00 (s, 2H), 6.94 (d, J = 8.4 Hz, 2H), 6.80 (d, J = 8.4 Hz, 2H), 5.10 (s, 2H), 4.23 (bs, 2H), 3.85 (s, 6H), 3.84 (s, 6H), 2.78 (bs, 2H), 2.45 (t, J = 11.9 Hz, 1H), 1.70-1.47 (m, 2H), 1.38-1.15 (m, 2H). MS (ESI+) m/z 444 (M+Na)+.
benzyl 4-(bis(4-(dimethylamino)phenyl)(hydroxy)methyl)piperidine-1-carboxylate (32d)
Prepared according to General Procedure C, using 4-bromo-N,N-dimethylaniline (890 mg, 4.5 mmol), 1-benzyl 4-ethyl piperidine-1,4-dicarboxylate (220 mg, 0.75 mmol), t-BuLi (1.7M in pentane, 2.5 ml, 4.3 mmol), and dry THF (20 ml). Purification of the crude oil by flash chromatography (4:1 Hex:EtOAc) gave 32d (210 mg, 57% yield): 1H NMR (CDCl3, 400 MHz) δ 7.39-7.19 (m, 9H), 6.65 (d, J = 8.8 Hz, 4H), 5.08 (s, 2H), 4.20 (bs, 2H), 2.90 (s, 12H), 2.81-2.71 (m, 2H), 2.45 (dd, J = 13.3, 10.4 Hz, 1H), 1.63 (bs, 2H), 1.34-1.18 (m, 2H). MS (ESI+) m/z 470 (M-H2O+H)+.
benzyl 4-(bis(4-chlorophenyl)(hydroxy)methyl)piperidine-1-carboxylate (32e)
Prepared according to General Procedure C, using 1-bromo-4-chlorobenzene (920 mg, 4.8 mmol), 1-benzyl 4-ethyl piperidine-1,4-dicarboxylate (220 mg, 0.75 mmol), t-BuLi (1.7M in pentane, 2.7 ml, 4.6 mmol), and dry ether (15 ml). Purification of the crude oil by flash chromatography (6:1 Hex:EtOAc) gave 32e (220 mg, 63% yield): 1H NMR (CDCl3, 400 MHz) δ 7.46-7.18 (m, 13H), 5.01 (d, J = 9.2 Hz, 2H), 4.19 (bs, 2H), 2.80-2.64 (m, 2H), 2.55-2.38 (m, 2H), 1.54-1.20 (m, 4H). MS (ESI+) m/z 452 (M-H2O+H)+.
benzyl 4-(hydroxydinaphthalen-2-ylmethyl)piperidine-1-carboxylate (32f)
Prepared according to General Procedure C, using 2-bromonaphthalene (300 mg, 1.45 mmol), 1-benzyl 4-ethyl piperidine-1,4-dicarboxylate (106 mg, 0.36 mmol), t-BuLi (1.7M in pentane, 0.6 ml, 1.1 mmol), and dry THF (6 ml). Purification of the crude oil by flash chromatography (4:1 Hex:EtOAc) gave 32f (79 mg, 44% yield): 1H NMR (CDCl3, 400 MHz) δ 8.04 (m, 2H), 7.74-7.84 (m, 6H), 7.56-7.43 (m, 6H), 7.31-7.25 (m, 5H), 5.08 (s, 2H), 4.24 (bs, 2H), 2.98-2.81 (m, 3H), 1.61-1.39 (m, 2H), 1.34-1.20 (m, 2H). MS (ESI+) m/z 524 (M+Na)+.
4-nitrophenyl 4-(hydroxybis(3-methoxyphenyl)methyl)piperidine-1-carboxylate (33a)
Prepared according to General Procedure B, using 32a (290 mg, 0.63 mmol), 10% Pd/C (70 mg), EtOH (4 ml) in the first step and CH2Cl2 (10 ml), triethylamine (1 ml, 7 mmol), and 4-nitrochloroformate (150 mg, 0.75 mmol) in the second step. Purification of the crude oil by flash chromatography (5:1 then 2:1 Hex:EtOAc) gave the product (220 mg, 73% yield over two steps): 1H NMR (CDCl3, 400 MHz) δ 8.22 (d, J = 9.1 Hz, 2H), 7.33-7.18 (m, 4H), 7.11-7.00 (m, 4H), 6.75 (dd, J = 8.2, 2.4 Hz, 2H), 4.29 (m, 2H), 3.79 (s, 6H), 3.01 (t, J = 12.0 Hz, 1H), 2.88 (t, J = 12.2 Hz, 1H), 2.59 (tt, J = 11.9, 3.3 Hz, 1H), 2.22-2.12 (m, 1H), 1.69-1.55 (m, 2H), 1.53-1.40 (m, 2H). HRMS calculated for C27H28N2NaO7 [M+Na]+ 515.1789, found 515.1829.
4-nitrophenyl 4-(hydroxybis(2-methoxyphenyl)methyl)piperidine-1-carboxylate (33b)
Prepared according to General Procedure B, using 32b (210 mg, 0.47 mmol), 10% Pd/C (50 mg), EtOH (5 ml) in the first step and CH2Cl2 (10 ml) and triethylamine (1 ml, 7 mmol) and 4-nitrochloroformate (170 mg, 0.85 mmol) in the second step. Purification of the crude oil by flash chromatography (8:1 then 2:1 Hex:EtOAc) gave the product (150 mg, 66% yield over two steps): 1H NMR (CDCl3, 400 MHz) δ 8.24 (d, J = 9.3 Hz, 2H), 7.65 (d, J = 6.0 Hz, 2H), 7.29 (d, J = 9.3 Hz, 2H), 7.21 (ddd, J = 8.1, 7.4, 1.7 Hz, 2H), 7.08-6.95 (m, 2H), 6.80 (dd, J = 8.2, 0.9 Hz, 2H), 4.28 (t, J = 10.8 Hz, 2H), 3.54 (s, 6H), 3.13-2.98 (m, 2H), 2.98-2.85 (m, 1H), 1.82-1.52 (m, 4H). HRMS calculated for C27H28N2NaO7 [M+Na]+ 515.1789, found 515.1837.
4-nitrophenyl 4-(bis(3,4-dimethoxyphenyl)(hydroxy)methyl)piperidine-1-carboxylate (33c)
Prepared according to General Procedure B, using 32c (50 mg, 0.1 mmol), 10% Pd/C (50 mg), EtOH:CH2Cl2 (4:1 v/v, 10 ml total) in the first step and CH2Cl2 (10 ml), triethylamine (1 ml, 7 mmol), and 4-nitrochloroformate (51 mg, 0.25 mmol) in the second step. Purification of the crude oil by flash chromatography (3:1 Hex:EtOAc) gave 33c (20 mg, 38% yield over two steps): 1H NMR (CDCl3, 400 MHz) δ 8.24 (d, J = 9.2 Hz, 2H), 7.35-7.19 (m, 2H), 7.02 (d, J = 1.8 Hz, 2H), 6.97 (d, J = 8.4 Hz, 2H), 6.83 (d, J = 8.4 Hz, 2H), 4.31 (bs, 1H), 3.87 (s, 6H), 3.86 (s, 6H), 3.02 (t, J = 12.4 Hz, 1H), 2.89 (t, J = 13.0 Hz, 1H), 2.54 (t, J = 11.7 Hz, 1H), 1.79-1.60 (m, 2H), 1.46-1.41 (m, J = 11.9 Hz, 2H). HRMS calculated for C29H32N2NaO9 [M+Na]+ 575.2000, found 575.2015.
4-nitrophenyl 4-(bis(4-(dimethylamino)phenyl)(hydroxy)methyl)piperidine-1-carboxylate (33d)
Prepared according to General Procedure B, using 32d (40 mg, 0.08 mmol), 10% Pd/C (50 mg), EtOH:CH2Cl2 (4:1 v/v, 5 ml total) in the first step and CH2Cl2 (8 ml), triethylamine (1 ml, 7 mmol), and 4-nitrochloroformate (40 mg, 0.2 mmol) in the second step. Purification of the crude oil by flash chromatography (6:1 Hex:EtOAc) gave 33d (14 mg, 35% yield over two steps): 1H NMR (CDCl3, 400 MHz) δ 8.22 (d, J = 9.2 Hz, 2H), 7.27 (m, 6H), 6.68 (d, J = 8.9 Hz, 4H), 4.27 (bs, 1H), 3.13-2.71 (m, 14H), 2.53 (t, J = 11.9 Hz, 1H), 1.72 (d, J = 11.9 Hz, 2H), 1.39 (q, J = 12.2 Hz, 2H). HRMS calculated for C29H33N4O4 [M-H2O+H]+ 501.2496, found 501.2543.
4-nitrophenyl 4-(bis(4-chlorophenyl)(hydroxy)methyl)piperidine-1-carboxylate (33e)
To a stirring solution of 32e (90 mg, 0.19 mmol) in water:EtOH (1:3 v/v, 20 ml total) was added KOH (1.0 g, 18 mmol) and the reaction was heated to reflux. After stirring overnight, TLC the next morning indicated complete consumption of the starting material. The reaction was diluted with CH2Cl2 and poured onto water. The organic layer was partitioned and the aqueous layer was extracted thrice with CH2Cl2. The combined organic layers were washed once with brine, dried over Na2SO4, and concentrated in vacuo. The crude product was taken up in CH2Cl2 (10 ml) and to the solution was sequentially added triethylamine (1 ml, 7 mmol) and 4-nitrochloroformate (90 mg, 0.45 mmol). After 1 h, TLC indicated complete consumption of the starting material and the reaction was diluted with CH2Cl2 and poured onto water. The organic layer was washed thrice with 2N NaOH, once with brine, dried over Na2SO4, and concentrated in vacuo. Purification of the crude oil by flash chromatography (2:1 Hex:EtOAc) gave 33e (50 mg, 54% yield over two steps): 1H NMR (CDCl3, 400 MHz) δ 8.23 (d, J = 8.2 Hz, 2H), 7.41-7.23 (m, 10H), 4.31 (bs, 2H), 3.02 (m, 1H), 2.88 (m, 1H), 2.57 (t, J = 2.9 Hz, 1H), 1.66-1.38 (m, 4H). HRMS calculated for C25H22Cl2N2NaO5 [M+Na]+ 523.0798, found 523.0823.
4-nitrophenyl 4-(hydroxydinaphthalen-2-ylmethyl)piperidine-1-carboxylate (33f)
Prepared according to General Procedure B, using 32f (50 mg, 0.1 mmol), 10% Pd/C (50 mg), EtOH:CH2Cl2 (4:1 v/v, 10 ml total) in the first step and CH2Cl2 (10 ml), triethylamine (1 ml, 7 mmol), and 4-nitrochloroformate (61 mg, 0.3 mmol) in the second step. Purification of the crude oil by flash chromatography (3:1 Hex:EtOAc) gave 33f (10 mg, 19% yield over two steps): 1H NMR (CDCl3, 400 MHz) δ 8.22 (d, J = 9.2 Hz, 2H), 8.06 (s, 2H), 7.85 (d, J = 7.4 Hz, 2H), 7.79 (d, J = 7.6 Hz, 4H), 7.57 (d, J = 8.7 Hz, 2H), 7.53-7.42 (m, 4H), 7.26 (dd, J = 4.9, 4.3 Hz, 2H), 4.32 (d, J = 13.5 Hz, 2H), 3.09 (t, J = 12.9 Hz, 1H), 3.03-2.85 (m, 2H), 1.75 (dd, J = 26.3, 13.0 Hz, 2H), 1.65-1.47 (m, 2H). HRMS calculated for C33H28N2NaO5 [M+Na]+ 555.1890, found 555.1916.
tert-butyl 4-(naphthalen-2-ylmethyl)piperazine-1-carboxylate (35a)
General Procedure D
To a stirring solution of N-Boc-piperazine (370 mg, 2.0 mmol) in CH2Cl2 (20 ml) was sequentially added 2-(bromomethyl)naphthalene (350 mg, 1.6 mmol) and K2CO3 (1 g, 7.2 mmol). The reaction was heated to reflux. After 10 h, TLC indicated complete consumption of the starting material, so the reaction was diluted with EtOAc and poured onto water. The organic layer was washed once with brine, dried over Na2SO4, and concentrated in vacuo. Purification of the crude oil by flash chromatography (5:1 to 3:1 Hex:EtOAc) gave 35a (420 mg, 76% yield): 1H NMR (CDCl3, 400 MHz) δ 7.82 (t, J = 7.6 Hz, 3H), 7.73 (s, 1H), 7.52-7.41 (m, 3H), 3.67 (s, 2H), 3.44 (bs, 4H), 2.43 (bs, 4H), 1.45 (s, 9H). MS (ESI+) m/z 327 [M+H]+.
tert-butyl 4-benzylpiperazine-1-carboxylate (35c)
Prepared according to General Procedure D, using N-Boc-piperazine (116 mg, 0.62 mmol), CH2Cl2 (6 ml), benzyl bromide (0.1 ml, 0.94 mmol), and K2CO3 (0.5 g, 3.6 mmol). Purification of the crude oil by flash chromatography (5:1 Hex:EtOAc) gave the 35c (120 mg, 69% yield): 1H NMR (CDCl3, 400 MHz) δ 7.35-7.20 (m, 5H), 3.49 (s, 2H), 3.46-3.37 (m, 4H), 2.43-2.29 (m, 4H), 1.45 (s, 9H). MS (ESI+) m/z 277 [M+H]+.
tert-butyl 4-(4-methoxybenzyl)piperazine-1-carboxylate (35d)
Prepared according to General Procedure D, using N-Boc-piperazine (116 mg, 0.62 mmol), CH2Cl2 (6 ml), 1-(bromomethyl)-4-methoxybenzene (0.2 ml, 1.4 mmol) and K2CO3 (0.5 g, 3.6 mmol). Purification of the crude oil by flash chromatography (5:1 Hex:EtOAc) gave 35d (134 mg, 71% yield): 1H NMR (CDCl3, 400 MHz) δ 7.22 (d, J = 8.6 Hz, 2H), 6.85 (d, J = 8.6 Hz, 2H), 3.79 (s, 3H), 3.45-3.40 (m, 6H), 2.43-2.31 (m, 4H), 1.45 (s, 9H). MS (ESI+) m/z 307 [M+H]+.
4-nitrophenyl 4-(naphthalen-2-ylmethyl)piperazine-1-carboxylate (36a)
General Procedure E
Compound 35a (420 mg, 1.1 mmol) was taken up in CH2Cl2:TFA (1:1 v/v, 6 ml total). After stirring for 2 h, TLC indicated complete consumption of the starting material. The crude was concentrated in vacuo and used without purification. The intermediate was then taken up in CH2Cl2 (5 ml) and to the solution was added triethylamine (2 ml, 14 mmol) and 4-nitrochloroformate (300mg, 1.5 mmol). After 3 h, TLC indicated complete consumption of the starting material. Purification of the crude oil by flash chromatography (5:1 Hex:EtOAc) gave 36a (102 mg, 20% yield over two steps): 1H NMR (CDCl3, 400 MHz) δ 8.27 (d, J = 9.3 Hz, 2H), 7.88-7.82 (m, 3H), 7.78 (s, 1H), 7.62-7.43 (m, 3H), 7.36-7.30 (m, 2H), 3.76 (s, 2H), 3.72 (bs, 2H), 3.64 (bs, 2H), 2.60 (bs, 2H). HRMS calculated for C22H22N3O4 [M+H]+ 392.1605, found 392.1638.
4-nitrophenyl 4-(quinolin-2-ylmethyl)piperazine-1-carboxylate (36b)
General Procedure F
To a stirring solution of N-Boc-piperazine (268 mg, 1.44 mmol) in dry MeOH (6 ml) was sequentially added quinoline-2-carbaldehyde (175 mg, 1.1 mmol), AcOH (60 μl), and Na(OAc)3BH (475 mg, 2.2 mmol). After 15 h, TLC indicated complete consumption of the starting material. The reaction was diluted with EtOAc and poured onto saturated aqueous NaHCO3. The organic layer was washed once with brine, dried over Na2SO4, and concentrated in vacuo. The crude oil was passed through a plug of silica and the intermediate was taken up in CH2Cl2:TFA (1:1 v/v, 10 ml total). After stirring for 2 h, TLC indicated complete consumption of the starting material. The crude was concentrated in vacuo and used without purification. The intermediate was then taken up in CH2Cl2 (5 ml) and to the solution was added triethylamine (1 ml, 7 mmol) and 4-nitrochloroformate (220 mg, 1.1 mmol). After 3 h, TLC indicated complete consumption of the starting material. The reaction was diluted with CH2Cl2 and poured onto saturated aqueous Na2CO3. The organic layer was washed thrice with 2N NaOH, once with brine, dried over Na2SO4, and concentrated in vacuo. Purification of the crude oil by flash chromatography (3:1 Hex:EtOAc) gave 36b (87 mg, 20% yield over three steps): 1H NMR (CDCl3, 400 MHz) δ 8.28-8.21 (m, 2H), 8.16 (d, J = 8.5 Hz, 1H), 8.09 (d, J = 8.5 Hz, 1H), 7.82 (d, J = 8.1 Hz, 1H), 7.72 (ddd, J = 8.4, 7.0, 1.3 Hz, 1H), 7.63 (d, J = 8.5 Hz, 1H), 7.57-7.51 (m, 1H), 7.33-7.27 (m, 2H), 3.91 (s, 2H), 3.72 (bs, 2H), 3.64 (bs, 2H), 2.76-2.52 (m, 4H). HRMS calculated for C21H21N4O4 [M+H]+ 393.1557, found 393.1590.
4-nitrophenyl 4-benzylpiperazine-1-carboxylate (36c)
Prepared according to General Procedure E, using 35c (113 mg, 0.5 mmol) and CH2Cl2:TFA (6 ml total) in the first step, and CH2Cl2 (5 ml), triethylamine (2 ml, 14 mmol) and 4-nitrochloroformate (160 mg, 0.8 mmol) in the second step. Purification of the crude oil by flash chromatography (4:1 Hex:EtOAc) gave 36c (61 mg, 69% yield): 1H NMR (CDCl3, 400 MHz) δ 8.24 (d, J = 9.3 Hz, 2H), 7.36-7.32 (m, 4H), 7.29 (d, J = 9.3 Hz, 3H), 3.73-3.65 (m, 2H), 3.62-3.56 (m, 4H), 2.57-2.47 (m, 4H). HRMS calculated for C18H20N3O4 [M+H]+ 342.1448, found 342.1470.
4-nitrophenyl 4-(4-methoxybenzyl)piperazine-1-carboxylate (36d)
Prepared according to General Procedure E, using 35d (110 mg, 0.36 mmol) and CH2Cl2:TFA (6 ml) in the first step and CH2Cl2 (5 ml), triethylamine (2 ml, 14 mmol), and 4-nitrochloroformate (160 mg, 0.8 mmol) in the second step. Purification of the crude oil by flash chromatography (4:1 Hex:EtOAc) gave 36d (43 mg, 32% yield): 1H NMR (CDCl3, 400 MHz) δ 8.24 (d, J = 9.3 Hz, 2H), 7.29 (d, J = 9.3 Hz, 2H), 7.24 (d, J = 8.7 Hz, 2H), 6.88 (d, J = 8.7 Hz, 2H), 3.81 (s, 3H), 3.70-3.64 (m, 2H), 3.62-3.55 (m, 2H), 3.51 (s, 2H), 2.58-2.41 (m, 4H). HRMS calculated for C19H22N3O5 [M+H]+ 372.1554, found 372.1581.
4-nitrophenyl 4-(biphenyl-4-ylmethyl)piperazine-1-carboxylate (36e)
Prepared according to General Procedure F, using N-Boc-piperazine (302 mg, 1.6 mmol), dry CH2Cl2 (10 ml), biphenyl-4-carbaldehyde (260 mg, 1.4 mmol), AcOH (0.1 ml), and Na(OAc)3BH (593 mg, 2.8 mmol) in the first step; CH2Cl2:TFA (10 ml) in the second step; and CH2Cl2 (5 ml), triethylamine (1 ml, 7 mmol), and 4-nitrochloroformate (280 mg, 1.4 mmol) in the third step. Purification of the crude oil by flash chromatography (5:1 Hex:EtOAc) gave 36e (300 mg, 51% yield over three steps): 1H NMR (CDCl3, 400 MHz) δ 8.25 (d, J = 9.2 Hz, 2H), 7.59 (dd, J = 13.0, 4.7 Hz, 4H), 7.51-7.25 (m, 7H), 3.70 (bs, 2H), 3.58 (bs, 4H), 2.65-2.36 (m, 4H). HRMS calculated for C24H24N3O4 [M+H]+ 418.1761, found 418.1802.
4-nitrophenyl 4-(biphenyl-2-ylmethyl)piperazine-1-carboxylate (36f)
Prepared according to general procedure F, using N-Boc-piperazine (242 mg, 1.3 mmol), dry MeOH (6 ml), biphenyl-2-carbaldehyde (208 mg, 1.1 mmol), AcOH (60 μl), and Na(OAc)3BH (467 mg, 2.2 mmol) in the first step; CH2Cl2:TFA (10 ml) in the second step; and CH2Cl2 (5 ml), triethylamine (1 ml, 7 mmol), and 4-nitrochloroformate (220 mg, 1.1 mmol) in the third step. Purification of the crude oil by flash chromatography (5:1 Hex:EtOAc) gave 36f (90 mg, 20% yield over three steps): 1H NMR (CDCl3, 400 MHz) δ 8.16 (d, J = 9.2 Hz, 2H), 8.08 (d, J = 9.1 Hz, 2H), 7.39-7.16 (m, 7H), 6.79 (d, J = 9.2 Hz, 2H), 3.54 (bs, 2H), 3.44 (bs, 4H), 2.34 (bs, 4H). HRMS calculated for C24H24N3O4 [M+H]+ 418.1761, found 418.1800.
4-nitrophenyl 4-(4-(benzyloxy)-2-methoxybenzyl)piperazine-1-carboxylate (36g)
Prepared according to general procedure F, using N-Boc-piperazine (345 mg, 1.9 mmol), dry MeOH (6 ml), 4-(benzyloxy)-3-methoxybenzaldehyde (364 mg, 1.5 mmol), AcOH (60 μl), and Na(OAc)3BH (575 mg, 2.7 mmol) in the first step; CH2Cl2:TFA (10 ml) in the second step; and CH2Cl2 (5 ml), triethylamine (1 ml, 7 mmol), and 4-nitrochloroformate (300 mg, 1.5 mmol) in the third step. Purification of the crude oil by flash chromatography (4:1 Hex:EtOAc) gave 36g (46 mg, 6% yield over three steps): 1H NMR (CDCl3, 400 MHz) δ 8.24 (d, J = 9.1 Hz, 2H), 7.44 (d, J = 7.3 Hz, 2H), 7.37 (dd, J = 11.2, 4.2 Hz, 2H), 7.33-7.23 (m, 3H), 6.91 (d, J = 1.4 Hz, 1H), 6.83 (d, J = 8.1 Hz, 1H), 6.77 (d, J = 8.1 Hz, 1H), 5.15 (s, 2H), 3.90 (s, 3H), 3.67 (bs, 2H), 3.59 (bs, 2H), 3.48 (s, 2H), 2.49 (bs, 2H). HRMS calculated for C26H28N3O6 [M+H]+ 478.1973, found 478.2014.
4-nitrophenyl 4-((5-phenylthiophen-2-yl)methyl)piperazine-1-carboxylate (36h)
Prepared according to general procedure F, using N-Boc-piperazine (373 mg, 2.0 mmol), dry THF (20 ml), 5-phenylthiophene-2-carbaldehyde (372 mg, 2.0 mmol), AcOH (0.2 ml), and Na(OAc)3BH (796 mg, 3.8 mmol) in the first step; CH2Cl2:TFA (10 ml) in the second step; and CH2Cl2 (15 ml), triethylamine (2 ml, 14 mmol), and 4-nitrochloroformate (400 mg, 2.0 mmol) in the third step. Purification of the crude oil by flash chromatography (6:1 Hex:EtOAc) gave 36h (250 mg, 30% yield over three steps): 1H NMR (CDCl3, 400 MHz) δ 8.23 (d, J = 9.1 Hz, 2H), 7.58 (d, J = 7.6 Hz, 2H), 7.36 (t, J = 7.6 Hz, 2H), 7.32-7.22 (m, 3H), 7.16 (d, J = 3.6 Hz, 1H), 6.89 (d, J = 3.5 Hz, 1H), 3.77 (s, 2H), 3.71 (bs, 2H), 3.62 (bs, 2H), 2.65-2.55 (m, 4H). HRMS calculated for C22H22N3O4S [M+H]+ 424.1325, found 424.1347.
4-nitrophenyl 4-((4-phenylthiophen-2-yl)methyl)piperazine-1-carboxylate (36i)
Prepared according to general procedure F, using N-Boc-piperazine (343 mg, 1.9 mmol), dry MeOH (10 ml), 4-phenylthiophene-2-carbaldehyde (260 mg, 1.4 mmol), AcOH (0.1 ml), and Na(OAc)3BH (738 mg, 3.48 mmol) in the first step; CH2Cl2:TFA (10 ml) in the second step; and CH2Cl2 (5 ml), triethylamine (1 ml, 7 mmol), and 4-nitrochloroformate (294 mg, 1.5 mmol) in the third step. Purification of the crude oil by flash chromatography (3:1 Hex:EtOAc) gave 36i (132 mg, 28% yield over three steps): 1H NMR (CDCl3, 400 MHz) δ 8.28-8.21 (m, 2H), 7.70-7.52 (m, 2H), 7.47-7.34 (m, 3H), 7.33-7.19 (m, 4H), 3.81 (d, J = 0.5 Hz, 2H), 3.72 (bs, 2H), 3.63 (bs, 2H), 2.75-2.52 (m, 4H). HRMS calculated for C22H22N3O4S [M+H]+ 424.1325, found 424.1345.
4-nitrophenyl 4-((5-phenylfuran-2-yl)methyl)piperazine-1-carboxylate (36j)
Prepared according to general procedure F, using N-Boc-piperazine (348 mg, 1.9 mmol), dry CH2Cl2 (10 ml), 5-phenylfuran-2-carbaldehyde (285 mg, 1.66 mmol), AcOH (1 ml), and Na(OAc)3BH (807 mg, 3.8 mmol) in the first step; CH2Cl2:TFA (10 ml) in the second step; and CH2Cl2 (5 ml), triethylamine (1 ml, 7 mmol), and 4-nitrochloroformate (372 mg, 1.85 mmol) in the third step. Purification of the crude oil by flash chromatography (3:1 Hex:EtOAc) gave 36j (196 mg, 29% yield over three steps): 1H NMR (CDCl3, 400 MHz) δ 8.33-8.11 (m, 2H), 7.67 (dd, J = 7.4, 0.9 Hz, 2H), 7.46-7.32 (m, 2H), 7.27 (ddd, J = 5.9, 3.5, 0.9 Hz, 3H), 6.61 (d, J = 3.2 Hz, 2H), 6.32 (d, J = 3.3 Hz, 2H), 3.70 (s, 4H), 3.63 (s, 2H), 2.61 (s, 4H). HRMS calculated for C22H21N3NaO5 [M+Na]+ 430.1373, found 430.1383.
4-nitrophenyl 4-(5-bromo-2-hydroxybenzyl)piperazine-1-carboxylate (36k)
Prepared according to general procedure F, using N-Boc-piperazine (367 mg, 2.0 mmol), dry THF (20 ml), 5-bromo-2-hydroxybenzaldehyde (366 mg, 1.8 mmol), AcOH (0.2 ml), and Na(OAc)3BH (799 mg, 3.8 mmol) in the first step; CH2Cl2:TFA (6 ml) in the second step; and CH2Cl2 (5 ml), triethylamine (2 ml, 14 mmol), and 4-nitrochloroformate (400 mg, 2.0 mmol) in the third step. Purification of the crude oil by flash chromatography (3:1 Hex:EtOAc) gave 36k (140 mg, 16% yield over three steps): 1H NMR (CDCl3, 400 MHz) δ 8.27 (d, J = 9.3 Hz, 2H), 7.42-7.24 (m, 3H), 7.15 (d, J = 2.4 Hz, 1H), 6.76 (d, J = 8.6 Hz, 1H), 3.82-3.51 (m, 6H), 2.67 (s, 4H). HRMS calculated for C18H19BrN3O5 [M+H]+ 436.0503, found 436.0528.
4-nitrophenyl 4-((5-(2-(trifluoromethoxy)phenyl)furan-2-yl)methyl)piperazine-1-carboxylate (36l)
Prepared according to general procedure F, using N-Boc-piperazine (427 mg, 2.3 mmol), dry MeOH (10 ml), 5-(2-(trifluoromethoxy)phenyl)furan-2-carbaldehyde (244 mg, 0.95 mmol), AcOH (0.1 ml), and Na(OAc)3BH (410 mg, 1.9 mmol) in the first step; CH2Cl2:TFA (10 ml) in the second step; and CH2Cl2 (5 ml), triethylamine (1 ml, 7 mmol), and 4-nitrochloroformate (200 mg, 1.0 mmol) in the third step. Purification of the crude oil by flash chromatography (4:1 Hex:EtOAc) gave 36l (110 mg, 24% yield over three steps): 1H NMR (CDCl3, 400 MHz) δ 8.24 (d, J = 9.3 Hz, 2H), 7.91 (ddd, J = 7.6, 1.7, 0.6 Hz, 1H), 7.52-7.15 (m, 5H), 6.84 (d, J = 3.3 Hz, 1H), 6.38 (d, J = 3.4 Hz, 1H), 3.76 (bs, 4H), 3.64 (bs, 2H), 2.63 (bs, 4H). HRMS calculated for C23H21F3N3O6 [M+H]+ 492.1377, found 492.1417.
4-nitrophenyl 4-(3-(p-tolyloxy)benzyl)piperazine-1-carboxylate (36m)
Prepared according to general procedure F, using N-Boc-piperazine (373 mg, 2.0 mmol), dry THF (20 ml), 3-(4-methylphenoxy)benzaldehyde (425 mg, 2.00 mmol), AcOH (0.2 ml), and Na(OAc)3BH (848 mg, 4.0 mmol) in the first step; CH2Cl2:TFA (15 ml) in the second step; and CH2Cl2 (6 ml), triethylamine (3 ml, 21 mmol), and 4-nitrochloroformate (588 mg, 2.92 mmol) in the third step. Purification of the crude oil by flash chromatography (2:1 Hex:EtOAc) gave 36m (456 mg, 51% yield over three steps): 1H NMR (CDCl3, 300 MHz) δ 8.26-8.21 (m, 2H), 7.32-6.84 (m, 8H), 6.86 (m, 2H), 3.67 (m, 2H), 3.58 (m, 2H), 3.53 (s, 2H), 2.50 (m, 4H), 2.34 (s, 3H). C25H26N3O5 [M+H]+ 448.1867, found 448.1911.
4-nitrophenyl 4-(3-(pyridin-2-yloxy)benzyl)piperazine-1-carboxylate (36n)
Prepared according to general procedure F, using N-Boc-piperazine (305 mg, 1.64 mmol), dry CH2Cl2 (10 ml), 3-(pyridin-2-yloxy)benzaldehyde (104 mg, 0.52 mmol), AcOH (0.1 ml), and Na(OAc)3BH (233 mg, 1.1 mmol) in the first step; CH2Cl2:TFA (5 ml) in the second step; and CH2Cl2 (5 ml), triethylamine (1 ml, 7 mmol), and 4-nitrochloroformate (100 mg, 0.50 mmol) in the third step. Purification of the crude oil by flash chromatography (2:1 Hex:EtOAc) gave the product (85 mg, 38% yield over three steps): 1H NMR (CDCl3, 400 MHz) δ 8.31-8.16 (m, 3H), 7.70 (ddd, J = 8.3, 7.2, 1.9 Hz, 1H), 7.37 (t, J = 7.7 Hz, 1H), 7.33-7.24 (m, 2H), 7.17 (d, J = 8.7 Hz, 2H), 7.09-7.03 (m, 1H), 7.01 (ddd, J = 7.2, 5.1, 0.8 Hz, 1H), 6.93 (dd, J = 8.3, 0.8 Hz, 1H), 3.68 (bs, 2H), 3.59 (bs, 4H), 2.53 (bs, 4H). HRMS calculated for C22H22N3O4 [M+H]+ 392.1605, found 392.1638. HRMS calculated for C23H23N4O5 [M+H]+ 435.1663, found 435.1699.
4-nitrophenyl 4-(3-(4-methoxyphenoxy)benzyl)piperazine-1-carboxylate (36o)
Prepared according to general procedure F, using N-Boc-piperazine (559 mg, 3.0 mmol), dry THF (20 ml), 3-(4-methoxyphenoxy)benzaldehyde (457 mg, 2.00 mmol), AcOH (0.2 ml), and Na(OAc)3BH (848 mg, 4.0 mmol) in the first step; CH2Cl2:TFA (15 ml) in the second step; and CH2Cl2 (6 ml), triethylamine (3 ml, 21 mmol), and 4-nitrochloroformate (653 mg, 3.24 mmol) in the third step. Purification of the crude oil by flash chromatography (2:1 Hex:EtOAc) gave 36o (343 mg, 37% yield over three steps): 1H NMR (CDCl3, 300 MHz) δ 8.26-8.21 (m, 2H), 7.32-7.20 (m, 3H), 7.05-6.80 (m, 7H), 3.80 (s, 3H), 3.67 (m, 2H), 3.58 (m, 2H), 3.52 (s, 2H), 2.49 (m, 4H). C25H26N3O6 [M+H]+ 464.1816, found 464.1882.
4-nitrophenyl 4-(3-(3-(trifluoromethyl)phenoxy)benzyl)piperazine-1-carboxylate (36p)
Prepared according to general procedure G, using N-Boc-piperazine (559 mg, 3.0 mmol), dry THF (20 ml), 3-(3-trifluoromethylphenoxy)benzaldehyde (532 mg, 2.00 mmol), AcOH (0.2 ml), and Na(OAc)3BH (848 mg, 4.0 mmol) in the first step; CH2Cl2:TFA (15 ml) in the second step; and CH2Cl2 (6 ml), triethylamine (3 ml, 21 mmol), and 4-nitrochloroformate (648 mg, 3.22 mmol) in the third step. Purification of the crude oil by flash chromatography (2:1 Hex:EtOAc) gave 36p (321 mg, 32% yield over three steps): 1H NMR (CDCl3, 300 MHz) δ 8.26-8.21 (m, 2H), 7.50-6.90 (m, 10H), 3.68 (m, 2H), 3.59 (m, 2H), 3.56 (s, 2H), 2.51 (m, 4H). C25H23F3N3O5 [M+H]+ 502.1584, found 502.1639.
4-nitrophenyl 4-(3-(3,5-dichlorophenoxy)benzyl)piperazine-1-carboxylate (36q)
Prepared according to general procedure G, using N-Boc-piperazine (373 mg, 2.0 mmol), dry THF (20 ml), 3-(3,5-dichlorophenoxy)benzaldehyde (534 mg, 2.00 mmol), AcOH (0.2 ml), and Na(OAc)3BH (848 mg, 4.0 mmol) in the first step; CH2Cl2:TFA (15 ml) in the second step; and CH2Cl2 (6 ml), triethylamine (3 ml, 21 mmol), and 4-nitrochloroformate (798 mg, 3.96 mmol) in the third step. Purification of the crude oil by flash chromatography (3:1 Hex:EtOAc) gave the 36q (422 mg, 42% yield over three steps): 1H NMR (CDCl3, 300 MHz) δ 8.27-8.22 (m, 2H), 7.38-6.93 (m, 7H), 6.86 (m, 2H), 3.70 (m, 2H), 3.61 (m, 2H), 3.54 (s, 2H), 2.51 (m, 4H). C24H22Cl2N3O5 [M+H]+ 502.0931, found 502.1004.
Preparations of mouse brain membrane proteomes
Tissues were Dounce-homogenized in PBS, pH 7.5, followed by a low-speed spin (1,400 × g, 5 min) to remove debris. The supernatant was then subjected to centrifugation (64,000 × g, 45 min) to provide the cytosolic fraction in the supernatant and the membrane fraction as a pellet. The pellet was washed and resuspended in PBS buffer by sonication. Total protein concentration in each fraction was determined using a protein assay kit (Bio-Rad). Samples were stored at −80°C until use.
Competitive ABPP experiments
Brain membrane proteomes were diluted to 1 mg/ml in PBS in a 50 μl total reaction volume. Inhibitors were added at the indicated concentration and the reactions were incubated for 30 min at 37°C. FP-rhodamine was then added at a final concentration of 1 μM. After an additional 30 min at 25°C, the reactions were quenched with 4x SDS-PAGE loading buffer, boiled for 5 min at 90°C, subjected to SDS-PAGE and visualized in-gel using a flatbed fluorescence scanner (Hitachi). Concentration-dependent inhibition curves were obtained from integrated gel band intensities (ImageJ) and were fit using Prism software (GraphPad) to obtain effector concentration for half-maximal response values (IC50). The IC50 values shown in Tables 1–6 are the means of at least triplicate measurements, and the standard error of the mean (SEM) is < 25% of the indicated value in all cases.
Acknowledgments
We thank the Cravatt lab for helpful discussion and critical reading of the manuscript. This work was supported by the US National Institutes of Health (DA017259, DA009789, DA025285, DA03672, DA005274, and T23DA07027, DA014277), the Helen L. Dorris Institute Child and Adolescent Neuro-Psychiatric Disorder Institute, and the Skaggs Institute for Chemical Biology.
Abbreviations List
- CB
cannabinoid receptor
- AEA
anandamide
- 2-AG
2-arachidonoylglycerol
- FAAH
fatty acid amide hydrolases
- MAGL
monoacylglycerol lipase
- ABPP
activity-based protein profiling
- ABHD6
alpha/beta hydrolases domain-containing 6
- NTE
neuropathy target esterase
- TRP
transient receptor potential
References
- 1.Ahn K, McKinney MK, Cravatt BF. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem Rev. 2008;108:1687–1707. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 3.Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci U S A. 2001;98:9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- 5.Ahn K, Johnson DS, Mileni M, Beidler D, Long JZ, McKinney MK, Weerapana E, Sadagopan N, Liimatta M, Smith SE, Lazerwith S, Stiff C, Kamtekar S, Bhattacharya K, Zhang Y, Swaney S, Van Becelaere K, Stevens RC, Cravatt BF. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem Biol. 2009;16:411–420. doi: 10.1016/j.chembiol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavon FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, Cravatt BF. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Long JZ, Nomura DK, Cravatt BF. Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem Biol. 2009;16:744–753. doi: 10.1016/j.chembiol.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Long JZ, Nomura DK, Vann RE, Walentiny DM, Booker L, Jin X, Burston JJ, Sim-Selley LJ, Lichtman AH, Wiley JL, Cravatt BF. Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc Natl Acad Sci U S A. 2009;106:20270–20275. doi: 10.1073/pnas.0909411106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahn K, Johnson DS, Fitzgerald LR, Liimatta M, Arendse A, Stevenson T, Lund ET, Nugent RA, Nomanbhoy TK, Alexander JP, Cravatt BF. Novel mechanistic class of fatty acid amide hydrolase inhibitors with remarkable selectivity. Biochemistry. 2007;46:13019–13030. doi: 10.1021/bi701378g. [DOI] [PubMed] [Google Scholar]
- 11.DeSimone RW, Currie KS, Mitchell SA, Darrow JW, Pippin DA. Privileged structures: applications in drug discovery. Comb Chem High Throughput Screen. 2004;7:473–494. doi: 10.2174/1386207043328544. [DOI] [PubMed] [Google Scholar]
- 12.Evans BE, Rittle KE, Bock MG, DiPardo RM, Freidinger RM, Whitter WL, Lundell GF, Veber DF, Anderson PS, Chang RS, et al. Methods for drug discovery: development of potent, selective, orally effective cholecystokinin antagonists. J Med Chem. 1988;31:2235–2246. doi: 10.1021/jm00120a002. [DOI] [PubMed] [Google Scholar]
- 13.Patricelli MP, Lovato MA, Cravatt BF. Chemical and mutagenic investigations of fatty acid amide hydrolase: evidence for a family of serine hydrolases with distinct catalytic properties. Biochemistry. 1999;38:9804–9812. doi: 10.1021/bi990637z. [DOI] [PubMed] [Google Scholar]
- 14.Karlsson M, Contreras JA, Hellman U, Tornqvist H, Holm C. cDNA cloning, tissue distribution, and identification of the catalytic triad of monoglyceride lipase. Evolutionary relationship to esterases, lysophospholipases, and haloperoxidases. J Biol Chem. 1997;272:27218–27223. doi: 10.1074/jbc.272.43.27218. [DOI] [PubMed] [Google Scholar]
- 15.Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S, Piomelli D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci U S A. 2002;99:10819–10824. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y, Patricelli MP, Cravatt BF. Activity-based protein profiling: the serine hydrolases. Proc Natl Acad Sci U S A. 1999;96:14694–14699. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alexander JP, Cravatt BF. Mechanism of carbamate inactivation of FAAH: implications for the design of covalent inhibitors and in vivo functional probes for enzymes. Chem Biol. 2005;12:1179–1187. doi: 10.1016/j.chembiol.2005.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tarzia G, Duranti A, Tontini A, Piersanti G, Mor M, Rivara S, Plazzi PV, Park C, Kathuria S, Piomelli D. Design, synthesis, and structure-activity relationships of alkylcarbamic acid aryl esters, a new class of fatty acid amide hydrolase inhibitors. J Med Chem. 2003;46:2352–2360. doi: 10.1021/jm021119g. [DOI] [PubMed] [Google Scholar]
- 19.Cravatt BF, Wright AT, Kozarich JW. Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu Rev Biochem. 2008;77:383–414. doi: 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- 20.Evans MJ, Cravatt BF. Mechanism-based profiling of enzyme families. Chem Rev. 2006;106:3279–3301. doi: 10.1021/cr050288g. [DOI] [PubMed] [Google Scholar]
- 21.Patricelli MP, Giang DK, Stamp LM, Burbaum JJ. Direct visualization of serine hydrolase activities in complex proteomes using fluorescent active site-directed probes. Proteomics. 2001;1:1067–1071. doi: 10.1002/1615-9861(200109)1:9<1067::AID-PROT1067>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 22.Jessani N, Niessen S, Wei BQ, Nicolau M, Humphrey M, Ji Y, Han W, Noh DY, Yates JR, 3rd, Jeffrey SS, Cravatt BF. A streamlined platform for high-content functional proteomics of primary human specimens. Nat Methods. 2005;2:691–697. doi: 10.1038/nmeth778. [DOI] [PubMed] [Google Scholar]
- 23.Li W, Blankman JL, Cravatt BF. A functional proteomic strategy to discover inhibitors for uncharacterized hydrolases. J Am Chem Soc. 2007;129:9594–9595. doi: 10.1021/ja073650c. [DOI] [PubMed] [Google Scholar]
- 24.Hoover HS, Blankman JL, Niessen S, Cravatt BF. Selectivity of inhibitors of endocannabinoid biosynthesis evaluated by activity-based protein profiling. Bioorg Med Chem Lett. 2008;18:5838–5841. doi: 10.1016/j.bmcl.2008.06.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leung D, Hardouin C, Boger DL, Cravatt BF. Discovering potent and selective reversible inhibitors of enzymes in complex proteomes. Nat Biotechnol. 2003;21:687–691. doi: 10.1038/nbt826. [DOI] [PubMed] [Google Scholar]
- 26.Bachovchin DA, Brown SJ, Rosen H, Cravatt BF. Identification of selective inhibitors of uncharacterized enzymes by high-throughput screening with fluorescent activity-based probes. Nat Biotechnol. 2009;27:387–394. doi: 10.1038/nbt.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bracey MH, Hanson MA, Masuda KR, Stevens RC, Cravatt BF. Structural adaptations in a membrane enzyme that terminates endocannabinoid signaling. Science. 2002;298:1793–1796. doi: 10.1126/science.1076535. [DOI] [PubMed] [Google Scholar]
- 28.Morphy R, Rankovic Z. Designed multiple ligands. An emerging drug discovery paradigm. J Med Chem. 2005;48:6523–6543. doi: 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- 29.Cavalli A, Bolognesi ML, Minarini A, Rosini M, Tumiatti V, Recanatini M, Melchiorre C. Multi-target-directed ligands to combat neurodegenerative diseases. J Med Chem. 2008;51:347–372. doi: 10.1021/jm7009364. [DOI] [PubMed] [Google Scholar]
- 30.Glynn P, Read DJ, Guo R, Wylie S, Johnson MK. Synthesis and characterization of a biotinylated organophosphorus ester for detection and affinity purification of a brain serine esterase: neuropathy target esterase. Biochem J. 1994;301 (Pt 2):551–556. doi: 10.1042/bj3010551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lush MJ, Li Y, Read DJ, Willis AC, Glynn P. Neuropathy target esterase and a homologous Drosophila neurodegeneration-associated mutant protein contain a novel domain conserved from bacteria to man. Biochem J. 1998;332 (Pt 1):1–4. doi: 10.1042/bj3320001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Glynn P. A mechanism for organophosphate-induced delayed neuropathy. Toxicol Lett. 2006;162:94–97. doi: 10.1016/j.toxlet.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 33.Casida JE, Nomura DK, Vose SC, Fujioka K. Organophosphate-sensitive lipases modulate brain lysophospholipids, ether lipids and endocannabinoids. Chem Biol Interact. 2008;175:355–364. doi: 10.1016/j.cbi.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson MK, Glynn P. Neuropathy target esterase (NTE) and organophosphorus-induced delayed polyneuropathy (OPIDP): recent advances. Toxicol Lett. 1995;82–83:459–463. doi: 10.1016/0378-4274(95)03495-1. [DOI] [PubMed] [Google Scholar]
- 35.Pan B, Wang W, Long JZ, Sun D, Hillard CJ, Cravatt BF, Liu QS. Blockade of 2-arachidonoylglycerol hydrolysis by selective monoacylglycerol lipase inhibitor 4-nitrophenyl 4-(dibenzo[d][1,3]dioxol-5-yl(hydroxy)methyl)piperidine-1-carboxylate (JZL184) Enhances retrograde endocannabinoid signaling. J Pharmacol Exp Ther. 2009;331:591–597. doi: 10.1124/jpet.109.158162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schlosburg JE, Carlson BL, Ramesh D, Abdullah RA, Long JZ, Cravatt BF, Lichtman AH. Inhibitors of endocannabinoid-metabolizing enzymes reduce precipitated withdrawal responses in THC-dependent mice. AAPS J. 2009;11:342–352. doi: 10.1208/s12248-009-9110-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kinsey SG, Long JZ, O’Neal ST, Abdullah RA, Poklis JL, Boger DL, Cravatt BF, Lichtman AH. Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. J Pharmacol Exp Ther. 2009;330:902–910. doi: 10.1124/jpet.109.155465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Straiker A, Hu SS, Long JZ, Arnold A, Wager-Miller J, Cravatt BF, Mackie K. Monoacylglycerol lipase limits the duration of endocannabinoid-mediated depolarization-induced suppression of excitation in autaptic hippocampal neurons. Mol Pharmacol. 2009;76:1220–1227. doi: 10.1124/mol.109.059030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nomura DK, Long JZ, Niessen S, Hoover HS, Ng S-W, Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell. 2010 doi: 10.1016/j.cell.2009.11.027. In Press, Accepted Manuscript. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Labar G, Bauvois C, Borel F, Ferrer JL, Wouters J, Lambert DM. Crystal Structure of the Human Monoacylglycerol Lipase, a Key Actor in Endocannabinoid Signaling. Chembiochem. 2009 doi: 10.1002/cbic.200900621. [DOI] [PubMed] [Google Scholar]
- 41.Bertrand T, Auge F, Houtmann J, Rak A, Vallee F, Mikol V, Berne PF, Michot N, Cheuret D, Hoornaert C, Mathieu M. Structural Basis For Human Mono-Glyceride Lipase Inhibition. J Mol Biol. 2009 doi: 10.1016/j.jmb.2009.11.060. [DOI] [PubMed] [Google Scholar]
- 42.Morera E, De Petrocellis L, Morera L, Moriello AS, Ligresti A, Nalli M, Woodward DF, Di Marzo V, Ortar G. Synthesis and biological evaluation of piperazinyl carbamates and ureas as fatty acid amide hydrolase (FAAH) and transient receptor potential (TRP) channel dual ligands. Bioorg Med Chem Lett. 2009;19:6806–6809. doi: 10.1016/j.bmcl.2009.09.033. [DOI] [PubMed] [Google Scholar]
- 43.Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di Marzo V, Julius D, Hogestatt ED. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]
- 44.Saghatelian A, McKinney MK, Bandell M, Patapoutian A, Cravatt BF. A FAAH-regulated class of N-acyl taurines that activates TRP ion channels. Biochemistry. 2006;45:9007–9015. doi: 10.1021/bi0608008. [DOI] [PubMed] [Google Scholar]
- 45.Karlsson M, Reue K, Xia YR, Lusis AJ, Langin D, Tornqvist H, Holm C. Exonintron organization and chromosomal localization of the mouse monoglyceride lipase gene. Gene. 2001;272:11–18. doi: 10.1016/s0378-1119(01)00559-5. [DOI] [PubMed] [Google Scholar]