

Abstract
Although the toxic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on T cells in vivo have been well characterized, attempts to reproduce these findings in vitro have not been successful. In the current study, we examined whether activation or the presence of dendritic cells (DCs) would make primary naive T cells from C57BL/6 mice susceptible to TCDD-induced apoptosis in vitro. Although nonactivated primary T cells cultured with 10 to 1000 nM TCDD were relatively resistant to apoptosis, they became sensitive to apoptosis upon activation with concanavalin A (ConA). Moreover, ConA-activated T cells cultured in the presence of DCs showed highest levels of TCDD-induced apoptosis. Likewise, primary T cells from OT.II.2a mice cultured with specific ovalbumin peptide and syngeneic DCs showed higher levels of apoptosis compared with similar nonactivated T cells. T-cell activation led to up-regulation of aryl hydrocarbon receptor (AhR), Fas, and Fas-ligand (FasL) expression. In addition, DC maturation and culture with TCDD caused significant induction of FasL. TCDD-mediated apoptosis in activated peripheral T cells was AhR-dependent. Analysis of why nonactivated T cells are more resistant, whereas activated T cells are sensitive to TCDD-induced apoptosis revealed that TCDD treatment of activated but not nonactivated T cells led to down-regulation of cellular FLICE inhibitory protein (c-FLIP), an inhibitor of apoptosis. Moreover, down-regulation of c-FLIP using small interfering RNA in nonactivated T cells made them sensitive to TCDD-induced apoptosis. The current study demonstrates for the first time that TCDD can induce apoptosis in vitro in peripheral T cells upon activation and in the presence of DCs and that this may be mediated by down-regulation of c-FLIP.
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) is an environmental pollutant that mediates toxicity through activation of the aryl hydrocarbon receptor (AhR), a ligand-dependent transcription factor and member of the basic helix-loop-helix–Per/Arnt/Sim (periodicity/aryl hydrocarbon receptor nuclear translocator/simple-minded) gene family (Marlowe and Puga, 2005; Bock and Kohle, 2006; Harper et al., 2006). TCDD binds and activates cytosolic AhR attached to 90-kDa heat shock protein; and upon activation, AhR dissociates from 90-kDa heat shock protein, migrates to the nucleus, dimerizes with AhR nuclear translocator (ARNT), and forms AhR/ARNT heterodimer. This in turn binds to dioxin-response elements (DRE), present as cis-acting elements in the regulatory regions of dioxin-responsive genes, and it up-regulates a large set of genes that directly or indirectly participate in TCDD-induced toxicity (Schmidt and Bradfield, 1996; Gonzalez and Fernandez-Salguero, 1998; Tian et al., 1999; Whitlock, 1999; Sulentic et al., 2000; Dertinger et al., 2001; Matikainen et al., 2001; Nazarenko et al., 2001; Mimura and Fujii-Kuriyama, 2003). Targeted disruption of the AhR in mice prevents induction of CYP1A1 and most forms of TCDD toxicity (Marlowe and Puga, 2005; Bock and Kohle, 2006; Harper et al., 2006).
Extensive previous studies have shown that the immune system is very sensitive to the toxic effects of TCDD (Kerkvliet, 2002). It is noteworthy that although TCDD triggers thymic atrophy in all species tested, it is less toxic to naive secondary lymphoid organs, including the spleen and lymph nodes. However, upon immunization of animals with an antigen, TCDD is known to suppress the antigen-specific T-cell response in secondary lymphoid organs (Kerkvliet, 2002). Yet another interesting feature about TCDD-induced immunotoxicity is that although TCDD has been shown to readily suppress antigen-specific T-cell responses as well as trigger thymic atrophy in vivo, attempts to demonstrate the direct toxic effects of TCDD in vitro against thymocytes and naive T cells have not been successful (Kerkvliet, 2002). Moreover, previous studies from our laboratory and elsewhere suggested that TCDD altered the functions of T cells activated through antigen priming in vivo, but it failed to affect resting T cells (Lundberg et al., 1992; Rhile et al., 1996; Pryputniewicz et al., 1998). Recent studies from our laboratory demonstrated that TCDD up-regulates FasL expression on thymic stromal cells and Fas on thymic T cells and that when such cells come in contact with each other, the latter cells undergo apoptosis (Camacho et al., 2005). In the current study, we speculated that a similar mechanism may be operative in the induction of apoptosis in antigen-activated primary T cells. Thus, we hypothesized that TCDD may act on antigen-presenting cells such as the DCs and increase FasL expression in such a way that when they come in contact with Fas+ antigen-specific T cells during antigen presentation, the T cells would undergo apoptosis. We also analyzed why activated peripheral T cells become susceptible to TCDD, and we noted that c-FLIP may play a crucial role in regulating apoptosis. The current study has identified certain key cells and molecules that regulate TCDD-induced apoptosis in activated peripheral T cells.
Materials and Methods
Mice
C57BL/6 (H-2b) mice were purchased from the National Institutes of Health (Bethesda, MD). AhR knockout (KO) (Chris Bradfield; on C57BL/6J background) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). OT.II.2a (C57BL/6-TgN(OT-II.2a)-RAG1tm1Mom) mice were purchased from Taconic Farms (Hudson, NY). All animals were housed in University of South Carolina Animal facility (Columbia, South Carolina). Care and maintenance of animals were carried out in accordance with the guide for the care and use of laboratory animals as adopted by Institutional and National Institutes of Health guidelines.
Reagents and Antibodies
TCDD was a generous gift from Dr. K. Chae (National Institute of Environmental Health Sciences (Research Triangle Park, NC). TCDD dissolved in DMSO was used in the in vitro studies.
l-Glutamine, HEPES, gentamicin, RPMI 1640 medium, Dulbecco’s modified Eagle’s medium, PBS, and fetal bovine serum were purchased from Invitrogen (Carlsbad, CA). ConA was purchased from Sigma-Aldrich (St. Louis, MO). The following monoclonal antibodies were purchased from BD Biosciences Pharmingen (San Diego, CA): anti-mouse IgG-PE, FcBlock, CD3-PE (chain)-purified anti-FasL (k-10), anti-FasL-PE (Kay-10), and anti-Fas-PE (Jo2). The following inhibitors against caspase-3 (Z-DEVD), caspase-8 (Z-IETD-FMK), and caspase-9 (Z-LEHD-FMK) were purchased from R&D Systems (Minneapolis, MN). The following primary antibodies for Western blots were used: c-FLIP (Cell Signaling Technology Inc., Danvers, MA) and β-actin (Sigma-Aldrich). HRP-conjugated secondary Abs was purchased from Cell Signaling Technology Inc. RNeasy Mini kit and iScript cDNA synthesis kit were purchased from QIAGEN (Valencia, CA). Epicenter’s PCR premix F and Platinum Taq Polymerase kits were purchased from Invitrogen. TUNEL kits were purchased from Roche Diagnostics (Indianapolis, IN). α-Naphthoflavone (ANF), an antagonist for AhR, was purchased from Sigma-Aldrich.
Assessment of TCDD-Induced Apoptosis in Primary T Cells
To determine TCDD-induced apoptosis in primary T cells, T cells from the spleens of three C57BL/6 mice were purified using nylon wool column (Polysciences, Warrington, PA), and the purity as determined by expression of CD3 and analysis by flow cytometry (Cytomics FC 500; Beckman Coulter, Fullerton, CA) was found to be >90%. Because ConA activation of T cells requires accessory cells such as macrophages or DCs, we did not attempt to purify T cells any further. Next, T cells were cultured with 2.5 μg/ml ConA for 24 h (referred to as activated) or without ConA (referred to as nonactivated), and then they were harvested, washed, and subsequently treated with different concentrations (10–1000 nM) of TCDD for an additional 24 h. Apoptosis in naive or activated T cells with or without DCs after TCDD or vehicle exposure was determined by performing TUNEL assays (FITC-dUTP nick-end labeling) using In Situ Cell-Death Detection kit (Roche Diagnostics) as described previously (Kamath et al., 1997, 1999). In brief, T cells were harvested after TCDD or vehicle treatment, and then they were washed twice with ice-cold PBS. Cells were then fixed in 4% paraformaldehyde for 30 to 45 min at room temperature. The cells were washed twice with ice-cold PBS, and then they were permeabilized using freshly prepared permeabilization solution and incubated at 4°C for 2 min. The cells were washed once with PBS, and then they were resuspended in 50 μl/tube TUNEL reaction mixture and incubated for 60 min at 37°C in a humidified atmosphere in the dark. The cells were washed twice with PBS, and finally they were suspended in 200 to 500 μl of ice-cold PBS and analyzed using flow cytometry (Cytomics FC 500; Beckman Coulter) and software.
Effect of DCs on TCDD-Induced Apoptosis in Nonactivated or Activated T Cells
In some experiments, purified T cells were cultured in the absence or presence of ConA for 24 h as described above. Next, mature syngeneic DCs, generated from bone marrow of C57BL/6 mice, were added to these cultures along with TCDD or vehicle, and the cultures were incubated for an additional 24 h. Apoptosis in T cells was determined by staining the cells with PE-anti-CD3 Abs and FITC-dUTP. DCs were generated from the bone marrows of C57BL/6 mice by culturing the bone marrow cells with granulocyte macrophage–colony-stimulating factor and interleukin-14 (immature) followed by additional incubation with LPS (mature) as described in our previous work (Do et al., 2004a,b). We also examined TCDD-induced apoptosis in antigen-specific activated T cells. To this end, we used purified T cells from OT.II.2a mice, and we cultured them in the absence or presence of mature syngeneic DCs pulsed with agonist ovalbumin peptide (Ova323-339: ISQAVHAAHAEINEAGR) for 3 days followed by addition of vehicle or TCDD to the cultures. Twenty-four hours later, the T-cell proliferation and apoptosis were studied as described previously.
Determination of Fas and FasL Expression in Dendritic Cells
Expression of Fas and FasL in immature and mature dendritic cells was analyzed by using flow cytometry and reverse transcriptase-polymerase chain reaction (RT-PCR). In brief, dendritic cells from C57BL/6 mice were generated as described previously (Do et al., 2004a,b). Immature DCs (without LPS) or mature DCs (pulsed with 100 nM LPS) for 48 h were cultured in the presence of vehicle or various concentrations (10–1000 nM) of TCDD for 24 h. Immature and mature DCs were harvested 24 h after vehicle or TCDD treatment, and then they were analyzed for Fas and FasL expression using flow cytometry or RT-PCR as described previously (Singh et al., 2007). For flow cytometry, FITC-labeled anti-mouse Fas and PE-labeled anti-mouse FasL antibodies were used to stain DCs. For RT-PCR, total RNA from immature and mature DCs was isolated using RNeasy Mini kit and following the protocol of the company (QIAGEN). First-strand cDNA synthesis was performed in a 20-μl reaction mix containing 2 μg of total RNA using iScript kit and following the protocol of the manufacturer (Bio-Rad Laboratories, Hercules, CA). PCR was performed using mouse FasL or Fas-specific sets of forward and reverse primers. To detect mouse FasL expression (435 bp), forward (5′-CGG TGG TAT TTT TCA TGG TTC TGG-3′) and reverse (5′-CTT GTG GTT TAG GGG CTG GTT GTT-3′) primers were used. Likewise, to detect the expression of mouse Fas (486 bp), forward (5′-TCT GGT GCT TGC TGG CTC AC-3′) and reverse (5′ CCA TAG GCG ATT TCT GGG AC-3′) primers were used. PCRs for both Fas and FasL were performed for 30 cycles using the following conditions: 30 s at 95°C (denaturing temperature), 40 s at 60°C (annealing temperature), and 60 s at 72°C (extension temperature), with a final incubation of 10 min at 72°C. The PCR products, generated from mouse Fas and FasL primer pairs, were normalized against PCR products generated from mouse 18S-specific forward 5′-GCC CGA GCC GCC TGG ATA C-3′ and reverse 5′-CCG GCG GGT CAT GGG AAT AAC-3′ primers after electrophoresis on 1.5% agarose gel and visualization with UV light. The band intensity of PCR products was determined using Chemi-Doc, a Bio-Rad Laboratories image analysis system.
RT-PCR to Determine the Expression of Fas and FasL in T Cells
For detection of mouse FasL (435-bp) and Fas (486-bp) expression, sets of forward and reverse primers specific to mouse Fas/FasL were used (as described above). The PCR products, generated from mouse Fas and FasL primer pairs, were normalized against PCR products generated from β-actin (427-bp) forward (5′-AAG GCC AAC CGT GAA AAG ATG ACC-3′) and reverse (5′-ACC GCT CGT TGC CAA TAG TGA TGA-3′) primers after electrophoresis on 1.5% agarose gel and visualization with UV light. In some experiments, we used 18S as a control. The band intensity of PCR products was determined using Chemi-Doc.
Blocking Assays to Determine the Role of FasL in TCDD-Mediated T-Cell Apoptosis
To investigate the role and participation of FasL in TCDD-mediated apoptosis in primary T cells, we used ConA-activated T cells as described above, and we cultured them in the absence or presence of antibody against mouse FasL (1–5 μg/ml) 1 h before 100 nM TCDD treatment. Apoptosis in T cells 24 h after TCDD treatment was determined as described previously. Data from three to four independent experiments were pooled, and they are depicted as mean fluorescence units ± S.E.M.
RT-PCR to Determine the Expression of AhR in T Cells
To detect the expression of AhR in nonactivated or ConA-activated primary T cells treated with vehicle (DMSO) or TCDD (482-bp), forward (5′-GCG GCC GCA GGA AGT GAG G-3′) and reverse (5′-GTG CCG TTG ATT TGC GTG TGCT-3′) primers specific to mouse AhR were used. PCR was performed for 30 cycles using the following conditions: 30 s at 95°C (denaturing temperature), 40 s at 60°C (annealing temperature), and 60 s at 72°C (extension temperature), with a final incubation of 10 min at 72°C. The PCR products, generated from mouse AhR primer pairs, were normalized against PCR products generated from β-actin as described above for Fas and FasL. The band intensity of PCR products was determined using ChemiDoc image analysis system (Bio-Rad Laboratories).
Role of AhR in TCDD-Induced Regulation of Fas and FasL Expression in T Cells
To determine the role of AhR in TCDD-induced up-regulation of Fas and FasL expression in T cells, we performed series of in vitro assays using nonactivated and ConA-activated T cells from wild-type (C57BL/6) and AhR KO mice. In brief, nonactivated and ConA-activated (2.5 μg/ml/24 h) purified T cells from wild-type or AhR KO mice were treated with vehicle (DMSO) or 10 to 1000 nM TCDD for 24 h. Cells were harvested 24 h after vehicle or TCDD treatment, and then they were stained using FITC-labeled anti-mouse Fas- and PE-labeled anti-mouse FasL antibodies. Cells were analyzed for Fas and FasL expression in T cells using flow cytometry (Cytomics FC 500; Beckman Coulter).
Role of AhR in TCDD-Induced T-Cell Apoptosis
To determine the role of AhR in TCDD-induced apoptosis, we performed a series of in vitro assays using T cells from wild-type (C57BL/6) and AhR KO mice. In brief, purified T cells from wild-type or AhR KO mice were activated with ConA (2.5 μg/ml) for 24 h, and then they were treated with vehicle (DMSO) or 100 to 1000 nM TCDD. Apoptosis in T cells 24 h after TCDD treatments was determined by performing TUNEL assays and using flow cytometry (Cytomics FC 500; Beckman Coulter) as described above. Furthermore, 1 μM ANF, an antagonist for AhR, was added in the culture of wild-type T cells 1 h before TCDD treatment, and apoptosis was determined. Data from three to four independent experiments were pooled, and they are depicted as mean fluorescence units ± S.E.M.
Analysis of Caspase-3/7, Caspase-8, and Caspase-9 Activity
Activities of caspase-3/7, -8, and -9 were measured in T cells exposed to TCDD using the Apo-ONE homogeneous caspase-3/7, caspase-8, and caspase-9 assays according to manufacturer’s instructions (Promega, Madison, WI). In brief, ConA-activated T cells were treated with various concentrations (1–1000 nM) of TCDD or vehicle (DMSO) for 24 h at 37°C, 5% CO2. The following day, the cells were collected and used for caspase assays. A Wallac 1420 multilabel counter, Victor2 (PerkinElmer Life and Analytical Sciences, Boston, MA) was used to measure the relative fluorescence units of each sample at an excitation wavelength of 485 nm and at an emission wavelength of 535 nm. Luminescence of caspase-8 and caspase-9 was also measured using Wallac 1420 multilabel counter, Victor2 (PerkinElmer Life and Analytical Sciences). Data from three to four independent experiments were pooled, and they are depicted as mean fluorescence units ± S.E.M.
Caspase Blocking Assays to Determine the Role of Various Caspases in TCDD-Induced T-Cell Apoptosis
To investigate the role and participation of various caspases in TCDD-mediated apoptosis in primary T cells, we performed in vitro assays as described above, and inhibitors specific to mouse caspase-3 (Z-DEVD), caspase-8 (Z-IETD-FMK), and caspase-9 (Z-LEHD-FMK) at a concentration of 20 μM were added in the culture. The cells were incubated with caspase inhibitor for at least 1 h before TCDD treatment. These inhibitors were purchased from R&D Systems. The cells were harvested 24 h after vehicle or TCDD treatment, and TUNEL assays were performed for apoptosis as described previously. At least three independent experiments were performed, and the data are shown represent one of the experiments.
Analysis of Mitochondrial Membrane Potential
Mitochondrial membrane potential (Δψm) of T cells after vehicle or TCDD exposure was determined using 3,3′-dihexyloxacarboeczyme (DiOC6; Sigma-Aldrich) as described previously (Singh et al., 2007). In brief, ConA-activated T cells were treated with TCDD or vehicle for 24 h at 37°C, 5% CO2. The cells were harvested, washed twice with ice-cold PBS, and then they were incubated in the presence of 40 nM DiOC6 for 30 min at 4°C. After washing several times with ice-cold PBS, the cells were suspended in 200 to 500 μl of PBS, and then they were analyzed using flow cytometry (Cytomics FC 500; Beckman Coulter) and software. Propidium iodide was used to differentiate the dead cells. At least three independent experiments were performed.
RT-PCR to Detect Expression of c-FLIP in T Cells after TCDD Treatment
To detect the expression of c-FLIP in T cells after vehicle or TCDD treatment, total RNA from various experimental samples was isolated, and cDNAs were synthesized as described above. To detect the expression of c-FLIP (518-bp), forward (5′-GTCCTGCTGATGGAGATTG-3′) and reverse (5′-GCTCCTTGGCT-GGAC TGGG-3′) primers specific to mouse c-FLIP were used. PCR was performed for 30 cycles using the following conditions: 30 s at 95°C (denaturing temperature), 40 s at 58°C (annealing temperature), and 60 s at 72°C (extension temperature), with a final incubation for 10 min at 72°C. The PCR products, generated from experimental samples of mouse T cells were normalized against PCR products, generated from β-actin (427 bp) forward (5′-AAG GCC AAC CGT GAA AAG ATG ACC-3′) and reverse (5′-ACC GCT CGT TGC CAA TAG TGA TGA-3′) primers after electrophoresis on 1.5% agarose gel and visualization of the PCR products with UV light. The band intensity of PCR products was determined using Chemi-Doc.
Immunoblot Analysis
Immunoblotting was performed as described previously (Singh et al., 2007). Cell lysates were prepared by freezing and thawing, and the protein concentration was measured using standard Bradford assay (Bio-Rad Laboratories). The proteins were fractionated in 12% SDS-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes using a DryBlot apparatus (Bio-Rad Laboratories). The membrane was incubated in blocking buffer for 1 h at room temperature, followed by incubation in mouse-specific c-FLIP (1:1000; Cell Signaling Technology Inc.) primary antibody, and β-actin (1:5000; Sigma-Aldrich) primary antibody at 4°C overnight. HRP-conjugated secondary Ab was used at 1:2000 dilutions (Cell Signaling Technology Inc.). The membrane was then washed three times (10–15 min) with washing buffer (PBS + 0.2% Tween 20), and then it was incubated for 1 h in HRP-conjugated secondary antibody (Cell Signaling Technology Inc.) in blocking buffer. The membrane was then washed several times and incubated in developing solution (equal volume of solution A and B; enhanced chemiluminescence Western blotting detection reagents; GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK), and signal was detected using ChemiDoc System (Bio-Rad Laboratories). Densitometric analyses of the Western blots were performed using ChemiDoc software (Bio-Rad Laboratories).
Transfection of Mouse Primary T Cells with Mouse c-FLIP siRNA
Primary T cells (5 × 106) purified from C57BL/6 mice were transfected with 1.7 μM mouse siGenome ON-TARGETplus-SMART pool duplex (5-8) small interfering RNAs (siRNAs) (Dharmacon RNA Technologies, Lafayette, CO) using nucleofection of primary T cells with mouse T-cell Nucleofector transfection reagent kit and Nucleo-factor II electroporation system following the protocols of the company (Amaxa Biosystems, Gaithersburg, MD). As a control, 5 × 106 mouse T cells were transfected with mouse-specific control SiGLO RISK-free siRNA (3 μg; designated control siRNA) unconjugated or conjugated with Cy-3 (pool D-001206-13-05; Dharmacon RNA Technologies) and pmaxGFP plasmid (2 μg). Transfected T cells were cultured for 48 h in complete medium at 37°C and 5% CO2. We observed >85% cell viability after transfection. To examine the efficiency of transfection, we observed pmaxGFP plasmid-transfected T cells under fluorescent microscope and observed >60% cells expressing green fluorescent protein. We also examined the expression of fluorescent protein by performing flow cytometry (Cytomics FC 500; Beckman Coulter). T cells, untransfected (nonactivated and ConA-activated) or transfected with control siRNA or mouse-specific c-FLIP siRNAs, were treated with vehicle or 100 nM TCDD. T cells 24 h after treatment were harvested and apoptosis was determined by performing TUNEL assay and using flow cytometry (Cytomics FC 500; Beckman Coulter).
Statistical Analysis
Results presented here represent at least four to five independent experiments, and they are presented as the mean ± S.E.M. Statistical analyses were performed using Prism software (GraphPad Software Inc., San Diego, CA). Student’s t test was used for paired observations if data followed a normal distribution to compare TCDD-induced apoptosis in T cells, expression and quantification of Fas, FasL, and AhR in DCs or in T cells, caspase assays, and quantification of proteins. The multiple comparisons were made using one-way analysis of variance (ANOVA) test and Tukey-Kramer multiple comparisons tests. A p value of ≤0.05 is considered to be statistically significant.
Results
TCDD-Mediated Apoptosis in Primary T Cells Required Their Activation and/or Their Interaction with Dendritic Cells
We first examined TCDD-induced apoptosis in activated and nonactivated primary peripheral T cells in vitro. To this end, T cells were purified from the spleens of C57BL/6 mice and cultured in the absence or presence of ConA for 24 h. Next, the cultures were incubated with TCDD or vehicle for an additional 24 h, and cell viability and apoptosis were determined. In some experimental settings, mature syngeneic DCs were added in both nonactivated and activated T cell cultures at the time of addition of TCDD. We observed a dose-dependent decrease in viability of T cells incubated with TCDD, and activated T cells were more sensitive compared with nonactivated T cells (Fig. 1A). Similar results were obtained using TUNEL assays in which we noted that activated primary T cells were more sensitive to apoptosis compared with nonactivated T cells. It is noteworthy that when we cultured the nonactivated T cells in the presence of syngeneic DCs and TCDD, we noted apoptosis in T cells (Fig. 1, B and C). However, we noted highest levels of apoptosis in T cells when activated T cells were cultured in the presence of syngeneic DCs and TCDD (Fig. 1, B and C). It should be noted that in these assays, the lowest concentration at which TCDD triggered significant levels of apoptosis was 10 nM. We did not observe apoptosis in nonactivated or activated T cells with or without DCs when 1 nM TCDD was used in the culture (data not presented). These data demonstrated that primary peripheral T cells become more susceptible to TCDD in vitro when they are activated with ConA and especially in the presence of mature syngeneic DCs.
Fig. 1.
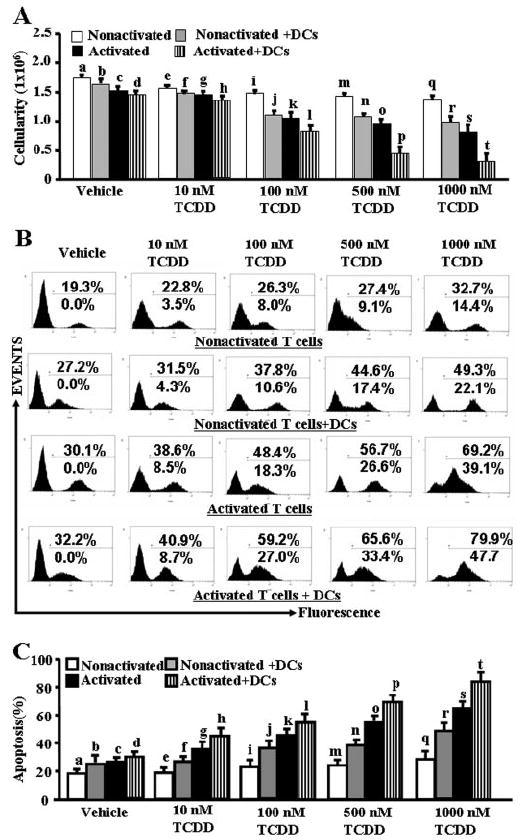
Effect of TCDD on induction of apoptosis in activated and nonactivated primary T cells in vitro. Purified primary T cells from C57BL/6 mice were cultured in the presence of ConA (activated) or absence (nonactivated) for 24 h followed by an additional culture for 24 h with TCDD. In some cultures, mature syngeneic DCs, generated from bone marrow of C57BL/6 mice, were added to these cultures along with 10 to 1000 nM TCDD or vehicle (DMSO), and the cultures were incubated for an additional 24 h. Apoptosis in T cells was determined by staining the cells with PE-anti-CD3 Abs and FITC-dUTP. The PE+ gated cells were analyzed for apoptosis using flow cytometry (Cytomics FC 500; Beckman Coulter). Data are depicted as viable cellularity (A) and apoptosis as determined by TUNEL assays (B and C). In A and C, the following comparisons made using ANOVA test and Tukey-Kramer multiple comparisons test between various groups were statistically significant (p < 0.05): vehicle versus TCDD-treated nonactivated T cells (a versus e, i, m, and q), vehicle versus TCDD-treated nonactivated T cells + DCs (b versus f, j, n, and r), vehicle versus TCDD-treated activated T cells (c versus g, k, o, and s), and vehicle versus TCDD-treated activated T cells + DCs (d versus h, l, p, and t). In addition, the following comparisons made using Student’s t test between different groups after 10 to 1000 nM TCDD treatment were also statistically significant (p < 0.05): nonactivated T cells versus nonactivated T cells + DCs (e versus f, i versus j, m versus n, and q versus r), activated T cells versus activated T cells + DCs (g versus h, k versus l, o versus p, and s versus t), nonactivated versus activated T cells (e versus g, i versus k, m versus o, and q versus s), and nonactivated T cells + DCs versus activated T cells + DCs (f versus h, j versus l, n versus p, and r versus t). In B, the top percentage data represent the actual percentage of apoptosis obtained in each histogram, and the bottom percentage data represent the percentage of apoptosis seen after subtracting the background apoptosis in vehicle-treated cells. In A and C, data from five independent experiments were analyzed and depicted as mean ± S.E.M. B is a representative experiment showing the histograms.
TCDD Caused Apoptosis in Antigen-Specific Activated T Cells
Normally, professional antigen-presenting cells (APCs) such as DCs present the processed antigens to T cells during an antigen-specific T-cell response. We therefore addressed whether during such T-cell DC interaction, presence of TCDD would promote apoptosis in T cells. To this end, we used purified T cells from OT.II.2a mice, and we cultured them in the presence of mature syngeneic DCs in the absence or presence of specific agonist ovalbumin peptide (Ova323-339: ISQAVHAAHAEINEAGR) for 3 days, and then we treated them with different concentrations (100–1000 nM) of TCDD for 24 h. In these experiments, we also included cultures in which nonactivated T cells from OT.II.2a mice were incubated with TCDD for an additional 24 h. The T-cell proliferation was measured by [3H]thymidine incorporation, and apoptosis in T cells was determined by gating CD3-positive T cells and detecting TUNEL-positive cells. We noted a dose-dependent decrease in the proliferative response of T cells to Ova323-339, upon TCDD treatment (Fig. 2A). We observed low levels of TCDD-induced apoptosis in nonactivated primary T cells (Fig. 2, B and C). It is noteworthy that Ova-specific T cells cultured with DCs in the absence of specific peptide also showed significant sensitivity to TCDD-induced apoptosis. However, highest levels of apoptosis were detected in T cells cultured with DCs + Ova peptide (Fig. 2, B and C). Together, these data demonstrated that DCs play a critical role in inducing apoptosis in T cells in the presence of TCDD.
Fig. 2.
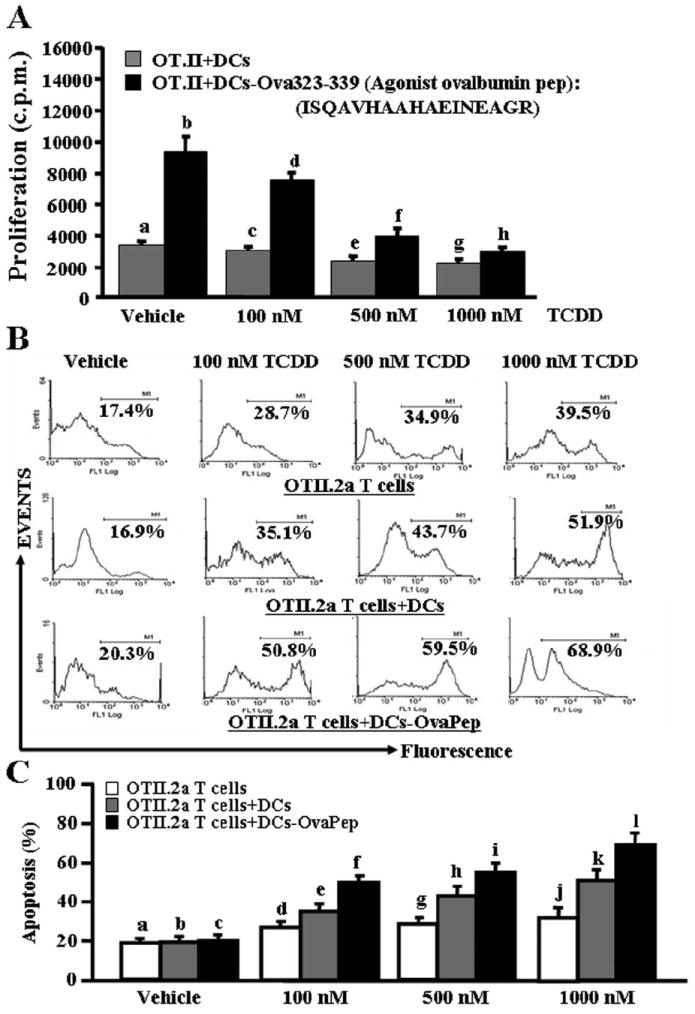
TCDD causes apoptosis in antigen-specific activated T cells in vitro. Purified primary T cells from OT.II.2a mice were activated with ovalbumin-specific peptide (ISQAVHAAHAEINEAGR) by coculturing T cells and peptide-pulsed mature DCs for 3 days, and then they were treated with various doses of TCDD (100–1000 nM; A–C). Proliferation of T cells was determined by [3H]thymidine incorporation (A) 24 h after TCDD exposure and apoptosis as determined by TUNEL assays (B and C). A represents mean of triplicate cultures ± S.E.M. The following comparisons made using ANOVA and Tukey-Kramer multiple comparisons test between various groups were statistically significant (p < 0.05): proliferation of vehicle versus TCDD-treated Ova-activated OT.II.2a T cells in the presence of DCs (b versus d, f, and h). No significant (p > 0.05) difference in proliferation was observed between vehicle and TCDD-treated nonactivated OT.II.2a T cells + DCs (a versus c, e, and g). B is representative of three independent TUNEL assays, and data are depicted as mean ± S.E.M. in C. Statistical analysis using ANOVA and Tukey-Kramer multiple comparisons test showed significant (p < 0.05) difference in apoptosis between the following groups: vehicle versus TCDD-treated nonactivated OT.II.2a T cells (a versus d, g, and j), vehicle versus TCDD-treated nonactivated OT.II.2a T cells + DCs (b versus e, h, and k), and vehicle versus TCDD-treated Ova-activated OT.II.2a T cells + DCs (c versus f, i, and l).
TCDD Up-Regulated Fas and FasL Expression in Mature DCs
Bone marrow-derived immature and mature DCs exposed to vehicle or various concentrations (10–1000 nM) of TCDD for 24 h were examined for expression of Fas and FasL by flow cytometry and RT-PCR using mouse Fas/FasL-specific primers. More than 90% of all DC cultures tested expressed Fas (Fig. 3A). Furthermore, immature DCs expressed low levels of Fas and upon maturation, they expressed significantly higher levels of Fas as indicated by an increase in mean fluorescent intensity (Fig. 3). Exposure of all DC cultures to TCDD did not cause a significant increase in Fas expression. Unlike Fas expression, only a small percentage of immature DCs expressed FasL. DC maturation did not increase the density of FasL but caused a significant increase in the percentage of cells expressing FasL in vehicle-treated groups (Fig. 3, B and C). Addition of TCDD to DC cultures increased the percentage of cells expressing FasL and the density of FasL particularly in mature DCs pulsed with the peptide (Fig. 3, B and C). RT-PCR data further corroborated data generated by flow cytometry (Fig. 3, D and E).
Fig. 3.
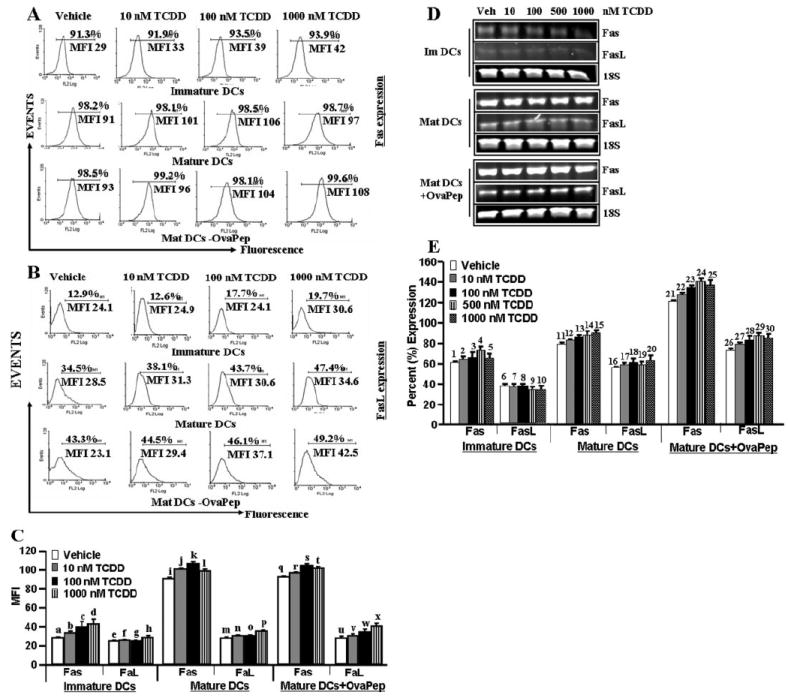
Mature DCs express up-regulated Fas and FasL upon TCDD exposure. Immature, mature, and mature DCs pulsed with ova peptides (OvaPep), generated from C57BL/6 mice were cultured in the presence of vehicle (DMSO) or 10 to 1000 nM TCDD for 24 h. Expression of Fas and FasL in immature and mature DCs was determined by flow cytometry (A–C) or RT-PCR (D) 24 h after TCDD treatments. Data in A and B are representatives of three independent assays, which are plotted in C as mean ± S.E.M. As summarized in C, Fas expression was noted to be increased (p < 0.0001 using ANOVA and Tukey-Kramer multiple comparisons test in mature DCs or in mature DCs + Ova peptide groups compared with immature DCs (a versus i and q, b versus j and r, c versus k and s, and d versus l and t). In the above-mentioned three groups, exposure of DCs to TCDD failed to cause a significant increase of Fas (p > 0.05; a versus f, g, and h; i versus j, k, and l; and q versus r, s, and t) and FasL (p > 0.05) in immature and mature DCs groups (b versus f, n, and v and c versus l, o, and w), but it caused significant increase (p < 0.05) in FasL in mature DCs + OvaPep group (u versus v, w, and x). Data in E represent semiquantitative RT-PCR. Expressions of Fas and FasL are presented as percentage of 18S expression on the y-axis, and expression of 18S was considered to be 100% for each experiment. Data in E represent mean ± S.E.M. of three independent experiments. As summarized in E, Fas expression was noted to be increased (p < 0.0001) using ANOVA and Tukey-Kramer multiple comparisons test in mature DCs or in mature DCs + Ova peptide groups compared with immature DCs (1 versus 11 and 21, 2 versus 12 and 22, 3 versus 13 and 23, 4 versus 14 and 24, and 5 versus 15 and 25). In the above-mentioned three groups, TCDD exposure caused a significant increase in FasL (p < 0.005) in mature DCs + Ova peptide groups (26 versus 27, 28, 29, and 30), 7 versus 17 and 27, 8 versus 18 and 28, 9 versus 19 and 29, and 10 versus 20 and 30), but it did not significantly (p > 0.05) increase in immature (6 versus 7, 8, 9, and 10) or mature DCs (16 versus 17, 18, 19, and 20). MIF, mean fluorescent intensity.
TCDD Up-Regulated the Expression of Fas, FasL, and AhR in ConA-Activated T Cells
Previous studies from our laboratory have shown that TCDD induces apoptosis in T cells involving Fas/FasL interactions in vivo (Kamath et al., 1999; Camacho et al., 2002, 2005). In the present study, therefore, we investigated whether the increased susceptibility of activated T cells to TCDD results from up-regulation of Fas, FasL, or AhR, in ConA-activated T cells. To this end, we determined the expression of Fas, FasL, and AhR in nonactivated and ConA-activated T cells exposed to TCDD or vehicle, by performing RT-PCR using mouse Fas-, FasL-, and AhR-specific sets of primers. We observed significant increase in expression of Fas and FasL in TCDD-treated activated T cells compared with similar cultures of nonactivated T cells (Fig. 4, A and B).
Fig. 4.
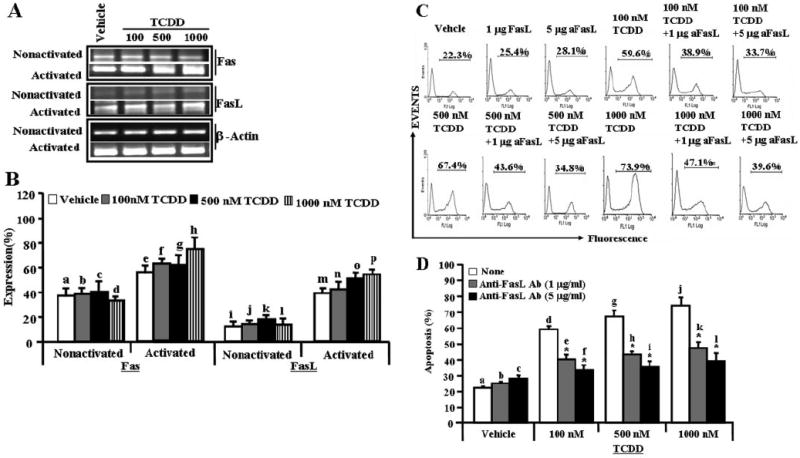
Role of Fas and FasL in TCDD-mediated apoptosis in T cells. Expression of Fas and FasL in nonactivated or ConA-activated T cells was determined by performing RT-PCR. A, expression of Fas and FasL in nonactivated and activated T cells (RT-PCR). β-Actin was used as an internal control to normalize the expression of Fas and FasL. The band intensity of PCR products was determined using a Bio-Rad Laboratories image analysis system. The quantity of PCR products was determined using ChemiDoc image analysis software (Bio-Rad Laboratories). B, semiquantitative RT-PCR and expressions of Fas and FasL are presented as percentage of β-actin expression on the y-axis, and expression of β-actin was considered to be 100% for each experiment. Data in B are depicted as mean ± S.E.M. of three independent experiments. Significant increase in expression of Fas in TCDD-treated activated T cells was noted compared with similar cultures of nonactivated T cells (p < 0.05) by Student’s t test (b versus f, c versus g, and d versus h). TCDD-treated activated T cells also showed an increase in FasL (p < 0.05) compared with nonactivated T cells (j versus n, k versus o, and l versus p). In C, ConA-activated T cells cultured with TCDD were incubated in the absence or presence of mouse-specific anti-FasL Ab, and apoptosis was studied by TUNEL assays. The data presented in C are representative of three independent experiments, and they are depicted as mean ± S.E.M. in D. Significant reduction in TCDD-mediated apoptosis of T cells cultured in the presence of anti-FasL Ab compared with T cells cultured in the absence of anti-FasL Ab (p < 0.0033 using ANOVA and Tukey-Kramer multiple comparisons test; d versus e and f, g versus h and i, and j versus k and l).
FasL Played a Significant Role in Initiating Death-Receptor Pathway during TCDD-Mediated T-Cell Apoptosis
To test whether FasL initiates death-receptor pathway to cause TCDD-mediated apoptosis in T cells, ConA-activated T cells were cultured in the absence or presence of various concentrations (1–5 μg/ml) of anti-mouse FasL mAb. FasL mAb was added to the culture 1 h before TCDD treatment. We observed significant (p < 0.0033) reduction in TCDD-mediated T-cell apoptosis when 1 to 5 μg/ml FasL mAb was added to the culture (Fig. 4, C and D). Addition of isotype control Abs failed to exhibit any significant effect on TCDD-mediated apoptosis (data not shown). The data obtained demonstrated that FasL plays a crucial role in TCDD-mediated apoptosis in primary peripheral T cells.
Role of AhR in TCDD-Induced Up-Regulation of Fas and FasL Expression in T Cells
Upon examination of TCDD-induced expression of AhR in T cells by RT-PCR, we observed significant (p < 0.05) increase in AhR expression in TCDD-treated nonactivated T cells compared with vehicle-treated nonactivated T cells (Fig. 5, A and B). We also observed significant increase in AhR expression (p < 0.05) in TCDD-treated activated T cells compared with vehicle-treated activated T cells (Fig. 5, A and B).
Fig. 5.

Role of AhR in TCDD-induced apoptosis in T cells. A, expression of AhR in nonactivated and ConA-activated T cells (RT-PCR). B, semiquantitative RT-PCR data and expressions of AhR are presented as percentage of β-actin expression on the y-axis, and expression of β-actin was considered to be 100% for each experiment. Data in B are depicted as mean ± S.E.M. of three independent experiments. TCDD, compared with the vehicle, increased the expression of AhR in both nonactivated T cells as determined by ANOVA test (p < 0.05; a versus b, c, and d) and activated T cells (p < 0.05; e versus f, g, and h), and in vehicle-treated activated T cells (p < 0.05; a versus e) compared with vehicle-treated nonactivated T cells. Data in C are summarized for three independent experiments, and they are represented as mean ± S.E.M. Significantly higher TCDD-induced expression of Fas as determined by Student’s t test (p < 0.0034; c versus m, e versus o, g versus q, and i versus s) and FasL (p < 0.0001; f versus p; h versus r, and j versus t) was seen in wild-type T cells compared with AhR KO nonactivated T cells. D, expression of Fas and FasL in TCDD- or vehicle-treated activated T cells from wild-type and AhR KO mice. Data presented in D are summarized for three independent experiments and represented as mean ± S.E.M. Significantly higher TCDD-induced expression of Fas (p < 0.0036; c versus m, e versus o, g versus q, and i versus s) and FasL (p < 0.0001; f versus p, h versus r, and j versus t) as determined by Student’s t test in wild-type T cells compared with AhR KO activated T cells. In E, ConA-activated T cells from wild-type and AhR KO mice were cultured with 100 and 1000 nM TCDD or vehicle (DMSO) for 24 h, and apoptosis in T cells was determined. Data presented in E represent one of three independent experiments, and vertical bars in F represent mean ± S.E.M. Significantly lower levels of apoptosis was noted in AhR KO mice compared with wild-type mice as determined by Student’s t test (p < 0.0001; a versus e, b versus f, c versus g, and d versus h). In G, ConA-activated T cells from wild-type mice were cultured with 100 and 1000 nM TCDD or vehicle (DMSO) for 24 h in the absence or presence of ANF in the culture, and apoptosis in T cells was determined. Data presented in G represent one of three independent experiments, and vertical bars represent mean ± S.E.M. Significantly lower TCDD-induced apoptosis was seen in T cells when ANF was used in the culture as determined by Student’s t test (p < 0.0001; c versus d, e versus f, and g versus h).
We performed a series of in vitro assays using nonactivated or ConA-activated primary T cells from wild-type (C57BL/6) or AhR KO mice to test the role of AhR in TCDD-induced up-regulation of Fas and FasL in T cells. Upon examination of whether TCDD-induced AhR participates in up-regulation of Fas and FasL in T cells, we observed significantly higher levels of Fas (p < 0.0034) and FasL (p < 0.0001) in wild-type T cells compared with AhR KO-nonactivated T cells (Fig. 5C); and similarly, significantly higher Fas (p < 0.0036) and FasL (p < 0.0001) in activated wild-type T cells compared with activated AhR KO T cells (Fig. 5D). The data clearly demonstrated that TCDD recruits AhR to up-regulate the expression of both Fas and FasL in T cells.
TCDD Recruited AhR to Kill Activated T Cells
We tested the role of AhR in TCDD-mediated apoptosis in activated T cells. To this end, we performed a series of in vitro assays using primary T cells from wild-type (C57BL/6) or AhR KO mice. We observed significantly less TCDD-mediated apoptosis in ConA-activated AhR KO T cells (Fig. 5E) compared with ConA-activated T cells from AhR wild-type mice at all TCDD doses tested (Fig. 5E). Likewise, we observed significant reduction in TCDD-mediated apoptosis in ConA-activated T cells in the presence of ANF, an antagonist for AhR, compared with activated T cells that were cultured in the absence of ANF (Fig. 5, F and G). These data together demonstrated that AhR plays a critical role in TCDD-mediated signaling and T-cell apoptosis. It should be noted that T cells from AhR KO mice did exhibit low levels of apoptosis, particularly at higher doses of TCDD (Fig. 5E), although this was significantly less than that seen in AhR wild-type T cells. These data suggested that higher doses of TCDD may induce apoptosis at least in part through an AhR-independent pathway.
Both Death-Receptor (Extrinsic) and Mitochondrial (Intrinsic) Pathways Were Involved in TCDD-Mediated T-Cell Apoptosis
To investigate whether TCDD-mediated apoptosis in activated T cells was caspase-dependent, we performed enzymatic assays for various caspases (caspase-3/7, caspase-8, and caspase-9). We observed significant increase in caspase enzymatic activities for all three caspases examined in ConA-activated and TCDD-treated T cells compared with vehicle-treated activated T cells (Fig. 6A). This activity was dose-dependent, and it was seen at TCDD concentrations of 10 nM and higher. The role of various caspases was further confirmed by blocking caspase activity using various caspases inhibitors (caspase-3, Z-DEVD; caspase-8, Z-IETD-FMK; and caspase-9, Z-LEHD-FMK). TUNEL assays performed on ConA-activated T cells that were cultured in the presence of various caspase inhibitors and treated with 100 nM TCDD demonstrated almost complete blocking (≈90%) of TCDD-mediated apoptosis in the presence of caspase-3 inhibitor (Fig. 6, B and C), significant blocking (>65%) in the presence of caspase-8 inhibitor (Fig. 6, B and C), and partial blocking (<50%) in the presence of caspase-9 inhibitor (Fig. 6, B and C). Furthermore, we observed similar results (Fig. 6, D and E) when we used inhibitors against caspase-8 in the culture of activated T cells, treated with various doses (100–1000 nM) of TCDD. Overall, these data demonstrated that TCDD-mediated apoptosis in activated T cells was caspase-dependent. To further corroborate the role of mitochondrial pathway, we studied Δψm loss using DIOC6 dye after TCDD or vehicle treatment. We observed significant reduction (<36%) in Δψm in activated and TCDD-treated T cells (Fig. 7, A and B) compared with minimal reduction in nonactivated T cells treated with TCDD (Fig. 7, A and B).
Fig. 6.
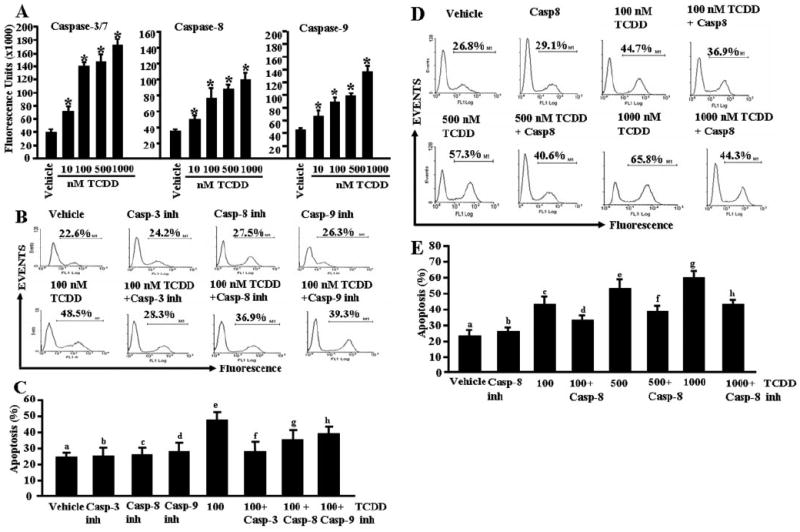
Role of caspases in TCDD-induced apoptosis in primary T cells. Enzymatic activities of caspase-3/7, -8, and -9 were determined in ConA-activated T cells 24 h after TCDD treatment. A, shows caspase-3/7, -8, and -9 activities, and the data represent mean ± S.E.M. of three independent experiments. Asterisks (*) represent statistically significant (p < 0.0002) increase in enzymatic activities of caspase-3, -8, and -9 in TCDD-treated T cells compared with vehicle-treated T cells. B and C, inhibitors against caspases block TCDD-mediated apoptosis in T cells. The data presented are representative of three independent experiments (B) summarized in C. There was statistically significant blocking of TCDD-induced apoptosis by various caspase inhibitors as determined by Student’s t test (caspase-3: p < 0.0001, e versus f, caspase-8: p < 0.0003, e versus g, and caspase-9: p < 0.0017, e versus h). In D and E, inhibitor against caspase-8 was used in the culture together with various concentrations of TCDD (100–1000 nM). Data presented represent one of the three independent experiments performed (D), and they are summarized in E. There was statistically significant (p < 0.0003) blocking of TCDD-induced apoptosis as determined by Student’s t test (c versus d, e versus f, and g versus h) when caspase-8 inhibitor was used in the culture.
Fig. 7.
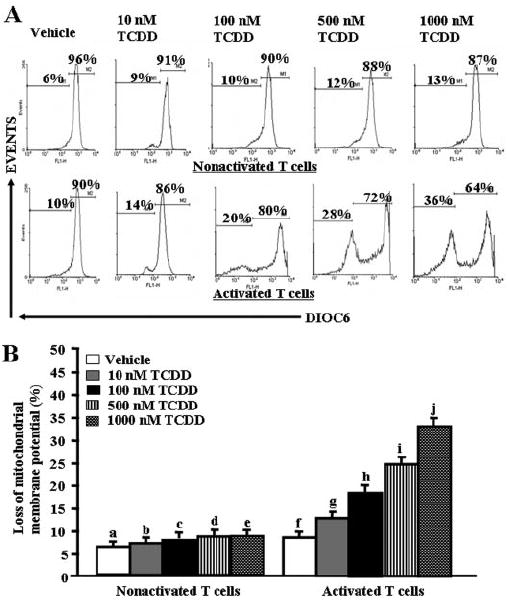
Effect of TCDD on Δψm in ConA-activated T cells. A, ConA-activated or nonactivated T cells were treated with vehicle (DMSO) or 100 to 1000 nM TCDD as described in Fig. 1, and then they were stained with DIOC6 to evaluate Δψm. Results are representative of three independent experiments, and they are summarized in B. There was a statistically significant (p < 0.0038) increase in Δψm in TCDD-treated activated T cells compared with vehicle-treated activated T cells (f versus g, h, i, and j) as determined by ANOVA and Tukey-Kramer multiple comparisons test. In nonactivated T cells, TCDD failed to increase the percentage of cells with loss of Δψm (p > 0.05; a versus b, c, d, and e).
TCDD Down-Regulated c-FLIP to Allow Apoptosis in T Cells
Presence of c-FLIP has been shown to block Fas/FasL-mediated death-receptor pathway (Golks et al., 2005; Lavrik et al., 2005a,b). In this context, expression of c-FLIP in TCDD-treated activated T cells was evaluated by performing RT-PCR and Western blot analysis. Nonactivated T cells showed high levels of c-FLIP; furthermore, TCDD treatment did not alter the expression of cFLIP (Fig. 8, A and B). Activated T cells also showed c-FLIP expression, but interestingly, TCDD treatment caused a significant decrease in c-FLIP in activated T cells (Fig. 8, A and B). Upon Western blot analysis, we also observed significant reduction in c-FLIP expression in TCDD-treated activated T cells compared with vehicle-treated T cells (Fig. 8, C and D). These data suggested that c-FLIP may play a crucial role in dictating the susceptibility of T cells to TCDD-induced apoptosis.
Fig. 8.
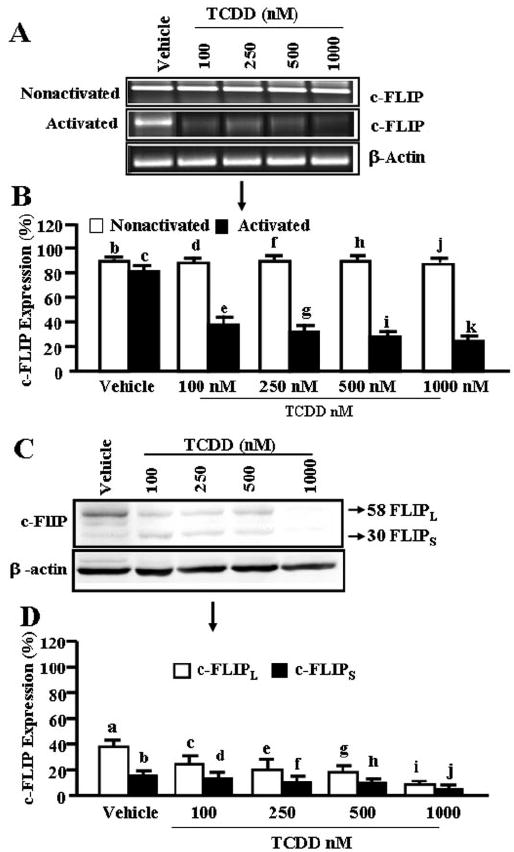
Expression of c-FLIP in primary T cells. A, nonactivated or ConA-activated T cells were treated with vehicle or 100 to 1000 nM TCDD for 24 h as described in Fig. 1 legend, and then they were evaluated for c-FLIP expression by RT-PCR. β-Actin was used as an internal control for normalization of c-FLIP expression. In B, results represent mean ± S.E.M. of three independent experiments. Expression of c-FLIP is presented as percentage of β-actin expression in the y-axis, and expression of β-actin was considered to be 100% for each experiment. There was significant (p < 0.0001) reduction in expression of c-FLIP in TCDD-treated activated T cells compared with nonactivated T cells as determined by Student’s t test (d versus e, f versus g, h versus i, and j versus k). There was no significant change in c-FLIP levels in nonactivated T cells incubated with vehicle or TCDD (p > 0.05; b versus d, f, h, and j) as well as between nonactivated versus activated T cells exposed to vehicle (p > 0.05; b versus c). C, immunoprecipitation of c-FLIP using primary antibody against anti-mouse c-FLIP in ConA-activated T cells. D, results represent mean ± S.E.M. of three independent experiments. Expression of c-FLIP is presented as percentage of β-actin expression on the y-axis, and expression of β-actin was considered to be 100% for each experiment. There was statistically significant (p < 0.0003) reduction in expression of c-FLIP in TCDD-treated T cells compared with vehicle-treated T cells, as determined by ANOVA and Tukey-Kramer multiple comparisons test (c-FLIPL: a versus c, e, and g, and i and c-FLIPs: b versus d, f, h, and j).
To further confirm the role of c-FLIP in TCDD-induced apoptosis of primary T cells in vitro, we used a RNA interference approach. siRNA consisting of 21-bp double-stranded RNA has been shown to mediate RNA interference effect in mammalian cells. We used mouse-specific c-FLIP siRNA (Dharmacon RNA Technologies) targeting four different sites of c-FLIP as described under Materials and Methods. As shown in Fig. 9A, we observed more than 59% of naive T cells were siRNA-transfected. Upon Western blot analysis, we observed that c-FLIP siRNAs were able to significantly reduce c-FLIP expression, whereas control siRNA had no significant effect (Fig. 9, B and C). We also observed that blocking of c-FLIP expression using c-FLIP siRNA in primary T cells allowed significant TCDD-induced apoptosis in primary T cells compared with T cells that were not transfected (Fig. 9D) or transfected with control siRNA (Fig. 9E). These data further corroborated our earlier results showing the crucial role of c-FLIP in TCDD-induced apoptosis in T cells in vitro.
Fig. 9.
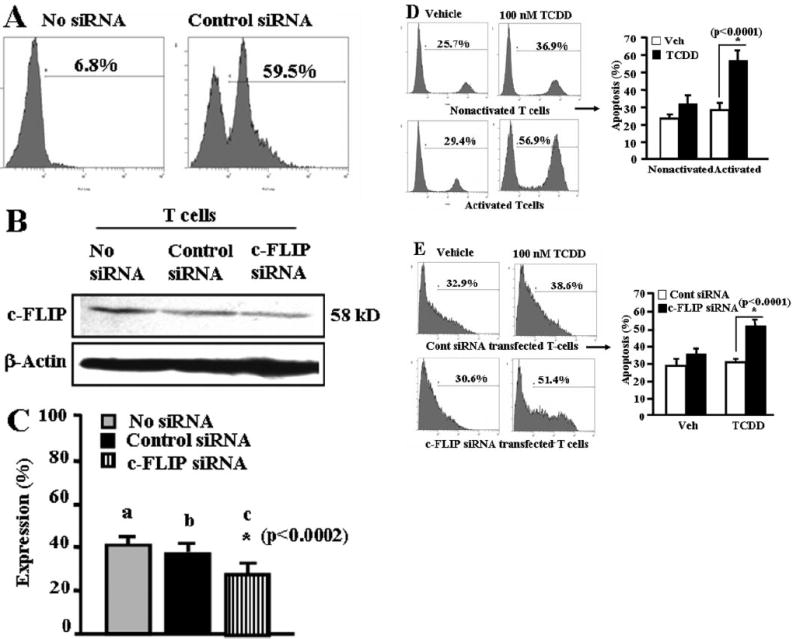
c-FLIP plays a critical role in TCDD-induced apoptosis in primary T cells in vitro. A, primary T cells were transfected with SiGLO RISK-free control siRNA. Efficiency of transfection was determined by performing flow cytometry (Cytomics FC 500; Beckman Coulter) analysis. B, primary T cells were transfected with mouse-specific c-FLIP siRNAs. c-FLIP expression level was analyzed by Western blotting using antibody against c-FLIP. C, signal intensity of c-FLIP expression compared with β-actin. Expressions of c-FLIP is presented as percentage of β-actin expression on the y-axis, and expression of β-actin was considered to be 100% for each experiment. Vertical bars represent mean ± S.E.M. of three independent experiments, and asterisks (*) represent statistically significant (p < 0.0002) reduction in expression of c-FLIP in T cells (b versus d) transfected with c-FLIP siRNA as determined by Student’s t test. D, primary T cells untransfected (nonactivated or ConA-activated) were treated with vehicle or 10 nM TCDD for 24 h, and then they were analyzed for apoptosis using TUNEL assay. Vertical bars on the right represent mean ± S.E.M. of three independent experiments, and asterisks (*) represent statistically significant (p < 0.0001) TCDD-induced apoptosis in activated T cells compared with vehicle-treated activated T cells as determined by Student’s t test. E, primary T cells transfected with control siRNA (top) or c-FLIP siRNAs (bottom) were cultured in the presence of vehicle or 100 nM TCDD for 24 h, and then apoptosis was determined using TUNEL assay. Vertical bars (right) represent mean ± S.E.M. of three independent experiments, and asterisks (*) represent statistically significant (p < 0.0001) TCDD-induced apoptosis in c-FLIP siRNA-transfected primary T cells compared with control siRNA-transfected primary T cells as determined by Student’s t test.
Discussion
TCDD-mediated immunotoxicity has been well characterized in various animal models (Faith and Luster, 1979; Nagarkatti et al., 1984; Greenlee et al., 1985; Fine et al., 1990; Lundberg et al., 1990a,b; Holladay et al., 1991; Esser, 1994; Kerkvliet et al., 2002). Currently, the consensus is that TCDD-mediated toxicity is initiated by TCDD-AhR interaction, AhR-ARNT interaction with DRE present in regulatory regions of TCDD-sensitive genes, their induction, and resultant immunotoxicity. However, the precise molecular mechanisms underlying the immunotoxicity are yet to be fully characterized, mainly due to lack of an appropriate in vitro model. Although thymocytes and antigen-activated T cells are highly sensitive to TCDD-mediated toxicity in vivo, it has been difficult to reproduce such findings in vitro. To this end, the present study demonstrates for the first time that activated but not naive T cells are more susceptible to TCDD-mediated apoptosis in vitro.
The data obtained from our present in vitro studies demonstrate that TCDD causes apoptosis in primary T cells in vitro primarily upon activation and triggers both death-receptor and mitochondrial pathways. These observations are based on the following findings: 1) TCDD triggered apoptosis to a greater extent in activated T cells compared with non-activated cells; 2) TCDD induced apoptosis in antigen-specific activated T cells; 3) TCDD-mediated early events/signals were initiated primarily through activation of AhR, leading to up-regulated expression of Fas and FasL; 4) interaction of FasL with Fas triggered death-receptor pathway by activating caspase-8 and caspase-3, culminating in apoptosis; and 5) loss of Δψm in activated and TCDD-treated T cells. It should be noted that the nonactivated T cells showed low levels of apoptosis. This can be explained because we had used T cells that were ~90% enriched. Thus, the 10% cells consisting of APCs may facilitate TCDD-induced apoptosis as evidenced by the addition of purified DCs to T-cell cultures further enhancing TCDD-induced apoptosis. In our studies, we did not use more enriched T cells because a small proportion of APCs are necessary to activate T cells using ConA (Fikri et al., 2002). We noted that if we use highly enriched primary nonactivated T cells (>95% purity), they are completely resistant to TCDD-induced apoptosis (data not shown).
We and others have previously shown that TCDD is not toxic to naive T cells, but it can suppress the immune response of T cells that are activated by an antigen in vivo (Camacho et al., 2001, 2002). This raises an important question as to why TCDD can suppress T-cell response to an antigen in vivo, whereas it is not directly toxic to T cells in vitro. Recent studies from our laboratory shed some light in this regard by demonstrating that TCDD-induced apoptosis in thymic T cells is dependent on their interactions with thymus stromal cells (Camacho et al., 2004a,b, 2005). We have demonstrated previously that in vivo administration of TCDD leads to up-regulation of Fas on T cells and FasL on thymic stromal cells and that interaction between these cells is critical for T cells to undergo apoptosis (Camacho et al., 2005). With respect to peripheral T cells, the current study demonstrates that T-cell activation or interaction between T cells and APCs such as DCs is critical for induction of TCDD-mediated apoptosis. The present study also demonstrates that activated T cells become more susceptible to TCDD-mediated apoptosis because they express higher levels of AhR, Fas, and FasL compared with nonactivated T cells. These molecules have been shown to play a critical role in TCDD-mediated apoptosis (Fernandez-Salguero et al., 1996; Hossain et al., 1998; Kamath et al., 1999; Camacho et al., 2001, 2002, 2004b, 2005). In addition to T-cell activation, we also noted that the presence of DCs in the culture increased the levels of apoptosis in T cells caused by TCDD. This observation on the role of DCs in inducing apoptosis in T cells by TCDD is somewhat similar to thymic stromal cells triggering apoptosis in thymic T cells, as described by us (Camacho et al., 2005). TCDD-mediated up-regulation of FasL in mature and Ova peptide-pulsed mature DCs explains the mechanism through which DCs may facilitate TCDD-mediated T-cell apoptosis. Our findings are consistent with previous reports demonstrating that specific sets of DCs express FasL (O’Connell et al., 2000). Based on these findings, we suggest that during T-cell-DC interactions, increased levels of FasL on DCs induced by TCDD may facilitate enhanced apoptosis in T cells.
We have also recently shown that TCDD regulates Fas and FasL promoters through DRE and/or nuclear factor-κB motifs via AhR (Camacho et al., 2005). The data obtained from the present study corroborates the critical role played by AhR in peripheral activated T-cell apoptosis by demonstrating 1) up-regulated expression of AhR, Fas, and FasL upon T-cell activation; 2) minimal apoptosis in activated T cells from AhR KO mice; and 3) complete block of TCDD-mediated apoptosis of T cells in the presence of ANF, an AhR antagonist. It should be noted that Park et al. (2003) have shown that TCDD triggered apoptosis in AhR-deficient EL4 cells through insulin-like growth-binding protein-6. In primary activated T cells from AhR KO mice, we also noticed that at higher concentrations of TCDD (1000 nM), there was a low level of apoptosis, thereby suggesting that TCDD at very high concentrations may mediate apoptosis through AhR-independent mechanisms.
Yet another reason as to why naive T cells are resistant to TCDD-mediated apoptosis could be the inability of TCDD to inhibit c-FLIP as shown in the current study. c-FLIP plays a critical role in the regulation of death receptor-induced apoptosis (Golks et al., 2005; Lavrik et al., 2005a,b). It is known that c-FLIP can prevent apoptosis triggered by death-inducing ligands by binding to the FAS-associated death domain protein and/or caspase-8 and -10 at the level of the death-inducing signaling complex. It has also been shown that down-regulation of c-FLIP leads to caspase-8 activation at the death-inducing signaling complex, and it allows death receptor-mediated pathway to proceed leading to apoptosis (Palao et al., 2004; Golks et al., 2005; Lavrik et al., 2005a,b; Salon et al., 2006). There are also reports demonstrating constitutive expression of c-FLIP in primary T cells (Strauss et al., 2003; Golks et al., 2005; Lavrik et al., 2005a,b; Salon et al., 2006) and its down-regulation in T cells allowed death receptor-mediated T-cell apoptosis (Suhara et al., 2001; Strauss et al., 2003; Tran et al., 2003; Uriarte et al., 2005). In the current study, we noted that nonactivated T cells expressed high levels of c-FLIP. In addition, the levels of c-FLIP expression remained unchanged in ConA-activated but vehicle-treated cells. It is noteworthy that TCDD treatment of activated but not naive T cells caused a dramatic decrease in c-FLIP. Based on previous reports and our findings, the presence of c-FLIP in primary T cells may be one of the important factors prohibiting TCDD-mediated apoptosis in naive T cells in vitro. Down-regulation of c-FLIP in activated T cells induced by TCDD as seen in the current study may play a critical role in induction of apoptosis. Moreover, down-regulation of c-FLIP expression by siRNA led to marked susceptibility of primary T cells to TCDD-induced apoptosis in vitro.
The current study demonstrates for the first time induction of apoptosis in T cells by TCDD in vitro. We have identified several mechanisms that explain this phenomenon. First, T-cell activation leads to increased AhR expression, which may make the T cells more susceptible to TCDD. Second, T-cell activation up-regulates Fas and FasL, molecules that play a critical role in TCDD-induced apoptosis. Third, TCDD down-regulates c-FLIP, an apoptosis inhibitory molecule in activated T cells. Fourth, DCs seem to play a significant role in facilitating TCDD-mediated apoptosis in activated T cells. The in vitro model will provide the basis for further investigating the molecular pathways through which TCDD triggers apoptosis in T cells.
Acknowledgments
We thank Daniel Sisco and Shweta Hegde for technical help.
This work was funded in part by National Institutes of Health Grants P01-AT03961, R01-ES09098, R01-DA016545, R01-AI058300, R01-AI053703, R01-HL058641, and R21-DA014885; A. D. Williams Trust Funds (to N.P.S.); and an American Cancer Society Institutional grant (to N.P.S.).
ABBREVIATIONS
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
- AhR
aryl hydrocarbon receptor
- ARNT
aryl hydrocarbon receptor nuclear translocator
- DRE
dioxin responsive elements
- c-FLIP
cellular FLICE inhibitory protein
- DC
dendritic cell
- AhR KO
aryl hydrocarbon receptor knockout
- Ova
ovalbumin
- DMSO
dimethyl sulfoxide
- PBS
phosphate-buffered saline
- PE
phycoerythrin
- FasL
Fas ligand
- HRP
horseradish peroxidase
- Ab
antibody
- PCR
polymerase chain reaction
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick-end labeling
- ANOVA
analysis of variance
- ANF
α-naphthoflavone
- ConA
concanavalin A
- FITC
fluorescein isothiocyanate
- LPS
lipopolysaccharide
- RT-PCR
reverse transcriptase-polymerase chain reaction
- bp
base pair(s)
- Δψm
mitochondrial membrane potential
- DiOC6
3′-dihexyloxacarboeczyme
- siRNA
small interfering RNA
- APC
antigen-presenting cell
- mAb
monoclonal antibody
- Z-
N-benzyloxycarbonyl-
- FMK
fluoromethyl ketone
References
- Bock KW, Kohle C. Ah receptor: dioxin-mediated toxic responses as hints to deregulated physiologic functions. Biochem Pharmacol. 2006;72:393–404. doi: 10.1016/j.bcp.2006.01.017. [DOI] [PubMed] [Google Scholar]
- Camacho IA, Hassuneh MR, Nagarkatti M, Nagarkatti PS. Enhanced activation-induced cell death as a mechanism of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced immunotoxicity in peripheral T cells. Toxicology. 2001;165:51–63. doi: 10.1016/s0300-483x(01)00391-2. [DOI] [PubMed] [Google Scholar]
- Camacho IA, Nagarkatti M, Nagarkatti PS. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) induces Fas-dependent activation-induced cell death in superantigen-primed T cells. Arch Toxicol. 2002;76:570–580. doi: 10.1007/s00204-002-0390-2. [DOI] [PubMed] [Google Scholar]
- Camacho IA, Nagarkatti M, Nagarkatti PS. Effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on maternal immune response during pregnancy. Arch Toxicol. 2004a;78:290–300. doi: 10.1007/s00204-003-0538-8. [DOI] [PubMed] [Google Scholar]
- Camacho IA, Nagarkatti M, Nagarkatti PS. Evidence for induction of apoptosis in T cells from murine fetal thymus following perinatal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) Toxicol Sci. 2004b;78:96–106. doi: 10.1093/toxsci/kfh048. [DOI] [PubMed] [Google Scholar]
- Camacho IA, Singh N, Hegde VL, Nagarkatti M, Nagarkatti PS. Treatment of mice with 2,3,7,8-tetrachlorodibenzo-p-dioxin leads to aryl hydrocarbon receptor-dependent nuclear translocation of NF-kappaB and expression of fas ligand in thymic stromal cells and consequent apoptosis in T cells. J Immunol. 2005;175:90–103. doi: 10.4049/jimmunol.175.1.90. [DOI] [PubMed] [Google Scholar]
- Dertinger SD, Nazarenko DA, Silverstone AE, Gasiewicz TA. Aryl hydrocarbon receptor signaling plays a significant role in mediating benzo[a]pyrene- and cigarette smoke condensate-induced cytogenetic damage in vivo. Carcinogenesis. 2001;22:171–177. doi: 10.1093/carcin/22.1.171. [DOI] [PubMed] [Google Scholar]
- Do Y, Hegde VL, Nagarkatti PS, Nagarkatti M. Bryostatin-1 enhances the maturation and antigen-presenting ability of murine and human dendritic cells. Cancer Res. 2004a;64:6756–6765. doi: 10.1158/0008-5472.CAN-03-4002. [DOI] [PubMed] [Google Scholar]
- Do Y, McKallip RJ, Nagarkatti M, Nagarkatti PS. Activation through cannabinoid receptors 1 and 2 on dendritic cells triggers NF-kappaB-dependent apoptosis: novel role for endogenous and exogenous cannabinoids in immunoregulation. J Immunol. 2004b;173:2373–2382. doi: 10.4049/jimmunol.173.4.2373. [DOI] [PubMed] [Google Scholar]
- Esser C. Dioxins and the immune system: mechanisms of interference. A meeting report. Int Arch Allergy Immunol. 1994;104:126–130. doi: 10.1159/000236719. [DOI] [PubMed] [Google Scholar]
- Faith RE, Luster MI. Investigations on the effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on parameters of various immune functions. Ann N Y Acad Sci. 1979;320:564–571. doi: 10.1111/j.1749-6632.1979.tb56634.x. [DOI] [PubMed] [Google Scholar]
- Fernandez-Salguero PM, Hilbert DM, Rudikoff S, Ward JM, Gonzalez FJ. Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol Appl Pharmacol. 1996;140:173–179. doi: 10.1006/taap.1996.0210. [DOI] [PubMed] [Google Scholar]
- Fikri Y, Pastoret PP, Nyabenda J. Costimulatory molecule requirement for bovine WC1+gammadelta T cells’ proliferative response to bacterial superantigens. Scand J Immunol. 2002;55:373–381. doi: 10.1046/j.1365-3083.2002.01069.x. [DOI] [PubMed] [Google Scholar]
- Fine JS, Gasiewicz TA, Fiore NC, Silverstone AE. Prothymocyte activity is reduced by perinatal 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure. J Pharmacol Exp Ther. 1990;255:128–132. [PubMed] [Google Scholar]
- Golks A, Brenner D, Fritsch C, Krammer PH, Lavrik IN. c-FLIPR, a new regulator of death receptor-induced apoptosis. J Biol Chem. 2005;280:14507–14513. doi: 10.1074/jbc.M414425200. [DOI] [PubMed] [Google Scholar]
- Gonzalez FJ, Fernandez-Salguero P. The aryl hydrocarbon receptor: studies using the AHR-null mice. Drug Metab Dispos. 1998;26:1194–1198. [PubMed] [Google Scholar]
- Greenlee WF, Dold KM, Osborne R. Actions of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on human epidermal keratinocytes in culture In Vitro. Cell Dev Biol. 1985;21:509–512. doi: 10.1007/BF02620843. [DOI] [PubMed] [Google Scholar]
- Harper PA, Riddick DS, Okey AB. Regulating the regulator: factors that control levels and activity of the aryl hydrocarbon receptor. Biochem Pharmacol. 2006;72:267–279. doi: 10.1016/j.bcp.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Holladay SD, Brownie CF, Corbett WT, Talley DD. Evaluation of an indirect enzyme-linked immunosorbent assay for screening antibody against aflatoxins. Am J Vet Res. 1991;52:222–223. [PubMed] [Google Scholar]
- Hossain A, Tsuchiya S, Minegishi M, Osada M, Ikawa S, Tezuka FA, Kaji M, Konno T, Watanabe M, Kikuchi H. The Ah receptor is not involved in 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated apoptosis in human leukemic T cell lines. J Biol Chem. 1998;273:19853–19858. doi: 10.1074/jbc.273.31.19853. [DOI] [PubMed] [Google Scholar]
- Kamath AB, Camacho I, Nagarkatti PS, Nagarkatti M. Role of Fas-Fas ligand interactions in 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced immunotoxicity: increased resistance of thymocytes from Fas-deficient (lpr) and Fas ligand-defective (gld) mice to TCDD-induced toxicity. Toxicol Appl Pharmacol. 1999;160:141–155. doi: 10.1006/taap.1999.8753. [DOI] [PubMed] [Google Scholar]
- Kamath AB, Xu H, Nagarkatti PS, Nagarkatti M. Evidence for the induction of apoptosis in thymocytes by 2,3,7,8-tetrachlorodibenzo-p-dioxin in vivo. Toxicol Appl Pharmacol. 1997;142:367–377. doi: 10.1006/taap.1996.8049. [DOI] [PubMed] [Google Scholar]
- Kerkvliet NI. Recent advances in understanding the mechanisms of TCDD immunotoxicity. Int Immunopharmacol. 2002;2:277–291. doi: 10.1016/s1567-5769(01)00179-5. [DOI] [PubMed] [Google Scholar]
- Kerkvliet NI, Shepherd DM, Baecher-Steppan L. T lymphocytes are direct, aryl hydrocarbon receptor (AhR)-dependent targets of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD): AhR expression in both CD4+ and CD8+ T cells is necessary for full suppression of a cytotoxic T lymphocyte response by TCDD. Toxicol Appl Pharmacol. 2002;185:146–152. doi: 10.1006/taap.2002.9537. [DOI] [PubMed] [Google Scholar]
- Lavrik IN, Golks A, Krammer PH. Caspases: pharmacological manipulation of cell death. J Clin Invest. 2005a;115:2665–2672. doi: 10.1172/JCI26252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavrik I, Golks A, Krammer PH. Death receptor signaling. J Cell Sci. 2005b;118:265–267. doi: 10.1242/jcs.01610. [DOI] [PubMed] [Google Scholar]
- Lundberg K, Dencker L, Gronvik KO. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) treatment in vivo on thymocyte functions in mice after activation in vitro. Int J Immunopharmacol. 1990a;12:459–466. doi: 10.1016/0192-0561(90)90029-m. [DOI] [PubMed] [Google Scholar]
- Lundberg K, Dencker L, Gronvik KO. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) inhibits the activation of antigen-specific T-cells in mice. Int J Immunopharmacol. 1992;14:699–705. doi: 10.1016/0192-0561(92)90133-6. [DOI] [PubMed] [Google Scholar]
- Lundberg K, Gronvik KO, Goldschmidt TJ, Klareskog L, Dencker L. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) alters intrathymic T-cell development in mice. Chem Biol Interact. 1990b;74:179–193. doi: 10.1016/0009-2797(90)90066-v. [DOI] [PubMed] [Google Scholar]
- Marlowe JL, Puga A. Aryl hydrocarbon receptor, cell cycle regulation, toxicity, and tumorigenesis. J Cell Biochem. 2005;96:1174–1184. doi: 10.1002/jcb.20656. [DOI] [PubMed] [Google Scholar]
- Matikainen T, Perez GI, Jurisicova A, Pru JK, Schlezinger JJ, Ryu HY, Laine J, Sakai T, Korsmeyer SJ, Casper RF, et al. Aromatic hydrocarbon receptor-driven Bax gene expression is required for premature ovarian failure caused by biohazardous environmental chemicals. Nat Genet. 2001;28:355–360. doi: 10.1038/ng575. [DOI] [PubMed] [Google Scholar]
- Mimura J, Fujii-Kuriyama Y. Xenobiotics and transcriptional regulation. Tanpakushitsu Kakusan Koso. 2003;48:2261–2266. [PubMed] [Google Scholar]
- Nagarkatti PS, Sweeney GD, Gauldie J, Clark DA. Sensitivity to suppression of cytotoxic T cell generation by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is dependent on the Ah genotype of the murine host. Toxicol Appl Pharmacol. 1984;72:169–176. doi: 10.1016/0041-008x(84)90261-8. [DOI] [PubMed] [Google Scholar]
- Nazarenko DA, Dertinger SD, Gasiewicz TA. In vivo antagonism of AhR-mediated gene induction by 3′-methoxy-4′-nitroflavone in TCDD-responsive lacZ mice. Toxicol Sci. 2001;61:256–264. doi: 10.1093/toxsci/61.2.256. [DOI] [PubMed] [Google Scholar]
- O’Connell PJ, Morelli AE, Logar AJ, Thomson AW. Phenotypic and functional characterization of mouse hepatic CD8 alpha+ lymphoid-related dendritic cells. J Immunol. 2000;165:795–803. doi: 10.4049/jimmunol.165.2.795. [DOI] [PubMed] [Google Scholar]
- Palao G, Santiago B, Galindo M, Paya M, Ramirez JC, Pablos JL. Down-regulation of FLIP sensitizes rheumatoid synovial fibroblasts to Fas-mediated apoptosis. Arthritis Rheum. 2004;50:2803–2810. doi: 10.1002/art.20453. [DOI] [PubMed] [Google Scholar]
- Park JH, Hahn EJ, Kong JH, Cho HJ, Yoon CS, Cheong SW, Oh GS, Youn HJ. TCDD-induced apoptosis in EL-4 cells deficient of the aryl hydrocarbon receptor and down-regulation of IGFBP-6 prevented the apoptotic cell death. Toxicol Lett. 2003;145:55–68. doi: 10.1016/s0378-4274(03)00259-5. [DOI] [PubMed] [Google Scholar]
- Pryputniewicz SJ, Nagarkatti M, Nagarkatti PS. Differential induction of apoptosis in activated and resting T cells by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and its repercussion on T cell responsiveness. Toxicology. 1998;129:211–226. doi: 10.1016/s0300-483x(98)00078-x. [DOI] [PubMed] [Google Scholar]
- Rhile MJ, Nagarkatti M, Nagarkatti PS. Role of Fas apoptosis and MHC genes in 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced immunotoxicity of T cells. Toxicology. 1996;110:153–167. doi: 10.1016/0300-483x(96)83962-x. [DOI] [PubMed] [Google Scholar]
- Salon C, Eymin B, Micheau O, Chaperot L, Plumas J, Brambilla C, Brambilla E, Gazzeri S. E2F1 induces apoptosis and sensitizes human lung adenocarcinoma cells to death-receptor-mediated apoptosis through specific downregulation of c-FLIP(short) Cell Death Differ. 2006;13:260–272. doi: 10.1038/sj.cdd.4401739. [DOI] [PubMed] [Google Scholar]
- Schmidt JV, Bradfield CA. Ah receptor signaling pathways. Annu Rev Cell Dev Biol. 1996;12:55–89. doi: 10.1146/annurev.cellbio.12.1.55. [DOI] [PubMed] [Google Scholar]
- Singh NP, Nagarkatti M, Nagarkatti PS. Role of dioxin response element and nuclear factor-kappaB motifs in 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated regulation of Fas and Fas ligand expression. Mol Pharmacol. 2007;71:145–157. doi: 10.1124/mol.106.028365. [DOI] [PubMed] [Google Scholar]
- Strauss G, Knape I, Melzner I, Debatin KM. Constitutive caspase activation and impaired death-inducing signaling complex formation in CD95-resistant, long-term activated, antigen-specific T cells. J Immunol. 2003;171:1172–1182. doi: 10.4049/jimmunol.171.3.1172. [DOI] [PubMed] [Google Scholar]
- Suhara T, Mano T, Oliveira BE, Walsh K. Phosphatidylinositol 3-kinase/ Akt signaling controls endothelial cell sensitivity to Fas-mediated apoptosis via regulation of FLICE-inhibitory protein (FLIP) Circ Res. 2001;89:13–19. doi: 10.1161/hh1301.092506. [DOI] [PubMed] [Google Scholar]
- Sulentic CE, Holsapple MP, Kaminski NE. Putative link between transcriptional regulation of IgM expression by 2,3,7,8-tetrachlorodibenzo-p-dioxin and the aryl hydrocarbon receptor/dioxin-responsive enhancer signaling pathway. J Pharmacol Exp Ther. 2000;295:705–716. [PubMed] [Google Scholar]
- Tian Y, Ke S, Denison MS, Rabson AB, Gallo MA. Ah receptor and NF-κB interactions, a potential mechanism for dioxin toxicity. J Biol Chem. 1999;274:510–515. doi: 10.1074/jbc.274.1.510. [DOI] [PubMed] [Google Scholar]
- Tran SE, Meinander A, Holmstrom TH, Rivero-Muller A, Heiskanen KM, Linnau EK, Courtney MJ, Mosser DD, Sistonen L, Eriksson JE. Heat stress downregulates FLIP and sensitizes cells to Fas receptor-mediated apoptosis. Cell Death Differ. 2003;10:1137–1147. doi: 10.1038/sj.cdd.4401278. [DOI] [PubMed] [Google Scholar]
- Uriarte SM, Joshi-Barve S, Song Z, Sahoo R, Gobejishvili L, Jala VR, Haribabu B, McClain C, Barve S. Akt inhibition upregulates FasL, downregulates c-FLIPs and induces caspase-8-dependent cell death in Jurkat T lymphocytes. Cell Death Differ. 2005;12:233–242. doi: 10.1038/sj.cdd.4401549. [DOI] [PubMed] [Google Scholar]
- Whitlock JP., Jr Induction of cytochrome P4501A1. Annu Rev Pharmacol Toxicol. 1999;39:103–125. doi: 10.1146/annurev.pharmtox.39.1.103. [DOI] [PubMed] [Google Scholar]