

Abstract
Background
Previous studies show that the potent, prototypical σ1-receptor agonist 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP) prevents cell death after oxygen-glucose deprivation (OGD) in primary cortical neuronal cultures. We tested the hypothesis that PPBP protects neurons by a mechanism involving activation of the transcription factor cyclic adenosine monophosphate response element-binding protein (CREB).
Methods
Primary cultured cortical neurons were exposed to 2 h of OGD and allowed to recover for 24 h, and PPBP treatment was initiated 15 min before the insult in the presence and absence of the σ1-receptor antagonist rimcazole and inhibitors against protein kinases known to activate signal transduction cascades that result in CREB phosphorylation, such as H89 (protein kinase A inhibitor), LY294002 (PI3K inhibitor), U0126 (MEK1/2 inhibitor), or KN62 calmodulin kinase II inhibitor). Neuronal cell death was assayed by lactate dehydrogenase measurement 24 h after OGD. CREB phosphorylation was measured by immunoblot analysis at 30 min, 1 h, and 3 h of reoxygenation. Blots were quantitatively analyzed using Quantity One image analysis software.
Results
PPBP increased CREB phosphorylation at 1 h after recovery from OGD, which was abolished by rimcazole (1.7 ± 0.2 in PPBP and 0.8 ± 0.1 in PPBP plus rimcazole with OGD compared with 0.9 ± 0.1 in OGD alone, p-CREB/CREB). The PPBP-induced increase in CREB phosphorylation was blocked by H89 (0.5 ± 0.07) but not U0126, KN62, or LY294002. PPBP treatment prevented OGD-induced cell death and pretreatment with H89 blocked this protection (0.18 ± 0.02 in PPBP and 0.27 ± 0.03 in PPBP plus H89 with OGD compared with 0.33 ± 0.02 in OGD alone, lactate dehydrogenase assay). Pretreatment with LY294002, UO126, or KN62 had no effect on neuronal protection by PPBP.
Conclusions
These data suggest that the mechanism of neuroprotection by PPBP may be linked to CREB phosphorylation.
The σ1 receptor is distributed in neural and nonneural tissues and is characterized by unique ligand specificity.1 Its ligand binding site is distinct from the phencyclidine receptor on the N-methyl-d-aspartate (NMDA) receptor channel complex.2 σ1 receptor agonists modulate the neuronal response to pharmacologic stimulation of the NMDA receptor3 and are neuroprotective in animal and neural culture models of cerebral ischemia.4–8 Mechanistically, the potent and prototypic σ1 receptor agonist, 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP), protects brain by attenuating ischemia- and NMDA-induced nitric oxide (NO) production.9,10 Furthermore, in primary cortical neuronal cultures treated with oxygen-glucose deprivation (OGD), we have shown that PPBP-linked neuroprotection is associated with increased Bcl-2 expression.4 Bcl-2 expression is regulated, in part, by the transcription factor cyclic adenosine monophosphate (AMP) response element binding protein (CREB).11 Accordingly, we hypothesized that CREB may be important to PPBPs mechanism of neuroprotection.
CREB12 is a member of the CREB/activating transcription factor family and is abundant in brain, particularly in neurons. CREB plays a principal role in neuronal plasticity processes underlying learning and memory.13 Phosphorylation of Ser-133 residue is necessary but not sufficient for activation of cyclic AMP response element (CRE)-mediated gene transcription.14 Several genes which have a CRE in the promoter region, including Bcl-2, c-fos, brain-derived neurotrophic factor, and NO synthase, are commonly induced after ischemia, suggesting that activation of CREB may be involved in determining neuron survival. Several studies have shown that the CREB protein can activate CRE-containing genes when phosphorylated on Ser133 by any of a variety of protein kinases, including protein kinase A (PKA), mitogen-activated protein kinase (MAPK), mitogen/extracellular signal-regulated kinase (MEK)/extracellular signal-regulated kinase, calcium/calmodulin kinase II (CaMKII), and phosphatidylinositol 3-kinase/AKT (PI3-K/AKT).15–20 In addition, inhibitors are available against protein kinases known to activate signal transduction cascades that result in CREB phosphorylation, such as H89 (N-[2-(p-bromocinnamy-lamino)ethyl]-5-isoquinolinesulfonamidedihydrochloride) (PKA inhibitor), LY294002 (2-(4-morpholinyl)-8-henyl-1(4H)-benzopyran-4-one hydrochloride) (PI3K inhibitor), U0126 (1,4- diamino-2,3-dicyano-1,4-bis(ominophenyl-mercapto)butadiene), (MEK1/2 inhibitor) or KN62 (1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-l-tyrosyl]-4-phenylpiperazine) (CaMKII inhibitor).
The relationship between CREB phosphorylation and σ1-receptor has not been previously evaluated. Here, we show that PPBP enhances CREB phosphorylation and that PPBPs ability to protect neurons and to increase CREB phosphorylation in vitro is blocked by PKA inhibitor H89 but not by U0126, KN62, or LY294002. These data suggest that PPBP reduces cell death in vitro by a mechanism linked to PKA-dependent CREB phosphorylation.
Methods
Experimental protocols were approved by the Institutional Animal Care and Use Committee and conform to the National Institutes of Health guidelines for the care and use of animals in research.
Chemicals
PPBP was obtained from Tocris (Ellisville, USA). The σ1 receptor antagonist rimcazole, the protein kinase a inhibitor H89, the calcium/calmodulin kinase inhibitor KN62, the MEK inhibitor U0126, and the phosphatidylinositol 3-kinase inhibitor LY294002 were obtained from Sigma (St. Louis, MO). Dimethyl-sulfoxide was used as vehicle at a final concentration of 0.02%. The antibody against p-CREB and CREB was obtained from Cell Signal Technology (Danvers, MA). Cytotoxicity detection kits were obtained from Roche Molecular Biochemicals (Mannheim, Germany). Neurobasal medium, B27, and Dulbecco phosphate-buffered saline were obtained from Invitrogen (Carlsbad, CA). Culture medium was purchased from Gibco (Grand island, NY).
Primary Neuronal Cell Cultures
Primary cortical neuronal cultures were derived from embryos (18-day gestation) of Sprague-Dawley rats (Charles River, MA). Cultures were prepared as described previously21 with modifications. Dissociated cells were plated onto precoated poly-l-ornithine plates (24-well plates, 2.5 × 105 cells/well and 6-well plates, 14 × 105 cells/well). Cells were maintained in cultivation medium (Neurobasal medium with supplement 2% B27, 2 mM Glutamax) at 37°C in 100% humidity and a 95% room air/5% CO2 atmosphere. All experiments were performed at 11 days in vitro (DIV).
Oxygen-Glucose Deprivation
The culture medium was replaced with OGD buffer (Dulbecco phosphate-buffered saline with 1 mM CaCl2, 0.8 mM MgCl2) and placed in a Coy anoxia chamber containing 90% N2, 5% H2, and 5% CO2 for 2 h. The OGD solution was prewarmed and bubbled with anaerobic gas mixture in the anoxia chamber (Coy) before the exchange. For reoxygenation, cells were removed from the anoxia chamber, the OGD medium was replaced with cultivating medium, and cells returned to normoxia. Control cells were maintained for 2 h in prewarmed, oxygenated phosphate buffered saline with glucose under normoxic conditions. In drug treatment studies, rimcazole 5 μM was added to the culture medium for 2 h; 10 μM of H89, LY294002, U0126, or Kn62 was added to the cells for 3 h before OGD and remained in the culture throughout OGD and 24 h of reoxygenation (Table 1). Just before OGD, growth media was replaced with OGD buffer and incubated with 10 μM PPBP for 20 min before OGD, during 2 h OGD, and for the 24 h of reoxygenation (for cell death assay) or 1 h recovery (for immunoblotting).
Table 1.
Experimental Groups and Reagents Used in the Experiments
| Reagent | Explanation | Concentration | Treatment | 2 h-OGD/recovery |
|---|---|---|---|---|
| PPBP | σ1 receptor agonist | 10 μM | 20 min before OGD | + |
| H89 | Protein kinase A inhibitor | 10 μM | 3 h preincubation | + |
| H89 + PPBP | 3 h preincubation + 20 min PPBP | + | ||
| KN62 | Calcium/calmodulin kinase inhibitor | 10 μM | 3 h preincubation | + |
| KN62 + PPBP | 3 h preincubation + 20 min PPBP | + | ||
| LY294002 | PI3K inhibitor | 10 μM | 3 h preincubation | + |
| LY29002 + PPBP | 3 h preincubation + 20 min PPBP | + | ||
| U0126 | MEK inhibitor | 10 μM | 3 h preincubation | + |
| U0126 + PPBP | 3 h preincubation + 20 min PPBP | + |
Cells were incubated with multiple protein kinases inhibitor 3 h, then subjected to 2-h OGD before cells were preincubated in 10-μM 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP) for 20 min, followed by reoxygenation for 24 h (for lactate dehydrogenase [LDH] assay) and 30 min, 1 h, or 3 h (for immunoblotting).
OGD = oxygen-glucose deprivation.
Assessment of Neuronal Cell Death
Cell death was assessed by lactate dehydrogenase (LDH) measurement. LDH release was measured in the culture medium using the cytotoxicity detection kit. Medium (100 μL) was transferred from culture wells to 96-well plates and mixed with 100 μL reaction solution provided by the kit. Optical density was measured at 492 nm 30 min later, using a microplate reading device (spectra Max plus, Molecular Devices). Background absorbance at 620 was subtracted. The maximal releasable LDH was obtained in each well by 10 min incubation with 1% triton x-100 at the end of each experiment. Cell death was measured in triplicates. Three measurements from one culture were then averaged and considered n = 1.
Immunoblotting
Six-well plate cultured cells were used for the experiment, with each condition being assessed in duplicate. After phosphate buffered saline was used to wash the plate, 100 μL boiled sample buffer (100 mM Tris pH 6.8, 2% sodium dodecyl sulfate, 20% glycerol, 10% β-mercaptoethanol, 0.025% bromphenol blue) was added to each well at room temperature. Cells were then scraped and transferred to 1.5 mL tube and boiled for 5 min. Collected protein samples were then frozen at −80°C for subsequent immunoblotting. Immunoblotting was conducted on the 30 μL of previously frozen protein sample using 12% SDS-PAGE and transferred onto a nitrocellulose membrane. Anti-p-CREB and CREB antibody were diluted 1:1000 and subjected to overnight incubation at 4°C with each sample. Membranes were incubated with anti-rabbit IgG conjugated to horseradish peroxidase (Jackson Immunoresearch, Plymouth, PA) for 1 h. Protein bands were visualized with Supersignal (Pierce, Rock-ford, IL) according to the manufacturer's instructions. Blots were quantitatively analyzed using Quantity One image analysis software (Biorad laboratory, Hercules, CA). CREB activation is expressed as the ratio of normalized phosphorylated CREB to total CREB.
Statistical Analysis
Results are presented as mean ± sem. Statistical significance was determined by one-way analysis of variance, followed by the Newman-Keuls post hoc test. P < 0.05 was considered statistically significant.
Results
PPBP Increased CREB Phosphorylation After 2-h OGD
To determine the mechanism for PPBP-induced neuroprotection, we measured the effect of PPBP on CREB phosphorylation by immunoblotting after 2 h OGD. PPBP upregulated CREB phosphorylation after 2 h OGD with 1 h reoxygenation, but there was no change in CREB phosphorylation at either 30 min or 3 h reoxygenation. PPBP-upregulated CREB phosphorylation at 1 h reoxygenation was prevented by pretreatment with the σ-1 receptor antagonist, rimcazole (Figure 1, n = 3). PPBP did not have an effect on CREB phosphorylation under baseline conditions (data not shown). Similarly, rimcazole alone and rimcazole plus PPBP had no effect on neuronal survival without OGD. Finally, consistent with our previous study,4 rimcazole alone had no effect on cell death as measured by LDH (data not shown).
Figure 1.
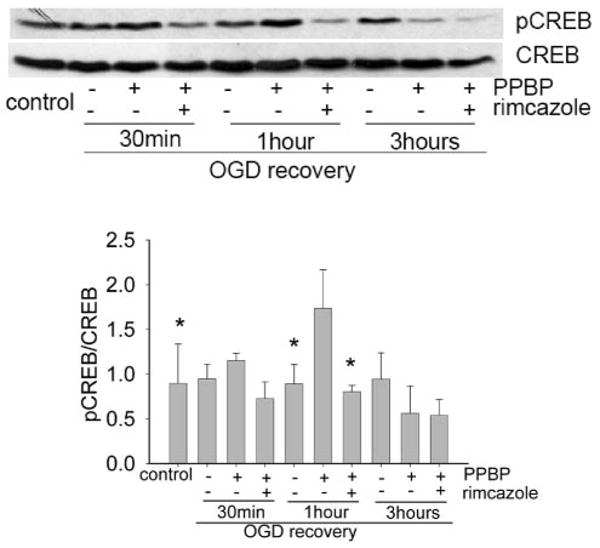
4-phenyl-1-(4-phenylbutyl) piperidine (PPBP) increases the level of cyclic adenosine monophosphate response element-binding protein (CREB) phosphorylation after 2 h oxygen-glucose deprivation (OGD). Cells were preincubated in 10 μM PPBP for 20 min before exposure to 2 h OGD and reoxygenation for 30 min, 1 h, 3 h. The Western blot shows an increase in the level of CREB phosphorylation afer 2-h OGD and 1-h recovery. The induced p-CREB can be reversed by rimcazole. *Denotes a significant difference from CREB phosphorylation by PPBP at 1-h recovery. *P < 0.05 (n = 3).
PKA Inhibitor H89 Blocks PPBP-Induced CREB Phosphorylation
To further understand the relationship between CREB phosphorylation and neuroprotection, we determined the effect of protein kinase inhibitors on PPBP-regulated CREB phosphorylation. Cells were preincubated with H89, KN62, U0126, or LY294002 for 3 h and then replaced with 10 μm PPBP for 20 min, followed by 2 h OGD and 1 h recovery. Western blot showed that PPBP-upregulated CREB phosphorylation after OGD was blocked by PKA inhibitor H89 but not by U0126, KN62, or LY294002 (Fig. 2, n = 4).
Figure 2.
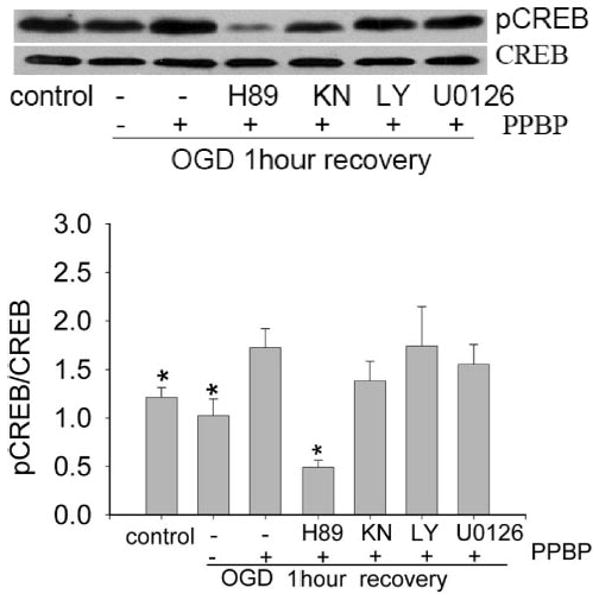
Protein kinase A (PKA) inhibitor H89 blocked 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP) upregulated the cyclic adenosine monophosphate response element-binding protein (CREB) phosphorylation. Cells were preincubated with H89 for 3 h and 10 μM PPBP for 20 min followed by 2-h oxygen-glucose deprivation (OGD) and 1-h recovery. The Western blot shows that the PKA inhibitor H89 inhibits OGD-induced p-CREB. *Denotes a significant difference from 2 h OGD + PBBP at 1-h recovery. *P < 0.05, n = 4).
The Effect of PKA Inhibition on PPBP-induced Neuronal Protection
CREB is a substrate for many protein kinases. To determine the specific protein kinase that mediated the neuroprotective effect of PPBP, we examined the effect of the PKA inhibitor H89 (10 μM), CaMKII inhibitor KN62 (10 μM), MEK inhibitor U0126 (10 μM), and PI3K inhibitor LY294002 (10 μM). Pretreatment with PKA inhibitor H89 completely prevented PPBP-mediated neuroprotection. (Fig. 3A, n = 6). However, pretreatment with KN62, LY294002, and U0126 under the same conditions had no effect on PPBP-conferred neuroprotection. (Figs. 3B–D, n = 4 each). H89 did not induce cell death by itself (without OGD), and it did not alter OGD-induced cell death in the absence of PPBP, suggesting that CREB is a neuroprotective pathway specifically activated by PPBP.
Figure 3.
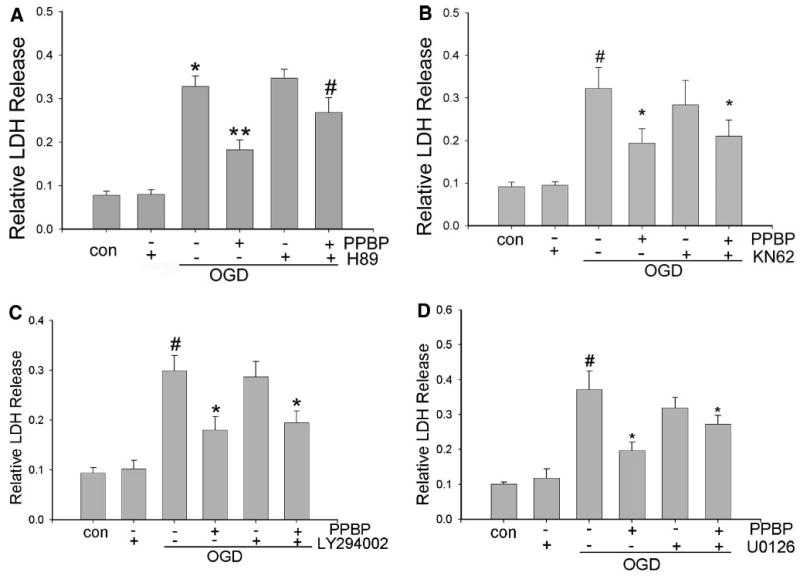
(A) The effect of protein kinase A on 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP) in neuronal protection. Cells were preincubated with H89 for 3 h and 10 μM PPBP for 20 min followed by 2-h oxygen-glucose deprivation (OGD) and 24-h recovery. The data show the inhibition of PPBP protection by protein kinase A inhibitor. *Denotes significant difference between 2 h OGD vs control. **Denotes a difference between 2 h OGD and 2 h OGD+PPBP. #Denotes a difference between 2 h OGD + PPBP vs 2 h OGD + PPBP + H89 (P < 0.05, n = 6). (B–D) Lack of inhibition of PBPP in neuronal protection by multiple protein kinase inhibitors. Cells were preincubated with KN62, U0126, or LY294002 for 3 h and 10 μM PPBP for 20 min followed by 2-h OGD and 24-h recovery. The data show no effect on PPBP neuronal protection by KN62, U0126, and LY294002. #Denotes a significant difference between 2 h OGD and control. *Denotes a significant difference from 2-h OGD (P < 0.05, n = 4).
Discussion
The present study demonstrates two important, novel findings. First, σ1 receptor agonist PPBP increases CREB phosphorylation after 2 h OGD, which is most apparent at 1 h of reoxygenation and is blocked by the selective σ1-receptor antagonist rimcazole. Second, PPBP neuroprotection and CREB phosphorylation after 2-h OGD was blocked by PKA inhibitor H89 but not by inhibitors of other protein kinases known to phosphorylate CREB. These data suggest that the σ1 receptor agonist PPBP protects neurons from ischemia-induced cell death via activation of CREB, which is dependent on PKA activity.
The σ receptor was originally proposed to be a subtype of the opioid receptor. However, it is now clear that σ receptors are unique nonopioid, nonphen-cyclidine brain protein. The σ receptors are classified into σ1 receptor and σ2 receptor subtypes. The σ1 receptors normally localize at the endoplasmic reticulum, activate a variety of signal transductions, including intracellular Ca2+ dynamics and neurotrophic factor, and regulate the activity of diverse ion channels via protein-protein interactions. It has neuronal protective effects and potential clinical utility in stroke and neuropsychiatric and neurodegenerative disorders.22
The transcription factor CREB has been identified as a target for several signaling pathways mediated by growth factors. In neuronal systems, a wide range of neuromodulators and neurotransmitters could converge on CREB, via various kinase pathways and regulate neuronal survival and plasticity. For example, activated CREB is important for survival of hippocampal neurons and other cell systems.23 CREB was previously shown to be involved in protecting neurons from ischemic cell death.24 In this investigation, PPBP was demonstrated to prevent neuronal cell death after 2 h OGD and 24 h recovery, by a mechanism that depends on CREB phosphorylation. The neuroprotective effect of PPBP and its dose-response relationship have been published.4 Previous studies have also demonstrated that σ1 receptor activation mediates neuroprotection by increasing Bcl-2 and reducing ischemia-induced NO production.
We here show that improved neuronal cell survival by PPBP is linked to an early and transient postischemic (post-OGD) CREB phosphorylation. PPBP increased CREB phosphorylation at 1 h after recovery from OGD, which was abolished by σ1 receptor blocker rimcazole and PKA inhibitor H89. More importantly, H89 (and rimcazole4) prevented the protection afforded by PPBP against OGD-induced cell death in primary cortical neurons, suggesting that PKA-dependent phosphorylation of CREB is an important mechanism of neuroprotection by σ1 receptor activation. Neither rimcazole nor H89 had an effect on cell death when applied alone without OGD, suggesting that they specifically block protection by PPBP.
Although this CREB phosphorylation appears critical in the mechanism of PPBP-induced neuroprotection in this model, further direct studies will be needed to determine whether CREB phosphorylation mediates the protective effect of PPBP against cerebral ischemic injury in vivo. Our data implicate PKA activation and CREB phosphorylation as downstream mediators of σ1 receptor stimulation. Whether σ1 receptor interacts directly or indirectly with PKA cannot be discerned from our data and is the subject of future studies. Although another PKA target could be responsible for the protection by PKA, CREB remains the strongest candidate, as its activation has been strongly implicated in neuroprotection and upregulation of neuroprotective genes after ischemia.11 It should be noted, however, that H89 may inhibit other kinases, at least in cell-free systems. Therefore, the evidence supporting a role for PKA in PPBP neuroprotection should be interpreted with caution until other evidence using different experimental approaches are available to corroborate this conclusion. Further studies are also needed to confirm the causal relationship between CREB phosphorylation and neuroprotection. CREB phosphorylation may represent a surrogate of protection, rather than causative neuroprotective mechanism.
Many neurotransmitters can lead to CREB phosphorylation through activation of several second messengers and kinases, including Akt, PKA, MAPK/extracellular signal-regulated kinase, and CamKII.25 Akt-mediated phosphorylation of cytosolic protein, such as glycogen synthase kinase-3 and Bad, plays a critical role in the regulation of metabolic pathways as well as prevention of cell death by insulin and growth factors. Akt can regulate gene expression at the transcription level26 and increases the expression of Bcl-2 in PC12 cells and BAF/3 cells.17 Growth factor-mediated signaling through PI3-kinase/Akt could also be involved in the induction of Bcl-2 expression via CREB phosphorylation.17 Akt (also known as protein kinase B) stimulates the phosphorylation of CREB on serine 133 and promotes the recruitment of the co-activator CREB-binding protein.27 Ischemia increases intracellular cyclic AMP levels and activates PKA, which can phosphorylate CREB.25 In addition, stimulation of Gs-protein-coupled receptors can also increase activity of the cAMP/PKA pathway and thereby increase phosphorylation of striatal CREB in vivo and in vitro.28 Interestingly, much like PPBP, activation of muscarinic receptors also induce phosphoCREB through a MEK-independent pathway.29 The cAMP content of brain tissue has been reported to increase shortly after the onset of ischemia and to be correlated with subsequent activation of PKA.30 Activated PKA then causes the phosphorylation of a critical serine residue on CREB (amino acid 133) with subsequent translocation of p-CREB to nucleus and binding to CRE and CREB binding protein.31 In an in vivo model of ischemia, PKA activity was enhanced during recirculation period, which could be closely associated with the sustained activation of CREB phosphorylation in this area. The enhanced phosphorylation of CREB was clearly sustained in the peri-ischemia area, where cresyl violet staining revealed almost no tissue damage, and the expression of p-CREB that was reduced in the ischemic core during 12–48 h recirculation.25
Mabuchi et al.32 have demonstrated that calcium-calmodulin-dependent protein kinase (CaMK) activation during ischemia is important in the mechanism of CREB phosphorylation. Neurons express at least five known CaMKs; the activation mechanisms of CaMKI and CaMKIV are phosphorylated by CaMK Kinase, and phosphorylation is essential for activation of CaMKI and CaMKIV.33,34 CaMKIV is translocated to nuclei and a major substrate for CaMKIV in the nuclei is CREB.35 Two MEK inhibitors (PD98059 and U0126) consistently blocked CREB phosphorylation.28 The MAPKs role is further supported by a number of reports showing the coupling of MAPK to CREB phosphorylation.28 Our data demonstrated that the PKA inhibitor H89 prevented both PPBP-induced neuronal protection and blocked PPBP-induced CREB phosphorylation. However, in our study the CaMK inhibition with KN62, blocking p42/p44 MAP kinase activation (via U0126) and Akt activation (via LY294002) had no effect on either PPBP-induced neuroprotection or CREB phosphorylation. This suggests that PPBP protects, in part, against neuronal cell death through a PKA pathway. Our data do not exclude the possibility that these inhibitors of alternate pathways for phosphorylation would be effective at a higher dose or different timing of administration. Furthermore, the specificity of H89 has been questioned.36 However, not unlike other pharmacological agents, the presumed specificity of H89 to PKA is limited to a dose range and experimental conditions, such as the adenosine triphosphate concentration used in the assay, and is usually determined in cell-free systems using purified enzymes. When tested in intact cells or in vivo, especially under conditions of low adenosine triphosphate as in OGD, specificity, and effectiveness, have to be reevaluated. Furthermore, even if the inhibitor is not absolutely specific for a particular enzyme, it might still provide useful information if the other crossreacting targets are not present in the system under investigation. For example, we found that PKC, a potential crossreacting enzyme, is not activated in neurons after OGD (data not shown), making any potential effect of H89 on PKC irrelevant to this particular study.
In conclusion, this study demonstrates that σ1-receptor agonist PPBP affords neuroprotection in vitro, via PKA-dependent phosphorylation of CREB.
Acknowledgments
Supported, in part, by US Public Health Service National Institutes of Health grants NS 20020 and NS 046379.
References
- 1.Maurice T, Phan VL, Urani A, Kamei H, Noda Y, Nabeshima T. Neuroactive neurosteroids as endogenous effectors for the sigma1 (sigma1) receptor: pharmacological evidence and therapeutic opportunities. Jpn J Pharmacol. 1999;81:125–55. doi: 10.1254/jjp.81.125. [DOI] [PubMed] [Google Scholar]
- 2.Walker JM, Bowen WD, Walker FO, Matsumoto RR, De CB, Rice KC. Sigma receptors: biology and function. Pharmacol Rev. 1990;42:355–402. [PubMed] [Google Scholar]
- 3.Lesage AS, De Loore KL, Peeters L, Leysen JE. Neuroprotective sigma ligands interfere with the glutamate-activated NOS pathway in hippocampal cell culture. Synapse. 1995;20:156–64. doi: 10.1002/syn.890200210. [DOI] [PubMed] [Google Scholar]
- 4.Yang S, Bhardwaj A, Cheng J, Alkayed NJ, Hurn PD, Kirsch JR. Sigma receptor agonists provide neuroprotection in vitro by preserving bcl-2. Anesth Analg. 2007;104:1179–84. doi: 10.1213/01.ane.0000260267.71185.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harukuni I, Bhardwaj A, Traystman RJ, Crain B, London ED, Kirsch JR. Neuroprotection from focal ischemia by 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP) is dependent on treatment duration in rats. Anesth Analg. 1998;87:1299–305. doi: 10.1097/00000539-199812000-00016. [DOI] [PubMed] [Google Scholar]
- 6.Takahashi H, Kirsch JR, Hashimoto K, London ED, Koehler RC, Traystman RJ. PPBP [4-phenyl-1-(4-phenylbutyl) piperidine], a potent sigma-receptor ligand, decreases brain injury after transient focal ischemia in cats. Stroke. 1995;26:1676–82. doi: 10.1161/01.str.26.9.1676. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi H, Kirsch JR, Hashimoto K, London ED, Koehler RC, Traystman RJ. PPBP [4-phenyl-1-(4-phenylbutyl) piperidine] decreases brain injury after transient focal ischemia in rats. Stroke. 1996;27:2120–3. doi: 10.1161/01.str.27.11.2120. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi H, Traystman RJ, Hashimoto K, London ED, Kirsch JR. Postischemic brain injury is affected stereospecifically by pentazocine in rats. Anesth Analg. 1997;85:353–7. doi: 10.1097/00000539-199708000-00020. [DOI] [PubMed] [Google Scholar]
- 9.Sugimoto K, Iadecola C. Effects of aminoguanidine on cerebral ischemia in mice: comparison between mice with and without inducible nitric oxide synthase gene. Neurosci Lett. 2002;331:25–8. doi: 10.1016/s0304-3940(02)00834-0. [DOI] [PubMed] [Google Scholar]
- 10.Bhardwaj A, Sawada M, London ED, Koehler RC, Traystman RJ, Kirsch JR. Potent sigma1-receptor ligand 4-phenyl-1-(4-phenylbutyl) piperidine modulates basal and N-methyl-d-aspartate-evoked nitric oxide production in vivo. Stroke. 1998;29:2404–10. [PubMed] [Google Scholar]
- 11.Meller R, Minami M, Cameron JA, Impey S, Chen D, Lan JQ, Henshall DC, Simon RP. CREB-mediated Bcl-2 protein expression after ischemic preconditioning. J Cereb Blood Flow Metab. 2005;25:234–46. doi: 10.1038/sj.jcbfm.9600024. [DOI] [PubMed] [Google Scholar]
- 12.Brindle PK, Montminy MR. The CREB family of transcription activators. Curr Opin Genet Dev. 1992;2:199–204. doi: 10.1016/s0959-437x(05)80274-6. [DOI] [PubMed] [Google Scholar]
- 13.Martin KC, Kandel ER. Cell adhesion molecules, CREB, and the formation of new synaptic connections. Neuron. 1996;17:567–70. doi: 10.1016/s0896-6273(00)80188-9. [DOI] [PubMed] [Google Scholar]
- 14.Hu BR, Fux CM, Martone ME, Zivin JA, Ellisman MH. Persistent phosphorylation of cyclic AMP responsive element-binding protein and activating transcription factor-2 transcription factors following transient cerebral ischemia in rat brain. Neuro-science. 1999;89:437–52. doi: 10.1016/s0306-4522(98)00352-2. [DOI] [PubMed] [Google Scholar]
- 15.Beitner-Johnson D, Millhorn DE. Hypoxia induces phosphorylation of the cyclic AMP response element-binding protein by a novel signaling mechanism. J Biol Chem. 1998;273:19834–9. doi: 10.1074/jbc.273.31.19834. [DOI] [PubMed] [Google Scholar]
- 16.Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–62. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- 17.Pugazhenthi S, Nesterova A, Sable C, Heidenreich KA, Boxer LM, Heasley LE, Reusch JE. Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J Biol Chem. 2000;275:10761–6. doi: 10.1074/jbc.275.15.10761. [DOI] [PubMed] [Google Scholar]
- 18.Sun P, Enslen H, Myung PS, Maurer RA. Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 1994;8:2527–39. doi: 10.1101/gad.8.21.2527. [DOI] [PubMed] [Google Scholar]
- 19.Walton M, Woodgate AM, Muravlev A, Xu R, During MJ, Dragunow M. CREB phosphorylation promotes nerve cell survival. J Neurochem. 1999;73:1836–42. [PubMed] [Google Scholar]
- 20.Zanassi P, Paolillo M, Feliciello A, Avvedimento EV, Gallo V, Schinelli S. cAMP-dependent protein kinase induces cAMP-response element-binding protein phosphorylation via an intracellular calcium release/ERK-dependent pathway in striatal neurons. J Biol Chem. 2001;276:11487–95. doi: 10.1074/jbc.M007631200. [DOI] [PubMed] [Google Scholar]
- 21.Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ, Simon RP. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–98. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 22.Hayashi T, Su TP. An update on the development of drugs for neuropsychiatric disorders: focusing on the sigma 1 receptor ligand. Expert Opin Ther Targets. 2008;12:45–58. doi: 10.1517/14728222.12.1.45. [DOI] [PubMed] [Google Scholar]
- 23.Walton MR, Dragunow I. Is CREB a key to neuronal survival? Trends Neurosci. 2000;23:48–53. doi: 10.1016/s0166-2236(99)01500-3. [DOI] [PubMed] [Google Scholar]
- 24.Walton M, Sirimanne E, Williams C, Gluckman P, Dragunow M. The role of the cyclic AMP-responsive element binding protein (CREB) in hypoxic-ischemic brain damage and repair. Brain Res Mol Brain Res. 1996;43:21–9. doi: 10.1016/s0169-328x(96)00144-1. [DOI] [PubMed] [Google Scholar]
- 25.Tanaka K, Nogawa S, Nagata E, Suzuki S, Dembo T, Kosakai A, Fukuuchi Y. Temporal profile of CREB phosphorylation after focal ischemia in rat brain. Neuroreport. 1999;10:2245–50. doi: 10.1097/00001756-199908020-00004. [DOI] [PubMed] [Google Scholar]
- 26.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 27.Du K, Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem. 1998;273:32377–9. doi: 10.1074/jbc.273.49.32377. [DOI] [PubMed] [Google Scholar]
- 28.Mao L, Tang Q, Samdani S, Liu Z, Wang JQ. Regulation of MAPK/ERK phosphorylation via ionotropic glutamate receptors in cultured rat striatal neurons. Eur J Neurosci. 2004;19:1207–16. doi: 10.1111/j.1460-9568.2004.03223.x. [DOI] [PubMed] [Google Scholar]
- 29.Greenwood JM, Dragunow M. Muscarinic receptor-mediated phosphorylation of cyclic AMP response element binding protein in human neuroblastoma cells. J Neurochem. 2002;82:389–97. doi: 10.1046/j.1471-4159.2002.00992.x. [DOI] [PubMed] [Google Scholar]
- 30.Kobayashi M, Lust WD, Passonneau JV. Concentrations of energy metabolites and cyclic nucleotides during and after bilateral ischemia in the gerbil cerebral cortex. J Neurochem. 1977;29:53–9. doi: 10.1111/j.1471-4159.1977.tb03923.x. [DOI] [PubMed] [Google Scholar]
- 31.Parker D, Ferreri K, Nakajima T, LaMorte VJ, Evans R, Koerber SC, Hoeger C, Montminy MR. Phosphorylation of CREB at Ser-133 induces complex formation with CREB-binding protein via a direct mechanism. Mol Cell Biol. 1996;16:694–703. doi: 10.1128/mcb.16.2.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mabuchi T, Kitagawa K, Kuwabara K, Takasawa K, Ohtsuki T, Xia Z, Storm D, Yanagihara T, Hori M, Matsumoto M. Phosphorylation of cAMP response element-binding protein in hippocampal neurons as a protective response after exposure to glutamate in vitro and ischemia in vivo. J Neurosci. 2001;21:9204–13. doi: 10.1523/JNEUROSCI.21-23-09204.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haribabu B, Hook SS, Selbert MA, Goldstein EG, Tomhave ED, Edelman AM, Snyderman R, Means AR. Human calcium-calmodulin dependent protein kinase I: cDNA cloning, domain structure and activation by phosphorylation at threonine-177 by calcium-calmodulin dependent protein kinase I kinase. EMBO J. 1995;14:3679–86. doi: 10.1002/j.1460-2075.1995.tb00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Selbert MA, Anderson KA, Huang QH, Goldstein EG, Means AR, Edelman AM. Phosphorylation and activation of Ca(2+)-calmodulin-dependent protein kinase IV by Ca(2+)-calmodulin-dependent protein kinase Ia kinase. Phosphorylation of threonine 196 is essential for activation. J Biol Chem. 1995;270:17616–21. doi: 10.1074/jbc.270.29.17616. [DOI] [PubMed] [Google Scholar]
- 35.Enslen H, Sun P, Brickey D, Soderling SH, Klamo E, Soderling TR. Characterization of Ca2+/calmodulin-dependent protein kinase IV. Role in transcriptional regulation. J Biol Chem. 1994;269:15520–7. [PubMed] [Google Scholar]
- 36.Lochner A, Moolman JA. The many faces of H89: a review. Cardiovasc Drug Rev. 2006;24:261–74. doi: 10.1111/j.1527-3466.2006.00261.x. [DOI] [PubMed] [Google Scholar]