

Summary
Dimethylglycine dehydrogenase (DMGDH) is a mitochondrial matrix flavoprotein that catalyses the demethylation of dimethylglycine to form sarcosine, accompanied by the reduction of the covalently bound FAD cofactor. Electron-transfer flavoprotein reoxidizes the reduced flavin and transfers reducing equivalents to the main mitochondrial respiratory chain through the enzyme ETF-ubiquinone oxidoreductase. DMGDH plays a prominent role in choline and 1-carbon metabolism. We have expressed the mature form of human DMGDH and the H109R variant identified in a DMGDH-deficient patient as N-terminally His6-tagged proteins in E. coli. The enzymes were purified to homogeneity by nickel affinity and anion exchange chromatography. The presence of FAD in the wild-type enzyme was confirmed by spectrophotometric analysis. The H109R variant, however, had only 47% of the wild-type level of bound flavin as expressed in E. coli, indicating its reduced affinity for FAD As previously described for rat enzyme studies, the wild-type human enzyme exhibited two Km values for N,N-dimethylglycine (Km1 = 0.039 ± 0.010 mmol/L and Km2 = 15.4 ± 1.2 mmol/L). The addition of 4 μmol/L tetrahydrofolate resulted in a slight decrease in specific activity and a substantial decrease in Km2 (1.10 ± 0.55 mmol/L). The flavinated H109R variant protein exhibited a 27-fold decrease in specific activity and a 65-fold increase in Km, explaining its pathogenicity. Additionally, the current expression system represents a significant improvement over a previously described rat DMGDH expression system and will enhance our ability to further study this important metabolic enzyme.
Introduction
Dimethylglycine dehydrogenase (DMGDH; EC 1.5.99.2) is a mitochondrial flavoenzyme involved in choline and 1-carbon metabolism (Abeles et al 1960; Mackenzie and Frisell 1958; Wittwer and Wagner 1981a). One methyl group is removed from dimethylglycine by DMGDH to form sarcosine, which is subsequently converted to glycine in an analogous reaction by the related enzyme sarcosine dehydrogenase (SDH). In each reaction, one methyl group is donated to tetrahydrofolate to form 5,10-CH2-H4PteGlu (active formaldehyde) (Scheme 1). Both DMGDH and SDH have been shown to be major folate-binding proteins in rats and humans. In the cases of DMGDH and SDH, the preferred folate cofactor is a pentaglutamate tetrahydrofolate (Wittwer and Wagner 1981a).
Scheme 1.

Reaction scheme for DMGDH
In addition to the folate cofactor, DMGDH and SDH have a covalently bound flavin–adenine dinucleotide (FAD) that is reduced to FADH2 (Cook et al 1984). The reduced flavin is in turn reoxidized by electron-transfer flavoprotein (ETF) (Hoskins and Mackenzie 1961). A homozygous 326A>G mutation in the DMGDH gene has been described in a patient who had extremely high levels of dimethylglycine excreted in his urine and presented with muscle weakness, chronic fatigue and a fish-like body odour (OMIM 605850) (Binzak et al 2001; Moolenaar et al 1999). This mutation leads to a predicted His-to-Arg (H109R) substitution near the flavin covalent attachment site (H91) in the amino acid sequence. An E. coli expression study with rat DMGDH H109R detected no activity; however, the assays were done with crude E. coli extracts and no further biochemical characterization was performed (Binzak et al 2001).
We have developed an E. coli expression system based on the pET-28a vector that allows high-level production of N-terminally His-tagged DMGDH (His6-DMGDH) and purification of wild-type and His-to-Arg (H109R) enzymes. Kinetic studies revealed that the H109R protein had significantly decreased specific activity and increased Km compared with wild-type DMGDH. In addition, there was a 2-fold decrease in percentage of the mutant protein flavinated (47%). This expression system represents the first human DMGDH expression system and a significant improvement in yield over previously reported rat enzyme system (Brizio et al 2004). It should provide an opportunity to study detailed characterization of this important metabolic enzyme.
Materials and methods
Plasmid
The first 100 bases of cDNA encoding the mature form of human DMGDH (Binzak et al 2001) were modified to reflect E. coli codon bias, then cloned into the pET-28a expression vector (Novagen, Madison, WI, USA). The resulting recombinant plasmid encoding the mature human DMGDH was fused to an N-terminal 6-His tag followed by a thrombin cleavage site. Site-directed mutagenesis for the production of the H109R construct was done with Quick Change Mutagenesis kit (Stratagene, La Jolla, CA, USA), with the following primers:
5′-ATAAACTTGAAGAAAATACGTTATGATA GCATCAAACTT -3′ (forward)
5′-AAGTTTGATGCTATCATAACGTATTTTCTT CAAGTTTAT -3′ (reverse)
Expression of recombinant His6-DMGDH
The pET-28a-DMGDH plasmid was transformed into chemically competent E. coli BL21-AI (Invitrogen, Carlsbad, CA, USA) by heat shock at 42°C for 30 s. Transformed colonies were selected by plating on LB-agar plates with 15 μg/ml kanamycin. E. coli BL21-AI transformed with the pET-28a-DMGDH plasmid was used to inoculate 10 ml of TB medium (12 g/L tryptone, 24 g/L yeast extract, 9.4 g/L K2HPO4, 2.2 g/L KH2PO4, 0.4% glycerol) with 15 mg/ml kanamycin overnight at 37°C in a rotary shaker (220 rpm). This was transferred to 1 L of fresh TB with 15 μg/ml kanamycin and was grown at 37°C to an OD600 of 0.6. The culture was then supplemented with 0.25 mmol/L IPTG and 0.2% l-arabinose to induce expression. Growth was continued for 16 h at 19°C. The bacterial cells were harvested by centrifugation at 5000g for 10 min and kept frozen until ready for use.
Measurement of DMGDH activity
DMGDH activity was measured at 25°C in 100 mmol/L Hepes pH 7.5, 0.1 mmol/L EDTA, 200 μmol/L ferricenium hexafluorophosphate, and 50 mmol/L dimethylglycine. The reduction of ferricenium ion (Δε = 4300 L/mol) was followed at 300 nm as previously described (Lehman et al 1990). All activity measurements are converted and expressed as nanomoles of dimethylglycine consumed as substrate. Measurements were made using the enzyme with the His tag cleaved off by treatment with thrombin. For steady-state kinetic analysis, enzymatic activity was measured in triplicate at each substrate concentration. Protein concentrations were measured by Bradford assay (Bradford 1976) and flavin contents were estimated by FAD absorption at 450 nm (Macheroux 1999). Steady-state kinetic parameters were obtained by nonlinear regression analysis using Graphpad Prism 4 software version 4.03 (San Diego, CA, USA).
Purification of recombinant His6-DMGDH and His6-DMGDH H109R
The bacterial pellet (9.2 g) was resuspended in 20 ml of lysis buffer (25 mmol/L Tris pH 8.2, 50 mmol/L NaCl, 10% glycerol, 1 mmol/L EDTA, and 0.5 mg/ml FAD) with 150 μl of protease inhibitor cocktail (P8849, Sigma-Aldrich, St. Louis, MO, USA). The cells were lysed by passing twice through a French press at 55 Mpa (8000 psi) (Aminco, SLM Instruments, Rochester, NY, USA). Soluble and insoluble fractions were separated by centrifugation at 100 000g for 1 h.
Twenty millilitres of the soluble fraction was loaded onto a 30 ml DEAE-Sepharose fast flow column equilibrated with sonication buffer. The column was washed with 60 ml of 10 mmol/L Tris pH 8.2, and the protein was eluted with a 50 ml gradient of 0–300 mmol/L KCl. Active fractions were pooled. With His6-DMGDH H109R, the fractions were pooled by SDS-gel visualization because of low activity. The pooled fractions were dialysed in 50 mmol/L Tris pH 7.5 and 50 mmol/L NaCl and concentrated to 10 ml. This was then loaded onto a Ni-NTA column (10 ml), which was equilibrated with dialysis buffer. The protein was eluted by applying 100 mmol/L imidazole to the column. Fractions with high DMGDH activity were pooled, concentrated, and stored at −80°C for later use. His6-DMGDH H109R was further purified on a 25 ml DEAE-Sepharose fast flow column equilibrated with 20 mmol/L Tris pH 7.5. The protein was eluted with a 0–300 mmol/L K2HPO4 pH 7.5 gradient. The fractions with the lowest A280/A450 absorbance ratios were pooled.
Purification of native DMGDH from pig liver mitochondria
Porcine DMGDH was purified from pig liver by a modification of methods previously described for rat DMGDH (Wittwer and Wagner 1981a). A mitochondrial matrix extract from 400 g of pig liver mitochondria was precipitated with 40% saturated ammonium sulfate and pelleted. The supernatant was precipitated with 70% saturated ammonium sulfate; the pellet following centrifugation was resuspended in 30 ml of 100 mmol/L potassium phosphate pH 8.0, 10 mmol/L EDTA, 10% glycerol, and 100 mg FAD/L; and the sample was dialysed overnight in 10 mmol/L dibasic potassium phosphate. The sample was applied to a 200 ml DEAE-Sepharose column equilibrated with dialysis buffer, washed with 200 ml of dialysis buffer, and eluted with a 500 ml gradient of 0–250 mmol/L KCl in 10 mmol/L Tris pH 7.5. The most active fractions were pooled, concentrated to 50 ml, and dialysed in 10 mmol/L Tris pH 7.0. The sample was applied to a 50 ml DEAE-Sepharose fast flow column, washed with dialysis buffer, and eluted with a 600 ml gradient of 0–300 mmol/L KCl in 10 mmol/L Tris pH 7.0. Fractions with maximum activity were pooled, concentrated to 25 ml, then applied to a 30 ml EAH Sepharose column (GE Health Care, Waukesha, WI, USA) conjugated with folinic acid as described previously (Wittwer and Wagner 1980). The column was washed with 120 ml of 20 mmol/L Tris pH 8.0 and 1 mol/L KCl and protein was eluted by the addition of 40 ml of 0.1 mol/L folic acid in the wash buffer. Fractions with maximum DMGDH protein as estimated by SDS-PAGE were pooled, concentrated to 20 ml, and dialysed in 10 mmol/L Tris pH 7.0. The sample was then applied to a 20 ml DEAE-Sepharose column equilibrated with the dialysis buffer. The column was washed with 20 ml of the dialysis buffer, and the protein was eluted with 200 ml of 0–300 mmol/L KCl in 10 mmol/L Tris pH 7.0. Fractions with maximum activity were pooled.
Results
Expression and purification of recombinant His6-DMGDH
The pET-28a expression vector typically gives high-level expression following induction of a T7 promoter, and the vector places a thrombin-cleavable N-terminal His tag on the expressed protein for ease of purification. Optimal expression of the DMGDH insert was achieved 16 h after induction with 0.25 mmol/L IPTG and 0.2% l-arabinose at 19°C. The yield of purified DMGDH protein was 12.7 mg/L cell culture with a percentage yield of 46.0% (Table 1). Antibodies raised against native pig DMGDH react with both the native pig and E. coli-expressed human enzyme, but not pig SDH (Fig. 1). The specific activity of the DMGDH was 165 nmol/min per mg, comparable to values obtained with native pig DMGDH (124 nmol/min per mg). The purified expressed protein shows an absorption spectrum characteristic of flavoprotein (Fig. 2) with flavin absorbance peaks at 375 and 450 nm (Frisell and Mackenzie 1962). The A280:A375:A450 ratio was 11.3:0.93:1.0, with ε280 = 120 L/mmol and ε450 = 10.6 L/mmol for human DMGDH.
Table 1.
Purification of 6His-DMGDH and 6His-DMGDH H109R
| Output | Protein (mg) | Total activity (nmol/min) | Specific activity (nmol/min per mg) | Yield (%) |
|---|---|---|---|---|
| His6-DMGDH | ||||
| Cells | 10 100 | |||
| Lysate | 814 | 4 558 | 5.6 | – |
| DEAE FF | 152 | 3 025 | 19.9 | 66.4 |
| Ni-NTA | 12.7 | 2 096 | 165 | 46.0 |
| His6-DMGDH H109R | ||||
| Cells | 9 210 | |||
| Lysate | 854 | – | – | |
| DEAE FF | 194 | – | – | 60a |
| Ni-NTA | 4.2 | 13 | 3.1 | 30a |
| DEAE FF - 2 | 0.8 | 5 | 6.8 | 5a |
Estimated from SDS-gel visualization.
Fig. 1.
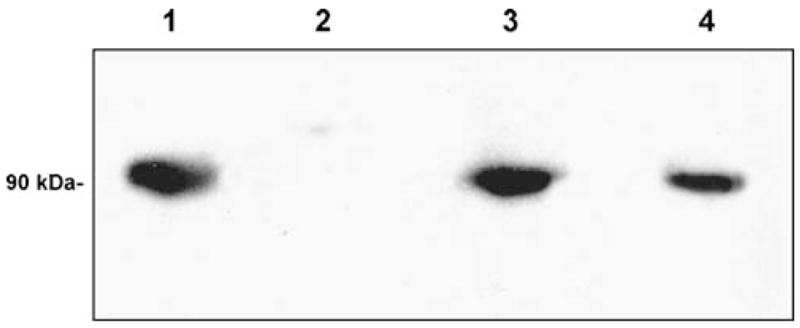
Western blot of purified His6-DMGDH. After separation by SDS-PAGE and blotting onto a nitrocellulose membrane, proteins were visualized with anti-DMGDH antibodies. Lanes: (1) purified porcine DMGDH (positive control); (2) purified porcine sarcosine dehydrogenase (negative control); (3) purified His6-DMGDH; and (4) purified flavinated 6His-DMGDH H109R
Fig. 2.

Absorption spectra of 20 μmol/L (by Bradford assay) of purified DMGDH (solid curve) and DMGDH H109R (dashed curve) recorded at 25°C in 25 mmol/L Tris pH 7.5 and 50 mmol/L NaCl. The lower A450 peak indicates the mutant is only 89% flavinated
Expression and purification of His6-H109R DMGDH
His6-DMGDH H109R was expressed and purified in the same manner as the wild-type protein, i.e. DEAE chromatography followed by Ni-NTA affinity chromatography. However, the A280/A450 ratio of the mutant protein was much higher than that of the wild-type enzyme and indicated that after the same purification steps only 47% of the enzyme was flavinated. Therefore, the protein was further purified and the resulting protein pool had an increased (89%) flavination from collecting the fractions with the lowest A280/A450 ratios. The non-flavinated fraction, which showed no flavoprotein peaks, was also pooled and saved. Purified mutant holoenzyme had a characteristic flavoprotein spectrum (Fig. 2) with absorbance peaks at 375 and 450 nm (Frisell and Mackenzie 1962). Flavinated DMGDH has previously been shown to be more resistant to trypsin digestion than is non-flavinated enzyme (Brizio et al 2000, 2002; Otto et al 1996). To examine this for the mutant enzyme 10 μmol/L of flavinated and non-flavinated DMGDH H109R were digested with 0.5 μmol/L trypsin at 37°C (Fig. 3). The non-flavinated enzyme was degraded within 20 min of trypsin treatment, while the flavin-containing enzyme was stable up to 120 min under the same conditions.
Fig. 3.

Trypsin sensitivity of unflavinated His6-DMGDH H109R. Purified His6-DMGDH H109R (10 μmol/L) with or without bound-FAD was incubated with 0.5 μmol/L trypsin at 37°C for 0 or 240 min. Reactions were stopped by the rapid addition of gel-loading buffer and boiling of the samples. Samples were separated by SDS-PAGE and stained with Coomassie blue dye
Steady-state kinetic parameters of wild-type and mutant DMGDH
Kinetic parameters were determined for wild-type and mutant DMGDH using dimethylglycine as substrate (see Fig. 5). An enzymatic turnover number of 18.2 ± 1.7 min−1 for wild-type is consistent with native porcine (12.7 ± 0.81 min−1) and rat DMGDH (8.4 min−1) (Porter et al 1985) as well as recombinantly expressed rat DMGDH (12.1 min−1) (Brizio et al 2004). Nonlinear regression analysis revealed two Km values, one (Km1) approximately 400-fold lower than the other (Km2) (Fig. 4, Table 2). The existence of two Km values and their magnitudes agree with previously published reports (Brizio et al 2004; Porter et al 1985). To test the effect of folate on the enzyme activation, kinetic experiments were repeated in the presence of 4 μmol/L monoglutamate tetrahydrofolate (THF), which is a 10-fold excess of the Km value (Wittwer and Wagner 1981b). A decrease of ~25% in Vmax was observed; however, the Km2 value was decreased dramatically from 15.4 ± 1.2 to 1.10 ± 0.55 mmol/L, suggesting a role for THF in enzyme activation.
Fig. 5.

Model of DMGDH built using the structure of dimethylglycine oxidase (PDB ID 1PJ5). Dimethylglycine has been modelled near the FAD to indicate the putative substrate binding location. The starting and ending residue numbers of the helices are indicated. H109 is near H67 (~4 Å), which is located in an α-helix that makes close interactions with the FAD. The substitution of H109 for a bulky, charged arginine (shown in grey carbons) would disrupt the position of H67 and the helix containing it, altering FAD binding. Additionally, the H109R may result in charge repulsion between R109 and K409 (~4 Å). The helix containing K409 also makes numerous interactions with FAD, and any effect on this helix position and alignment would also have a concomitant effect on FAD binding. The mutation site (H109) is far from the flavin attachment site, H91 (~22 Å). The folate binding site is located in a separate domain, ~40 Å from the dimethylglycine binding site. The effects of the H109R mutation are not expected to extend beyond the dimethylglycine binding domain
Fig. 4.

Plot of the specific activity of DMGDH (0.1 nmol) with dimethylglycine as a substrate, with (squares) or without (diamonds) 4 μmol/L folate. Inset is an Eadie–Hofstee transformation of the data to show the two Km values. DMGDH activity was assayed under standard conditions as described under Materials and Methods. Vertical bars indicate error ranges from triplicate measurements
Table 2.
Kinetic data obtained by measuring wild-type and mutant DMGDH under standard conditions
| Km1 (mmol/L) | Km2 (mmol/L) | Turnover number (min−1) | |
|---|---|---|---|
| DMGDH | 0.039 ± 0.010 | 15.4 ± 1.2 | 18.2 ± 1.7 |
| DMGDH + 4 μmol/L THF | 0.045 ± 0.011 | 1.10 ± 0.55 | 13.5 ± 1.8 |
| DMGDH H109R | 2.55 ± 1.7 | – | 0.600 ± 0.55 |
For the recombinant mutant DMGDH H109R, a Km of 2.6 ± 1.7 mmol/L was observed, which is 65-fold higher than wild-type. A second Km was not evident in the analysis. The specific activity of the mutant enzyme was 0.600 ± 0.58 min−1, nearly 30-fold lower than wild-type. Since the purified mutant protein has about 10% non-flavinated protein, the specific activity of the mutant is about 27-fold lower than that of wild-type (the Km value need not be corrected for the non-flavinated protein contaminant). This represents a decrease in catalytic efficiency of approximately 1800-fold. This is consistent with patient fibroblast studies and also with the previous prokaryotic expression study, in which no detectable activity was observed for rat DMGDH H109R in E. coli crude cellular extracts despite the detection of antigen by western blot (Binzak et al 2001).
Discussion
We report a high level E. coli expression system leading to successful production of sufficient mature human dimethylglycine dehydrogenase for biochemical and kinetic analysis. The yield of 13 mg of pure protein per litre of bacterial culture was significantly higher than yields achieved with previously reported systems (Brizio et al 2004), allowing for the production of large quantities of protein for further biochemical and biophysical studies. Spectrophotometric analysis and trypsin digestion studies confirmed the presence of bound FAD. In agreement with previous reports, two Km values were evident on kinetic analysis; however, the physiological role of the second substrate binding remains unclear. There appears to be an activation of enzyme activity at substrate concentrations greater than 20 mmol/L, although this is unlikely to be physiologically significant since this is well above the in vivo concentration of dimethylglycine.
THF is the acceptor of the methyl group that is abstracted from the substrate, dimethylglycine. Addition of THF to purified wild-type DMGDH led to a decrease of ~25% in specific activity, a phenomenon previously reported with dimethylglycine oxidase (DMGO), a related bacterial enzyme. It has been hypothesized that binding of folate in the folate-binding tunnel hinders transfer of dimethylglycine to the active site (Leys et al 2003). Interestingly, the presence of THF also reduced Km2 from 15.4 mmol/L to 1.10 mmol/L. In the absence of THF, the DMGDH reaction has been shown to lead to the formation of free formaldehyde (Abeles and Mackenzie 1956; Mackenzie and Frisell 1958; Wittwer and Wagner 1981b). Therefore, THF appears to play a regulatory role in DMGDH, functioning to keep free formaldehyde to a minimum within the mitochondria.
Expression and purification of human DMGDH afforded us the opportunity to examine the effects of a mutation identified in a patient who suffered chronic muscle fatigue and excreted elevated levels of dimethylglycine in his urine. The mutation, 326A>G transition in the DMGDH gene, leads to a His-to-Arg (H109R) substitution near the flavin attachment site. Purified DMGDH H109R showed a 2-fold decrease in flavination as compared to wild-type, suggesting that the mutant has less affinity for FAD. Flavinated mutant enzyme revealed a 65-fold increase in Km and a 27-fold decrease in activity (corrected for 89% flavination determined by spectrophotometric analysis). These findings substantiate the role of the mutation as causing a significant loss of function in vivo. The ferricenium enzyme activity assay used in this study does not require the presence of the physiological electron acceptor ETF, indicating that the catalytic impairment in mutant enzyme is not due to a change in its ability to interact with ETF. Examination of the published x-ray crystal structure of the highly homologous bacterial enzyme DMGO provides some insight into the possible effects of this mutation on enzyme function (Leys et al 2003). Figure 5 shows a structural model of DMGDH built from the crystal structure of DMGO (PDB 1PJ5) (Leys et al 2003). H109 is relatively distant (~18 Å from the H109 Cα to the N5 of the FAD) from the putative substrate-binding site and even farther from the folate-binding site (~40 Å). However, H109 is about 4 Å from H67, which is on an α-helix composed of residues 59–71. This helix and the preceding loop make numerous interactions with the bound FAD. The steric clash caused by substitution of a bulky arginine residue for H109 would affect the position of H67 and possibly perturb the overall position of the α-helix that contains H67. This would cause a weaker binding and/or misalignment of the cofactor. Additionally, K409 on the α-helix, which includes residues 401–417, is close enough for a potential charge repulsion when H109 is substituted for an arginine. This helix also makes numerous interactions with FAD, and any influence on its position would result in a decreased FAD affinity and/or misaligned/unstable FAD binding. These subtle but significant structural changes affect the conformation of the substrate-binding site, resulting in a diminished substrate affinity, reflected by increased Km. Our investigations demonstrated a decreased flavination of mutant enzyme and the flavinated fraction of the mutant showed an increased Km and decreased Vmax. This suggests that substitution of an Arg for H109 does indeed have an effect on the attachment and alignment of bound FAD. In addition, as was demonstrated by sensitivity to in vitro trypsin digestion, the under-flavinated enzyme may also be less stable in vivo than the wild-type enzyme, a possibility consistent with the observation that DMGDH antigen in patient fibroblasts was decreased (Binzak et al 2001). Yield of the recombinant DMGDH H109R polypeptide is less than one-third of that of the wild-type enzyme. It is possible that the deflavinated enzyme may account for a greater portion of the enzyme pool, some of which is degraded prior to purification. Conversely, it is possible that the H109R mutation causes a global instability in the protein that results in deflavination. This may in turn lead to degradation in vivo, which is also consistent with decreased antigen in patient fibroblasts. Thus, there are at least three potential sources for decreased DMGDH activity in the patient with the H109R mutation: (1) the mutant protein is misfolded due to the mutation and rapidly degraded upon translation; (2) even after ‘semi-stable’ folding, only less than 50% of the protein can be flavinated due to the decreased affinity for FAD and the unflavinated protein is degraded; and (3) even the flavinated protein has 1800-fold less catalytic efficiency compared to that of wild-type. The biochemical and structural bases for this inefficiency must await further studies of wild type and mutant DMGDH.
While our findings unequivocally substantiate the inactivating nature of this mutation and indicate that the patient likely had substantially reduced DMGDH activity in vivo, the true physiological function/role of this enzyme remains in question. Given the importance of methyl groups in a number of metabolic reactions, a disruption of their homeostasis would be predicted to have serious physiological sequelae. In contrast, the patient had relatively mild clinical symptoms consisting of muscular weakness and an unusual urine odour, indicating that there are other sources of methyl groups when DMGDH is deficient. Sarcosine can still be produced directly from glycine by glycine methyl-transferase, which uses S-adenosylmethionine as a methyl donor (Blumenstein and Williams 1960; Kerr 1972; Mitchell and Benevenga 1978). However, the patient with the DMGDH mutation exhibited chronic elevation of serum creatine kinase levels, which indicates ongoing muscle breakdown consistent with muscle weakness, presumably to supply additional S-adenosylmethionine to compensate for insufficient amount of methyl groups originated from choline metabolism. Since no additional DMGDH-deficient patients have been identified subsequent to the original one, either the mutation must be extremely rare, or have an as yet undefined alternative clinical phenotype, or in fact be most frequently lethal. An answer to this conundrum awaits identification of additional DMGDH-deficient humans or an appropriate mouse model.
Acknowledgments
This work was supported, in part, by NIH grants GM29076 (J.-J.P.K) and DK54936 (J.V.).
Abbreviations
- DMGDH
dimethylglycine dehydrogenase
- DMGO
dimethylglycine oxidase
- ETF
electron-transfer flavoprotein
- FAD
flavin–adenine dinucleotide
- SDH
sarcosine dehydrogenase
- THF
tetrahydrofolate
Footnotes
Competing interests: None declared
References to electronic databases: Dimethylglycine dehydrogenase: EC 1.5.99.2. Dimethylglycine dehydrogenase deficiency: OMIM 605850. Dimethylglycine oxidase structure: PDB 1PJ5.
References
- Abeles RH, Mackenzie CG. Production of active formaldehyde in the mitochondrial oxidation of sarcosine-CD3. J Biol Chem. 1956;222:145–150. [PubMed] [Google Scholar]
- Abeles RH, Frisell WR, Mackenzie CG. A dual isotope effect in the enzymatic oxidation of deuteromethyl sarcosine. J Biol Chem. 1960;235:853–856. [PubMed] [Google Scholar]
- Binzak BA, Wevers RA, Moolenaar SH, et al. Cloning of dimethylglycine dehydrogenase and a new human inborn error of metabolism, dimethylglycine dehydrogenase deficiency. Am J Hum Gen. 2001;68:839–847. doi: 10.1086/319520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenstein J, Williams GR. The enzymic N-methylation of glycine. Biochem Biophys Res Commun. 1960;3:259–263. doi: 10.1016/0006-291X(60)90235-7. [DOI] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Brizio C, Otto A, Brandsch R, Passarella S, Barile M. A protein factor of rat liver mitochondrial matrix involved in flavinylation of dimethylglycine dehydrogenase. Eur J Bioch. 2000;267:4346–4354. doi: 10.1046/j.1432-1327.2000.01464.x. [DOI] [PubMed] [Google Scholar]
- Brizio C, Barile M, Brandsch R. Flavinylation of the precursor of mitochondrial dimethylglycine dehydrogenase by intact and solubilised mitochondria. FEBS Lett. 2002;522:141–146. doi: 10.1016/S0014-5793(02)02927-7. [DOI] [PubMed] [Google Scholar]
- Brizio C, Brandsch R, Bufano D, Pochini L, Indiveri C, Barile M. Over-expression in Escherichia coli, functional characterization and refolding of rat dimethylglycine dehydrogenase. Protein Expr Purif. 2004;37:434–442. doi: 10.1016/j.pep.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Cook RJ, Misono KS, Wagner C. Identification of the covalently bound flavin of dimethylglycine dehydrogenase and sarcosine dehydrogenase from rat liver mitochondria. J Biol Chem. 1984;259:12475–12480. [PubMed] [Google Scholar]
- Frisell WR, Mackenzie CG. Separation and purification of sarcosine dehydrogenase and dimethylglycine dehydrogenase. J Biol Chem. 1962;237:94–98. [PubMed] [Google Scholar]
- Hoskins DD, Mackenzie CG. Solubilization and electron transfer flavoprtein requirement of mitochondrial sarcosine dehydrogenase and dimethylglycine dehydrogenase. J Biol Chem. 1961;236:177–183. [PubMed] [Google Scholar]
- Kerr SJ. Competing methyltransferase systems. J Biol Chem. 1972;247:4248–4252. [PubMed] [Google Scholar]
- Lehman TC, Hale DE, Bhala A, Thorpe C. An acyl-coenzyme A dehydrogenase assay utilizing the ferricenium ion. Anal Biochem. 1990;186:280–284. doi: 10.1016/0003-2697(90)90080-S. [DOI] [PubMed] [Google Scholar]
- Leys D, Basran J, Scrutton NS. Channelling and formation of ‘active’ formaldehyde in dimethylglycine oxidase. EMBO J. 2003;22:4038–4048. doi: 10.1093/emboj/cdg395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macheroux P. UV-visible spectroscopy as a tool to study flavoproteins. Methods Mol Biol. 1999;131:1–7. doi: 10.1385/1-59259-266-X:1. [DOI] [PubMed] [Google Scholar]
- Mackenzie CG, Frisell WR. The metabolism of dimethylglycine by liver mitochondria. J Biol Chem. 1958;232:417–427. [PubMed] [Google Scholar]
- Mitchell AD, Benevenga NJ. The role of transamination in methionine oxidation in the rat. J Nutr. 1978;108:67–78. doi: 10.1093/jn/108.1.67. [DOI] [PubMed] [Google Scholar]
- Moolenaar SH, Poggi-Bach J, Engelke UF, et al. Defect in dimethylglycine dehydrogenase, a new inborn error of metabolism: NMR spectroscopy study. Clin Chem. 1999;45:459–464. [PubMed] [Google Scholar]
- Otto A, Stoltz M, Sailer HP, Brandsch R. Biogenesis of the covalently flavinylated mitochondrial enzyme dimethylglycine dehydrogenase. J Biol Chem. 1996;271:9823–9829. doi: 10.1074/jbc.271.16.9823. [DOI] [PubMed] [Google Scholar]
- Porter DH, Cook RJ, Wagner C. Enzymatic properties of dimethylglycine dehydrogenase and sarcosine dehydrogenase from rat liver. Arch Biochem Biophys. 1985;243:396–407. doi: 10.1016/0003-9861(85)90516-8. [DOI] [PubMed] [Google Scholar]
- Wittwer AJ, Wagner C. Identification of folate binding protein of mitochondria as dimethylglycine dehydrogenase. Proc Natl Acad Sci U S A. 1980;77:4484–4488. doi: 10.1073/pnas.77.8.4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittwer AJ, Wagner C. Identification of the folate-binding proteins of rat liver mitochondria as dimethylglycine dehydrogenase and sarcosine dehydrogenase. Flavoprotein nature and enzymatic properties of the purified proteins. J Biol Chem. 1981a;256:4109–4115. [PubMed] [Google Scholar]
- Wittwer AJ, Wagner C. Identification of the folate-binding proteins of rat liver mitochondria as dimethylglycine dehydrogenase and sarcosine dehydrogenase. Purification and folate-binding characteristics. J Biol Chem. 1981b;256:4102–4108. [PubMed] [Google Scholar]