

A 60-year old patient with a clinical history of peripheral vascular disease and joint pains presented with a slowly progressive painful mass in the left hip region. Twenty-five years previously, he had been diagnosed with calcifications in the left hip region, and several operations had been performed. His family history was negative for tumoral calcinosis. Laboratory investigation showed a normal serum calcium level, elevated serum phosphate level, normal renal function, normal parathyroid hormone (PTH) level, and a slightly elevated level of 1,25-dihydroxy-vitamin D. Urinary calcium excretion was low, phosphate excretion was low. Tubular maximum phosphate was elevated [2.33 nmol/l (normal 0.8–1.3nmol/l)].
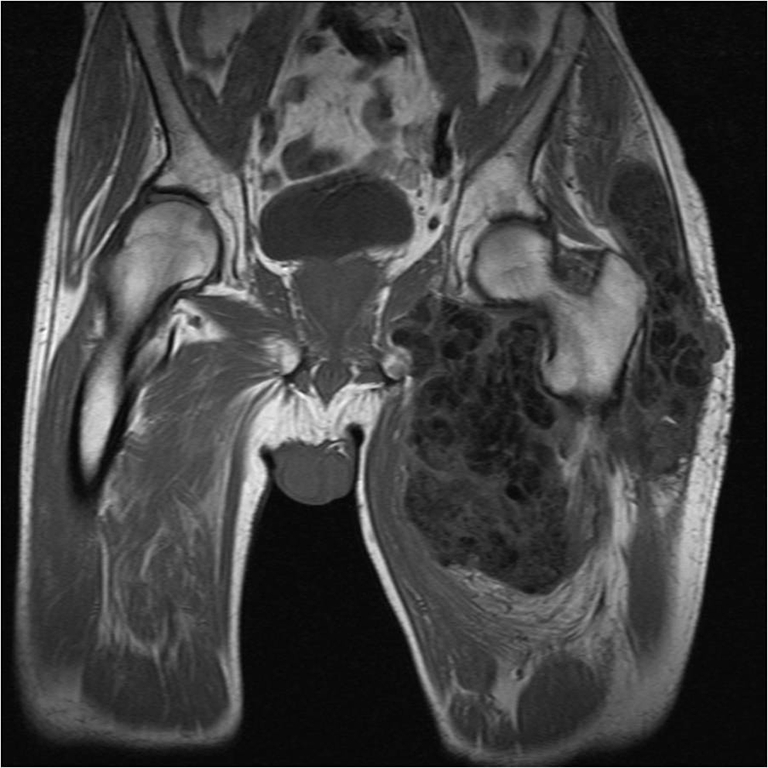
Pelvic X-ray and magnetic resonance imaging (MRI) showed a giant calcified mass (Figures 1 and 2). Surgical biopsy showed a bloody, milky, fluid, with calcifications. Histological evaluation was characteristic for tumoral calcinosis.
Fig. 1.
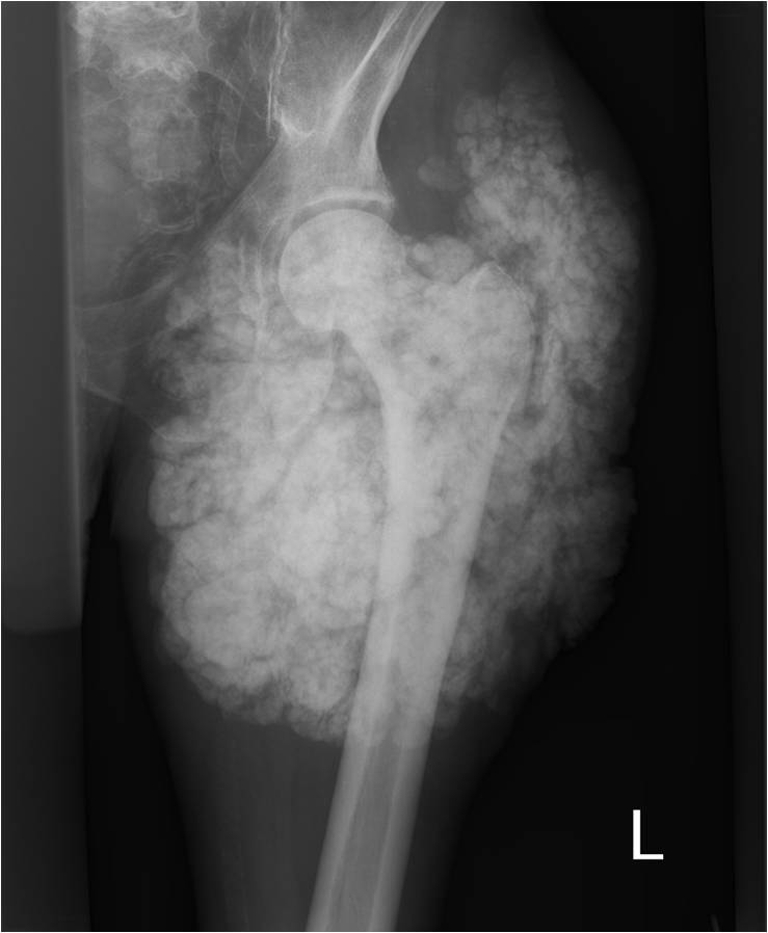
Fig. 2.
Tumoral calcinosis is a rare autosomal recessive disorder, characterized by hyperphosphataemia due to an increase in proximal tubular phosphate transport. The underlying defect might be a mutation in the GALNT3 gene, but mutations in the fibroblast growth factor (FGF)23 gene have also been implicated. The inactivation of FGF23 and GALNT3 mutations both result in lack of FGF23 and result in hyperphosphatic tumoral calcinosis with enhanced renal phosphate reabsorption. Serum calcium and PTH concentrations are typically within the normal range. The tendency of phosphate to complex with calcium and cause hypocalcaemia appears to be counterbalanced by increased production of calcitriol and, perhaps, hyperphosphataemia-induced stimulation of calcium reabsorption in the distal nephron. The combination of hyperphosphataemia and normocalcaemia results in a high calcium phosphate product and calcium phosphate deposition in the skin and subcutaneous tissues. Younger male patients and black people are more frequently affected by this disease. Calcinosis is often associated with diseases such as chronic renal failure, primary hyperparathyroidism, hypervitaminosis D, milk–alkali syndrome and massive osteolysis.
Complete surgical excision has been recommended, but recurrences are common. Phosphate depletion has proved to have variable success. Therapy for tumoral calcinosis consists of lowering the phosphate level with dietary restriction, antacids that impair phosphate absorption and/or phosphate binders. If this is ineffective, increasing urinary phosphate excretion by the chronic administration of acetazolamide might be beneficial. If patients are informed of the familial risks, early stages of the disease might be detected and treated.
In our patient, mutation analysis was not performed. He was treated by protein restriction and with phosphate binders and aluminium hydroxide, which resulted in a reduction of the tumoral mass of 8 cm in 2 years.
Acknowledgments
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.