

Abstract
Elevation in intracellular cAMP level has been associated with increased endothelial barrier integrity and linked to the activation of protein kinase A (PKA). Recent studies have shown a novel mechanism of cAMP-mediated endothelial barrier regulation via cAMP-dependent nucleotide exchange factor Epac1 and Rap1 GTPase. This study examined a contribution of PKA-dependent and PKA-independent pathways in the human pulmonary endothelial (EC) barrier protection by cAMP. Synthetic cAMP analog, 8-Bromoadenosine-3′,5′-cyclic monophosphate (Br-cAMP), induced dose-dependent increase in EC transendothelial electrical resistance which was associated with activation of PKA, Epac/Rap1, and Tiam/Vav/Rac cascades, and significantly attenuated thrombin-induced EC barrier disruption. Both, specific Epac/Rap1 activator 8CPT-2Me-cAMP (8CPT) and specific PKA activator N6-Benzoyladenosine-3′,5′-cyclic monophosphate (6Bnz) enhanced EC barrier, suppressed thrombin-induced EC permeability, and independently activated small GTPase Rac. SiRNA-induced Rac knockdown suppressed barrier-protective effects of both PKA and Epac signaling in pulmonary EC. Intravenous administration of either 6Bnz, or 8CPT, significantly reduced lung vascular leak in the murine model of lung injury induced by high tidal volume mechanical ventilation (HTV, 30 ml/kg, 4 hrs), whereas combined treatment with 6Bnz and 8CPT showed no further additive effects. This study dissected for the first time PKA and Epac pathways of lung EC barrier protection caused by cAMP elevation and identified Rac GTPase as a hub for PKA and Epac signaling leading to enhancement of lung vascular barrier.
Keywords: small GTPases, permeability, cytoskeleton, endothelium, acute lung injury
Introduction
Clinical observations and animal studies show beneficial effects of elevated intracellular cAMP levels in various pathological settings such as ischemia/reperfusion, acute lung injury, pulmonary hypertension and asthma (Matthay et al., 2005; Olschewski et al., 2002). Beneficial effects of cAMP are in part due to improvement of pulmonary vascular barrier function and increased fluid reabsorbtion from alveolar compartment (McAuley et al., 2004; Perlman et al., 1986).
Studies using pulmonary endothelial cells show that elevation of intracellular cAMP levels promotes endothelial barrier properties and attenuates thrombin-, LPS- and pertussis toxin-induced EC barrier dysfunction (Birukova et al., 2007b; Essler et al., 2000; Garcia et al., 2002). Effects of cAMP-elevating agents on EC monolayers have been mostly associated with activation of cAMP-dependent protein kinase (PKA)-dependent mechanisms of endothelial barrier protection (Patterson et al., 2000; Qiao et al., 2003). Studies using PKI, an endogenous PKA competitor, demonstrated that inhibition of PKA by PKI overexpression abolishes the cAMP protective effect against increased endothelial permeability and significantly increases the formation of actomyosin stress fibers and paracellular gaps indicating barrier compromise (Lum et al., 1999). It is also possible that the PKA pathway functions through interactions with other signaling systems.
Elevation of cAMP may trigger cAMP-dependent, PKA-independent regulation of EC barrier by small GTPases. The nucleotide exchange proteins directly activated by cAMP (Epac or cAMP-GEFs) (de Rooij et al., 1998) contain a cAMP-binding domain and may activate small GTPase Rap1 in a cAMP-dependent, PKA-independent manner (Bos, 2003). The Rap proteins are members of Ras family of small GTPases. Rap1 plays a key role in the regulation of adhesion-dependent signals during formation of cell-cell junctions, and also functions as signal transducer activated by cAMP-elevating agonists (Bos, 2005; Fukuhara et al., 2005).
Another small GTPase Rac is involved in endothelial barrier protective responses induced by mechanical forces (laminar shear stress and physiological cyclic stretch) and bioactive molecules, i.e. prostacyclin, oxidized phospholipids, hepatocyte growth factor, and sphingosine 1-phosphate (Birukova et al., 2006; Birukova et al., 2007b; Dudek et al., 2004; Tzima et al., 2002; Vouret-Craviari et al., 2002). However, the impact of PKA and Rap1 in regulation of Rac signaling and endothelial barrier is not completely understood. Development of a cAMP analog 8-(4-Chlorophenylthio)-2′-O-methyl-adenosine-3′,5′-cyclic monophosphate (8CPT), that specifically activates Epac, but not PKA (Christensen et al., 2003), allowed separation of PKA- and Epac-mediated pathways normally induced by cAMP elevation. In turn, cAMP analog N6-Benzoyl-adenosine-3′,5′-cyclic monophosphate (6Bnz) selectively activates PKA (Christensen et al., 2003).
PKA and Epac1 pathways may play distinct roles in various biologic processes. For example, anti-proliferative effects caused by activation of prostanoid receptors are mediated by Epac1, but not PKA signaling (Haag et al., 2008). PKA exclusively mediated inhibitory effects of PGE2 on expression of collagen I, while inhibition of fibroblast proliferation was exclusively mediated by Epac1 (Huang et al., 2008). Specific PKA activation by cAMP analog N(6)-Benzoyl- (6-Bnz-) cAMP inhibited contractile activity of lung fibroblasts, while activation of Epac by 8CPT had no effect (Kamio et al., 2007). These data show that in different cell types Epac and PKA may act in parallel or play distinct physiological roles. Therefore, specific contribution of Epac1 and PKA signaling in the pulmonary EC barrier regulation deserves special attention.
This study investigated the impact of PKA and Epac/Rap activation on basal and agonist-induced permeability in human endothelial cell cultures and animal models of ventilator induced lung injury and vascular leak. Using siRNA approach we further examined a role of Rac signaling in the protective effects exhibited by PKA or Epac/Rap activators.
Materials and Methods
Cell culture and reagents
Human pulmonary artery endothelial cells (HPAEC) were obtained from Lonza Inc (Allendale, NJ), cultured according to the manufacturer's recommendations and used for experiments at passages 5-9. Unless specified, biochemical reagents were obtained from Sigma (St. Louis, MO). All reagents used for immunofluorescence were purchased from Molecular Probes (Eugene, OR). Rac antibody was purchased from BD Transduction Laboratories (San Diego, CA); Tiam1, phospho-Vav2, and phospho-Rho antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA); HRP-linked anti-mouse and anti-rabbit IgG, diphospho-MLC, phospho-PKA substrate, and phospho-PAK1 antibodies were obtained from Cell Signaling (Beverly, MA); phospho-tyrosine and phospho-MYPT1 antibodies were purchased from Upstate Biotechnology (Lake Placid, NY). 8-Bromo-adenosine-3′,5′-cyclic monophosphate (Br-cAMP), 8-(4-Chlorophenylthio)-2′-O-methyl-adenosine-3′,5′-cyclic monophosphate (8CPT) and PKA inhibitory peptide (PKI) were purchased from Calbiochem (La Jolla, CA); N6-Benzoyl-adenosine-3′,5′-cyclic monophosphate (6Bnz) was purchased from Biolog (Hayward, CA). TNFα, IL-6, and IL-6 soluble receptor (SR) were obtained from R&D Systems (Minneapolis, MN).
Si-RNA-based knockdown of Epac1 and Rac in pulmonary EC
To reduce the content of endogenous Epac1 or Rac cells were treated with gene-specific siRNA duplexes. Pre-designed standard purity Stealth™ Select RNAi sets were ordered from Invitrogen (Carlsbad, CA), and transfection of EC with siRNA was performed as previously described (Birukov et al., 2004). Nonspecific, non-targeting siRNA was used as a control treatment. After 48 or 72 hours of transfection cells were used for experiments or harvested for western blot verification of specific protein depletion.
Measurement of transendothelial electrical resistance
Measurements of transendothelial electrical resistance (TER) across confluent HPAEC monolayers were taken using the electrical cell-substrate impedance sensing system (ECIS) (Applied Biophysics, Troy, NY) as previously described (Birukov et al., 2004).
Measurements of protein kinase A activity
were performed using non-radioactive HitHunter Chemiluminescence assays (Amersham Bioscience, Piscataway, NJ), and PepTag non-radioactive assays (Promega, Madison, WI) according to the manufacturer's instructions.
GTPase activation assays
were performed using commercially available assay kits purchased from Upstate Biotechnology (Lake Placid, NY), as described elsewhere (Birukov et al., 2004).
Immunoblotting
After stimulation cells were lysed, and protein extracts were separated by SDS-PAGE, transferred to nitrocellulose membrane and probed with specific antibodies as previously described (Birukov et al., 2003).
Immunofluorescence staining
Endothelial monolayers plated on glass cover slips were treated with the agonist of interest, fixed in 3.7% formaldehyde and permeabilized with 0.1% triton X-100 in PBS-Tween. Actin filaments were stained with Texas Red-conjugated phalloidin, adherens junctions were detected using VE-cadherin antibodies. After immunostaining, slides were analyzed using a Nikon video imaging system (Nikon Instech Co., Tokyo, Japan) as described elsewhere (Birukov et al., 2004).
Mechanical ventilation protocol
Adult male C57BL/6J mice, 8-10 weeks old, with average weight 20-25 grams (Jackson Laboratories, Bar Harbor, ME) were bred at University of Chicago animal care center. All experimental protocols involving the use of animals were approved by the University of Chicago Institutional Animal Care & Use Committee for the humane treatment of experimental animals. C57BL/6J mice were anesthetized with an intraperitoneal injection of ketamine (75 mg/kg) and acepromazine (1.5 mg/kg). Tracheotomy was performed and the trachea was cannulated with a 20-gauge one inch catheter (Penn-Century Inc., Philadelphia, PA), which was tied into place to prevent air leak. The animals have been placed on mechanical ventilator (Harvard Apparatus, Boston, MA) for 4 hours with high tidal volume (30 ml/kg, HTV) ventilation. Mice were randomized to concurrently receive sterile saline solution or agonist of interest (8CPT 0.4 μM/mouse, i/v, 6Bnz 0.6 μM/mouse, i/v, or combination of 8CPT and 6Bnz) at the onset of mechanical ventilation. After the experiment, animals were sacrificed by exsanguination under anesthesia. BAL was performed using 1 ml of sterile Hanks Balanced Salt Buffer. The BAL cell count and protein concentration was determined previously described by a modified Lowry colorimetric assay using a Bio-Rad DC protein assay kit (Bio-Rad Laboratories, Hercules, CA). BAL cell count was performed with a hemacytometer as described elsewhere (Fu et al., 2009).
Assessment of pulmonary vascular leak by Evans blue
Evans blue dye (EBD, 30 ml/kg) was injected into the external jugular vein 2 hrs before termination of ventilation to assess vascular leak as described previously (Moitra et al., 2007). In brief, at the end of experiment, thoracotomy was performed, and the lungs were perfused free of blood with PBS containing 5 mM EDTA. Both left lung and right lung were excised and imaged by Kodak digital camera. After imaging lungs were blotted dry, weighed and homogenized in PBS (1ml/100 μg tissue). Homogenized tissue was incubated with 2 volume formamide (18 h, 60°C), centrifuged at 12,000 g for 20 min. Optical density of the supernatant was determined by spectrophotometry at 620 nm and 740nm. Extravasated EBD concentration (micrograms of Evans blue dye per g lung) in lung homogenates was calculated against a standard curve.
Statistical analysis
Results are expressed as means ± SD of three to ten independent experiments. Stimulated samples were compared to the controls using the unpaired Student's t-test. For multiple-group comparisons, a one-way variance analysis (ANOVA) and post hoc multiple comparisons tests were used. P<0.05 was considered statistically significant.
Results
cAMP elevation activates PKA, Rap1, and Rac-dependent signaling and attenuates EC barrier dysfunction
Activation of Rac GTPase triggers downstream cytoskeletal mechanisms leading to enhancement of EC barrier function (Birukova et al., 2007b; Tzima et al., 2002; Vouret-Craviari et al., 2002). This study tested involvement of cAMP-dependent signaling in the Rac activation and EC barrier protection against edemagenic and inflammatory stimuli. Pulmonary EC were treated with stable cAMP analog 8Br-cAMP. Elevation of cAMP increased transendothelial electrical resistance in a dose-dependent manner, reflecting EC barrier enhancement (Figure 1A). Elevation of cAMP by prostacyclin and its stable analog beraprost activates both, PKA-dependent and Epac/Rap1-dependent signaling pathways (Birukova et al., 2007b; Fukuhara et al., 2005). Consistently with this notion, 8Br-cAMP treatment caused time-dependent activation of both, PKA and Rap1 GTPase (Figure 1B and C). Challenge of HPAEC with 8Br-cAMP caused rapid activation of Rac (Figure 1D) and Rac-specific GEFs Tiam1 and Vav2 detected by increased association of Tiam1 with activated Rac (Figure 1E), and Vav2 phosphorylation (Figure 1F). In addition, cAMP elevation induced phosphorylation and activation of Rac downstream target PAK1 (Figure 1F), and increased total protein tyrosine phosphorylation (Figure 1G).
Figure 1. Effect of cAMP elevation on pulmonary EC barrier function.
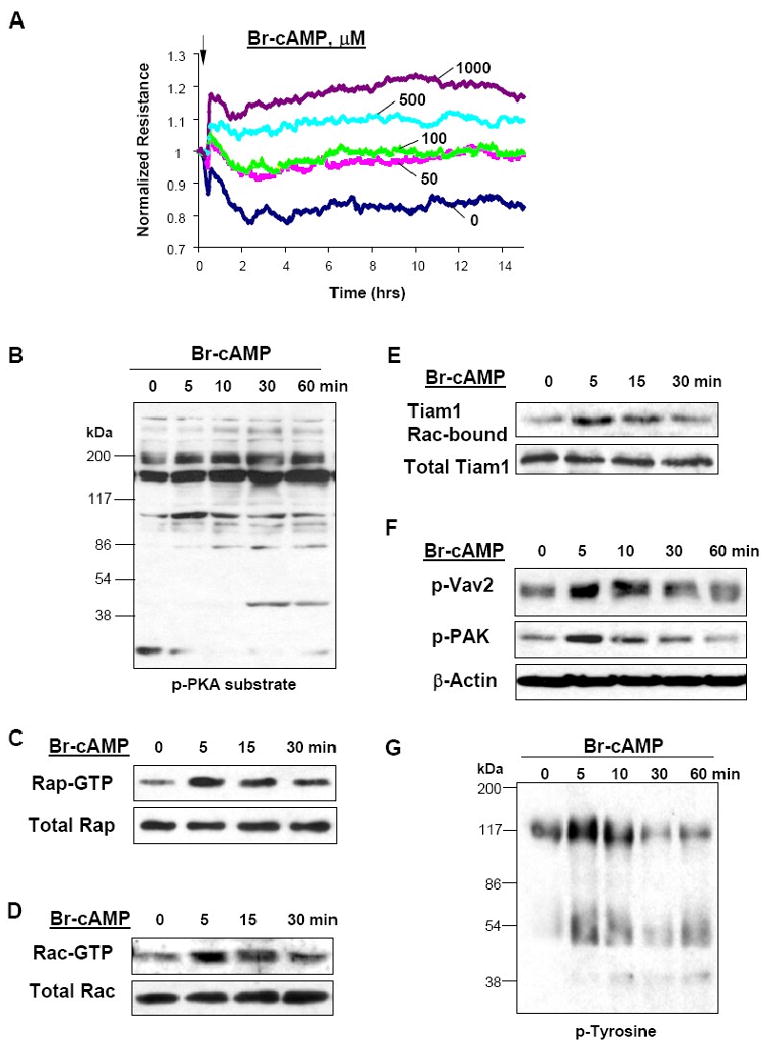
A - Endothelial cells were plated onto gold microelectrodes. At the time indicated by arrow, EC were treated with either vehicle or Br-cAMP (50 ng/ml, 100 ng/ml, 500 ng/ml, or 1000 ng/ml) and used for measurements of transendothelial electrical resistance. B - G - Cells were treated with Br-cAMP (500 ng/ml) for various periods of time followed by sample collection and western blot analysis of signaling pathway activation. Cyclic AMP-mediated phosphorylation of endogenous substrates was monitored by immunoblotting with anti-phospho-PKA substrate antibodies (B). Effects of cAMP on activation of Rap1 (C) and Rac (D) were evaluated using GTPase pulldown assays and normalized to the total GTPase content in cell lysates. Rac-GTP-bound Tiam1 was detected by probing of membranes with Tiam1 antibodies (E). Phosphorylation of Vav2 and PAK1 in control and cAMP-stimulated EC was determined in the total lysates using corresponding phospho-specific antibodies (F). Levels of tyrosine phosphorylated proteins in the total lysates were determined by western blot (G).
Elevation of intracellular cAMP by EC treatment with Br-cAMP protected against pulmonary EC hyper-permeability induced by inflammatory agonists LPS, IL-6, and TNFα (Figure 2A-C). In addition, cAMP elevation attenuated thrombin-induced permeability increase (Figure 2D) and suppressed activation of Rho-dependent pathway indicated by decreased phosphorylation of downstream Rho targets, myosin regulatory light chain (MLC) and myosin-binding subunit of myosin-associated phosphatase type 1 (MYPT1) (Figure 2E).
Figure 2. Effect of cAMP and Rac on agonist-induced EC barrier disruption.
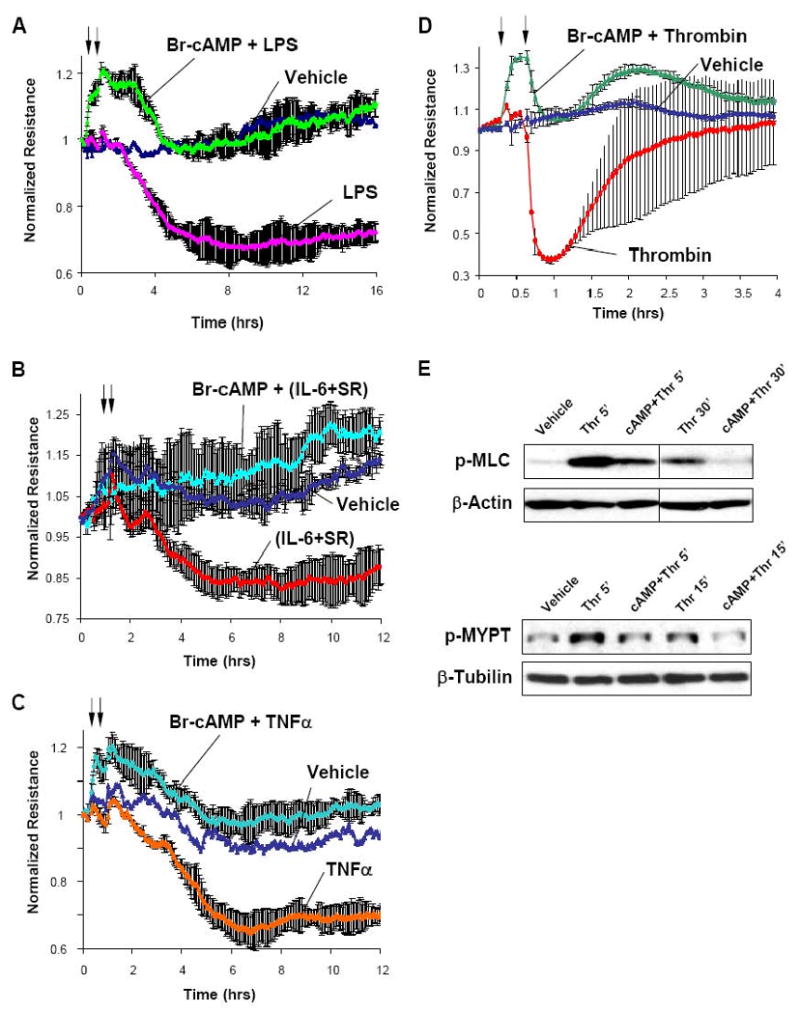
A - D - Human pulmonary EC were treated with Br-cAMP (500 ng/ml, first arrow) followed by stimulation with LPS (200 ng/ml) (A), combination of IL-6 (25 ng/ml) and its soluble co-receptor IL-6-SR (10o ng/ml) (B), TNFα (20 ng/ml) (C), or thrombin (0.1 U/ml) (D) as outlined by second arrow, and transendothelial electrical resistance was measured over the time. E - Cells were treated with thrombin for indicated periods of time with or without pre-incubation with Br-cAMP. Levels of phosphorylated MLC and MYPT1 were determined by western blot analysis.
Rac is involved in PKA-mediated EC barrier protective effects
Rac is one of multiple PKA targets (Abe et al., 2000; O'Connor and Mercurio, 2001). However, the role of Rac in PKA-mediated endothelial barrier protection is not clear. The next experiments tested the involvement of Rac pathway in the EC barrier preservation by PKA. Treatment of pulmonary EC monolayers with selective PKA activator N6-Benzoyl-adenosine-3′,5′-cyclic monophosphate (6Bnz) (Holz et al., 2006) increased TER, reflecting EC barrier enhancement (Figure 3A) and rapidly stimulated Rac activity in pulmonary EC with maximal activation observed at 5 min (Figure 3B). Since at high concentrations cAMP analogs designed to specifically activate particular targets may loose their selectivity, the following experiments were performed to confirm that at concentration used in this study (100 μM), 6Bnz did not activate Epac/Rap pathway. Knockdown of Rap1-specific GEF Epac1 using specific siRNA did not affect TER increases caused by 6Bnz. In contrast, Epac1 depletion inhibited TER increase induced by specific Epac activator 8CPT (Figure 3C).
Figure 3. Effects of PKA activator 6Bnz-cAMP on EC barrier enhancement.
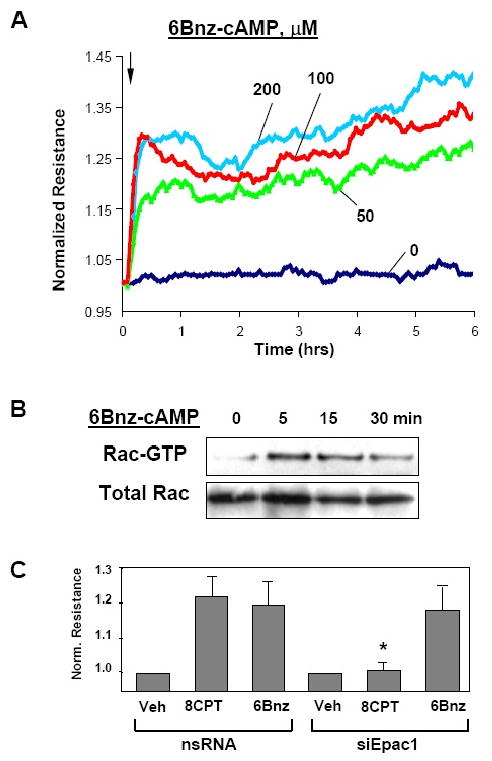
A - At the time indicated by arrow, confluent pulmonary EC were stimulated with 50, 100, or 200 μM 6Bnz, and TER changes were monitored over the time. B - Rac activity after 6Bnz challenge was measured using the Rac-GTP pull-down assay. The lower panel shows total Rac content in EC lysates. C - HPAEC were transfected with Epac1-specific or non-specific RNA duplexes. Seventy two hours after transfection, EC were treated with 6Bnz (100 μM) or 8CPT (100 μM), and TER was measured after 30 min of stimulation (*p<0.05; n= 3).
In the next experiments we used a model of thrombin-induced EC barrier dysfunction and found that activation of PKA pathway by 6Bnz significantly attenuated thrombin-induced permeability increase in pulmonary EC monolayers (Figure 4A) and suppressed Rho kinase-dependent phosphorylation of MYPT1 and MLC (Figure 4B). Previous report shows that PKA may directly suppress Rho activity via direct phosphorylation of Rho at Ser-188 (Lang et al., 1996). Immunofluorescence staining showed marked increase in phospho-Rho-Ser-188 immunoreactivity in 6Bnz-treated EC (Figure 4C). To test involvement of Rac pathway in PKA-mediated protective responses, endogenous Rac was depleted from pulmonary EC using siRNA approach. Controls cells and EC with depleted Rac were pretreated with 6Bnz followed by thrombin challenge. Rac knockdown suppressed PKA protective effects against thrombin-induced cell contraction, as detected by MLC phosphorylation (Figure 4D). Taken together, these results strongly suggest the essential role for Rac signaling in the EC barrier enhancement by PKA, but also indicate direct PKA-mediated phosphorylation of Rho leading to reduction of its activity as an additional mechanism of EC protection against thrombin-induced barrier disruption.
Figure 4. Effect of PKA activation on thrombin-induced EC barrier dysfunction and Rho signaling.
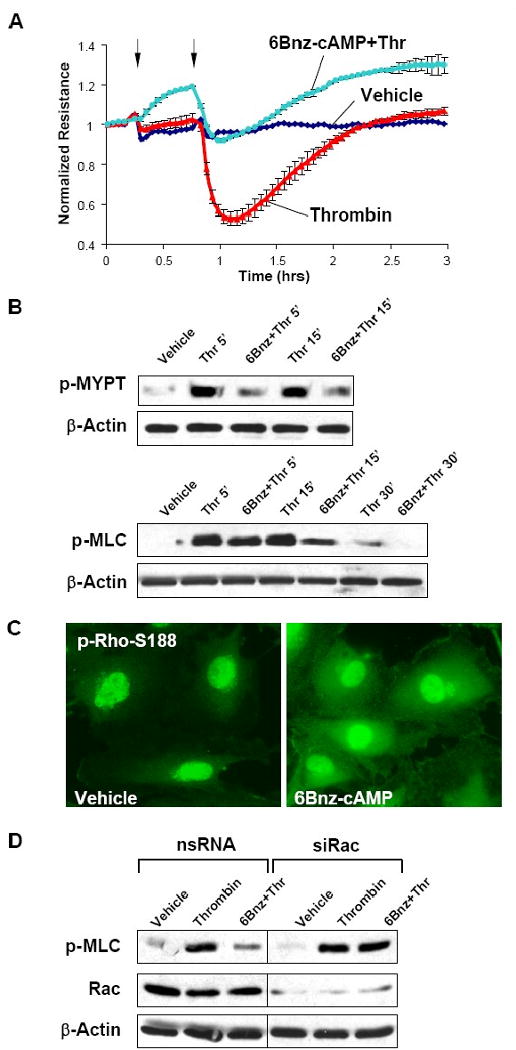
A - Endothelial monolayers were pre-treated with 6Bnz (100 μM, indicated by first arrow) and stimulated with thrombin (0.1 U/ml, indicated by second arrow). TER measurements were performed over 3 hrs. B - Site-specific phosphorylation of MYPT1 and MLC in lung EC challenged with thrombin for 5, 15, or 30 min with or without 6Bnz pretreatment was detected by western blot. C - Rho phosphorylation was determined in control and 6Bnz-treated (100 μM, 30 min) cells by immunofluorescence with phospho-Ser188-specific antibodies. D - HPAEC transfected with Rac-specific or non-specific RNA for 48 hrs were stimulated with thrombin with or without 6Bnz pretreatment. Levels of phosphorylated MLC were determined by western blot analysis.
Rac is involved in Epac-mediated EC barrier protective effects
Recent studies described a novel mechanism of cAMP-dependent regulation of Rac activity by cAMP-activated Epac-Rap1 pathway (Birukova et al., 2007b; Bos, 2003). The following experiments tested the involvement of Rac in barrier protection induced by activation of Epac/Rap1. Treatment of pulmonary EC with selective Epac activator 8CPT (Christensen et al., 2003) caused sustained TER increase in a dose-dependent manner (Figure 5A). EC barrier protective response to 8CPT was accompanied by dramatic cytoskeletal remodeling and enhancement of peripheral F-actin rim (Figure 5B). Functional and morphological changes in EC monolayers in response to Epac/Rap1 activation by 8CPT were associated with marked activation of Rac (Figure 5C).
Figure 5. Effects of Epac/Rap1 activation on EC barrier enhancement.
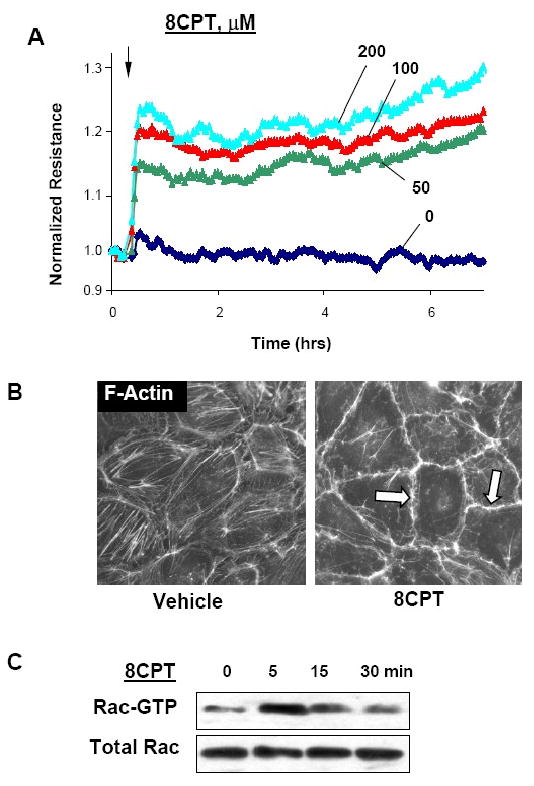
A - At the time point indicated by arrow, EC were treated with 50, 100, or 200 μM 8CPT followed by TER measurements reflecting EC monolayer barrier properties. B - EC monolayers grown on glass coverslips were treated with 8CPT (100 μM, 30 min) followed by immunofluorescence staining with Texas-Red phalloidin to detect F-actin. C - Effects of 8CPT on Rac activation were evaluated using GTPase pulldown assays and normalized to the total Rac content in cell lysates.
8CPT at high concentrations may activate PKA (Holz et al., 2006). The following experiments were designed to test potential effect of 8CPT at the concentration used in our study (100 μM) on PKA activity. Non-radioactive PKA kinase assay shows that in contrast to 6Bnz (100 μM), EC stimulation with 8CPT (100 μM) did not induce phosphorylation of PKA substrate kemptide (Figure 6A). Phosphorylation of other PKA targets also was not detected, as shown by western blot analysis of lysates obtained from 8CPT-stimulated EC with antibodies which recognize phosphorylated PKA protein substrates (Figure 6B). Finally, TER measurements showed that PKA inhibitory peptide PKI completely attenuated EC barrier enhancement in response to 6Bnz, but did not affect TER increase induced by 8CPT (Figure 6C).
Figure 6. Effect of 8CPT on PKA activation.
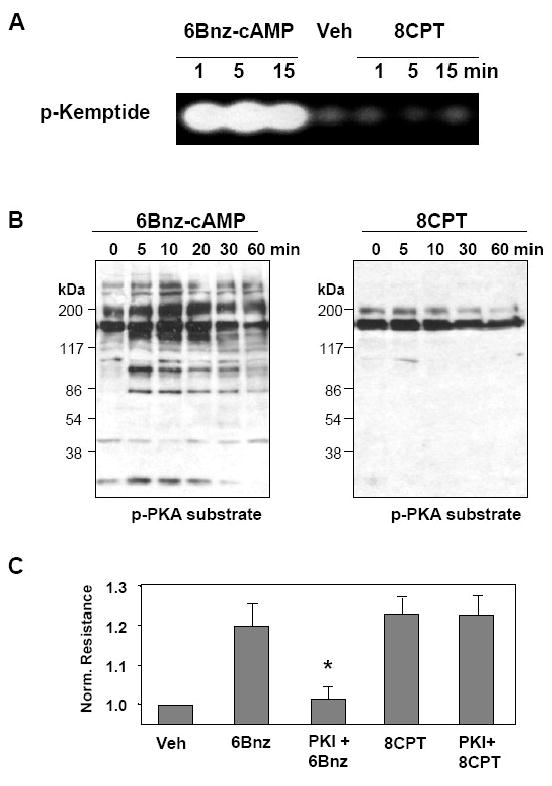
A - Cell lysates were analyzed for PKA activity by non-radioactive in vitro PKA assay. EC were stimulated with 6Bnz (100 μM) or 8CPT (100 μM) for the indicated times periods followed by determination of PKA activity by fluorescent phosphorylation of PKA substrate kemptide separated from non-phosphorylated form by 0.8% agarose gel electrophoresis. B - EC were stimulated with 6Bnz or 8CPT for indicated time periods, and phosphorylation of PKA endogenous substrates was monitored by immunoblotting with anti-phospho-PKA substrate antibodies. C - HPAEC were stimulated with 6Bnz or 8CPT with or without pretreatment with PKA peptide inhibitor PKI (10 μM, 30 min), and TER was measured after 30 min of stimulation (*p<0.05; n= 3).
The next experiments tested involvement of Epac mechanism in EC protection against thrombin-induced permeability. Pulmonary EC were pretreated with 8CPT followed by thrombin challenge. 8CPT exhibited prominent barrier protective effects against thrombin-mediated EC hyper-permeability (Figure 7A) and thrombin-increased MYPT1 and MLC phosphorylation (Figure 7B). To delineate the role of Rac in EC barrier preservation by 8CPT, cells were transfected with siRNA specific for Rac or nonspecific siRNA. Rac depletion significantly attenuated inhibitory effects of 8CPT on MLC phosphorylation, a hallmark of EC contraction and barrier dysfunction (Figure 7C).
Figure 7. Effect of Epac/Rap1 activation on thrombin-induced EC barrier dysfunction.
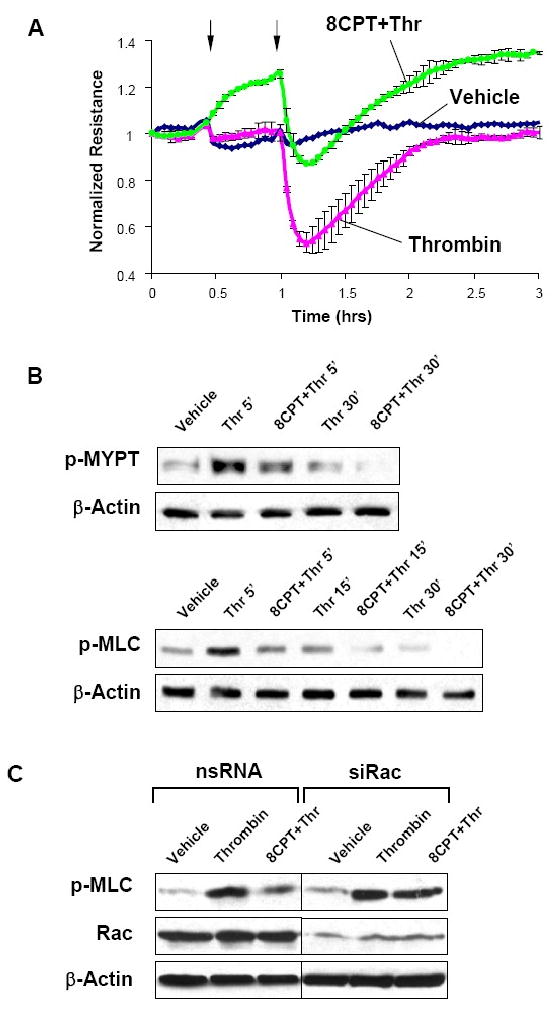
A - Human pulmonary EC were treated with 8CPT (100 μM, marked by first arrow). At the time point indicated by second arrow, cells were stimulated with thrombin (0.1 U/ml), and TER was monitored over 3 hrs. B - Phosphorylation of MYPT1 and MLC in EC pretreated with 8CPT followed by thrombin (5, 15, or 30 min) challenge was detected by western blot with specific antibodies. C - HPAEC were transfected with Rac-specific or non-specific siRNA. After 48 hrs cells were harvested, and effects of 8CPT on thrombin-induced MLC phosphorylation was analyzed in control and Rac-depleted cells.
Rac as a convergence point of PKA- and Epac- induced EC barrier protection
Our results show Rac activation by both PKA and Rap1 signaling and EC barrier protection by activation of both signaling pathways. We further tested the effects of combined PKA and Rap1 activation on the thrombin-induced EC barrier compromise. Similar to results obtained with PKA or Rap1 activation alone, combination of 6Bnz and 8CPT significantly attenuated thrombin-induced EC hyper-permeability (Figure 8A). Of note, when used at low concentrations (25 μM 6Bnz and 25 μM 8CPT), protective effect of combined pretreatment with 6Bnz and 8CPT prior to thrombin challenge was additive, as compared to effects exhibited by either 6Bnz or 8CPT alone (Figure 8A, lower panel). In contrast, combined treatment of EC with higher concentrations of PKA and Epac/Rap activators (100 μM 6Bnz and 100 μM 8CPT, respectively), which alone induced maximal TER increase, did not show further suppression of thrombin-induced permeability (Figure 8A, upper panel). These results may be explained by sub-maximal activation of Rac by lower doses of 6Bnz or 8CPT when added alone, whereas combined treatment with same individual 6Bnz and 8CPT concentrations allows additional Rac activation, which causes stronger protective response. In turn, high 6Bnz and 8CPT concentrations induce maximal Rac activation and therefore do not lead to additional EC barrier enhancement.
Figure 8. Role of Rac in PKA- and Epac-mediated EC barrier protection.
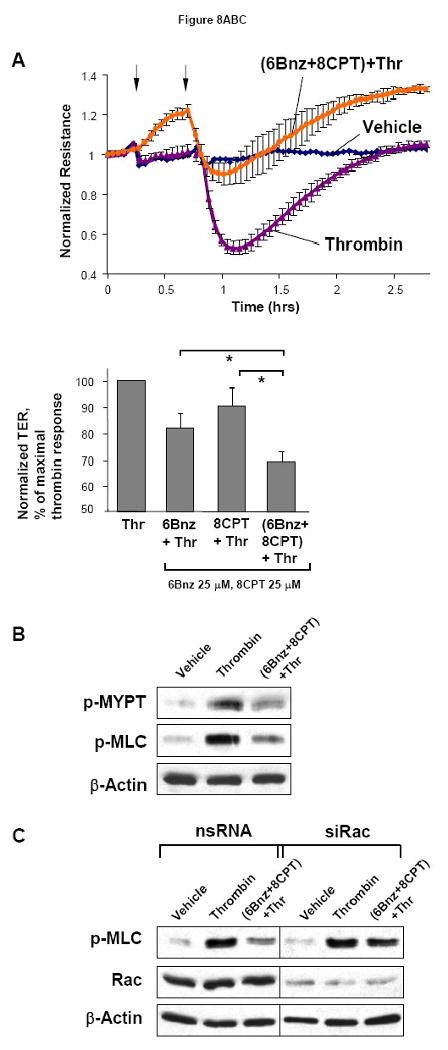
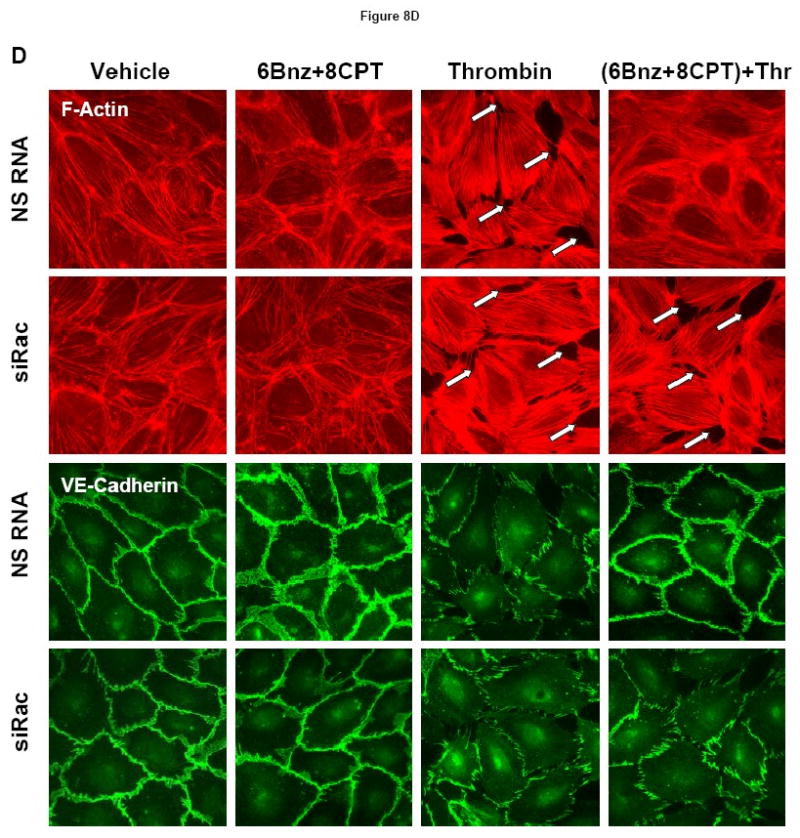
A - At the time point indicated by first arrow, cells were treated with a mixture of 6Bnz (100 μM) and 8CPT (100 μM), followed by thrombin (0.1 U/ml) challenge indicated by second arrow and measurements of TER (upper panel). Bar graphs (lower panel) represent statistical analysis of TER measurements performed in EC pretreated with either 6Bnz (25 μM), 8CPT (25 μM), or a combination of 6Bnz (25 μM) and 8CPT (25 μM) followed by thrombin challenge (0.1 U/ml, 15 min). TER decline induced by thrombin was taken as 100%; n=3-5 per condition; *p<0.05. B - Phosphorylation of MYPT1 and MLC after thrombin stimulation (0.1 U/ml, 5 min) with or without combined 6Bnz and 8CPT pretreatment was detected by western blot with specific antibodies. C - Cells were transfected with Rac-specific or non-specific siRNA for 48 hours. Effects of 6Bnz and 8CPT on thrombin-induced MLC phosphorylation were detected in control and Rac-depleted EC by immunoblotting with specific antibodies. D - Cytoskeletal remodeling was analyzed by double inmmunifluorescence staining using Texas Red phalloidin to detect F-actin and VE-cadherin antibodies to visualize adherens junctions. Paracellular gaps are marked by arrows.
Combined treatment with 6Bnz (100 μM) and 8CPT (100 μM) suppressed thrombin-induced MYPT1 and MLC phosphorylation (Figure 8B), which was attenuated by siRNA-induced molecular inhibition of Rac (Figure 8C). The role of Rac mechanism in the barrier protective effects exhibited by combined treatment with PKA and Epac activators was further evaluated by morphological analysis of endothelial monolayer integrity in experiments where HPAEC were transfected with non-specific or Rac-specific siRNA (Figure 8D). Combined stimulation with PKA and Epac activators dramatically reduced formation of stress fibers and paracellular gaps and prevented adherens junction disruption after thrombin challenge in HPAEC treated with control non-specific RNA. In contrast, these protective effects of 6Bnz and 8CPT co-treatment were abolished in HPAEC with depleted Rac.
PKA and Epac reduce lung dysfunction in vivo
Relative impact of PKA- and Epac-Rap1-dependent signaling in the lung protective mechanisms was further investigated in the murine model of ventilator-induced lung injury (VILI). Mice were treated with vehicle, 8CPT, 6Bnz, or a mixture of 8CPT and 6Bnz at the onset of high tidal volume mechanical ventilation (30 ml/kg, HTV). At 4 hours, bronchoalveolar lavage (BAL) was performed, and lung injury was evaluated by measurements of protein concentration and cell counts in BAL fluid. The results demonstrate that HTV significantly increased BAL cell counts, as compared to non-treated controls. The increase in cell count was attenuated by treatment with either 8CPT, or 6Bnz, or a combination of 8CPT and 6Bnz, as compared to HTV alone (p<0.05 for all groups) (Figure 9A). HTV significantly increased protein content in BAL fluid reflecting increased lung vascular permeability. All three treatments (8CPT, 6Bnz, and their combination) markedly decreased BAL protein concentration (Figure 9B). We noticed that protective effects of 8CPT and 6Bnz were not additive, and combined treatment with PKA and Epac/Rap1 activators did not lead to further lung protection. These data suggest that PKA and Epac/Rap1 pathways of lung vascular barrier protection may converge on a common downstream mechanism. Cell culture studies described above strongly suggest Rac GTPase as a potential “hub” of PKA and Epac/Rap1 induced lung protective signaling.
Figure 9. Effect of PKA and Epac activators on ventilator-induced lung injury.
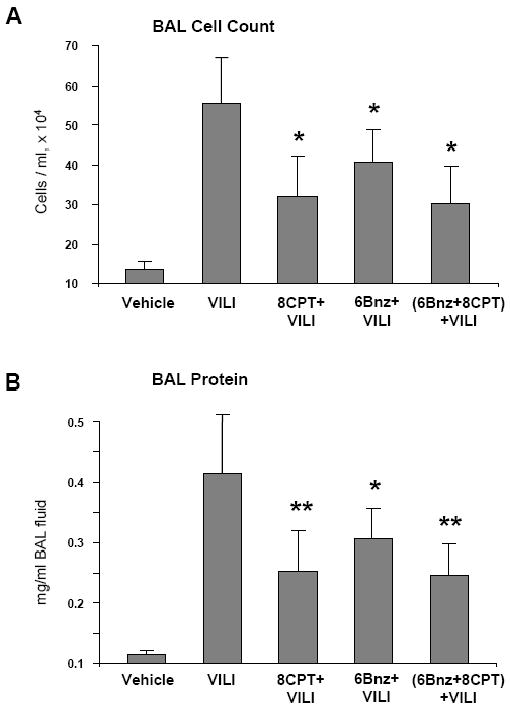
A and B - Mice were subjected to mechanical ventilation at high tidal volume (HTV, 30 ml/kg, 4 hrs) or left spontaneously ventilated. Intravenous administration of 8CPT (0.4 μM/mouse), 6Bnz (0.6 μM/mouse), or combination of 8CPT and 6Bnz was performed at the onset of mechanical ventilation. Cell count (A) and measurement of protein concentration (B) were performed in bronchoalveolar lavage fluid taken from control and experimental animals (n=4-7 per condition; *p < 0.05; **p < 0.02).
The protective effects of PKA and Epac activators against HTV-induced lung vascular dysfunction were further assessed by measurement of Evans blue leakage into the lung tissue. HTV caused significant Evans blue accumulation in the lung parenchyma, which was markedly reduced by combined treatment with 8CPT and 6Bnz (Figure 10A). These results were confirmed by quantitative analysis of Evans blue-labeled albumin accumulation in the lung tissue samples (Figure 10B), which showed significantly decreased Evans blue accumulation in the lungs from HTV-treated animals after a single intravenous injection of 8CPT and 6Bnz combination (37.66 ± 8.35 μg/g wet weight lung), as compared to HTV-treated animals injected with vehicle control (56.67 ± 11.35 μg/g wet weight lung, data expressed as mean ± SD; p<0.05). Evans blue accumulation in lung tissues from untreated controls was 14.85 ± 3.16 μg/g wet weight.
Figure 10. Effect of PKA and Epac activators on ventilator-induced lung vascular leak.

A and B - Effects of combined treatment with 8CPT (0.4 μM/mouse) and 6Bnz (0.6 μM/mouse) on the HTV-induced vascular leak were assessed by measurements of Evans blue leakage into the lung tissue (A). The quantitative analysis of Evans blue labeled albumin extravasation was performed by spectrophotometric analysis of Evans blue extracted from the lung tissue samples (B) (n=3-4 per condition; *p < 0.05).
Discussion
This study shows that direct intracellular cAMP elevation by stable cell permeable cAMP analog Br-cAMP causes sustained, dose-dependent barrier protective response in human pulmonary EC, which was associated with activation of PKA, tyrosine phosphorylation, and components of Epac signaling cascade: Rap1 GTPase and Rac-specific guanosine exchange factors, Tiam1, and Vav2.
Our results also show that direct elevation of cAMP was protective against EC barrier compromise induced by thrombin, as well as by inflammatory mediators LPS, IL-6 and TNFα. Endothelial barrier dysfunction in this case may be mediated by several pathways. One mechanism leading to increased permeability is activation of stress-induced MAP kinases p38 and JNK, weakening of adherens junctions and remodeling of actin cytoskeleton controlled by p38 and JNK MAPK cytoskeletal effectors (Becker et al., 2001; Garcia et al., 2002; Kiemer et al., 2002). The other critical mechanism is activation of Rho pathway leading to stress fiber formation, actomyosin contraction and paracellular gap formation (Birukova et al., 2007a; Essler et al., 2000; Tasaka et al., 2005). Protective effects of cAMP elevation in pulmonary EC stimulated with inflammatory mediators were at least in part associated with inhibition of Rho kinase-dependent phosphorylation of MLC phosphatase. Inhibition of Rho signaling caused by cAMP elevation was dependent on activation of Rac (this study and (Birukova et al., 2007b)). This mechanism is further supported by our data showing that Rac knockdown abolished inhibitory effects of cAMP on Rho signaling. However, involvement of more than one mechanism in EC barrier dysfunction triggered by inflammatory mediators explains partial attenuation of EC barrier compromise by PKA and Epac activators.
The major finding of this study is convergence of PKA and Epac pathways on Rac activation suggesting Rac pathway as a common barrier protective mechanism shared by PKA and Epac. It is important to note in the context of this study that Rac may be activated via PKA-dependent mechanism. PKA-induced activation of Rac may occur without direct phosphorylation (Feoktistov et al., 2000; Fleming et al., 1999), but via activation of nucleotide exchange activities of Rac-specific GEFs Tiam1 and Trio, which have consensus PKA phosphorylation sites (O'Connor and Mercurio, 2001). In contrast, activated PKA inhibits Rho signaling. Our results show increased Rho phosphorylation at Ser188 in pulmonary EC with activated PKA. This phosphorylation decreases Rho association with Rho-kinase and attenuates Rho signaling (Lang et al., 1996). Another potential Rho inhibitory mechanism is PKA-mediated phosphorylation of a Rho negative regulator, Rho-GDP dissociation inhibitor (RhoGDI) (Qiao et al., 2003). This study shows that knockdown of Rac activity attenuated inhibitory effects of PKA and Epac activators on thrombin-induced Rho signaling. These results indicate a key role of Epac and PKA dependent Rac activation in suppression of Rho-dependent cascade of EC hyperpermeability. However, exact mechanisms of Rac-induced suppression of Rho in each particular condition remain to be investigated.
Besides suppression of Rho and activation of Rac signaling, PKA may also regulate endothelial barrier via other mechanisms. PKA reduces endothelial MLC kinase activity leading to decreased MLC phosphorylation, reduced EC contraction (Birukova et al., 2004; Yuan et al., 1997), and strengthening of cell-matrix adhesions (Lampugnani et al., 1990). PKA-induced phosphorylation of actin-binding vasodilator-stimulated phosphoprotein (VASP) leads to structural relaxation of the actin cytoskeleton, its linkage with tight junctions via VASP/ZO-1 complex, and EC barrier enhancement (Comerford et al., 2002). In vivo, PKA-induced inhibition of leukocyte adhesion to the vascular endothelium may play additional role in endothelial barrier preservation during acute lung injury (Yuan, 2002).
PKA-independent mechanisms of EC barrier enhancement by elevated cAMP levels include activation of cAMP-activated nucleotide exchange factor Epac leading to activation of Epac/Rap1/Tiam1-Vav2/Rac cascade of barrier protection (Birukova et al., 2007b; Cullere et al., 2005; Fukuhara et al., 2005). Our data further support these findings and show that Epac-induced Rac activation is a part of Rac-Rho crosstalk mechanisms stimulated by cAMP elevation and critical for downregulation of thrombin-induced Rho signaling. Thus, combination of Epac and PKA activators also inhibited agonist induced EC permeability via Rac-dependent suppression of Rho signaling. Co-treatment with maximal barrier protective doses of Epac and PKA specific activators did not show further enhancement of lung protection in the animal model of VILI in comparison to administration of either activator alone. These results were surprising given the role of PKA in both Rac-dependent and independent barrier protective mechanisms. On the other hand, these results strongly suggest that Rac is a convergence point of barrier protective signaling by PKA and Epac. Our results also do not exclude other pathways triggered by PKA such as direct attenuation of MLC kinase and Rho activities discussed above. In addition, Epac-induced activation of Rap1 may directly trigger barrier protective mechanisms, such as Rap1-dependent assembly of adherens junction and tight junction complexes observed in epithelial cell lines (Takai and Nakanishi, 2003). Whether these mechanisms are active in pulmonary EC and dependent on Rac activity remains to be investigated.
Thus, although both pathways exhibit barrier protective effects in the models of acute lung vascular barrier dysfunction, their utilization alone or in combination may depend on particular type of ALI. We speculate that Epac activators may be efficient in acute aseptic lung injury due to: a) direct stimulation of Rac-dependent peripheral cytoskeletal enhancement and enlargement of adherens junction complexes in pulmonary endothelial monolayers; and b) Rap1-dependent formation of novel nectin-based cell junctions during EC monolayer recovery, which precede assembly of major endothelial adherens junction complexes containing VE-cadherin. In turn, PKA activators may be more efficient in treatment of more prolonged septic or inflammatory ALI due to sustained attenuation of EC contractile mechanisms leading to barrier compromise and additional inhibitory effects on inflammatory pathways (Hata and Breyer, 2004; Ueno et al., 1997). Further studies are needed to test efficacy of single or combined administration of PKA or Epac activators in specific types of ALI. This approach may be important to avoid undesirable side effects of cAMP-elevating agonists caused by dual activation of Epac and PKA pathways.
Acknowledgments
Supported by the National Heart, Lung, and Blood Institute grants HL087823, HL58064, HL76259 for KGB; HL89257 and the American Lung Association Biomedical Research Grant for AAB
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe K, Rossman KL, Liu B, Ritola KD, Chiang D, Campbell SL, Burridge K, Der CJ. Vav2 is an activator of Cdc42, Rac1, and RhoA. J Biol Chem. 2000;275:10141–9. doi: 10.1074/jbc.275.14.10141. [DOI] [PubMed] [Google Scholar]
- Becker PM, Verin AD, Booth MA, Liu F, Birukova A, Garcia JG. Differential regulation of diverse physiological responses to VEGF in pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1500–11. doi: 10.1152/ajplung.2001.281.6.L1500. [DOI] [PubMed] [Google Scholar]
- Birukov KG, Bochkov VN, Birukova AA, Kawkitinarong K, Rios A, Leitner A, Verin AD, Bokoch GM, Leitinger N, Garcia JG. Epoxycyclopentenone-containing oxidized phospholipids restore endothelial barrier function via Cdc42 and Rac. Circ Res. 2004;95:892–901. doi: 10.1161/01.RES.0000147310.18962.06. [DOI] [PubMed] [Google Scholar]
- Birukov KG, Jacobson JR, Flores AA, Ye SQ, Birukova AA, Verin AD, Garcia JG. Magnitude-dependent regulation of pulmonary endothelial cell barrier function by cyclic stretch. Am J Physiol Lung Cell Mol Physiol. 2003;285:L785–97. doi: 10.1152/ajplung.00336.2002. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Chatchavalvanich S, Rios A, Kawkitinarong K, Garcia JG, Birukov KG. Differential regulation of pulmonary endothelial monolayer integrity by varying degrees of cyclic stretch. Am J Pathol. 2006;168:1749–61. doi: 10.2353/ajpath.2006.050431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova AA, Fu P, Chatchavalvanich S, Burdette D, Oskolkova O, Bochkov VN, Birukov KG. Polar head groups are important for barrier protective effects of oxidized phospholipids on pulmonary endothelium. Am J Physiol Lung Cell Mol Physiol. 2007a;292:L924–35. doi: 10.1152/ajplung.00395.2006. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Liu F, Garcia JG, Verin AD. Protein kinase A attenuates endothelial cell barrier dysfunction induced by microtubule disassembly. Am J Physiol Lung Cell Mol Physiol. 2004;287:L86–93. doi: 10.1152/ajplung.00441.2003. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Zagranichnaya T, Alekseeva E, Fu P, Chen W, Jacobson JR, Birukov KG. Prostaglandins PGE2 and PGI2 promote endothelial barrier enhancement via PKA- and Epac1/Rap1-dependent Rac activation. Exp Cell Res. 2007b;313:2504–20. doi: 10.1016/j.yexcr.2007.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos JL. Epac: a new cAMP target and new avenues in cAMP research. Nat Rev Mol Cell Biol. 2003;4:733–8. doi: 10.1038/nrm1197. [DOI] [PubMed] [Google Scholar]
- Bos JL. Linking Rap to cell adhesion. Curr Opin Cell Biol. 2005;17:123–8. doi: 10.1016/j.ceb.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Christensen AE, Selheim F, de Rooij J, Dremier S, Schwede F, Dao KK, Martinez A, Maenhaut C, Bos JL, Genieser HG, Doskeland SO. cAMP analog mapping of Epac1 and cAMP kinase. Discriminating analogs demonstrate that Epac and cAMP kinase act synergistically to promote PC-12 cell neurite extension. J Biol Chem. 2003;278:35394–402. doi: 10.1074/jbc.M302179200. [DOI] [PubMed] [Google Scholar]
- Comerford KM, Lawrence DW, Synnestvedt K, Levi BP, Colgan SP. Role of vasodilator-stimulated phosphoprotein in PKA-induced changes in endothelial junctional permeability. Faseb J. 2002;16:583–5. doi: 10.1096/fj.01-0739fje. [DOI] [PubMed] [Google Scholar]
- Cullere X, Shaw SK, Andersson L, Hirahashi J, Luscinskas FW, Mayadas TN. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood. 2005;105:1950–5. doi: 10.1182/blood-2004-05-1987. [DOI] [PubMed] [Google Scholar]
- de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–7. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- Dudek SM, Jacobson JR, Chiang ET, Birukov KG, Wang P, Zhan X, Garcia JG. Pulmonary endothelial cell barrier enhancement by sphingosine 1-phosphate: roles for cortactin and myosin light chain kinase. J Biol Chem. 2004;279:24692–700. doi: 10.1074/jbc.M313969200. [DOI] [PubMed] [Google Scholar]
- Essler M, Staddon JM, Weber PC, Aepfelbacher M. Cyclic AMP blocks bacterial lipopolysaccharide-induced myosin light chain phosphorylation in endothelial cells through inhibition of Rho/Rho kinase signaling [In Process Citation] J Immunol. 2000;164:6543–9. doi: 10.4049/jimmunol.164.12.6543. [DOI] [PubMed] [Google Scholar]
- Feoktistov I, Goldstein AE, Biaggioni I. Cyclic AMP and protein kinase A stimulate Cdc42: role of A(2) adenosine receptors in human mast cells. Mol Pharmacol. 2000;58:903–10. doi: 10.1124/mol.58.5.903. [DOI] [PubMed] [Google Scholar]
- Fleming IN, Elliott CM, Buchanan FG, Downes CP, Exton JH. Ca2+/calmodulin-dependent protein kinase II regulates Tiam1 by reversible protein phosphorylation. J Biol Chem. 1999;274:12753–8. doi: 10.1074/jbc.274.18.12753. [DOI] [PubMed] [Google Scholar]
- Fu P, Birukova AA, Xing J, Sammani S, Murley JS, Garcia JG, Grdina DJ, Birukov KG. Amifostine reduces lung vascular permeability via suppression of inflammatory signalling. Eur Respir J. 2009;33:612–24. doi: 10.1183/09031936.00014808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuhara S, Sakurai A, Sano H, Yamagishi A, Somekawa S, Takakura N, Saito Y, Kangawa K, Mochizuki N. Cyclic AMP potentiates vascular endothelial cadherin-mediated cell-cell contact to enhance endothelial barrier function through an Epac-Rap1 signaling pathway. Mol Cell Biol. 2005;25:136–46. doi: 10.1128/MCB.25.1.136-146.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JG, Wang P, Schaphorst KL, Becker PM, Borbiev T, Liu F, Birukova A, Jacobs K, Bogatcheva N, Verin AD. Critical involvement of p38 MAP kinase in pertussis toxin-induced cytoskeletal reorganization and lung permeability. Faseb J. 2002;16:1064–76. doi: 10.1096/fj.01-0895com. [DOI] [PubMed] [Google Scholar]
- Haag S, Warnken M, Juergens UR, Racke K. Role of Epac1 in mediating anti-proliferative effects of prostanoid EP(2) receptors and cAMP in human lung fibroblasts. Naunyn Schmiedebergs Arch Pharmacol. 2008;378:617–30. doi: 10.1007/s00210-008-0334-3. [DOI] [PubMed] [Google Scholar]
- Hata AN, Breyer RM. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther. 2004;103:147–66. doi: 10.1016/j.pharmthera.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Holz GG, Kang G, Harbeck M, Roe MW, Chepurny OG. Cell physiology of cAMP sensor Epac. J Physiol. 2006;577:5–15. doi: 10.1113/jphysiol.2006.119644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SK, Wettlaufer SH, Chung J, Peters-Golden M. Prostaglandin E2 inhibits specific lung fibroblast functions via selective actions of PKA and Epac-1. Am J Respir Cell Mol Biol. 2008;39:482–9. doi: 10.1165/rcmb.2008-0080OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamio K, Liu X, Sugiura H, Togo S, Kobayashi T, Kawasaki S, Wang X, Mao L, Ahn Y, Hogaboam C, Toews ML, Rennard SI. Prostacyclin analogs inhibit fibroblast contraction of collagen gels through the cAMP-PKA pathway. Am J Respir Cell Mol Biol. 2007;37:113–20. doi: 10.1165/rcmb.2007-0009OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiemer AK, Weber NC, Furst R, Bildner N, Kulhanek-Heinze S, Vollmar AM. Inhibition of p38 MAPK activation via induction of MKP-1: atrial natriuretic peptide reduces TNF-alpha-induced actin polymerization and endothelial permeability. Circ Res. 2002;90:874–81. doi: 10.1161/01.res.0000017068.58856.f3. [DOI] [PubMed] [Google Scholar]
- Lampugnani MG, Giorgi M, Gaboli M, Dejana E, Marchisio PC. Endothelial cell motility, integrin receptor clustering, and microfilament organization are inhibited by agents that increase intracellular cAMP. Lab Invest. 1990;63:521–31. [PubMed] [Google Scholar]
- Lang P, Gesbert F, Delespine-Carmagnat M, Stancou R, Pouchelet M, Bertoglio J. Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. Embo J. 1996;15:510–9. [PMC free article] [PubMed] [Google Scholar]
- Lum H, Jaffe HA, Schulz IT, Masood A, RayChaudhury A, Green RD. Expression of PKA inhibitor (PKI) gene abolishes cAMP-mediated protection to endothelial barrier dysfunction. Am J Physiol. 1999;277:C580–8. doi: 10.1152/ajpcell.1999.277.3.C580. [DOI] [PubMed] [Google Scholar]
- Matthay MA, Robriquet L, Fang X. Alveolar epithelium: role in lung fluid balance and acute lung injury. Proc Am Thorac Soc. 2005;2:206–13. doi: 10.1513/pats.200501-009AC. [DOI] [PubMed] [Google Scholar]
- McAuley DF, Frank JA, Fang X, Matthay MA. Clinically relevant concentrations of beta2-adrenergic agonists stimulate maximal cyclic adenosine monophosphate-dependent airspace fluid clearance and decrease pulmonary edema in experimental acid-induced lung injury. Crit Care Med. 2004;32:1470–6. doi: 10.1097/01.ccm.0000129489.34416.0e. [DOI] [PubMed] [Google Scholar]
- Moitra J, Sammani S, Garcia JG. Re-evaluation of Evans Blue dye as a marker of albumin clearance in murine models of acute lung injury. Transl Res. 2007;150:253–65. doi: 10.1016/j.trsl.2007.03.013. [DOI] [PubMed] [Google Scholar]
- O'Connor KL, Mercurio AM. Protein kinase A regulates Rac and is required for the growth factor-stimulated migration of carcinoma cells. J Biol Chem. 2001;276:47895–900. doi: 10.1074/jbc.M107235200. [DOI] [PubMed] [Google Scholar]
- Olschewski H, Simonneau G, Galie N, Higenbottam T, Naeije R, Rubin LJ, Nikkho S, Speich R, Hoeper MM, Behr J, Winkler J, Sitbon O, Popov W, Ghofrani HA, Manes A, Kiely DG, Ewert R, Meyer A, Corris PA, Delcroix M, Gomez-Sanchez M, Siedentop H, Seeger W. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347:322–9. doi: 10.1056/NEJMoa020204. [DOI] [PubMed] [Google Scholar]
- Patterson CE, Lum H, Schaphorst KL, Verin AD, Garcia JG. Regulation of endothelial barrier function by the cAMP-dependent protein kinase. Endothelium. 2000;7:287–308. doi: 10.3109/10623320009072215. [DOI] [PubMed] [Google Scholar]
- Perlman MB, Lo SK, Malik AB. Effect of prostacyclin on pulmonary vascular response to thrombin in awake sheep. J Appl Physiol. 1986;60:546–53. doi: 10.1152/jappl.1986.60.2.546. [DOI] [PubMed] [Google Scholar]
- Qiao J, Huang F, Lum H. PKA inhibits RhoA activation: a protection mechanism against endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2003;284:L972–80. doi: 10.1152/ajplung.00429.2002. [DOI] [PubMed] [Google Scholar]
- Takai Y, Nakanishi H. Nectin and afadin: novel organizers of intercellular junctions. J Cell Sci. 2003;116:17–27. doi: 10.1242/jcs.00167. [DOI] [PubMed] [Google Scholar]
- Tasaka S, Koh H, Yamada W, Shimizu M, Ogawa Y, Hasegawa N, Yamaguchi K, Ishii Y, Richer SE, Doerschuk CM, Ishizaka A. Attenuation of endotoxin-induced acute lung injury by the Rho-associated kinase inhibitor, Y-27632. Am J Respir Cell Mol Biol. 2005;32:504–10. doi: 10.1165/rcmb.2004-0009OC. [DOI] [PubMed] [Google Scholar]
- Tzima E, Del Pozo MA, Kiosses WB, Mohamed SA, Li S, Chien S, Schwartz MA. Activation of Rac1 by shear stress in endothelial cells mediates both cytoskeletal reorganization and effects on gene expression. Embo J. 2002;21:6791–6800. doi: 10.1093/emboj/cdf688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno Y, Koike H, Annoh S, Nishio S. Anti-inflammatory effects of beraprost sodium, a stable analogue of PGI2, and its mechanisms. Prostaglandins. 1997;53:279–89. doi: 10.1016/s0090-6980(97)89601-3. [DOI] [PubMed] [Google Scholar]
- Vouret-Craviari V, Bourcier C, Boulter E, van Obberghen-Schilling E. Distinct signals via Rho GTPases and Src drive shape changes by thrombin and sphingosine-1-phosphate in endothelial cells. J Cell Sci. 2002;115:2475–84. doi: 10.1242/jcs.115.12.2475. [DOI] [PubMed] [Google Scholar]
- Yuan SY. Protein kinase signaling in the modulation of microvascular permeability. Vascul Pharmacol. 2002;39:213–23. doi: 10.1016/s1537-1891(03)00010-7. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Huang Q, Wu HM. Myosin light chain phosphorylation: modulation of basal and agonist-stimulated venular permeability. Am J Physiol. 1997;272:H1437–43. doi: 10.1152/ajpheart.1997.272.3.H1437. [DOI] [PubMed] [Google Scholar]