

Abstract
TGFβ Inducible Early Gene-1 (TIEG) is a member of the Krüppel-like family of transcription factors (KLF10) that plays an important role in TGFβ mediated Smad signaling. In order to better understand the role of TIEG in bone, we generated TIEG knockout (KO) mice. Calvarial osteoblasts (OBs) isolated from these mice exhibit a reduced ability to support osteoclastogenesis in vitro. Gene expression studies revealed decreased receptor activator of NF-kB ligand (RANKL) and increased osteoprotegerin (OPG) expression in TIEG KO OBs, suggesting a potential role for TIEG in regulating the expression of these genes. Since OPG and RANKL are two important regulators of osteoclast (OC) differentiation, we sought to determine if TIEG directly regulates their expression. Luciferase constructs, containing fragments of either the mouse OPG promoter (−1486 to +133 bp) or the RANKL promoter (−2000 to +1 bp) were each cloned into the pGL3 basic reporter vector and transiently transfected into TIEG KO calvarial OBs with and without a TIEG expression vector. No significant changes in the activity of the RANKL promoter were detected in the presence of TIEG. However, OPG promoter activity was inhibited in the presence of TIEG protein suggesting that TIEG directly represses the expression of OPG in OBs. In order to determine the region of this promoter through which TIEG acts, sequential 5′-deletion constructs were generated. Transient transfection of these constructs revealed that the TIEG regulatory element(s) reside within a 200 bp region of the OPG promoter. Transient ChIP analyses, using a TIEG-specific antibody, revealed that TIEG binds to this region of the OPG promoter. Since we have previously shown that TIEG regulates target gene expression through Sp-1 sites, we examined this region of the OPG promoter for potential TIEG binding elements and identified four potential Sp-1 binding sites. Site directed mutagenesis was used to determine if TIEG utilizes these Sp-1 elements to regulate the activity of the OPG promoter. The data demonstrate that two Sp-1 sites are likely to be involved in TIEG’s repression of the OPG promoter. Taken together, these results confirm that TIEG directly binds to and inhibits OPG promoter activity in OBs, partially explaining the inability of TIEG KO OBs to fully support osteoclast differentiation.
Keywords: osteoprotegerin (OPG), TGFβ inducible early gene-1 (TIEG1), Krüppel-like transcription factor-10 (KLF10), osteoblasts (OBs)
Introduction
TGFβ inducible early gene-1 (TIEG), also referred to as KLF10, was originally cloned from human osteoblasts (OBs) as a primary response gene following TGFβ treatment [1]. Following the initial discovery, we demonstrated that TIEG over-expression in MG63 human osteosarcoma cells mimicked TGFβ treatment by inducing alkaline phosphatase mRNA and inhibiting cell proliferation and osteocalcin secretion [2]. Over-expression of TIEG in pancreatic carcinoma and hepatocarcinoma cells also inhibited cell proliferation and induced apoptosis similar to that of TGFβ [3]. TIEG also plays an important role in mediating the TGFβ Smad signaling pathway. More specifically, we have shown that TIEG over-expression enhances Smad binding element (SBE) reporter activity that is dependent on Smad 4 expression [4]. Further, TIEG over-expression in target cells enhances the induction of TGFβ regulated genes p21 and PAI-1 [4].
Because TIEG was originally cloned from human OBs, and TIEG plays an important role in mediating OB growth and differentiation, we generated TIEG KO mice in order to gain a better understanding of TIEG’s action in bone [5]. Characterization of the skeleton of TIEG KO mice revealed that bone content, density, and size were significantly decreased in female animals relative to their wild-type (WT) littermates [6]. Interestingly, male mice displayed no significant bone phenotype, suggesting a gender-specific mechanism of action in these animals [7]. Three point bending tests revealed that the femurs of female TIEG KO mice were also weaker than WT littermates [6]. Micro-CT analysis of the femoral head and vertebrae revealed increases in femoral head separation, including reductions in several other cortical parameters [6]. Histomorphometric analysis of the distal femur revealed that female TIEG KO mice display a decrease in cancellous bone area, which is primarily due to decreased trabecular number [7]. At the cellular level, female TIEG KO mice also have reduced bone formation rates, as indicated by double-labeled perimeter [7]. In order to further characterize the functions of TIEG in OBs, calvarial OBs were isolated and their growth properties, mineralization rates and expression levels of OB-specific marker genes were compared to that of calvarial OBs isolated from WT littermates. The OBs isolated from TIEG KO mice were defective in regard to two important OB functions, mineralization and OB support of osteoclast (OC) differentiation [5]. To address the important function of OB support of OC differentiation, and to better understand the molecular mechanism(s) behind this specific defect, additional gene expression studies were performed. In this manuscript, we report that OBs isolated from TIEG KO mice exhibit decreased RANKL expression (an activator of osteoclastogenesis) and increased OPG expression (an inhibitor of osteoclastogenesis). Further, we have demonstrated that TIEG regulates the OPG expression by binding to Sp-1 sites in the upstream region of the OPG promoter to repress its activity. This explains the increased OPG expression in TIEG KO osteoblasts, which likely contributes to the decreased support of OC differentiation.
Materials and Methods
Calvarial osteoblast cell culture
Calvarial OBs were isolated from 1-3 day old neonatal pups obtained from TIEG KO and WT mice, as described previously [5]. Calvarial OBs were routinely maintained in α-MEM containing 10% fetal bovine serum and antibiotics/antimycotics. Cells at passages 1 and 2 were used for all experiments.
RNA isolation and real-time PCR analysis
Calvarial OBs isolated from TIEG KO and WT pups were plated in 12 well plates. To rule out the individual variability of these primary osteoblasts, cells isolated from three individual pups derived from both TIEG KO and WT animals were used in this study. When the cells were approximately 90% confluent, they were washed with 1× PBS and lysed in Trizol (Invitrogen, CA) for isolation of total RNA as specified by the manufacturer. The cDNA synthesis and real-time PCR analyses were performed as described earlier [8]. The following gene specific primers were used to amplify RANKL and OPG. All data were normalized to tubulin controls.
- RANKL:
- F 5′-GCTGGGACCTGCAAATAAGT-3′
- R 5′-TTGCACAGAAAACATTACACCTG-3′
- OPG:
- F 5′-CCAAGAGCCCAGTGTTTCTT-3′
- R 5′-CCAAGCCAGCCATTGTTAAT-3′
- Tubulin
- F 5′-CTGCTCATCAGCAAGATCAGAG-3′
- R 5′-GCATTATAGGGCTCCACCACAG-3′
RANKL and OPG promoter constructs
The mouse OPG promoter, which spans from −1486 to +133 bp relative to the transcription start site, was PCR amplified using mouse genomic DNA as a template and cloned into the pGL3 basic luciferase reporter construct. The RANKL promoter used in this study was a gift from Dr. S. V. Reddy, Medical University of South Carolina. The RANKL promoter consisting of −2000 to +1 bp relative to the transcription site was subcloned into the pGL3 basic luciferase reporter construct as previously described [9].
Transient transfection and luciferase assays
A calvarial OB cell line derived from a TIEG KO pup, that spontaneously immortalized, was used for all transient transfection assays. The use of this cell line eliminates the potential problems related to endogenous TIEG expression that is present in WT calvarial OBs and other cell lines. Immortalized TIEG KO calvarial OBs were plated in 12 well plates at approximately 50% confluence. The following day, cells were transfected with 500 ng of the promoter reporter constructs along with 500 ng of either pcDNA4/TO empty vector or 500 ng of pcDNA4/TO TIEG expression plasmid using Fugene-6 transfection reagent (Roche, NJ) as specified by the manufacturer.
Creation of various Sp-1 mutations in the −1483 to +133 OPG promoter
Mutations in the identified Sp-1 binding sites were created using a Quick Change kit (Stratagene, CA) as specified by the manufacturer. The native and the mutated sequences of each of the Sp-1 sites are shown below:
- Site-1
- WT 5′-TGCA TGCAC GTGT-3′
- mut-1 5′-TGCA aatta GTGT-3′
- Site-2
- WT 5′-TTTC TGCAC TTCT-3′
- mut-2 5′-TTTC aaata TTCT-3′
- Site-3
- WT 5′-GGTT CTGCCC ACCA-3′
- mut-3 5′-GGTT aaaata ACCA-3′
- Site-4
- WT 5′-ACAA TGGGCTGAGT CTTC-3′
- mut-4 5′-ACAA aaataaatta CTTC-3′
Transient Chromatin Immunoprecipitation (ChIP) assays
Immortalized TIEG KO calvarial OBs were plated in 100 mm plates and transfected with 5 μg of the OPG promoter reporter construct and 5 μg of either the empty vector or TIEG expression construct as specified above. Following 48 hours of transfection, cells were fixed in collection buffer, pelleted and resuspended in lysis buffer. The transient ChIP was performed following the previously described protocol [8] using a TIEG-specific antibody. Immunoprecipitated chromatin samples were amplified by PCR using a forward primer that was specific for pGL3 vector, PGL3 ChIP-F 5′-TCCCCAGTGCAAGTGCAGGTGCC-3′ and a reverse primer specific for the OPG promoter, mOPG ChIP-R 5′-TGTAACGTGATGTCAGGGTGCAGT-3′.
Results
OPG and RANKL mRNA levels in TIEG KO and wild-type calvarial osteoblasts
Previously, we have shown that calvarial OBs isolated from TIEG KO mice are not able to support OC differentiation in vitro [5]. It has also been well established that OPG and RANKL, produced by OBs, are critical regulators of osteoclastogenesis. To determine if the expression levels of OPG and RANKL are altered in TIEG KO OBs, real-time PCR analysis was performed. Calvarial OBs isolated from three individual WT and KO pups were used for this study. As shown in Figure 1A, calvarial OBs derived from TIEG KO mice exhibit an approximately 3.5-fold increase in OPG expression and an approximately 3-fold decrease in RANKL expression relative to WT controls.
Figure 1.

A) Increased OPG and decreased RANKL mRNA expression levels in TIEG KO osteoblasts. Calvarial osteoblasts were isolated from three individual WT and KO littermate pups and cultured in vitro. Total RNA was isolated from these cells and real-time PCR analysis was performed to measure OPG and RANKL mRNA levels. The results are expressed as fold change relative to WT cells. The results were repeated in three independent experiments and a representative data set is shown. * indicates significance at the P<0.05 level (ANOVA) compared to WT cells. B) Transient transfection analysis of TIEG mediated OPG and RANKL promoter activity in calvarial osteoblasts. TIEG KO calvarial osteoblasts were transiently transfected with 500 ng of either the OPG or RANKL promoter construct in combination with 500 ng of either the pcDNA4/TO empty vector or pcDNA4/TO-TIEG expression construct. Twenty-four hours following transfection, cells were lysed and analyzed for luciferase activity. The results were normalized to empty vector controls. The results were repeated in three independent experiments and a representative data set is shown. * indicates significance at the P<0.05 level (ANOVA) compared to vector controls.
TIEG regulation of the OPG, but not the RANKL, promoter in osteoblasts
As shown in Figure 1A, TIEG KO OBs display increased OPG and decreased RANKL expression levels compared to WT cells suggesting that TIEG is involved in the regulation of these two genes. To determine if TIEG directly regulates the promoters of these genes, luciferase reporter constructs containing the OPG and RANKL promoters were transiently transfected into TIEG KO OBs with and without a TIEG expression construct. As shown in Figure 1B, TIEG expression significantly inhibited OPG promoter activity by approximately 80% compared to the empty vector transfected cells. However, RANKL promoter activity was not affected by the presence of TIEG (Figure 1B). These data suggest that TIEG protein can directly regulate the upstream region of the OPG promoter but not the RANKL promoter. Since TIEG does not directly regulate this region of the RANKL promoter, the increased expression levels of RANKL in WT OBs is likely due to a secondary (indirect) event, or regulation of another region of the promoter.
TIEG-specific regulation of the OPG promoter occurs through regulatory elements located in the first 200 bp of this promoter fragment
To identify the location of potential regulatory elements for TIEG regulation of the OPG promoter, deletion analysis was performed. As can be seen in Figure 2, a 200 bp deletion of the upstream region completely blocks the ability of TIEG to repress OPG promoter activity, suggesting that the TIEG binding site(s) reside within this 200 bp region.
Figure 2. The regulatory elements for TIEG repression of OPG promoter activity are located within a 200 bp fragment of the promoter.

Deletion constructs were created from the “full length” OPG promoter (−1486 to +133) and cloned into the pGL3 basic luciferase construct. Promoter constructs were transfected into TIEG KO calvarial OBs with or without a TIEG expression construct. Deletion of the first 200 bps of the OPG promoter completely abolishes TIEG’s ability to repress promoter activity; therefore additional deletion constructs are not shown. The results were repeated in three independent experiments and a representative data set is shown. * indicates significance at the P<0.05 level (ANOVA) compared to vector controls.
Transient chromatin immunoprecipitation analysis reveals that TIEG directly binds to elements located in the first 200 bp region of the OPG promoter
To determine if TIEG directly binds to this 200 bp region of the OPG promoter, transient ChIP analysis was performed. As shown in Figure 3, the OPG promoter was immunoprecipitated with a TIEG specific antibody in KO cells transfected with a TIEG expression construct, but not in cells transfected with empty vector. Input DNA was used as an internal loading control. These data suggest that TIEG directly binds to the OPG promoter in OBs to repress its activity.
Figure 3. Transient chromatin immunoprecipitation (ChIP) analysis demonstrates that TIEG protein binds to the OPG promoter.
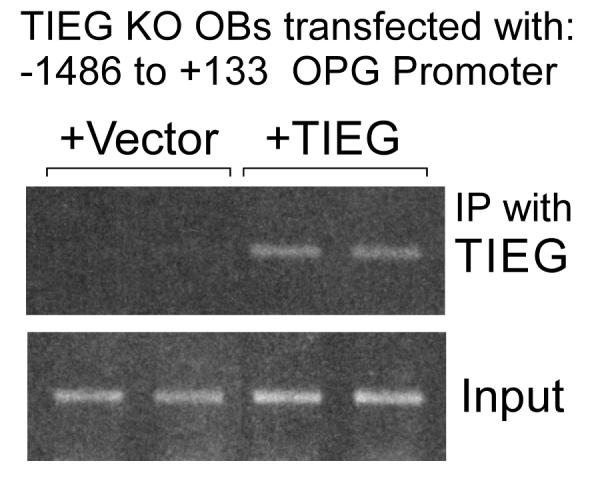
TIEG KO calvarial osteoblasts were transiently transfected with the full-length OPG promoter (5 μg) in combination with either pcDNA4/TO empty vector (5 μg) or pcDNA4/TO-TIEG expression vector (5 μg). Twenty-four hours following transfection, cells were cross-linked and processed for ChIP analysis. Chromatin samples were immunoprecipitated with a TIEG-specific antibody and standard PCR reactions were performed using primers surrounding the first 200 bps of the OPG promoter. Input DNA is shown as a loading control.
Identification of potential Sp-1 binding sites within the first 200 bps of OPG promoter
Previously, we have shown that TIEG protein can bind to Sp-1 like binding sites to down-regulate Smad 7 promoter activity [4]. Others have also shown that TIEG binds to Sp-1 sites in the promoters of other genes to regulate their activity [10, 11]. Therefore, we searched for and identified four potential Sp-1 sites located in the first 200 bps of the OPG promoter (Figure 4A).
Figure 4. Identification of Sp-1 sites important for TIEG regulation of OPG promoter activity.

A) Four potential Sp-1 binding sites were identified within the first 200 bps of the OPG promoter. B) Site-directed mutagenesis was used to alter each of the identified Sp-1 binding sites. pGL3 basic luciferase constructs harboring the full length OPG promoter with or without individually mutated Sp-1 sites were transfected into TIEG KO calvarial osteoblasts in the presence or absence of a TIEG expression construct. Twenty-four hours following transfection, cells were lysed and analyzed for luciferase activity. The results were repeated in three independent experiments and a representative data set is shown. * indicates significance at the P<0.05 level (ANOVA) compared to vector controls. # indicates significance at the P<0.05 level (ANOVA) compared to the full-length OPG promoter.
Mutation of specific Sp-1 binding sites reduces TIEG’s ability to regulate OPG promoter activity
To determine which of the identified Sp-1 sites are critical for TIEG regulation of OPG promoter activity, we created mutation constructs for each of the four Sp-1 sites using a Quick Change mutagenesis kit (Stratagene, CA) in the context of the full length promoter (−1486 to +133). These constructs were transiently transfected into TIEG KO OBs and analyzed for TIEG repression. As shown in Figure 4B, mutation of sites 2 and 3 did not alter the ability of TIEG to repress promoter activity. Mutation of sites 1 and 4 significantly reduced the ability of TIEG to repress OPG promoter activity (Figure 4B). These data indicate that Sp-1 sites 1 and 4 play important roles in mediating the repression of OPG promoter activity by TIEG.
Discussion
In this study, we provide evidence that calvarial OBs isolated from TIEG KO mice express increased levels of OPG and decreased levels of RANKL relative to OBs isolated from WT littermates. Although we observed decreased RANKL expression in TIEG KO OBs, transient transfection studies using the RANKL promoter and a TIEG expression construct did not reveal any significant alterations in promoter activity, suggesting that TIEG regulation of RANKL is either indirect, or occurs through regulatory elements located outside of the cloned fragment of the RANKL promoter (−2000 to +1). However, our results do demonstrate that TIEG expression in OBs can directly inhibit OPG promoter activity. We also demonstrate that TIEG protein directly binds to sites located in the first 200 bp of the cloned OPG promoter. This domain was found to contain four potential Sp-1/TIEG binding sites. By performing site directed mutagenesis of these sites, we have provided evidence that two sites are critical for TIEG repression of OPG promoter activity.
OPG belongs to a tumor necrosis factor receptor (TNFR) superfamily which was originally identified as a secreted glycoprotein [12, 13]. OPG acts as a decoy receptor for RANKL thereby blocking the interaction between RANKL and RANK which in turn inhibits OC differentiation [13, 14]. OPG KO mice demonstrate severe early onset of osteoporosis [14], whereas transgenic mice that over-express OPG exhibit an osteopetrotic phenotype [13]. OPG plays an important role in the regulation of bone turnover and several studies have linked OPG to a number of bone-related diseases such as osteoporosis and rheumatoid arthritis [15].
Studies have also correlated serum OPG levels to post-menopausal osteoporosis. One study reported that serum OPG levels were increased in osteoporosis and another reported a correlation with bone mineral density [16-18]. Using Japanese women as cohorts, Yano et al [16] presented evidence that serum OPG levels increased significantly in post-menopausal osteoporotic women. The results of this study parallel our model system in that TIEG KO mice have increased OPG levels and display a gender-specific, osteopenic bone phenotype.
TIEG belongs to the Krüppel-like family of transcription factors which possess three C2H2 type zinc finger DNA binding domains that can bind to GC-rich Sp-1 sites in DNA. Previously, we have shown that TIEG can bind to GC-rich elements in the Smad 7 promoter to down-regulate its activity [4]. In another independent study, Alvarez-Rodriguez et al [10], have shown evidence that TIEG binds to Sp-1 sites in the N-myc promoter to block its transcriptional activity. Through the use of transient transfection and EMSA assays, Noti et al [11], have shown that TIEG protein competes with Sp-1 and Sp-3 proteins for binding to sites in the CD11d promoter. In this same study, they demonstrate that during myeloid cell differentiation, TIEG binding to Sp-1 sites of the CD11d promoter increases during differentiation of these cells [11]. Our findings that TIEG protein also binds to the OPG promoter via Sp-1 sites provide further evidence for this mechanism of TIEG action.
It has been shown that TGFβ inhibits OC differentiation by increasing OPG and decreasing RANKL mRNA in murine bone marrow precursor cells [19]. In these same studies, the authors demonstrate that TGFβ induces OPG promoter activity in UMR 106 cells. Deletion analysis of the promoter led them to identify Cfa-1 and Smad-binding elements through which TGFβ mediates its effect on OPG promoter activity. Although we have previously demonstrated that TIEG can mimic TGFβ action, it does not appear to do so with regard to regulation of OPG expression in osteoblasts. This could be explained by variations between cell types and/or the expression of specific co-regulators. In order to further determine the role of TIEG in mediating osteoclastogenesis, one would need to generate a mouse model in which TIEG is suppressed only in osteoclasts. Such a model system (TIEG floxed mice) is currently being developed in our laboratory to address these important questions.
In summary, we have provided evidence that OBs isolated from TIEG KO mice exhibit increased OPG and decreased RANKL expression. Our data provide evidence that TIEG directly binds to and regulates OPG expression through specific Sp-1 sites in the OPG promoter. The increased expression of OPG in TIEG KO OBs is likely to explain, at least in part, their decreased ability to support osteoclastogenesis as we have shown previously [5]. This decreased support of OC differentiation is likely to disrupt the cross-talk between OB and OC which is critical for normal bone formation and maintenance. In addition to the previously described OB defects in TIEG KO mice [5], disruption of cross-talk between OBs and OCs in TIEG KO animals is likely to play a role in the development of a gender-specific osteopenic bone phenotype [6, 7]
Acknowledgments
This work was supported by NIH grants R01 DE14036 (TCS), AR52004 (MJO), and the Mayo Foundation. We would like to thank Dr. S. V. Reddy for providing the RANKL promoter used in this study, Jacquelyn House for excellent clerical assistance, and Kenneth Peters for data analysis and graphics generation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Subramaniam M, Harris SA, Oursler MJ, Rasmussen K, Riggs BL, Spelsberg TC. Identification of a novel TGF-beta-regulated gene encoding a putative zinc finger protein in human osteoblasts. Nucleic Acids Res. 1995;23:4907–4912. doi: 10.1093/nar/23.23.4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hefferan TE, Reinholz GG, Rickard DJ, Johnsen SA, Waters KM, Subramaniam M, Spelsberg TC. Overexpression of a nuclear protein, TIEG, mimics transforming growth factor-beta action in human osteoblast cells. J. Biol. Chem. 2000;275:20255–20259. doi: 10.1074/jbc.C000135200. [DOI] [PubMed] [Google Scholar]
- [3].Tachibana I, Imoto M, Adjei PN, Gores GJ, Subramaniam M, Spelsberg TC, Urrutia R. Overexpression of the TGFbeta-regulated zinc finger encoding gene, TIEG, induces apoptosis in pancreatic epithelial cells. J. Clin. Invest. 1997;99:2365–2374. doi: 10.1172/JCI119418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Johnsen SA, Subramaniam M, Janknecht R, Spelsberg TC. TGFbeta inducible early gene enhances TGFbeta/Smad-dependent transcriptional responses. Oncogene. 2002;21:5783–5790. doi: 10.1038/sj.onc.1205681. [DOI] [PubMed] [Google Scholar]
- [5].Subramaniam M, Gorny G, Johnsen SA, Monroe DG, Evans GL, Fraser DG, Rickard DJ, Rasmussen K, van Deursen JM, Turner RT, Oursler MJ, Spelsberg TC. TIEG1 null mouse-derived osteoblasts are defective in mineralization and in support of osteoclast differentiation in vitro. Mol. Cell. Biol. 2005;25:1191–1199. doi: 10.1128/MCB.25.3.1191-1199.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bensamoun SF, Hawse JR, Subramaniam M, Ilharreborde B, Bassillais A, Benhamou CL, Fraser DG, Oursler MJ, Amadio PC, An KN, Spelsberg TC. TGFbeta inducible early gene-1 knockout mice display defects in bone strength and microarchitecture. Bone. 2006a;39:1244–1251. doi: 10.1016/j.bone.2006.05.021. [DOI] [PubMed] [Google Scholar]
- [7].Hawse JR, Iwaniec UT, Bensamoun SF, Monroe DG, Peters KD, Ilharreborde B, Rajamannan NM, Oursler MJ, Turner RT, Spelsberg TC, Subramaniam M. TIEG-null mice display an osteopenic gender-specific phenotype. Bone. 2008;42:1025–1031. doi: 10.1016/j.bone.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hawse JR, Subramaniam M, Monroe DG, Hemmingsen AH, Ingle JN, Khosla S, Oursler MJ, Spelsberg TC. Estrogen receptor beta isoform-specific induction of transforming growth factor beta-inducible early gene-1 in human osteoblast cells: an essential role for the activation function 1 domain. Mol. Endocrinol. 2008;22:1579–1595. doi: 10.1210/me.2007-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sundaram K, Mani SK, Kitatani K, Wu K, Pestell RG, Reddy SV. DACH1 negatively regulates the human RANK ligand gene expression in stromal/preosteoblast cells. J. Cell. Biochem. 2008;103:1747–1759. doi: 10.1002/jcb.21561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Alvarez-Rodriguez R, Barzi M, Berenguer J, Pons S. Bone morphogenetic protein 2 opposes Shh-mediated proliferation in cerebellar granule cells through a TIEG-1-based regulation of Nmyc. J. Biol. Chem. 2007;282:37170–37180. doi: 10.1074/jbc.M705414200. [DOI] [PubMed] [Google Scholar]
- [11].Noti JD, Johnson AK, Dillon JD. The zinc finger transcription factor transforming growth factor beta-inducible early gene-1 confers myeloid-specific activation of the leukocyte integrin CD11d promoter. J. Biol. Chem. 2004;279:26948–26958. doi: 10.1074/jbc.M310634200. [DOI] [PubMed] [Google Scholar]
- [12].Tsuda E, Goto M, Mochizuki S, Yano K, Kobayashi F, Morinaga T, Higashio K. Isolation of a novel cytokine from human fibroblasts that specifically inhibits osteoclastogenesis. Biochem. Biophys. Res. Commun. 1997;234:137–142. doi: 10.1006/bbrc.1997.6603. [DOI] [PubMed] [Google Scholar]
- [13].Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, Shimamoto G, DeRose M, Elliott R, Colombero A, Tan HL, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Sander S, Van G, Tarpley J, Derby P, Lee R, Boyle WJ. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- [14].Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, Boyle WJ, Simonet WS. osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12:1260–1268. doi: 10.1101/gad.12.9.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Reid P, Holen I. Pathophysiological roles of osteoprotegerin (OPG) Eur. J. Cell Biol. 2009;88:1–17. doi: 10.1016/j.ejcb.2008.06.004. [DOI] [PubMed] [Google Scholar]
- [16].Yano K, Tsuda E, Washida N, Kobayashi F, Goto M, Harada A, Ikeda K, Higashio K, Yamada Y. Immunological characterization of circulating osteoprotegerin/osteoclastogenesis inhibitory factor: increased serum concentrations in postmenopausal women with osteoporosis. J. Bone Miner. Res. 1999;14:518–527. doi: 10.1359/jbmr.1999.14.4.518. [DOI] [PubMed] [Google Scholar]
- [17].Rogers A, Saleh G, Hannon RA, Greenfield D, Eastell R. Circulating estradiol and osteoprotegerin as determinants of bone turnover and bone density in postmenopausal women. J. Clin. Endocrinol. Metab. 2002;87:4470–4475. doi: 10.1210/jc.2002-020396. [DOI] [PubMed] [Google Scholar]
- [18].Mezquita-Raya P, de la Higuera M, Garcia DF, Alonso G, Ruiz-Requena ME, de Dios Luna J, Escobar-Jimenez F, Munoz-Torres M. The contribution of serum osteoprotegerin to bone mass and vertebral fractures in postmenopausal women. Osteoporos. Int. 2005;16:1368–1374. doi: 10.1007/s00198-005-1844-1. [DOI] [PubMed] [Google Scholar]
- [19].Thirunavukkarasu K, Miles RR, Halladay DL, Yang X, Galvin RJ, Chandrasekhar S, Martin TJ, Onyia JE. Stimulation of osteoprotegerin (OPG) gene expression by transforming growth factor-beta (TGF-beta). Mapping of the OPG promoter region that mediates TGF-beta effects. J. Biol. Chem. 2001;276:36241–36250. doi: 10.1074/jbc.M104319200. [DOI] [PubMed] [Google Scholar]