

The intramolecular Schmidt reaction is a useful method for the synthesis of lactams.1 Most commonly, this reaction affords fused lactams bearing a nitrogen atom at a ring fusion position by preferential migration of the carbon bearing the azide-containing side chain. We have recently shown that certain substitution types lead to the alternative regioisomeric product containing a bridged ring system incorporating nitrogen at a bridgehead position (Scheme 1).2 Such compounds are noteworthy since they contain distorted amide bond linkages, which are of interest from theoretical, mechanistic, and reactivity perspectives.3 However, these methods are substrate dependent and provide mixtures of bridged and fused lactams, even in the most favorable cases. We now show that intramolecular Schmidt reactions can be reliably steered toward bridged heterocycles containing orthoamides in high yields. The method is broad in scope and allows for systematic study of compounds that are analogous to elusive tetrahedral intermediates of amide addition reactions.4
Scheme 1.
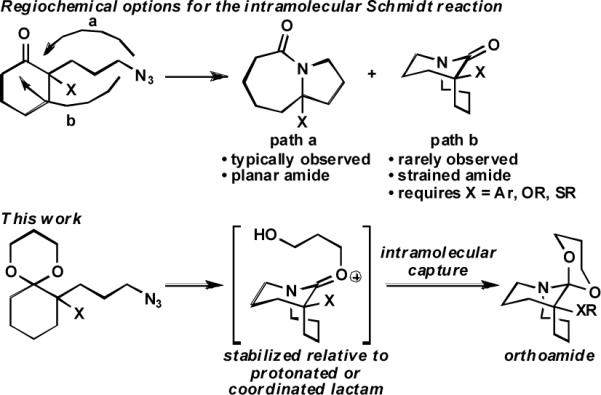
We hypothesized that (1) the formation of bridged lactams using the Schmidt reaction of ketones and azides was hampered by the necessity of forming a strained non-planar amide, (2) that this could be mitigated by relying instead on formation of a relatively stable iminium ether intermediate, and (3) that intramolecular capture would provide an easy road to a stable – and unusual – orthoamide product. That we had previously demonstrated that ketals also participate in azido-Schmidt chemistry5a combined with a single report of a bridged ring system formed in this way5b suggested that reactions of these species might favor bridged products to a greater degree.
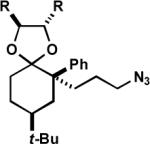
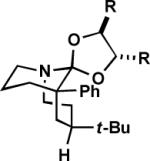
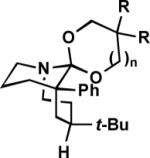
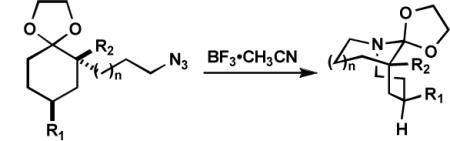
To maximize the chance for success, we focused first on alkyl azides that had already been shown to react preferentially by path b (Scheme 1).2 Thus, Lewis or protic acid treatment of a series of ketals derived from 1,3-dioxolane afforded bridged orthoamides in excellent yield (Table 1). The products were stable to the reaction and chromatographic isolation conditions. As hypothesized, the transformation proceeded with efficient control of regiochemistry, while the ketones corresponding to 1a–f provided a ca. 5:2 mixture of bridged and fused lactams.2a These results demonstrate that this approach accommodates a variety of ketal types. In contrast, the behavior of enol ether 1f shows that success depends upon intramolecular capture of the intermediate oxonium ion and that the [4.3.1] ring system containing a hemiaminal is unstable.
Table 1.
Formation of bridged orthoamides from ketals.
| entry | azide | product | conditionsa | yield (%)b |
|---|---|---|---|---|
 |
 |
|||
| 1 | (1a) R, R = H | (2a) | A | 88 |
| 2 | (1b) R, R = -(CH2)4- | (2b) | A | 71 |
 |
 |
|||
| 3 | (1c) n = 1, R, R = H | (2c) | B | 77 |
| 4 | (1d) n = 1, R, R = Me | (2d) | B | 75 |
| 5 | (1e) n = 2, R, R = H | (2e) | B | 70c |
| 6 | 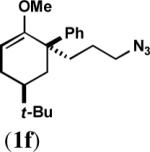 |
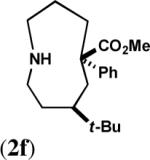 |
C | 63 |
A: BF3•CH3CN, 5.0 equiv, 0 °C to rt; B: BF3•CH3CN, 3.0 equiv, −78 °C to rt; C: TfOH, 5.0 equiv, 0 °C.
Isolated after chromatography
Determined by 1H NMR, see SI (Supporting Information) for details.
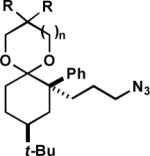
Subsequent work showed that a range of conformationally locked azido alkyl 1,3-dioxolanes substituted with aromatic rings in the 2 position could easily be transformed into analogous products (Table 2, entries 1–4). Throughout, these reactions proceeded with complete control of regiochemistry.6 Even the conformationally flexible azide 1k (entry 5) gave exclusively bridged product, in sharp contrast to the corresponding ketone, which only afforded fused lactam. A number of substrates with heteroatoms in the 2-position underwent the reaction in good to excellent yields (entries 6–8). Finally, a conformationally flexible azide with a shorter azide chain cleanly furnished the [4.2.1] bridged scaffold (entry 9). It is likely that in this case the fused product did not form due to strain developing en route to the corresponding a four-membered-lactam containing fused product.
Table 2.
Azide scope.a
| entry | azide | R1 | R2 | n | ketal | yield (%) |
|---|---|---|---|---|---|---|
| 1 | (1g) | t-Bu | 4-(OMe)C6H4 | 1 | (2g) | 92 |
| 2 | (1h) | t-Bu | 3,4,5-(OMe)3C6H2 | 1 | (2h) | 86 |
| 3 | (1i) | t-Bu | 3,5-(OMe)2C6H3 | 1 | (2i) | 86 |
| 4 | (1j) | t-Bu | 4-(NO2)C6H4 | 1 | (2j) | 94 |
| 5 | (1k) | H | C6H5 | 1 | (2k) | 59 |
| 6 | (1l) | H | OMe | 1 | (2l) | 52b |
| 7 | (1m) | H | SMe | 1 | (2m) | 50 |
| 8 | (1n) | H | SPh | 1 | (2n)7 | 78b |
| 9 | (1o) | H | C6H5 | 0 | (2o) | 91 |
BF3•CH3CN, 3.0–5.0 equiv in CH2Cl2
TMSOTf.
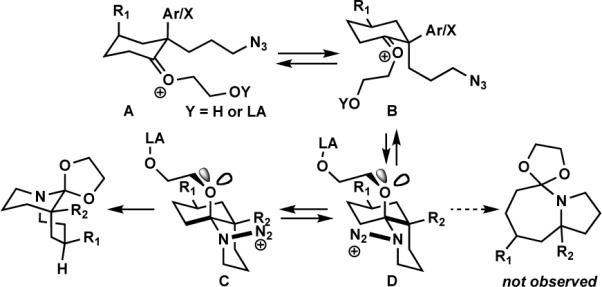
We have proposed that chairlike cyclohexanones afford bridged products only when the azide-containing side chain and the diazonium cation are both pseudoaxial (Scheme 2, intermediate C).5c Here, the N2+ leaving group is axial, leading to migration of the antiperiplanar alkyl group. We propose that the enhanced regioselectivity observed with ketal-containing vs. ketone-containing azides results from steric and electronic repulsion between the diazonium cation in the D conformation relative to C. In both cases, the most stable conformation of the O-alkyl side chain is likely to be as drawn; the N2+ leaving group in D experiences a disfavorable syn-1,5 interaction with the O-alkyl group that is absent in C. In the analogous reactions of ketones, where the OR group is replaced by either a proton or a Lewis acid, such effects are minimized relative to the present case (and the role of attractive non-bonded stabilization between N2+ and R2 is even more important). Interestingly, the Deslongchamps principle of maximal overlap could also come into play here. Thus, if the side chain orientation proposed predominates in the transition state leading to product, it is only possible for an oxygen electron lone pair to be antiperiplanar to the migrating bond in C but not in D (where the O-alkyl group is instead antiperiplanar to the darkened bond in D).4b Finally, the success of cases lacking an alkyl group at R2 indicates that the reactive conformation in these examples is exclusively B and not A.
Scheme 2.
The α-amino ketal functionality is stabilized by a non-planar arrangement of atoms. The X-ray crystal structure of 2n reveals that the N1–C1 bond (1.448 Å) as well as the C1-O1 (1.405 Å) and C1–O2 (1.416 Å) bonds are shorter than typical N–Csp3 (1.469 Å) and Csp3–O (1.432 Å) bonds (Figure 1).4a The C1-C2 bond length of 1.543 Å is slightly longer than the average Csp3-Csp3 bond (1.530 Å).4a The torsion angles between Nlp and C1-O1 of 69.5° and between Nlp and C1-O2 of 46.1° are consistent with the absence of that Nlp→σ*C-O interactions in this system. By contrast, there exists a reasonably good arrangement between O1lp and N1-C1 bond (~148°) and between O2lp and N1-C1 bond (~158°). While the lengths of C1-O1, C1-O2 and C1-C2 bonds are in agreement with an anomeric effect resulting from Olp→σ*C1-N1 and Olp→σ*C1-C2 interactions, the shortened N1-C1 bond seems to be characteristic to the tetrahedral intermediate stabilized by scaffolding effects of a medium-sized ring.4
Figure 1.
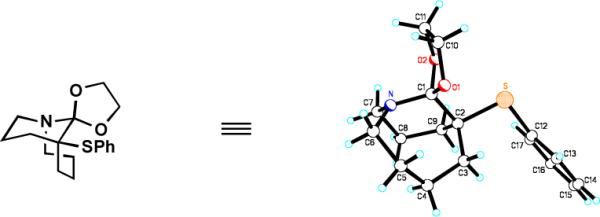
X-ray structure of 2n. Selected bond lengths [Å] and angles [°]: N1-C1 1.448, C1-O1 1.405, C1-O2 1.416, C1-C2 1.543, C6-N1-C1-O1 −38.1, C7-N1-C1-O1 177.0, C7-N1-C1-O2 61.5, C6-N2-C1-O1 −153.7
We begun to examine the reactivity of these α-amino ketals, (Scheme 3). For example, we found that acid hydrolysis affords 9-membered ring amino esters (these can be converted into the bridged lactams by treatment with base; see SI). In contrast, chromatography-stable hemiaminal ethers are formed following reduction (DIBAL-H), while reaction with Me3Al delivers a “twisted enamine”. As expected, α-amino ketals also undergo reactions at nitrogen.
Scheme 3.
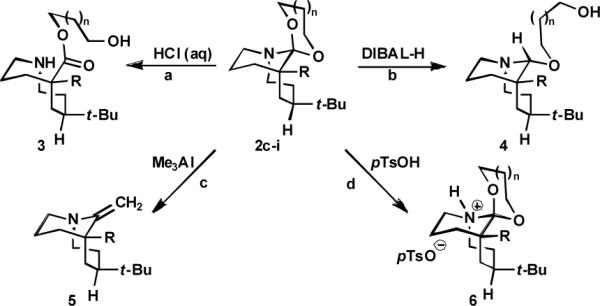
Conditions: (a) 2i, THF, 60 °C, 12 h, 89%.(b) 2c, PhH, 80 °C, 15 h, 67%. (c) 2d, CH2Cl2, 40 °C, 15 h, 56%. (d) 2h, rt, 20 h, 96%.
In summary, we have established a direct synthesis of orthoamides, which are analogs of tetrahedral intermediates derived from amide addition reactions. Continuing efforts to study this class of compounds are underway.
Supplementary Material
Acknowledgements
This work was supported by the National Institute of General Medical Sciences (GM-49093). We thank Victor Day for X-ray crystallography, Ruzhang Liu and Angelica Meyer for helpful discussions.
Footnotes
Supporting Information Available: Experimental details characterization data for new compounds, and the .cif file for 2n. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).For recent reviews, see: Nyfeler E, Renaud P. Chimia. 2006;60:276–284. Lang S, Murphy JA. Chem. Soc. Rev. 2006;35:146–156. doi: 10.1039/b505080d. Grecian S, Aubé J. In: Organic Azides: Syntheses and Applications. Bräse S, Banert K, editors. John Wiley and Sons; 2009.
- (2).(a) Yao L, Aubé J. J. Am. Chem. Soc. 2007;129:2766–2767. doi: 10.1021/ja068919r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Szostak M, Yao L, Aubé J. Org. Lett. 2009;11:4386–4389. doi: 10.1021/ol901771b. [DOI] [PubMed] [Google Scholar]
- (3).For recent reviews, see: Greenberg A, Breneman CM, Liebman JF. Amide Linkage: Selected Structural Aspects in Chemistry, Biochemistry, and Materials Science. Wiley; New York: 2000. . Clayden J, Moran WJ. Angew. Chem., Int. Ed. 2006;45:7118–7120. doi: 10.1002/anie.200603016. For selected examples, see: Kirby AJ, Komarov IV, Feeder N. J. Chem. Soc., Perkin Trans. 2001;2:522–529. Tani K, Stoltz BM. Nature. 2006;441:731–734. doi: 10.1038/nature04842. Lei Y, Wrobleski AD, Golden JE, Powell DR, Aubé J. J. Am. Chem. Soc. 2005;127:4552–4553. doi: 10.1021/ja050214m.
- (4).(a) Adler M, Adler S, Boche G. J. Phys. Org. Chem. 2005;18:193–209. [Google Scholar]; (b) Deslongchamps P. Stereoelectronic Effects in Organic Chemistry. Pergamon Press; 1983. [Google Scholar]; (c) Evans DA, Borg G, Scheidt KA. Angew. Chem., Int. Ed. 2002;41:3188–3191. doi: 10.1002/1521-3773(20020902)41:17<3188::AID-ANIE3188>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]; (d) Kirby AJ, Komarov IV, Feeder N. J. Am. Chem. Soc. 1998;120:7101–7102. [Google Scholar]; (e) Szostak M, Aubé J. J. Am. Chem. Soc. 2009;131:13246–13247. doi: 10.1021/ja906471q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Mossman CJ, Aubé J. Tetrahedron. 1996;52:3403–3408. [Google Scholar]; (b) Iyengar R, Schildknegt K, Morton M, Aubé J. J. Org. Chem. 2005;70:10645–10652. doi: 10.1021/jo051212n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Milligan GL, Mossman CJ, Aubé J. J. Am. Chem. Soc. 1995;117:10449–10459. [Google Scholar]
- (6).Only bridged orthoamides were observed in the analysis of the crude reaction mixtures by 1H NMR. See SI for details.
- (7).Interestingly, 2n was originally reported in Reference 5a as a fused isomer. We now reassign 2n as a bridged orthoamide. See, SI for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.