

Abstract
Gene and cell therapies of cardiac arrhythmias are nascent fields whose raison d’etre derives from (1) the problematic state of arrhythmia treatment today (especially atrial and ventricular tachyarrhythmias for which drugs, devices and ablation remain more stopgaps then optimal interventions), and (2) the opportunity to learn and potentially treat and cure by exploring new technologies. The state of antiarrhythmic therapy and new directions being taken are reviewed.
Keywords: Ventricular tachycardia and fibrillation, atrial fibrillation, reentry, myocardial infarction, stem cells, viral vectors
Ventricular tachycardia (VT) and/or fibrillation (VF) lead to 200,000–400,000 sudden cardiac deaths/year in the US.1 Atrial fibrillation (AF) afflicts about 2.3 million Americans and may reach 12–16 million by 2050.2 Reentry accounts for nearly all AF and approximately 85% of arrhythmias in ischemic heart disease.3 In one conceptualization the reentrant waveform follows a well-defined anatomic pathway, reaching its point of origin after the standing wave that is the action potential has ended and the tissue at that site is no longer refractory. For this to happen a number of changes in conduction must occur, including unidirectional block and slow retrograde propagation.4,5 Another concept is functional reentry,6 in which the length and/or position of the reentrant path changes over time.
Therapeutic approaches to reentry use drugs to prolong the effective refractory period (ERP) and/or to slow/block conduction, and/or surgery or catheter ablation to interrupt the pathway. For example, for AF, flecainide, dofetilide, sotalol, amiodarone are representative drugs having therapeutic roles.8 Regrettably, clinical trials have demonstrated virtually all antiarrhythmics tested to be proarrhythmic.7 A different pharmacological approach, uses ACE inhibitors or AT-1 receptor blockers to reduce paroxysmal AF recurrences.e.g. 9 Radiofrequency ablation terminates AF due to triggered foci in pulmonary veins, prevents recurrences and appears more effective than antiarrhythmic drugs. However, ablation manifests variable success rates: recurrence is more frequent than anticipated.
No antiarrhythmic drug is acceptable primary or sole therapy for VT/VF in the setting of ischemic heart disease.7 In contrast, several clinical device trialsreviewed in 7 have shown survival benefit in treating potentially lethal arrhythmias in patients with ischemic heart disease. But device and combined drug/device approaches to VT/VF carry a high economic price. Emotional and physical tolls comprise a different and important cost. All told, new approaches to prevention/termination would be welcome.
NOVEL APPROACHES TO PREVENTING/TERMINATING REENTRY
Nearly 100 years ago Mines4,5 conceptualized and experimentally demonstrated reentry and communicated its potential role in clinical arrhythmias. He postulated the relationship between path length, propagation velocity and refractoriness determines whether reentry will evolve and persist. Much subsequent discovery derives from Mines’ observations: in the process we have learned of the multiple wavelet and leading circle concepts of reentry, of anisotropy and of rotors.
The actions of clinically-used IKr- and INa-blocking antiarrhythmic drugs dovetail with Mines’ concepts: they prolong ERP and slow conduction to interrupt reentry.10 Yet Mines’ concepts avail us of much more information that has not been applied to antiarrhythmic advantage. Figure 1 (from Schmitt and Erlanger’s 1928 adaptation of Mines5) explores this: A waveform activates the peripheral conducting system, but if there is conduction block (grey), antegrade propagation may fail (Panels A–C) and reentry may occur (Panels D–E). We can terminate the reentrant loop by slowing conduction until it fails bidirectionally and/or by prolonging ERP. Both are outcomes of antiarrhythmic drug therapy: the conundrum in prolonging ERP is that we usually prolong repolarization as well and encounter proarrhythmia.10 We can cut the loop surgically or using catheter ablation, in the process perhaps creating a scar that may then serve as a nidus for further reentry.
Figure 1.

Panels A–E Schmitt and Erlanger’s5 concept of reentry sees an impulse propagating through the conducting system, with antegrade conduction blocked in one limb and then conducting retrogradely through to reactivate the proximal portion of the system. Panel F shows the antegrade activation that would be expected to occur if one could facilitate conduction through depressed regions.
Mines recognized therapeutic options other than prolonging ERP or slowing conduction.4,5 We should be able to speed conduction such that a waveform “catches its tail,” thereby encountering refractory tissue and failing to propagate further. We also should be able to prolong ERP relative to repolarization, but without prolonging repolarization, itself.10 To date, neither approach has achieved consistent success. Moreover, a clear derivative of Mines’ model is the concept of regional therapies that might modify a portion of a pathway. For example if the Figure 1 pathway were made to propagate normally (Panel F) then reentry would not commence.
CURRENT STATUS OF ANTIARRHYTHMIC GENE AND CELL THERAPIES
Antiarrhythmic gene and cell therapies have (1) resulted in biological pacemakers for treating heart block;11 (2) utilized regional delivery of viral constructs for treating VT or AF,12 and (3) developed alternatives to surgery for ablating the AV junction in AF.13–15 General approaches to gene and cell therapy are summarized in Figure 2. To modify cardiac rhythm directly we can use gene therapy carried via viral vectors. These include (1) adenovirus, which expresses briefly and episomally and is used largely for proof-of-concept experiments, (2) adeno-associated virus which shows long persistence in expression but whose genomic incorporation is uncertain, and (3) lentivirus which results in genomic incorporation. Despite persisting questions regarding long-term impact on human subjects, but these viruses are being used in clinical trials. Cells loaded via electroporation or viral vectors have also been used to carry gene constructs. These include fibroblasts and the hMSCs in Figure 2. Finally (not depicted) administration of plasmids in the absence of a carrier has shown poor incorporation although electroporation is being attempted as a means to improve this approach. The other antiarrhythmic approach (Figure 2, right) is to repair/regenerate myocardium using embryonic or human mesenchymal stem cells (hMSCs) or more refined precursor cells (reviewed elsewhere in this issue). Reduction of arrhythmias is an expected secondary outcome of this strategy.
Figure 2.

Approaches to gene and cell therapy of arrhythmias. (Left) To modify cardiac function directly, we can use gene or cell therapy (Right) Cardiac repair/regeneration strategies. See text for discussion.
The leading edge of gene/cell arrhythmia research has focused on bradyarrhythmias and creation of biological pacemakers or AV bridges. Biological pacing strategies have included overexpressing β2-adrenergic receptors, transfecting a dominant negative construct to reduce IK1, overexpressing the HCN gene family to increase pacemaker current and using mutagenesis to create designer pacemakers based on HCN or K channel genes.reviewed in 11 Cell therapy has seen human embryonic stem cells coaxed into a pacemaker line,16 adult hMSCs used as platforms to carry pacemaker genes17,18 and fibroblasts used to carry pacemaker genes to myocytes with which they have been fused.19 Finally, in AV block cell-engineered bypass tracts have carried sinus impulses to the ventricles.20
The treatment of tachyarrhythmias has been more challenging than bradyarrhythmias. Issues include design of therapeutic constructs and whether to administer treatment globally or locally, as discussed below.
GLOBAL VERSUS LOCAL ADMINISTRATION
Permeabilizing agents, vasodilators and VEGF have been used to facilitate gene delivery to large or localized regions of the heart.21,22 Cooling and aortic cross-clamping have been employed to improve gene delivery through the distribution of a coronary artery or the flooding of a chamber or chambers.21,22 Not only do these approaches appear excessive for clinical application but the best success to date has seen about 50% of cells in any region transfected, with viral transfer being diffusion-limited and especially problematic in the ventricles.22
Tempering interest in some viral vectors are concerns about inflammation, chronic illness or neoplasia. These issues led us to explore hMSCs as platforms for gene delivery. That hMSCs can be loaded with specific gene constructs17,18 and delivered to the heart without eliciting inflammation or rejection and not differentiating into other cell typesreviewed in 23 is exciting. But long-term stability of hMSC therapies raises concern (e.g. migration to other sites, differentiation into other cell types, and duration of expression of genes of interest).11 The use of various markers to trace cell location should facilitate our understanding of the extent of hMSC localization to sites of administration.11
Hence, viral vector-based therapies are not yet applied clinically to arrhythmia management but have been effective in proof-of-concept experiments (see below), suggesting that gene therapy can be of use. Cell therapies generally have been intended to regenerate and repair myocardium rather than to be specifically antiarrhythmic. While we have found hMSCs to be adequate delivery platforms for ion channel generated currents, we have followed them for only 6 weeks.18 The question of long-term applicability will await long-term studies of hMSC survival as well as comparison with genomically-incorporated viral constructs.
NOVEL ION CHANNEL CONSTRUCTS AS ANTIARRHYTHMIC INTERVENTIONS
Given the feasibility of gene/cell therapy approaches, a potential advantage is that unlike drugs they do not limit us to the channels and transporters expressed by native cardiac myocytes. Instead, channels resident in other tissues or man-made mutant or chimeric channels with more favorable biophysical properties can be employed. Such a unique arsenal of antiarrhythmic tools allows a “rational” approach to antiarrhythmic therapy in which the biophysical properties of an ideal therapeutic agent are defined, synthesized and delivered. A general approach to administering gene therapy constructs is in Figure 3. Theoretically, we might employ novel Na channels or connexins to speed conduction, and/or alter the properties of inward or repolarizing currents to produce post-repolarization refractoriness. We might alternatively deliver small interfering RNA to specifically inhibit channels or block conduction at localized sites.
Figure 3.

Top: Two myocytes and their ion channels. Na and K channels are transmembrane structures carrying, respectively, inward (red arrow) Na current resulting in depolarization and outward (yellow arrow) K current resulting in repolarization. Connexins (Cx) populate the intercellular gap junctions and permit electron flow from one cell to another facilitating propagation of the cardiac impulse. Bottom: Schematic of action potentials from normal and depolarized (infarcted) ventricular myocytes. Upper traces are the action potentials and lower traces are the maximal upstroke velocity of phase 0 (V̇max) of the action potential reflecting rapid Na entry. Note the depolarized cell has a low V̇max and would be expected to result in slowed propagation. (Created using Servier technology; action potentials obtained from Lue and Boyden: Circulation: 1992: 85: 1175–1188, by permission).
Atrial fibrillation
A major goal of gene therapy experiments on atrial fibrillation has been to induce atrioventricular block. To this end, AF G-αi2 overexpression via AV nodal artery injection in pig was used to suppress basal adenylyl cyclase activity and via amplified vagal tone to indirectly reduce Ca current.13 During sinus rhythm AV conduction slowed and ERP prolonged, and during AF there was a 20% reduction in ventricular rate.13 Other strategies reported are creating an AV nodal site of Ca channel blockade,14 or implanting fibroblasts to induce AV nodal scarring and block.15 All these approaches exemplify local gene delivery whose therapeutic intent is to produce rate control. Whether they will be a practical alternative to radiofrequency ablation is uncertain.
Another experimental AF therapy aimed at rhythm control uses an ion channel mutation Q9E-hMiRP1 (a contributor to long QT syndrome induced by IKr-blocking drugs). Levy et al administered the construct into the atrial epicardium of pigs: about 15% of cells manifested uptake. Clarithromycin was then infused and profoundly blocked IKr. This led the investigators to hypothesize that in AF they might achieve regional atrial IKr blockade without prolonging the QT interval.24
Ventricular tachycardia/fibrillation
Whereas myocardial infarct-induced arrhythmias might respond to local therapy, variations in anatomy from patient to patient require extensive mapping to determine sites at which to localize therapy. For example, Reddy et al demonstrated that mapping to identify sites for local radiofrequency ablation reduced the need for defibrillation in patients who had devices implanted for secondary prevention.25 Using mapping to identify the border zone of an infarct in a canine model we have replaced ablation with intramyocardially-administered gene therapy in preliminary studies and - without destroying tissue - achieved a reduction in VT/VF incidence.26
Specific gene therapies for ischemic arrhtyhmias
1. Speeding conduction via Na channels or connexins
At least 10 different Na channel genes encode alpha subunits in the mammalian genome and these have been cloned from brain, spinal cord, skeletal and cardiac muscle, uterus, and glia.6 Since slow conduction is an essential feature of reentrant cardiac arrhythmias we sought other mammalian Na channels that might have more favorable properties than the cardiac Na channel in circumstances that favor slow conduction (Figure 4).26 One such circumstance is membrane depolarization, as in myocardial infarction (as in Figure 3). Here, the voltage dependence of steady state Na channel inactivation is of interest. The midpoint of the cardiac Na channel (SCN5A) is negative to −73mV. This is important because in infarcted tissue when myocytes are depolarized to −65mV virtually all SCN5A-derived cardiac Na channels are inactivated. In contrast, skeletal muscle (SkM1) Na channels have an inactivation midpoint of −68mV and almost half of these channels would be available to open during an action potential in a depolarized cell. This suggests that Na channels such as SkM1 with more favorable biophysical properties than SCN5A might be a useful antiarrhythmic therapy (Figure 4). Data from our laboratory have demonstrated the effectiveness of this approach in a canine model in which the incidence of inducible polymorphic VT was 75% of controls and 17% of SkM1-administered dogs 5 days post-infarction.26 Moreover, as shown in Figure 5, SkM1 administration reduced electrogram fragmentation and increased V̇max of phase 0 (consistent with more rapid conduction), as had been predicted for SkM1.
Figure 4.

Comparison of the sodium channels SCN5A and SkM1 as inward charge carriers in normal and depolarized cells (the latter mimicking an epicardial border zone in a myocardial infarct). Depicted are the sequence of channel states from resting through open (depolarized) and repolarizing. The Na channel is shown as are the m and h gates (in open or closed configurations). Top: At −90 mV SCN5A (and SkM1) carries inward Na current (yellow arrow) during open channel state. Middle: At −60 mV the h gate is closed and there is no inward current via SCN5A. Bottom: At −60mV there is inward Na current through SkM1 (yellow arrow). Hence, SkM1 is capable of carrying robust current at membrane potentials at which SCN5A is inactivated and non-functional. (created using Servier technology, from an idea of Ira Cohen and Jia Lu).
Figure 5.
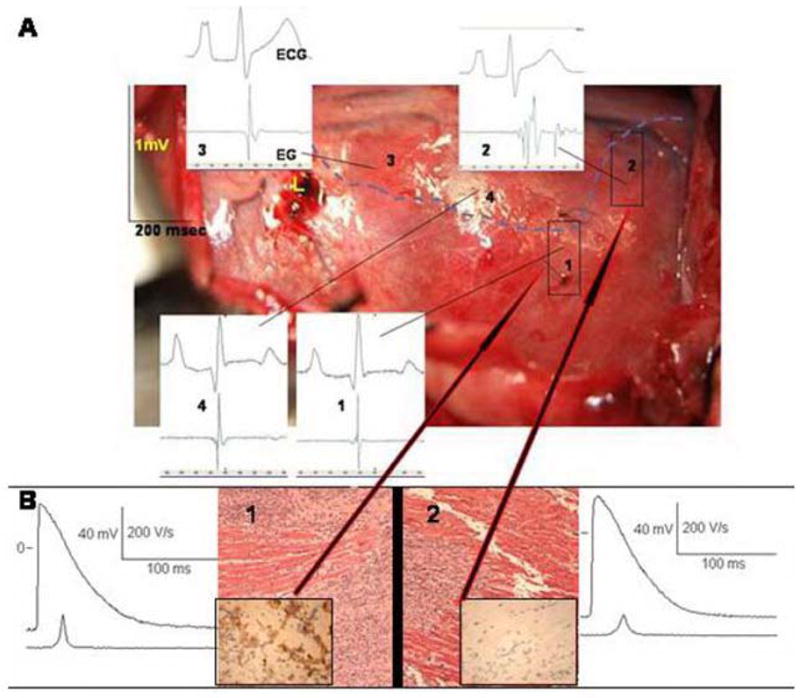
Effect of SkM1 adenoviral infection of epicardial site. The canine heart was infarcted and injected with SkM1+GFP adenovirus 1 week before this experiment. Panel A: Photo of LV epicardial surface. Each panel displays a surface ECG (upper) and a bipolar local electrogram (lower). Broken line demarcates infarcted (lower) from non-infarcted (upper) myocardium. Note the local electrogram in non-injected infarcted site 2 is markedly fragmented. Infarcted site 1 (injected with SkM1) shows a normal EG as do non-infarcted sites 3 and 4. Panel B: Hematoxylin and eosin stain of tissues from sites 2 (no SkM1) and 1 (+SkM1) show infarcted myocardium (×200). Inset in site 1 is GFP positive; that in site 2 is GFP negative (×400). Representative action potential recorded from site 1 has higher V̇max and amplitude than that from site 2. (modified after reference 25, by permission).
Several studies have lent credence to the importance of connexins and hence gap junctions in arrhythmias. For example, overexpression of Cx45 results in ventricular tachycardia in mice27 while mutations of Cx40 are associated with atrial fibrillation in humans.28 Studies of the epicardial border zone of healing canine myocardial infarcts have demonstrated altered connexin distribution and density in regions important to the generation of reentrant ventricular tachycardia.29
The modulation of gap junctions as an antiarrhythmic strategy initially initially attempted to block conduction. However, the gap junctional blockers used to date have not been channel-specific or isoform-specific and in disrupting coupling between cells have been found to cause potentially fatal arrhythmias. On the positive side, antiarrhythmic peptides have been used to increase junctional conductance. One such peptide, rotigaptide, appears to target Cx43 specifically,30 and is purportedly antiarrhythmic.
2. Targeting diastolic membrane potential
In VT in the setting of a partially healed infarct, the viable but depolarized tissue in the border zone provides the substrate for a reentrant arrhythmia (Figure 3).6 A logical approach to enhance conduction in these circumstances is to hyperpolarize diastolic membrane potential, thereby making more Na current available. In normal myocytes the diastolic membrane potential is largely set by the inward rectifier IK1 (generated by Kir2.1 with some contribution from Kir2.2).31 Studies overexpressing these channels are in progress.
3. Enhancing rate responsiveness and/or refractoriness
Reentrant arrhythmias require reexcitation of tissue by a propagating waveform. Here, an intervention that facilitates recovery of excitability in the pathway may restore antegrade activation and forestall retrograde invasion of that path by the reentering waveform. Alternatively, it may speed propagation of the reentering waveform such that it encounters tissue that remains refractory. Hua et al32 showed that 6-fold overexpression of native hERG eliminates T wave alternans in isolated canine ventricular myocytes and in computer simulations.
Using a different approach, Sasano et al delivered a dominant negative HERG mutant (HERG-G628S) via vascular infusion to a peri-infarct zone of pigs.12 Monomorphic VT had been consistently inducible in infarcted animals before gene transfer, but one week later all HERG-G628S-transferred pigs showed no such arrhythmia. This result emphasizes the therapeutic potential of yet a different local approach to VT therapy in chronic infarcts.
CONCLUSIONS
How best to prevent/treat arrhythmias that confer significant morbidity and/or are life-threatening is a question that forty years of targeted pharmacologic therapy and many more years of empirical drug therapy have not answered. While surgery, ablation and cardioverter-defibrillators are newer, robust alternatives, all appear draconian when compared to a targeted therapy that might be administered via catheter and doesn’t destroy tissue. Gene and cell therapies meld principles for rational drug design with a new approach to treatment. The approach starts by simulating an optimal change in an ion current present in the heart, then selects constructs independent of their tissue origins based on their close mimicry of the proposed optimal change. Additionally, use of novel genes not normally found in the heart expands our therapeutic universe.
Also innovative are the focus on regionally-delivering these novel ion channel constructs, the use of hMSCs as platforms for therapeutic intervention and the harnessing of these new therapies to mechanistically test old, but to-date largely untestable concepts (e.g. speed conduction, increase ERP/repolarization ratio without prolonging repolarization).
These approaches should not be interpreted as providing a “quick fix.” However, the ability to prepare constructs and to apply them based on an understanding of arrhythmogenic mechanisms makes it highly likely that (1) definitive answers - whether positive or negative - will be obtained to the questions we have regarding improvement of antiarrhythmic therapy, and (2) we will understand why, mechanistically, an approach has succeeded or failed - or, as a worst case, been proarrhythmic. Negative answers will be as important as positive: of signal importance is that as accurately and rapidly as possible we find the proper route, vector and/or platform and construct for reducing the threat to the population of the arrhythmias of concern.
Acknowledgments
The authors gratefully acknowledge the assistance of Eileen Franey in the preparation of this manuscript. Research was supported in part by USPHS-NHLBI grants HL-28958 and HL-67101.
References
- 1.Myerburg RJ, Feigal DW, Jr, Lindsay BD. Life-threatening malfunction of implantable cardiac devices. N Engl J Med. 2006;354:2309–2311. doi: 10.1056/NEJMp068112. [DOI] [PubMed] [Google Scholar]
- 2.Miyasaka Y, Barnes ME, Gersh BJ, et al. Secular trends in incidence of atrial fibrillation in Olmsted County, Minnesota, 1980 to 2000, and implications on the projections for future prevalence. Circulation. 2006;114:119–125. doi: 10.1161/CIRCULATIONAHA.105.595140. [DOI] [PubMed] [Google Scholar]
- 3.Wit AL, Janse MJ. Experimental models of ventricular tachycardia and fibrillation caused by ischemia and infarction. Circulation. 1992;85:I32–142. [PubMed] [Google Scholar]
- 4.Mines GR. On circulating excitations in heart muscles and their possible relation to tachycardia and fibrillation. Trans R Soc Can. 1914;IV:43–52. [Google Scholar]
- 5.Schmitt FO, Erlanger J. Directional differences in the conduction of the impulse through heart muscle and their possible relations to extrasystolic and fibrillary contractions. Am J Physiol. 1928;87:326–347. [Google Scholar]
- 6.Allessie MA, Bonke FIM, Schopman FJG. Circus movement in rabbit atrial muscle as a mechanism of tachycardia. III. The “leading circle” concept: a new model of circus movement in cardiac tissue without the involvement of an anatomical obstacle. Circ Res. 1977;41:9–18. doi: 10.1161/01.res.41.1.9. [DOI] [PubMed] [Google Scholar]
- 7.Spooner PM, Rosen MR. Perspectives on arrhythmogenesis, antiarrhythmic strategies and sudden cardiac death. In: Spooner PM, Rosen MR, editors. Foundations of Cardiac Arrhythmias. New York: Marcel Dekker Inc; 2000. pp. 1–20. [Google Scholar]
- 8.Fuster V, Ryden LE, Cannom DS, et al. American College of Cardiology. American Heart Association Task Force on Practice Guidelines. European Society of Cardiology Committee for Practice Guidelines. ACC/AHA/ESC 2006 guidelines for the management of patients with atrial fibrillation--executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol. 2006;48:854–906. doi: 10.1016/j.jacc.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 9.Ducharme A, Swedberg K, Pfeffer MA, et al. CHARM Investigators. Prevention of atrial fibrillation in patients with symptomatic chronic heart failure by candesartan in the Candesartan in Heart failure: Assessment of Reduction in Mortality and morbidity (CHARM) program. Am Heart J. 2006;152:86–92. [PubMed] [Google Scholar]
- 10.Members of the Sicilian Gambit. New approaches to antiarrhythmic therapy. Emerging therapeutic applications of the cell biology of cardiac arrhythmias. Eur Heart J. 2001;22:2148–2163. doi: 10.1053/euhj.2001.3036. [DOI] [PubMed] [Google Scholar]
- 11.Rosen M. Biological pacemaking: In our lifetime? Heart Rhythm. 2005;2:418–428. doi: 10.1016/j.hrthm.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 12.Sasano T, McDonald AD, Kikuchi K, Donahue JK. Molecular ablation of ventricular tachycardia after myocardial infarction. Nature Med. 2006;12:1256–1258. doi: 10.1038/nm1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bauer A, McDonald AD, Nasir K, et al. Inhibitory G protein overexpression provides physiologically relevant heart rate control in persistent atrial fibrillation. Circulation. 2004;110:3115–3120. doi: 10.1161/01.CIR.0000147185.31974.BE. [DOI] [PubMed] [Google Scholar]
- 14.Murata M, Cingolani E, McDonald AD, Donahue JK, Marban E. Creation of a genetic calcium channel blocker by targeted gem gene transfer in the heart. Circ Res. 2004;95:398–405. doi: 10.1161/01.RES.0000138449.85324.c5. [DOI] [PubMed] [Google Scholar]
- 15.Bunch TJ, Mahapatra S, Bruce GK, et al. Impact of transforming growth factor-β1 on atrioventricular node conduction modification by injected autologous fibroblasts in the canine heart. Circulation. 2006;113:2485–2494. doi: 10.1161/CIRCULATIONAHA.105.570796. [DOI] [PubMed] [Google Scholar]
- 16.Kehat I, Khimovich L, Caspi O, et al. Electromechanical integration of cardiomyocytes derived from human embryonic stem cells. Nature Biotechnol. 2004;22:1282–1289. doi: 10.1038/nbt1014. [DOI] [PubMed] [Google Scholar]
- 17.Potapova I, Plotnikov A, Lu Z, et al. Human mesenchymal stem cell as a gene delivery system to create cardiac pacemakers. Circ Res. 2004;94:841–959. doi: 10.1161/01.RES.0000123827.60210.72. [DOI] [PubMed] [Google Scholar]
- 18.Plotnikov AP, Shlapakova I, Szabolcs MJ, et al. Xenografted adult human mesenchymal stem cells provide a platform for sustained biological pacemaker function in canine heart. Circulation. 2007;116:706–713. doi: 10.1161/CIRCULATIONAHA.107.703231. [DOI] [PubMed] [Google Scholar]
- 19.Cho HC, Kashiwakura Y, Marban E. Creation of a biological pacemaker by cell fusion. Circ Res. 2007;100:1112–1115. doi: 10.1161/01.RES.0000265845.04439.78. [DOI] [PubMed] [Google Scholar]
- 20.Choi YH, Stamm C, Hammer PE, et al. Cardiac conduction through engineered tissue. Am J Pathol. 2006;169:72–85. doi: 10.2353/ajpath.2006.051163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lehnart SE, Donahue JK. Coronary perfusion cocktails for in vivo gene transfer. Methods Mol Biol. 2003;219:213–218. doi: 10.1385/1-59259-350-x:213. [DOI] [PubMed] [Google Scholar]
- 22.Roth DM, Lai NC, Gao MH, et al. Indirect intracoronary delivery of adenovirus encoding adenylyl cyclase increases left ventricular contractile function in mice. Am J Physiol Heart Circ Physiol. 2004;287:H172–177. doi: 10.1152/ajpheart.01009.2003. [DOI] [PubMed] [Google Scholar]
- 23.Zimmett JM, Hare JM. Emerging role for bone marrow derived mesenchymal stem cells in myocardial regenerative therapy. Basic Res Cardiol. 2005;100:471–481. doi: 10.1007/s00395-005-0553-4. [DOI] [PubMed] [Google Scholar]
- 24.Perlstein I, Burton DY, Ryan K, et al. Posttranslational control of a cardiac ion channel transgene in vivo: clarithromycin-hMiRP1-Q9E interactions. Hum Gene Ther. 2005;16:906–910. doi: 10.1089/hum.2005.16.906. [DOI] [PubMed] [Google Scholar]
- 25.Reddy VY, Reynolds MR, Neuzil P, et al. Prophylactic catheter ablation for the prevention of defibrillator therapy. N Engl J Med. 2007;357:2657–65. doi: 10.1056/NEJMoa065457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lau DH, Clausen C, Sosunov EA, et al. Epicardial border zone overexpression of skeletal muscle sodium channel, SkM1, normalizes activation, preserves conduction and suppresses ventricular arrhythmia: an in silico, in vivo, in vitro study. Circulation. doi: 10.1161/CIRCULATIONAHA.108.809301. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Betsuyaku T, Nnebe NS, Sundset R, Patibandla S, Krueger CM, Yamada KA. Overexpression of cardiac connexin45 increases susceptibility to ventricular tachyarrhythmias in vivo. Am J Physiol Heart Circ Physiol. 2006;290(1):H163–H171. doi: 10.1152/ajpheart.01308.2004. [DOI] [PubMed] [Google Scholar]
- 28.Gollob MH, Jones DL, Krahn AD, et al. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N England J Med. 2006;354(25):2677–2688. doi: 10.1056/NEJMoa052800. [DOI] [PubMed] [Google Scholar]
- 29.Peters NS, Coromilas J, Severs NJ, Wit AL. Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation. 1997;95(4):988–996. doi: 10.1161/01.cir.95.4.988. [DOI] [PubMed] [Google Scholar]
- 30.Dhein S, Larsen BD, Petersen JS, Mohr FW. Effects of the new antiarrhythmic peptide ZP123 on epicardial activation and repolarization pattern. Cell Commun Adhes. 2003;10(4–6):371–378. doi: 10.1080/cac.10.4-6.371.378. [DOI] [PubMed] [Google Scholar]
- 31.Zaritsky JJ, Redell JB, Tempel BL, Schwarz TL. The consequences of disrupting cardiac inwardly rectifying K+ current (IK1) as revealed by the targeted deletion of the murine Kir2.1 and Kir2.2 genes. J Physiol. 2001;533.3:697–710. doi: 10.1111/j.1469-7793.2001.t01-1-00697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hua F, Johns DC, Gilmore RF., Jr Suppression of electrical alternans by overexpression of HERG in canine ventricular myocytes. Am J Physiol Heart Circ Physiol. 2004;286:H2342–H2352. doi: 10.1152/ajpheart.00793.2003. [DOI] [PubMed] [Google Scholar]