

Abstract
Recent studies suggest that statins can function to protect the vasculature in a manner that is independent of their lipid-lowering activity. We show here that statins rapidly activate the protein kinase Akt/PKB in endothelial cells. Accordingly, simvastatin enhanced phosphorylation of the endogenous Akt substrate endothelial nitric oxide synthase (eNOS), inhibited apoptosis and accelerated vascular structure formation in vitro in an Akt-dependent manner. Similar to vascular endothelial growth factor (VEGF) treatment, both simvastatin administration and enhanced Akt signaling in the endothelium promoted angiogenesis in ischemic limbs of normocholesterolemic rabbits. Therefore, activation of Akt represents a mechanism that can account for some of the beneficial side effects of statins, including the promotion of new blood vessel growth.
Statins inhibit the activity of 3-hydroxyl-3-methyl coenzyme A (HMG-CoA) reductase, which catalyzes the rate-limiting step in cholesterol biosynthesis1. Statins are widely prescribed to lower cholesterol in hyperlipidemic patients at risk of cardiovascular disease2,3. More recently, it has been recognized that the protective effects of these drugs extend to myocardial infarction patients with average cholesterol concentrations4, and that lipid reduction alone cannot entirely account for the benefits of statin therapy5. In normocholesterolemic animals, statin therapy has been shown to protect against stroke6, ischemia-reperfusion injury of the heart7 and vascular inflammatory responses8, through mechanisms that may be mediated by an increase in endothelium-derived nitric oxide (NO) production. Endothelium-derived NO is the primary relaxing factor of the large blood vessels and its production is impaired in atherogenic vessels9 and following ischemia-reperfusion injury10. Similarly, statin therapy in patients has been found to rapidly improve vasomotor response to endothelium-dependent agonists11 and enhance coronary blood flow12. Collectively, these data suggest that statins can act to improve endothelial function through NO-dependent mechanisms that may operate independently of their lipid-lowering action.
The protein kinase Akt serves as a multifunctional regulator of cell survival, growth and glucose metabolism13. With respect to its cardiovascular function, Akt acts downstream of the angiogenic growth factors vascular endothelial growth factor (VEGF) (refs. 14,15) and angiopoietin 16–18 to confer endothelial cell survival and ensure proper blood vessel development19. Constitutive activation of Akt signaling also protects cardiomyocytes from apoptosis following ischemia-reperfusion injury in vivo20. In addition to this cytoprotective role, Akt functions as an activator of endothelial cell NO production in response to VEGF and shear flow through its ability to phosphorylate endothelial nitric oxide synthase (eNOS) on serine 1179 or 1177 (refs. 21 and 22), thereby controlling vasomotor activity 23. Akt is also essential for directed endothelial cell migration towards VEGF (ref. 24). The ability of Akt to mediate VEGF-induced endothelial cell survival, NO production and migration suggests that Akt signaling may mediate the response of the endothelium to angiogenic stimuli.
Because both statins and Akt signaling are implicated in NO regulation, we examined the ability of simvastatin to regulate the PI 3-kinase/Akt pathway in endothelial cells. Simvastatin activation of Akt signaling was examined for its ability to elicit features of the cellular response to angiogenic growth factors including endothelial tube formation, eNOS phosphorylation, NO production and the inhibition of endothelial cell apoptosis. Finally, we evaluated whether constitutive Akt signaling in the vascular endothelium or simvastatin administration could promote blood vessel growth in ischemic tissue.
Results
To test whether the beneficial actions of statins on the endothelium could be mediated by the Akt signaling pathway, cultured human umbilical vein endothelial cells (HUVEC) were incubated with simvastatin and Akt phosphorylation was assessed at amino acid residue 473 (serine). Simvastatin treatment led to a dose-dependent increase in serine 473 phosphorylation within 30 minutes, with maximal Akt phosphorylation occurring at 1.0 μM simvastatin (Fig. 1a). Statin treatment did not affect Akt protein concentrations, nor did it affect the concentrations of the Akt substrate eNOS. Increased Akt phosphorylation was detected as early as 15 minutes following exposure to 1.0 μM simvastatin and peaked at about 1 hour (Fig. 1b). The concentrations of Akt phosphorylation declined by 3 hours and was further reduced at 21 hours, but remained elevated relative to non-stimulated HUVEC. No changes in total Akt or eNOS protein concentrations were detected over this time course. Treatment with L-mevalonate, the product of the HMG-CoA reductase reaction, blocked activation of Akt phosphorylation by simvastatin (Fig. 1c). However, mevalonate had no effect on the robust activation of Akt phosphorylation by 100 ng/ml VEGF. Simvastatin-activation of Akt phosphorylation was also blocked by incubation with the phosphoinositide 3-kinase (PI 3-kinase) inhibitors wortmannin (Fig. 1d) and LY294002 (Fig. 1e). Akt phosphorylation also increased following treatment with 1.0 μM pravastatin (Fig. 1e). Like statin treatment, incubation with mevalonate, wortmannin or LY294002 had no effect on eNOS protein concentrations or on total Akt concentrations (Fig. 1c, d and e). Finally, neither simvastatin nor pravastatin had a stimulatory effect on Akt phosphorylation in either human saphenous vein smooth muscle cells, rat aorta smooth muscle cells or neonatal rat cardiomyocytes (data not shown).
Fig. 1.

Statins promote Akt phosphorylation in endothelial cells. Representative western immunoblots of the effects of simvastatin on Akt phosphorylation concentration are shown under various conditions as follows: a, Dose-dependent phosphorylation of Akt by simvastatin. b, Time-dependent changes in Akt phosphorylation following stimulation by simvastatin (1 μM). c, Reversal of simvastatin-induced Akt phosphorylation by mevalonate (200 μM). d, Sensitivity of simvastatin-induced Akt phosphorylation to wortmannin. e, Sensitivity of simvastatin-, pravastatin- and VEGF-induced Akt phosphorylation to LY294002. The extent of Akt phosphorylation was detected by anti-phosphorylated serine 473 residue of Akt1 specific antibody (phospho-Akt (Ser473)). HUVEC were treated with simvastatin for 30 min except for time course experiment shown in (a). (c, d, and e), HUVEC were pretreated with mevalonate (200 μM), wortmannin (500 nM) or LY294002 (10 μM) for 1 h, before 30 min stimulation with 1 μM simvastatin, 1 μM pravastatin or 100 ng/ml VEGF.
To examine the effect of simvastatin on Akt protein kinase activity in HUVEC cultures, lysates prepared from treated cells were immunoprecipitated with anti-Akt antibody and assessed for their ability to phosphorylate histone H2B (Fig. 2a). Akt protein kinase activity was increased in lysates of HUVEC treated with 1.0 μM simvastatin or 100 ng/ml VEGF. The specificity of the kinase assay was demonstrated by infecting HUVEC cultures with an adenoviral vector carrying a dominant-negative form of Akt, (Ad-dnAkt) which blocked the detectable increases in kinase activity. Activation of Akt kinase activity was also observed when lysates prepared from simvastatin-treated cells were tested for their ability to phosphorylate a peptide containing the Akt phosphorylation site of eNOS (amino acids 1174 to 1194)(Fig. 2b). Treatment of cells with wortmannin abolished simvastatin-induced kinase activity towards the eNOS peptide. A peptide with alanine substituted for serine at position 1179 did not incorporate appreciable phosphate under any condition tested, demonstrating that the simvastatin-activated phosphorylation occurs at the Akt consensus site within the peptide.
Fig. 2.

Simvastatin stimulates Akt protein kinase activity. a, Representative autoradiogram of phosphorylated histone H2B (2 μg) by immunoprecipitated Akt. Akt was immunoprecipitated from lysate prepared from HUVEC treated with simvastatin (SIM) or 100 ng/ml VEGF for 30 min. Under some conditions, cells were infected with adenovirus construct carrying a dominant-negative form of Akt1 (Ad-dnAkt) 24 h before drug treatment. Immunoprecipitated Akt was incubated with histone H2B (2 μg) for 30 min at 30°C and reaction was terminated by adding SDS-sample buffer. Proteins were separated by SDS–PAGE and the extent of histone H2B phosphorylation was visualized by autoradiography. b, Analysis of Akt protein kinase activity towards eNOS peptides. Akt was immunoprecipitated and incubated with 25 μg eNOS peptide (wild: eNOS peptide corresponding to positions including its functional site, serine 1179 residue. S1179A: the mutant peptide where the serine residue phosphorylated by Akt changed to alanine). Treatment with wortmannin (WM, 500 nM) was performed 1 h before addition of simvastatin. Results are presented as mean ± S.E.M. (n = 5–7, * P < 0.05).
The ability of simvastatin to stimulate Akt-mediated phosphorylation of eNOS in intact HUVEC was examined by incubating cultures with inorganic 32P and assessing endogenous eNOS phosphorylation by immunoprecipitation and autoradiography. Treatment with simvastatin or VEGF led to an increase in phosphate incorporation in eNOS, which was blocked when cells were treated with wortmannin or infected with Ad-dnAkt before stimulation (Fig. 3a and b). In contrast, adenovirus-mediated delivery of constitutively active Akt (myrAkt) led to the incorporation of phosphate into eNOS in the absence of stimulation with simvastatin or VEGF (Fig. 3a).
Fig. 3.

Simvastatin induces Akt-mediated phosphorylation of eNOS in intact cells. a, Simvastatin (SIM) increases phosphorylation of endogenous eNOS, which is abrogated by overexpression of dominant negative Akt1. HUVEC were radiolabeled with 32P-orthophosphate and stimulated with simvastatin (1 μM) or VEGF (100 ng/ml) for 30 min. Alternatively, HUVEC were infected with adenovirus expressing constitutively-active Akt (myrAkt) 24 h before radiolabeling and assayed for endogenous eNOS phosphorylation in the absence of Akt agonist. Parallel cultures were mock infected or infected with Ad-dnAkt 24 h before simvastatin or VEGF activation. b, Simvastatin-induced eNOS phosphorylation is sensitive to wortmannin. After 1 h pre-treatment with 500 nM wortmannin, HUVEC were stimulated with simvastatin or VEGF in the presence or absence of 500 nM wortmannin. Wortmannin was added to cultures 1 h before stimulation with simvastatin or VEGF. Endogenous eNOS protein was immunoprecipitated, separated by SDS–PAGE (7.5%), and its phosphorylation was visualized by autoradiography. c, COS-7 cells co-transfected with eNOS (wt: wild-type eNOS, S1179A: serine 1179 mutant) and Akt (HA-Akt) expression plasmids. Following transfection, cells were incubated in serum-depleted media for 48 h and then subjected to radiolabeling with 32P-orthophosphate and treatment with wortmannin and simvastatin. Immunoprecipitates of wild-type or mutant eNOS were examined for 32P-orthophosphate incorporation by autoradiography following SDS–PAGE (7.5%).
COS cells were transfected with wild-type or mutant eNOS expression plasmids to examine simvastatin-stimulated phosphorylation in greater detail. In this reconstituted system, simvastatin treatment induced phosphate incorporation into wild-type eNOS protein (Fig. 3c). Phosphorylation of eNOS was blocked when cells were treated with wortmannin. Simvastatin treatment did not induce phosphate incorporation into eNOS when serine 1179 was mutated to an alanine residue, consistent with the in vitro peptide phosphorylation data in Fig. 2b. These data indicate that simvastatin induces phosphate incorporation into eNOS at the Akt phosphorylation site that has been shown to be essential for stimulating catalytic activity in response to VEGF and shear flow21,22. Consistent with this observation, treatment of bovine aortic endothelial cells with simvastatin for 1 hour induced NO release 1.7-fold (from 0.28 ± 0.05 to 0.48 ± 0.07 nmol NO2−/ml; P < 0.05), as determined by NO-specific chemiluminescence (n = 4–6). Treatment with wortmannin blocked simvastatin-stimulated NO release (0.26 ± 0.05 nmol NO2−/ml), but had no effect on basal NO synthesis (0.29 ± 0.07 nmol NO2−/ml). Under these conditions, there was no detectable change in the concentration of eNOS protein (Figs. 1, 3 and data not shown), suggesting that simvastatin stimulated eNOS enzymatic activity through phosphorylation of serine 1179.
Akt has been shown to mediate growth factor- and anchorage-dependent survival signals in endothelial cells 15. Thus, we reasoned that simvastatin might protect endothelial cells from apoptosis through its ability to activate Akt. HUVEC incubated in serum-free media undergo apoptosis as assessed by annexin-V staining (Fig. 4a). Under these conditions a substantial fraction of the annexin-V positive cells are also positive for propidium iodide staining, a marker for later stages of cell death. Incubation with 1.0 μM simvastatin decreased the number of endothelial cells that were positive for annexin-V or propidium iodide staining. Apoptosis in HUVEC cultures was also scored by assessing the frequency of pyknotic nuclei observed by staining with Hoechst 33342 (Fig. 4b). Simvastatin promoted endothelial cell survival in a manner similar to VEGF. Incubation with wortmannin or infection with an adenovirus expressing dominant-negative Akt blocked both statin- and VEGF-induced endothelial cell survival. The downstream metabolite mevalonate also blocked the effect of simvastatin on endothelial cell survival. However, mevalonate had no effect on VEGF-induced survival, consistent with its effect on HMG-CoA reductase. In the absence of statin or VEGF stimulation, wortmannin and mevalonate had no detectable effect on survival, whereas infection with adeno-dnAkt promoted apoptosis slightly (1.2-fold, data not shown). Finally, higher doses of simvastatin (≥10 μM) decreased HUVEC viability (data not shown).
Fig. 4.

Simvastatin promotes endothelial cell survival through an Akt-dependent pathway. a, Representative images showing the cell-survival effects by simvastatin as detected by double-staining with annexin-V (green) and propidium iodide (red). HUVEC cultures were plated on chamber slides at a density of 4 × 104 cells/well. HUVEC were incubated in serum-depleted media for 3 h and subjected to stimulation with simvastatin (1 μM) for an additional 5 h. Parallel cultures were infected with Ad-dnAkt 24 h before the change to serum-free media. Some cultures were treated with 500 nM wortmannin (WM) for 1 h before each stimulation with simvastatin. b, Quantitative analysis of simvastatin-promoted endothelial survival by counting pyknotic nuclei stained by Hoechst 33342. HUVEC were examined in serum-free media as described in (a) to assess the effects of 1 μM simvastatin (SIM) or VEGF (100μg/ml) on survival. Data are shown as the mean ± S.E.M. (n = 4–6, *P < 0.05).
To examine the effect of statins on endothelial cell differentiation into vascular structures in vitro, a Matrigel tube formation assay was performed in the absence or presence of simvastatin, pravastatin or VEGF. Like VEGF, both simvastatin and pravastatin treatment promoted the formation of capillary-like tubes, and this process was blocked when cells were infected with Ad-dnAkt (Fig. 5a). Statin- or VEGF-induced tube formation was also blocked by incubation with the PI 3-kinase inhibitor LY294002, whereas infection of cells with constitutively active Akt was sufficient to promote vascular structure formation in the absence of statins or growth factor (Fig. 5b).
Fig. 5.

Statins promote vascular structure formation in an in vitro Matrigel assay. HUVEC were seeded on growth factor-reduced Matrigel in the presence of simvastatin (SIM, 0.1 μM), pravastatin (PRV, 0.1 μM), VEGF (100 ng/ml) or no addition (control) in the absence of serum. Parallel HUVEC cultures were infected with adenoviral vectors (50 MOI) carrying either (dnAkt) or (myrAkt) or treated with LY294002 (10μM) at the time of seeding. a, Cultures were photographed after 8 h. b, Quantitation assessment of the extent of tube formation. *P < 0.01 relative to control.
Because both endothelial cell survival 25,26 and NO production 27,28 are features of the cellular response to angiogenic growth factors in vivo, we tested whether simvastatin could promote revascularization of ischemic tissue. Normocholesterolemic rabbits underwent unilateral resection of their femoral arteries and their main branches, resulting in a marked decrease in hindlimb perfusion 29. To first demonstrate that Akt signaling is sufficient to promote angiogenesis in this model, a method of adenovirus-mediated gene transfer to endothelial cells of the ischemic hindlimb was established (Fig. 6a). Infusion of these limbs with an adenovirus construct expressing β-galactosidase (Ad-βgal) revealed that transgene expression was restricted to the vascular endothelium (Fig. 6b). Infusion of Ad-myrAkt in this model enhanced collateral vessel formation (Fig. 6c), and improved tissue perfusion as indicated by an increase in calf blood pressure ratio (Table 1). Constitutive Akt signaling in endothelial cells also increased capillary density in the adductor muscle of the ischemic limb (Fig. 6d). In contrast, infusion with adeno-βgal did not promote vessel formation or tissue perfusion relative to untreated ischemic hindlimbs (control) or vessels infused with saline (Fig. 6c and d and Table 1).
Fig. 6.
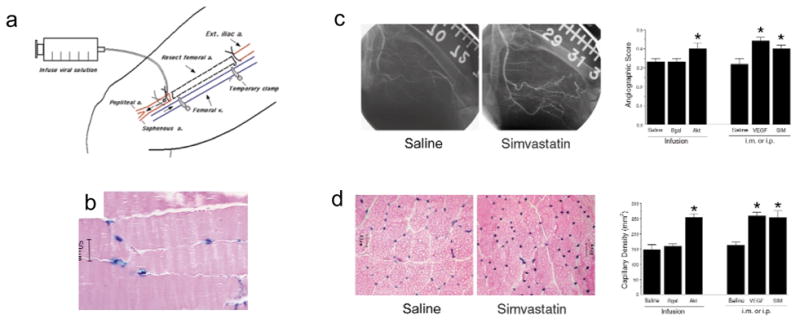
Statin administration or enhanced Akt signaling in endothelial cells promotes blood vessel formation in the rabbit hindlimb in response to unilateral femoral artery resection. a, The femoral artery and its main branches were dissected. Adenovirus-mediated gene transfer to the endothelium was achieved by infusing saline containing Ad-Bgal or Ad-myrAkt through the distal end of the femoral artery and incubating for 15 min in the limb by temporarily clamping the femoral vein. b, Gastrocnemius muscle was excised 3 days after surgery and perfusion with Ad-βgal, and stained with X-gal to determine transgene distribution in hematoxylin and eosin-stained tissue. c, Internal iliac angiography was performed on the different treatment groups to assess collateral vessel formation. Angiograms at 40 days after femoral artery resection showing enhanced collateral vessel formation in animals that received 0.1 mg/kg/d of simvastatin by intraperitoneal injection (i.p.) relative to control animals that underwent surgery but received saline. Quantitative measurements of collateral vessels were performed on the control group, the simvastatin-treated group and a group that received an intramuscular injection (i.m.) of Ad-VEGF. Angiographic score was also assessed in the experimental groups receiving infusions of saline, Ad-βgal or Ad-myrAkt at 31 days after surgery. d, Alkaline phosphatase staining of the adductor muscle from ischemic limbs showing greater capillary density in the simvastatin-treated animals than in control animals at 40 days post-surgery. Average capillary density for all experimental groups is reported. Data in each experiment are presented as mean ± S.E.M. (n = 6 rabbits for each treatment group, *P < 0.05 relative to the saline-infused or saline-injected groups compared by one-way analysis of variance).
Table 1.
Simvastatin treatment and enhanced Akt signaling improves calf blood pressure in ischemic limbs
| Treatment | Blood Pressure Ratio |
|---|---|
| Saline infusion | 0.53 ± 0.04 |
| Ad-βgal infusion | 0.52 ± 0.03 |
| Ad-myrAkt infusion | 0.67 ± 0.05* |
| Saline i.p. injection | 0.58 ± 0.06 |
| Simvastatin i.p. injection | 0.75 ± 0.04* |
| Ad-VEGF i.m. injection | 0.81 ± 0.05* |
Calf blood pressure ratio was calculated as the systolic pressure of the surgically-treated limb divided by that of the normal limb for each animal. Data are presented as mean ± S.E.M. (n = 6).
P < 0.05 relative to the saline-infused or saline-injected groups compared to one-way analysis of variance.
i.p., intraperitoneal, i.m., intramuscular.
To test the effects of simvastatin on limb revascularization, a dose of 0.1 mg/kg was administered daily by intraperitoneal injection after femoral artery resection. Animals receiving statin treatment displayed more detectable collateral vessels with characteristic corkscrew morphology than the untreated control group at 40 days following femoral artery resection (Fig. 6c). Correspondingly, the limbs of the simvastatin-treated animals displayed reduced hemodynamic deficit determined by ratio of the systolic pressure of the ischemic limb to that of the normal limb (Table 1). Simvastatin administration also promoted capillary formation in the ischemic limb (simvastatin = 253 ± 23 capillaries/mm2; control = 163 ± 9 capillaries/mm2 in adductor muscle; P < 0.01; Fig. 6d). For comparison, some animals received an intramuscular injection of an adenovirus encoding VEGF (adeno-VEGF) into the thigh of the ischemic limb. Like simvastatin, VEGF treatment enhanced collateral and capillary vessel formation (Fig. 6) and increased calf blood pressure (Table 1).
Discussion
This study identifies endothelial cell Akt as a new biological target for statin action. These data show that simvastatin and pravastatin rapidly induce the phosphorylation of Akt at serine residue 473, which increases its protein kinase activity. Simvastatin induces Akt-mediated phosphorylation of eNOS, leading to NO production, and promotes endothelial cell survival in an Akt-dependent manner. Because the production of NO by viable endothelium serves several protective functions including the inhibition of platelet adhesion30, inflammation31 and smooth muscle cell proliferation32,33, modulation of Akt activity by statins may help to explain the improvement in endothelial function, enhanced tissue perfusion and reduction in cardiovascular events that are seen in patients who receive these drugs.
Though the mechanism of Akt activation by statins is not known, it seems that PI 3-kinase signaling is involved because this process was blocked by wortmannin or LY294002. In addition, inhibition of the HMG-CoA reductase reaction is implicated because simvastatin-induced Akt activation was reversed when cultures were incubated with the downstream metabolite L-mevalonate. In addition to steroid biosynthesis, mevalonate is required for the production of ubiquinone, dolichols and isoprenoids that are essential for diverse cellular processes. It has been reported that statins stabilize eNOS mRNA through changes in isoprenoid synthesis34. However, no changes in eNOS protein concentrations could be detected at any time point analyzed in this study. Of note, the reported increase in eNOS transcript concentration occurs at later time points (24 hours) than is seen with simvastatin activation of eNOS phosphorylation by Akt (15 minutes). The rapid time course of Akt-mediated eNOS activation by simvastatin is also consistent with the acute changes in NO production and vasorelaxation observed in aortic rings following statin treatment ex vivo35.
Recent studies have shown that Akt functions downstream of angiogenic growth factors to promote endothelial cell survival14–18, NO synthesis21 and migration24; cellular responses that contribute to new blood vessel growth and stabilization of the vascular network. Here, we provide evidence showing that Akt signaling is essential and sufficient for the differentiation of endothelial cells into vascular structures in vitro. We also show that enhanced Akt signaling in vascular endothelium is sufficient to promote blood vessel growth in a rabbit model of vascular insufficiency. Consistent with this concept, we demonstrate that simvastatin induces vascular structure formation in vitro and promotes blood vessel formation in the ischemic tissue of normocholesterolemic rabbits in a manner similar to treatment with VEGF.
The results from this study indicate that statins may have utility for ‘therapeutic angiogenesis’ of ischemic tissue. These findings suggest that statin therapy could be important in patient populations with occlusive vessel disease, yet normal cholesterol concentrations. Such may be the case in some patients with peripheral ischemic disease where, unlike coronary ischemic disease, hyperlipidemia is not recognized as a strong risk factor36. If this holds true, statin therapy may provide an additional strategy to angiogenic growth factor therapies (protein and gene) that are in early clinical trials. In some cases, statins may be advantageous because these drugs are orally administered and known to be safe in humans.
Methods
Cell culture, transfection, adenoviral infection and immunoblot analysis
HUVEC and bovine aortic endothelial cells (BAEC) were cultured in endothelial cell growth medium (EGM, Clontech, Palo Alto, California). Before each experiment, cells were placed in serum-depleted medium (endothelial cell basal medium, EBM, Clontech, Palo Alto, California) for 4 or 24 h for HUVEC or 24 h for BAEC. Experiments were initiated by the addition of the indicated amount of alkaline hydrolysis-activated simvastatin7, 100 ng/ml VEGF (R&D Systems, Minneapolis, Minnesota), pravastatin or vehicle. In some experiments, HUVEC were infected with adenoviral constructs encoding dominant-negative or constitutively-active form of Akt1 (dnAkt or myrAkt, respectively) at multiplicity of infection (MOI) of 50 or 100 for 24 h in EGM, which results in 95% effeciency of infection15. COS-7 cells were plated on 60-mm dish in Dulbecco’s minimum essential medium (DMEM) plus 10% fetal bovine serum and transfected with 3 μg of wild type or mutant (S1179A) eNOS expression plasmid21 and 1 μg of wild-type hemagglutinin (HA)-tagged Akt1 expression plasmid using the lipofection (Life Technologies, Gaithersburg, Maryland). Following transfection and incubation in DMEM without serum for 48 h, COS-7 cells were stimulated with 1 μM simvastatin. In some experiments, cells (HUVEC, BAEC or COS-7) were pretreated with wortmannin (500 nM), LY294002 (10 μM) or mevalonate (200 μM) for 1 h before stimulation with simvastatin or VEGF. Mevalonate was activated by alkaline hydrolysis7,8. Cell lysates (30 μg total protein) were resolved by SDS–PAGE (10%), subjected to western immunoblot analysis by using rabbit polyclonal anti-phosphorylated serine 473 residue of Akt1 antibody (New England Biolabs, Beverly, Massachusetts). To verify the amount of loaded proteins, blots were reprobed with goat polyclonal anti-Akt1 antibody (Santa Cruz, Santa Cruz, California), mouse monoclonal anti-eNOS antibody (Transduction Laboratories, Lexington, Kentucky), or mouse monoclonal anti-tubulin antibody (Oncogene, Cambridge, Massachusetts). Simvastatin was provided by Merck & Co. (West Point, Pennsylvania) and activated by alkaline hydrolysis 7,8. Pravastatin was of pharmaceutical grade and it was solubilized in saline.
Akt protein kinase assay
For assessment of Akt protein kinase activity in vitro, substrate (2 μg histone H2B or 25 μg eNOS peptide) was incubated with Akt immunoprecipitated from cell lysate using goat polyclonal anti-Akt1 antibody (Santa Cruz) as described previously 15. Kinase reactions were initiated following the addition of reaction components to a final concentration of ATP (50 μM) containing 10 μCi of 32P-γATP, dithiotreitol (1 mM), HEPES buffer (20 mM, pH 7.4), MnCl2 (10 mM), MgCl2 (10 mM). After incubation for 30 min at 30 °C, phosphorylated histone H2B was visualized after SDS–PAGE (15%) and autoradiography. To estimate the extent of 32P incorporation into eNOS peptides, each reaction mixture was measured by spotting onto phosphocellulose disc filter (Amersham-Pharmacia, Chicago, Illinois) and the amount of phosphate incorporated was measured by Cerenkov counting21. The wild-type peptide sequence was 1174-RIRTQSFSLQERHLRGAVPWA-1194, and the mutant eNOS peptide was identical except that serine 1179 was substituted by alanine.
Phosphorylation of eNOS in intact cells
HUVEC or COS-7 cells were placed into phosphate-free DMEM supplemented with 100 μCi/ml of 32P-orthophosphate for 4 h before stimulation with simvastatin or VEGF. In some samples, cells were infected with adenovirus encoding dominant-negative Akt1 at an MOI of 100 for 24 h followed by incubation with wortmannin (500 nM) for 1 h before stimulation with simvastatin or VEGF. After 30 min stimulation with simvastatin or VEGF, cells were washed with TBS and collected with lysis buffer (RIPA buffer plus 20 mM NaF, 10 mM Na4P2O7, 1 mM NaVO4). The lysate was subjected to immunoprecipitation using mouse monoclonal anti-eNOS antibody (Transduction Laboratories). Radiolabeled phosphate incorporation into each protein was visualized after SDS–PAGE (7.5%) by autoradiography.
Apoptosis assays
Fluorescence-labeled annexin-V-FLUOS staining of HUVEC was performed according to manufacturer’s instructions (Roche Molecular Biochemicals, Indianapolis, Indiana). Briefly, cells were plated on to chamber slides (Nunc) and placed in EBM for 3 h. After stimulation for 5 h with simvastatin, double-staining was performed with annexin-V-FLUOS (0.1 μg/ml) and propidium iodine (10 μg/ml). For detection of pyknotic nuclei, cells were stained with Hoechst 33342. Where indicated, incubation with wortmannin was initiated 1 h before stimulation with simvastatin or VEGF. Infection with adenovirus encoding dominant-negative Akt1 was initiated 24 h before incubation in EBM media.
NO release from endothelial cells
Nitric oxide release was measured in the cell supernatant by chemiluminescent detection of nitrate (NO2−), the stable breakdown product of NO (ref. 21). In brief, BAEC were incubated in EBM for 24 h following stimulation with simvastatin for 1 h. In some samples, wortmannin was added 1 h before stimulation. Cell supernatants were collected, deproteinized and refluxed in glacial acetic acid containing sodium iodide. Processed specimens were measured with a NO analyzer (Sievers).
Tube formation assays
The formation of vascular-like structures by HUVEC was assessed on the basement membrane matrix preparation, growth factor-reduced Matrigel (Becton Dickinson, Bedford, Massachusetts). Two-well chamber slides were coated with Matrigel (10 mg/ml) according to the manufacturer’s instructions. HUVEC were seeded on coated plates at 4–5 × 104 cells/well in EBM and incubated at 37 °C for 60 minutes. The media were supplemented with the agents (statins, VEGF, etc. or adenoviral constructs) and incubated at 37 °C for 8–12 h. Tube formation image was observed using an inverted phase contrast microscope (Nikon Diaphot). Images were captured with a video graphic system (DEI-750 CE Digital Output Camera, Optronics, Goleta, California). The degree of tube formation was quantified by measuring the length of tubes in random fields from each well using the National Institutes of Health (NIH) Image Program.
Limb revascularization studies
Male New Zealand white rabbits, weighing 3.0–3.5 kg and fed a normal diet, were used to examine the effects of Akt signaling and simvastatin-mediated stimulation of vessel growth. For the infusion model, the left femoral artery and main side branches were excised from their proximal origin to within 2 cm of the bifurcation into the saphenous and popliteal arteries. After 10 days, to permit post-operative recovery, the distal femoral artery was re-exposed and, after temporary clamping of the femoral vein, 50 ml of saline, saline with 3.5 × 1010 viral particles of Ad-βgal, or saline with 3.5 × 1010 viral particles of Ad-myrAkt was infused through the distal femoral artery and incubated for 15 min. After clamp removal, the distal femoral artery was ligated. Two animals infused with Ad-βgal were killed 3 days after surgery to determine β-galactosidase expression in the gastrocnemial muscle. The remainder of the animals (n = 6) were analyzed for limb revascularization at 31 days after femoral artery resection. For the intramuscular injection of Ad-VEGF or the intraperitoneal injection of simvastatin, the left femoral artery and side branches were completely excised from their proximal origin to the point distally where bifurcation occurs. After 10 days, to permit post-operative recovery, a total of 3.5 × 1010 viral particles of Ad-VEGF in 2.5 ml of saline was injected through a 27-gauge needle at a depth of 3 to 5 mm in the adductor (2 sites), medial large (2 sites) and semimembranous (1 site) muscle (500 μl per injection site). Alternatively, simvastatin (0.1 mg/kg/day) or saline was given intraperitoneally (1 ml) from the day after surgery until one day before sacrifice. Animals in these groups (n = 6) were analyzed for limb revascularization 40 days after surgery. No adverse events, including death, edema or angioma formation, were noted with any treatment regimen. Calf blood pressure was measured in both limbs by Doppler flow meter (model 1059, Parks Medical Electronics, Aloha, Oregon). The calf blood pressure is defined as the ratio of the left calf to right calf systolic pressure. Collateral arteries were evaluated by internal iliac angiography. A 3-F infusion catheter (Tracker-18, Target Therapeutic, San Jose, California) was introduced into the common carotid artery and advanced to the internal iliac artery of the ischemic limb using a 0.014-inch guide wire under fluoroscopic guidance. Non-ionic contrast media (Isovue-370, Squibb Diagnostics, New Brunswick, New Jersey) was injected at a rate of 1 ml/sec and serial images of the ischemic hindlimb were recorded at a rate of 1 film/sec for 10 sec. Quantitative angiographic analysis of collateral vessels were performed by an investigator blinded to the outcome using a grid overlay composed of 2.5-mm diameter circles arranged in rows spaced 5 mm apart placed over the 4-sec angiogram. An angiographic score was calculated as the number of circles crossed by visible arteries divided by the total number of circles in the medial thigh. Capillary density was evaluated by investigator blinded to the outcome using light microscopic sections taken from the adductor muscle of the ischemic limb at the time of euthanasia. Muscle samples were embedded in OCT compound (Miles, Elkhart, IN) and snap-frozen in liquid nitrogen. Frozen sections (5 μm in thickness) with muscle fibers oriented in a transverse fashion were stained for alkaline phosphatase using indoxyltetrazolium, and then counterstained with 0.5% eosin. The capillary density was calculated as capillaries/mm2 averaged from 10 randomly selected fields.
Statistical analysis
Data are shown as mean ± S.E.M. All data were evaluated with a two tailed, unpaired Student’s t test or compared by one-way analysis of variance.
Acknowledgments
This work was support by National Institutes of Health grants RO1-AR40197, RO1-HL50692, RO1-AG15052 and PO1-HD23681 to K.W. We thank Neil Tritman for supplying the human vascular cells.
References
- 1.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 2.Scandinavian Simvastatin Survival Study Group. Randomized trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S) Lancet. 1994;344:1383–1389. [PubMed] [Google Scholar]
- 3.Levine GN, Keaney JF, Jr, Vita JA. Cholesterol reduction in cardiovascular disease. Clinical benefits and possible mechanisms. N Engl J Med. 1995;332:512–521. doi: 10.1056/NEJM199502233320807. [DOI] [PubMed] [Google Scholar]
- 4.Sacks FM, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N Engl J Med. 1996;335:1001–1009. doi: 10.1056/NEJM199610033351401. [DOI] [PubMed] [Google Scholar]
- 5.West of Scotland Coronary Prevention Study Group. Influence of pravastatin and plasma lipids of clinical events in the West of Scotland Coronary Prevention Study (WOSCOPS) Circulation. 1998;97:1440–1445. doi: 10.1161/01.cir.97.15.1440. [DOI] [PubMed] [Google Scholar]
- 6.Endres M, et al. Stroke protection by 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 1998;95:8880–8885. doi: 10.1073/pnas.95.15.8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lefer AM, et al. Simvastatin preserves the ischemic-reperfused myocardium in normocholesterolemic rat hearts. Circulation. 1999;100:178–184. doi: 10.1161/01.cir.100.2.178. [DOI] [PubMed] [Google Scholar]
- 8.Pruefer D, Scalia R, Lefer AM. Simvastatin inhibits leukocyte-endothelial cell interactions and protects against inflammatory processes in normocholesterolemic rats. Arterioscler Thromb Vasc Biol. 1999;19:2894–2900. doi: 10.1161/01.atv.19.12.2894. [DOI] [PubMed] [Google Scholar]
- 9.Osborne JA, Siegman MJ, Sedar AW, Mooers SU, Lefer AM. Lack of endothelium-dependent relaxation in coronary resistance arteries of cholesterol-fed rabbits. Am J Physiol. 1989;256(3 Pt 1):C591–C597. doi: 10.1152/ajpcell.1989.256.3.C591. [DOI] [PubMed] [Google Scholar]
- 10.Lefer AM, Lefer DJ. The role of nitric oxide and cell adhesion molecules on the microcirculation in ischaemia-reperfusion. Cardiovasc Res. 1996;32:743–751. [PubMed] [Google Scholar]
- 11.Dupuis J, Tardif JC, Cernacek P, Théroux P. Cholesterol reduction rapidly improves endothelial function after acute coronary syndromes: the RECIFE (Reduction of Cholesterol in Ischemia and Function of the Endothelium) trial. Circulation. 1999;99:3227–3233. doi: 10.1161/01.cir.99.25.3227. [DOI] [PubMed] [Google Scholar]
- 12.Baller D, et al. Improvement in coronary flow reserve determined by positron emission tomography after 6 months of cholesterol-lowering therapy in patients with early stages of coronary atherosclerosis. Circulation. 1999;99:2871–2875. doi: 10.1161/01.cir.99.22.2871. [DOI] [PubMed] [Google Scholar]
- 13.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 14.Gerber HP, et al. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway: Requirement for Flk-1/KDR activation. J Biol Chem. 1998;273:30336–30343. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- 15.Fujio Y, Walsh K. Akt mediates cytoprotection of endothelial cells by vascular endothelial growth factor in an anchorage-dependent manner. J Biol Chem. 1999;274:16349–16354. doi: 10.1074/jbc.274.23.16349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim I, et al. Angiopoietin-1 regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Circ Res. 2000;86:24–29. doi: 10.1161/01.res.86.1.24. [DOI] [PubMed] [Google Scholar]
- 17.Kontos CD, et al. Tyrosine 1101 of Tie2 is the major site of association of p85 and is required for activation of phosphatidylinositol 3-kinase and Akt. Mol Cell Biol. 1998;18:4131–4140. doi: 10.1128/mcb.18.7.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Papapetropoulos A, et al. Angiopoietin-1 inhibits endothelial cell apoptosis via the Akt/Survivin pathway. J Biol Chem. 2000;275:9102–9105. doi: 10.1074/jbc.275.13.9102. [DOI] [PubMed] [Google Scholar]
- 19.Carmeliet P, et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 1999;98:147–157. doi: 10.1016/s0092-8674(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 20.Fujio Y, Nguyen T, Wencker D, Kitsis RN, Walsh K. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation. 2000;101:660–667. doi: 10.1161/01.cir.101.6.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fulton D, et al. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dimmeler S, et al. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 23.Luo Z, et al. Acute modulation of endothelial Akt/PKB activity alters NO-dependent vasomotor activity in vivo. J Clin Invest. 2000 doi: 10.1172/JCI9419. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morales-Ruiz M, et al. Vascular endothelial growth factor-stimulated actin reorganization and migration of endothelial cells is regulated via the serine/threonine kinase Akt. Circ Res. 2000;86:892–896. doi: 10.1161/01.res.86.8.892. [DOI] [PubMed] [Google Scholar]
- 25.Benjamin LE, Golijanin D, Itin A, Pode D, Keshet E. Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J Clin Invest. 1999;103:159–165. doi: 10.1172/JCI5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alon T, et al. Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nature Med. 1995;1:1024–1028. doi: 10.1038/nm1095-1024. [DOI] [PubMed] [Google Scholar]
- 27.Ziche M, et al. Nitric oxide synthase lies downstream from vascular endothelial growth factor-induced but not basic fibroblast growth factor-induced angiogenesis. J Clin Invest. 1997;99:2625–2634. doi: 10.1172/JCI119451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murohara T, et al. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest. 1998;101:2567–2578. doi: 10.1172/JCI1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pu LQ, et al. Angiogenic growth factor and revascularization of the ischemic limb: evaluation in a rabbit model. J Surg Res. 1993;54:575–583. doi: 10.1006/jsre.1993.1088. [DOI] [PubMed] [Google Scholar]
- 30.Simon DI, et al. Antiplatelet properties of protein S-nitrosothiols derived from nitric oxide and endothelium-derived relaxing factor. Arterioscler Thromb. 1993;13:791–799. doi: 10.1161/01.atv.13.6.791. [DOI] [PubMed] [Google Scholar]
- 31.Qian HS, Neplioueva V, Shetty GA, Channon KM, George SE. Nitric oxide synthase gene therapy rapidly reduces adhesion molecule expression and inflammatory cell infiltration in carotid arteries of cholesterol-fed rabbits. Circulation. 1999;99:2979–2982. doi: 10.1161/01.cir.99.23.2979. [DOI] [PubMed] [Google Scholar]
- 32.von der Leyen HE, et al. Gene therapy inhibiting neointimal vascular lesion: in vivo transfer of endothelial cell nitric oxide synthase gene. Proc Natl Acad Sci USA. 1995;92:1137–1141. doi: 10.1073/pnas.92.4.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo K, Andrés V, Walsh K. Nitric oxide-induced downregulation of cdk2 activity and cyclin A gene transcription in vascular smooth muscle cells. Circulation. 1998;20:2066–2072. doi: 10.1161/01.cir.97.20.2066. [DOI] [PubMed] [Google Scholar]
- 34.Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem. 1998;273:24266–24271. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- 35.Kaesemeyer WH, Caldwell RB, Huang J, Caldwell RW. Pravastatin sodium activates endothelial nitric oxide synthase independent of its cholesterol-lowering actions. J Amer Coll Cardiol. 1999;33:234–241. doi: 10.1016/s0735-1097(98)00514-2. [DOI] [PubMed] [Google Scholar]
- 36.TASC Working Group. Investigation of patients with intermittent claudication. J Vasc Surg. 2000;31(Suppl):S62–S66. [Google Scholar]