

Abstract
The mixed A1/A2a-adenosine agonist AMP579 given at reperfusion is protective in animal models of myocardial infarction. Receptor-blocking studies have indicated that the protection came from an adenosine receptor (AR), but neither A1- nor A2a-selective agonists could duplicate its protection. We recently found that A2b-selective agonists given at reperfusion are protective, and, therefore, tested whether AMP579 might also be an A2b agonist. We used human embryonic kidney cells overexpressing human A2b receptors as an assay system. In these cells, A2b receptor occupancy causes phosphorylation of ERK. AMP579 induced ERK phosphorylation with an EC50 of 250 nM and this phosphorylation could be blocked by MRS1754 or PSB1115, two highly selective blockers of human A2b receptors. We attempted to confirm our A2b hypothesis in a rabbit heart model of ischemia–reperfusion. AMP579 (500 nM) for 1 h starting at reperfusion reduced infarct size in isolated rabbit hearts exposed to 30 min of regional ischemia and 2 h of reperfusion (12.9 ± 2.2% infarction of risk zone vs. 32.0 ± 1.9% in untreated hearts). PSB1115 (500 nM) given for the first 15 min of reperfusion blocked AMP579’s protection (32.2 ± 3.1% infarction) which is consistent with an A2b mechanism. We conclude that AMP579 is a non-selective, but potent A2b-AR agonist, and that its protection against infarction is through that receptor.
Keywords: Adenosine, A2b receptor, AMP579, Cardioprotection, Myocardial infarction
Introduction
Adenosine is a purine nucleoside that is ubiquitous in the body. Its concentration may increase to 100-fold during periods of oxygen depletion and ischemia when there is net dephosphorylation of ATP. There are four known adenosine receptor (AR) subtypes: A1AR and A3AR are Gi-coupled while the high-affinity A2aAR and low-affinity A2bAR are Gs-coupled. ARs are involved in the signaling by which ischemic preconditioning (IPC) makes the ischemic heart resistant to infarction. IPC involves a trigger phase prior to ischemia during which transient activation of the trigger pathway puts the heart into a protected state that persists even after the trigger stimulus has been withdrawn. We have shown that the A1AR [18] and A3AR [17] participate in triggering of the protected state of IPC, but A1-selective agonists offer no protection from infarction when given just prior to reperfusion [1]. Although these ARs are physiological triggers of IPC, opioid, and bradykinin receptors also participate in parallel. Because of this redundancy, loss of the ability of one or two receptors to bind agonists does not block the ability of ischemia to precondition the heart. Rather, their absence only raises the threshold of ischemia required to trigger protection [6]. Accordingly knocking out either A1, A2a, or A3 AR in mice failed to block the ability of IPC to protect their hearts [5]. The A2bAR knockout mice, however, seemed to be incapable of being protected by IPC indicating that the A2b receptor plays a critical and unique role in this protection.
The mediator phase of IPC’s protection is initiated after the ischemic heart is reperfused when a series of signal transduction events acts to inhibit permeability transition pore formation in the mitochondria [8]. Transition pores form in the first few minutes of reperfusion and kill cells by destroying mitochondria. IPC’s signaling pathways make the heart resistant to transition pore formation, but the signal transduction pathways must be activated in the critical first seconds of reperfusion. We have recently shown that the A2bAR plays a key role in this protective pathway [23]. The A2bAR normally has a low affinity for adenosine. IPC through PKC activation sensitizes the A2bAR such that endogenous adenosine released by the ischemic heart can activate the protective pathway early in the reperfusion period [14, 15].
The adenosine A1/A2a-receptor agonist AMP579 has also been reported to protect the ischemic heart in animal models of myocardial infarction when it was administered at reperfusion [2, 19, 24, 30]. Protection from AMP579 requires activation of an AR [30]. However, neither adenosine nor A1- or A2a-selective agonists infused at reperfusion can duplicate preconditioning’s protection [1, 2, 7, 12, 26–28]. This was indeed a puzzling observation. Because little was known about the A2bAR at the time AMP579 was being investigated, the drug was not tested for A2bAR binding. Furthermore, because the A2bAR has such a low affinity for adenosine (EC50~5 μM) [20, 22], it was largely dismissed as being unimportant because adenosine concentration in the heart seldom approaches that value even in deep ischemia.
Because protection in IPC hearts seems to depend on a change in the A2bAR’s sensitivity to adenosine, administration of an A2b agonist at a dose high enough to occupy the low-affinity receptors should protect even a non-preconditioned heart. We indeed confirmed that prediction using the highly selective A2bAR agonist BAY 60-6583 [14]. 5′-(N-ethylcarboxamido) adenosine (NECA) is a potent, although not selective, A2bAR agonist with an EC50 for raising cAMP in cells expressing A2bAR of ~3.1 μM [4]. NECA at reperfusion is as protective as AMP579, and the protection is dependent on the involvement of A2b receptors [23]. Moreover, the structure of AMP579 is very similar to that of NECA. Therefore, we hypothesized that AMP579 is also an A2b agonist and that it protects, like NECA, by binding to the A2bAR to mimic IPC’s signaling. To test that hypothesis, we used both HEK293 cells that had been transfected with human A2bAR to overexpress them as an assay system for A2b activity and an isolated rabbit heart model of myocardial infarction.
Materials and methods
Cell culture and biochemical studies
We used human embryonic kidney (HEK) 293 cells that had been stably transfected with human adenosine A2b receptors by Anna Robeva at the University of Virginia who generously shared them with us [16]. In brief, a plasmid for recombinant A2b receptors was introduced into HEK 293 cells by lipofectin. The construct included a gentamycin-resistant gene so that successfully transfected cells could be selected by growing them in a medium containing G418, which resulted in a stable cell line that overexpresses A2bAR. The stably transfected cells were grown in laminin-coated dishes at 37°C in 5% CO2/95% air and in Dulbecco’s modified Eagle’s medium [0.3 mg/ml G418, 50 U/ml penicillin, 50 μg/ml streptomycin, 2 mM L-glutamine, and 10% fetal calf serum (FCS)]. All cells were split three times a week at a ratio of 1:5. These cells overexpress A2bAR making them a sensitive and selective assay system for A2bAR-active ligands.
Prior to their use, HEK-A2b cells were maintained overnight in medium deprived of serum. They were washed two times with calcium-free Tyrode’s solution, and then stimulated with AMP579, BAY 60-6583, 2-chloro-N6-cyclopentyladenosine (CCPA), CGS 21680, and 2-Cl-N6-(iodobenzyl) adenosine-5′-N-methyluronamide (2-CI-IB-MECA) at 37°C in calcium-free Tyrode’s solution for 10 min. When used, the selective A2b antagonists MRS1754 or PSB1115 were added for 20 min before stimulation by agonists. After two washes in ice-cold phosphate-buffered saline, cells were lysed in lysis buffer (70 mM β-glycerophosphate, 0.5% Triton X-100, 2 mM MgCl2, 1 mM dithiothreitol, 1 mM NaF, 1 mM Na3VO4, 20 μg/ml aprotinin, 5 μg/ml leupeptin) and cellular debris were removed by centrifugation. Samples were denatured with Laemmli buffer and protein content analyzed using the Bradford method. Fifty microgram of protein was loaded on each lane of a 10% polyacrylamide gel. Proteins were separated with electrophoresis and then transferred to polyvinylidene difluoride membranes. To analyze ERK1/2 phosphorylation membranes were blocked in 5% dry milk powder in PBST and incubated with mouse monoclonal phospho-specific anti-phospho Thr202/Tyr204 ERK1/2 antibodies (Cell Signaling 9106) overnight on a shaker at 4°C. After incubation with anti-mouse horseradish peroxidase-coupled secondary antibody, bands were visualized with Immobilon™ western chemiluminescent HRP substrate according to the manufacturer’s instructions. Equal loading of the lanes was confirmed by Ponceau stain of the membranes and by stripping the membranes and reprobing them with an anti-ERK1/2 antibody (Cell Signaling 9107).
We measured the expression of A2bAR in the plasma membranes of transfected and non-transfected HEK cells. Cultured cells were lysed in Tris–HCl (5 mM, pH 7.4) by freezing and thawing three times and then centrifuged for 1 h at 100,000 g. The pellet was re-suspended in Tris–HCl and used for western blot analysis as described above by incubating with a primary anti-A2bAR antibody (SC-28996f, Santa Cruz, CA), and 50 μg of protein was loaded on each lane.
Immunofluorescence
Stably transfected HEK cells seeded on coverslips were washed once in PBS and then fixed in 4% paraformaldehyde and permeabilized by incubation in 0.1% Triton X-100/PBS. After washing, cells were blocked for 1 h at room temperature in 5% FCS in PBS. They were then incubated with either the above-mentioned primary anti-A2bAR antibody (1:100) or affinity-purified rabbit IgG in a corresponding concentration (negative control) overnight at 4°C. After washing with PBS, cells were incubated with a secondary antibody conjugated to Alexa Fluor 488 for an additional hour at room temperature. After extensive washing, coverslips were mounted on microscope slides (DAKO mounting medium, Carpinteria, CA, USA) and observed with a confocal laser scanning microscope (Chromaphor Analysen Technik, Duisburg, Germany). A CCD camera and VoxCell software from VisiTech International (Sunderland, UK) were used for analysis.
Isolated heart model
All animal care satisfied published guidelines [21], and procedures were approved by institutional committees. New Zealand White rabbits of either sex weighing 2–3 kg were anesthetized with sodium pentobarbital (30 mg/kg) and ventilated with 100% oxygen. Hearts were exposed through a left thoracotomy, and a suture was passed around a branch of the left coronary artery. The heart was removed and perfused on a Langendorff apparatus with modified Krebs–Henseleit bicarbonate buffer that contained (mM) 118.5 NaCl, 24.7 NaHCO3, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 2.5 CaCl2, and 10.0 glucose. The buffer was gassed with 95% O2 and 5% CO2. A fluid-filled latex balloon was inserted into the left ventricle to measure pressure. All hearts were allowed to equilibrate for 20 min before the protocol was started.
Protocol for infarct studies
Six groups of hearts were studied (Fig. 1). All hearts were subjected to 30 min of regional ischemia and 120 min of reperfusion. Control hearts received no treatment. The second group of hearts was treated with AMP579 (500 nM) starting 5 min before reperfusion and continuing for 60 min. The third group of hearts was treated with NECA (100 nM) for an equal amount of time. In the fourth group, PSB1115 (500 nM), a highly selective A2b antagonist, was infused for 20 min beginning 5 min before the release of the coronary occlusion. In the fifth and sixth groups, PSB1115 infusion was combined with either AMP579 or NECA (100 nM) administered for 60 min from 5 min before the onset of reperfusion.
Fig. 1.

Experimental protocols
Measurement of infarct size
At the end of the experiment, the coronary artery was re-occluded, and 2–9 μm fluorescent microspheres (Micro-genics, Fremont, CA, USA) were infused to delineate the ischemic zone (region at risk) as the area of tissue without fluorescence. The heart was cut into 2-mm thick slices which were incubated in 1% triphenyltetrazolium chloride in sodium phosphate buffer (pH 7.4) at 37°C for 10 min. The slices were immersed in 10% formalin to preserve the tissue. The risk zone was identified by illuminating slices with ultraviolet light. The areas of infarct and risk zone were determined by planimetry of each slice, and volumes were calculated by multiplying each area by slice thickness and summing them for each heart. Infarct size is expressed as a percentage of the risk zone.
Materials
Cell culture media and FCS were obtained from Sigma-Aldrich. AMP579 was a gift from Aventis Pharm. MRS1754, PSB1115, CCPA, CGS 21680, and 2-Cl-IB-MECA were purchased from Tocris Bioscience. Mouse monoclonal anti-phospho Thr202/Tyr204 ERK1/2, anti-ERK1/2, and anti-mouse horseradish peroxidase-coupled antibodies were obtained from Cell Signaling Technology. Immobilon™ western chemiluminescent HRP substrate was purchased from Millipore. The anti-A2bAR antibody was purchased form Santa Cruz.
Statistics
All data are expressed as mean ± SEM. One-way analysis of variance (ANOVA) with Student–Newman–Keuls post hoc test was performed on baseline hemodynamic variables, risk zone, infarct size, and western blot band densities. P < 0.05 was considered significant.
Results
HEK-A2b cell studies
Figure 2a shows the stably transfected HEK cells subjected to immunofluorescence with an anti-A2bAR primary antibody. Transfected cells show intense membrane staining. No fluorescence staining could be observed in cells overexpressing A2b receptors but not exposed to A2b antibody, and, therefore, without bound A2b-receptor–antibody–fluorochrome complex indicating that the fluorescence did reflect antibody binding (Fig. 2b). We then subjected a membrane fraction of transfected and non-transfected cells to western blotting with the anti-A2bAR antibody. A single band was observed at the expected molecular weight of 36 kDa in the transfected cells (Fig. 2c). No band was detected in the wild-type cells.
Fig. 2.

Transfected HEK cells overexpressing A2b receptors treated with A2b antibodies coupled to a fluorochrome show intense fluorescence of the plasma membrane (a), while no fluorescence is seen in negative control cells (b) where IgG was substituted for the primary anti-A2b antibody. c Representative western blot of A2b-transfected HEK and unaltered HEK cells showing detection of adenosine A2bAR only in the transfected cells
Ischemic preconditioning increases phosphorylation and activation of both ERK isoforms in the first minutes of reperfusion [9]. Since inhibiting ERK at reperfusion blocks the protection of IPC, it has been assumed that this kinase is involved in the protective signaling [9]. In our preliminary investigations, we had observed that A2b agonists induced phosphorylation of ERK 1/2 in HEK cells that had been stably transfected with human A2b receptors. We, therefore, used ERK phosphorylation as an A2b assay system to test whether AMP579 is an A2b-receptor agonist. Because the cells express human receptors, the selectivity and potency of the ligands we used are well known.
To test for other surface ARs the cells were incubated with highly selective agonists for all four AR subtypes: CCPA (1 nM) for A1 receptors, CGS 21680 (300 nM) for A2a receptors, BAY 60-6583 (100 nM) for A2b receptors [14], and 2-CI-IB-MECA (20 nM) for A3 receptors. The doses of agonists were approximately 10-fold higher than their published Kds. Figure 3a shows there was an increase in phosphorylation of both isoforms of ERK after exposure of cells to all four agonists, although the increase in phosphorylation was much higher after BAY 60-6583 which would be expected since the cells are overexpressing A2bAR. Therefore, although A2b receptors are dominant in the HEK-A2b cells, the other three AR subtypes appear to be expressed at a small, but detectable level. Therefore, we also had to show that A2b-selective antagonists would block a response to confirm A2b binding.
Fig. 3.

a Agonists of A1 (CCPA), A2a (CGS), A2b (Bay 60), and A3 (MECA) adenosine receptors induced ERK1/2 phosphorylation, but the A2b agonist BAY 60-6583 was the most potent. b Phosphorylation of ERK1/2 by either CCPA or CGS 21680 was not affected by the A2b-selective antagonist MRS 1754. c PSB 1115 and MRS 1754 could both dramatically attenuate phosphorylation of ERK1/2 induced by Bay 60. d AMP579-induced phosphorylation of ERK 1/2 could also be attenuated by both A2b-selective antagonists MRS 1754 and PSB 1115. In b–d total ERK 1/2 was unchanged by any intervention. Con control
We tested the specificity of the antagonist MRS1754 by administering it prior to adding the agonist. The increases in ERK phosphorylation induced by CCPA and CGS 21680 (Fig. 3b) were not affected by MRS1754 (20 nM); neither were those triggered by 2-CI-IB-MECA (data not shown). But the increased phosphorylation from the highly selective A2bAR agonist BAY60-6583 was strongly attenuated (Fig. 3c). PSB1115 (500 nM), another selective A2bAR antagonist, also blocked BAY 60-6583-induced phosphorylation (Fig. 3c).
AMP579 could also induce ERK1/2 phosphorylation and both A2b-selective antagonists, MRS1754, and PSB 1115, dramatically attenuated the phosphorylation (Fig. 3d). This is strong evidence that AMP579 is an agonist of human A2b ARs. Figure 4 shows that the application of increasing concentrations of AMP579 induces a dose-dependent increase in ERK1/2 phosphorylation with an EC50 of about 250 nM.
Fig. 4.

a Dose-dependent increase in ERK1/2 phosphorylation with increasing concentrations of AMP579 in an individual experiment. Total ERK 1/2 is unchanged as AMP concentrations are increased. b Summary of ERK 1/2 phosphorylation data following increasing doses of AMP579. The plot of blot density against concentration indicates the EC50 for ERK1/2 phosphorylation by AMP579 is about 250 nM (n = 4)
Hemodynamics in isolated hearts
Having established that AMP579 is an A2bAR-potent agonist, we next tested whether PSB1115 could block protection from AMP579 in an isolated rabbit heart undergoing ischemia/reperfusion. No group differences in heart rate, developed pressure, or coronary flow were observed at baseline (Table 1). Coronary branch occlusion caused an expected decrease in left ventricular developed pressure and coronary flow in all groups. Both AMP579 and NECA significantly increased coronary flow during the last few minutes of the coronary occlusion following their addition to the perfusate, and this increase was attenuated by PSB1115. There was a partial recovery of both left ventricular developed pressure and coronary flow following reperfusion. The increased coronary flow caused by NECA was also seen during reperfusion. PSB1115 had no significant independent effect on coronary flow.
Table 1.
Hemodynamic data
| Baseline |
25′ Occlusion |
30′ Occlusion |
15′ Reperfusion |
30′ Reperfusion |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HR (bpm) | LVDP (mmHg) | CF (ml/min/g) | HR (bpm) | LVDP (mmHg) | CF (ml/min/g) | HR (bpm) | LVDP (mmHg) | CF (ml/min/g) | HR (bpm) | LVDP (mmHg) | CF (ml/min/g) | HR (bpm) | LVDP (mmHg) | CF (ml/min/g) | |
| Control | 204 ± 12 | 114 ± 6 | 9.3 ± 0.5 | 192 ± 13 | 38 ± 5* | 5.2 ± 0.3* | 188 ± 14 | 73 ± 3* | 5.8 ± 0.5* | ||||||
| AMP579 | 215 ± 10 | 110 ± 3 | 8.4 ± 0.2 | 202 ± 14 | 37 ± 9* | 4.9 ± 0.3* | 230 ± 0 | 52 ± 7* | 7.2 ± 0.0* | 227 ± 3 | 83 ± 4* | 6.5 ± 0.3* | |||
| NECA | 213 ± 13 | 114 ± 6 | 9.4 ± 0.2 | 208 ± 18 | 36 ± 3* | 4.2 ± 0.3* | 204 ± 4 | 41 ± 2* | 7.8 ± 0.8* | 211 ± 7 | 86 ± 6* | 9.4 ± 0.2 | |||
| PSB1115 | 218 ± 12 | 114 ± 7 | 9.1 ± 0.2 | 225 ± 19 | 53 ± 1* | 4.5 ± 0.2* | 213 ± 16 | 51 ± 2* | 4.3 ± 0.2* | 203 ± 13 | 70 ± 12* | 6.1 ± 0.2* | 208 ± 16 | 66 ± 14* | 6.0 ± 0.1* |
| AMP579 + PSB1115 | 215 ± 11 | 113 ± 3 | 9.2 ± 0.4 | 207 ± 11 | 50 ± 6* | 5.6 ± 0.6* | 218 ± 17 | 47 ± 8* | 7.0 ± 0.7* | 246 ± 8 | 7.3 ± 7* | 7.1 ± 0.7* | 244 ± 8 | 78 ± 3* | 7.6 ± 0.6* |
| NECA + PSB1115 | 199 ± 6 | 111 ± 4 | 7.9 ± 0.4 | 191 ± 6 | 45 ± 5* | 4.9 ± 0.1* | 172 ± 6 | 52 ± 6* | 5.5 ± 0.4* | 184 ± 16 | 73 ± 6* | 6.7 ± 0.5* | 180 ± 20 | 75 ± 5* | 6.5 ± 0.6* |
Values are mean ± SEM
bpm beats per minute, CF coronary flow, HR heart rate, LVDP left ventricular developed pressure, NECA 5′-(N-ethylcarboxamido) adenosine
Statistical significance:
P < 0.05 between experimental points and baseline
Infarct size in isolated hearts
There was no significant difference in body weight, heart weight, or risk zone volume among the groups (Table 2). Control hearts undergoing 30 min of regional ischemia and 2 h of reperfusion had 32.0 ± 1.9% infarction of the risk zone (Fig. 5). AMP579 started 5 min before reperfusion and continued for 60 min decreased infarction to 12.9 ± 2.2% (P < 0.05 vs. control). Because we have previously shown that AMP579 limits infarct size in this model [28], we studied only four additional hearts in this group (white symbols in Fig. 5) to verify that our drug was still active. The previous data points are included in Fig. 5 for comparison (gray symbols). The non-selective, but A2b-potent, AR agonist NECA has been shown to limit infarct size when administered just before reperfusion through an A2bAR-dependent mechanism [31]. We confirmed NECA’s infarct-sparing effect in four additional hearts (white symbols in Fig. 5). Again, previous data points (gray symbols) are included for comparison. We tested whether protection from NECA, structurally very similar to AMP579, could also be blocked by a highly selective A2b antagonist PSB1115. PBS1115 blocked the protective effect of both AMP579 and NECA (32.2 ± 3.1 and 38.7 ± 2.4% infarction, respectively). PSB1115 administered alone at reperfusion had no significant effect on infarction (32.6 ± 1.8%).
Table 2.
Infarct size data
| n | Body weight (kg) | Heart weight (g) | Risk zone volume (cm3) | Infarct volume (cm3) | I/R (%) | |
|---|---|---|---|---|---|---|
| Control | 6 | 2.3 ± 0.1 | 7.2 ± 0.2 | 1.24 ± 0.06 | 0.47 ± 0.03 | 38.0 ± 2.1 |
| AMP579 | 4 | 2.5 ± 0.0 | 8.5 ± 0.2 | 1.22 ± 0.13 | 0.16 ± 0.04* | 12.9 ± 2.2* |
| PSB1115 | 4 | 2.4 ± 0.0 | 7.8 ± 0.4 | 1.48 ± 0.10 | 0.49 ± 0.05 | 32.6 ± 1.8 |
| NECA | 4 | 2.4 ± 0.2 | 7.5 ± 0.4 | 1.45 ± 0.10 | 0.21 ± 0.05* | 14.3 ± 0.3* |
| AMP579 + PSB1115 | 6 | 2.5 ± 0.1 | 8.1 ± 0.2 | 1.47 ± 0.18 | 0.49 ± 0.09 | 32.2 ± 3.1 |
| NECA + PSB1115 | 6 | 2.5 ± 0.0 | 8.5 ± 0.2 | 1.43 ± 0.14 | 0.55 ± 0.07 | 38.7 ± 2.4 |
Values are mean ± SEM
see Table 1; I/R infarction as a percentage of risk zone, n number of animals
Statistical significance of difference between experimental and control groups:
P < 0.05
Fig. 5.

Myocardial infarct size expressed as a percentage of risk zone in isolated rabbit hearts treated with AMP579 alone or in addition to the selective A2b receptor blocker PSB1115. Open and gray circles represent individual experiments while black circles depict group mean ± SEM. Gray circles depict previously obtained data with AMP579 [28] or NECA [31] and are presented for comparison. Infusion of AMP579’s at reperfusion was protective. PSB115 blocked both AMP579s and 5′-(N-ethylcarboxamido) adenosine’s (NECA) protection. PSB1115 alone had no effect on infarct size. Thus, A2b receptors are involved in the AMP579 signaling that leads to protection. *P < 0.05 versus control
Discussion
The present study demonstrated that the limitation of infarct size by AMP579 administered just prior to reperfusion of ischemic rabbit hearts is dependent on adenosine A2b receptors. Furthermore, using phosphorylation of ERK in HEK 293 cells that were stably transfected with human A2b receptors as our end-point, we could demonstrate that AMP579 is a potent agonist of human adenosine A2b receptors with an EC50 of about 250 nM. The conclusion that AMP579 protects ischemic hearts from infarction by activation of adenosine A2bAR is consistent with our previous findings that A2b receptors control the protective signal transduction pathway in ischemically preconditioned hearts in the early reperfusion period [25]. As a result, A2b-selective agents mimic IPC’s anti-infarct effect when infused at reperfusion [14].
We measured ERK phosphorylation in HEK-A2b cells because activation of ERK through phosphorylation in the reperfusion period is believed to be central to the mechanism of preconditioning’s protection [9]. In addition, A2b agonists given at reperfusion not only protect against infarction, but also increase ERK phosphorylation in the heart [14]. ERK and Akt are often referred to as “survival kinases” since their activation opposes infarction [9]. The studies in HEK cells indicate that this pathway also exists in cell types other than heart muscle. It is likely that AMP579 protects by triggering activation of these survival kinases, but we did not measure phosphorylation of these kinases in the rabbit hearts to confirm this hypothesis since this has already been demonstrated for other A2b agonists, i.e., NECA [31] and BAY 60-6583 [14]. In support of our hypothesis, Kis et al. [12] showed ERK inhibitors blocked AMP579’s anti-infarct effect.
AMP579 limits infarct size in pigs [24], rabbits [30], and dogs [2] when administered just before reperfusion. MRS1754 infused at reperfusion blocks protection from IPC [25], thus supporting the involvement of the A2b receptor. The A2bAR has been proposed to mediate preconditioning’s protection at the time of reperfusion [3]. Kuno et al. [14] reported that IPC sensitizes the heart to A2b-selective agonists through a PKC-dependent pathway. They concluded that preconditioning allows endogenous adenosine to populate the normally low-affinity A2bAR at reperfusion and initiate signaling leading to the activation of the survival kinases. Those activated kinases likely protect by preventing opening of the mitochondrial permeability transition pore in the first minutes of reperfusion [10]. This high conductance pore spans the inner and outer mitochondrial membrane and uncouples mitochondria, which stops ATP production at a time when the heart needs it the most. If enough mitochondria are affected, the cell will die in the first minutes of reperfusion. Eckle et al. [5] recently evaluated the ability of IPC to protect mice with selective deletion of either A1, A2a, A2b, or A3 receptors. Protection from IPC persisted in the A1, A2a, and A3 knockout strains of mice. Only in A2b-knockout mice was IPC’s protection aborted.
In a previous study, we found that AMP579 needed to be administered immediately on reperfusion; delaying administration of AMP579 for 10 min after the onset of reperfusion aborted its protection [29]. Similarly, the drug had to be present for an extended period of time. A 60-min infusion of AMP579 was protective, but stopping the infusion after only 30 min of reperfusion resulted in loss of protection. Finally, transiently blocking either survival kinases or the A2bAR for only 15 min at any time during the first hour of reperfusion aborted protection from IPC [25]. We suggested forces are trying to open the transition pores from the onset of reperfusion and that a 60-min convalescence period is required before those forces subside and protection from either a drug or the survival kinases can be withdrawn. Accordingly, we had to interrupt the protection from A2bAR’s signaling for only 15 min to cause irreversible injury, presumably because of pore opening.
It is interesting that AMP579 was tested in a small-scale clinical trial in patients with acute myocardial infarction [13]. In that study, an infusion of AMP579 was started several minutes after an open artery was confirmed. Because a loading dose was not used a protective blood level would not have been achieved until about 30 min after reperfusion. Clearly, they failed to establish the drug’s schedule requirements before implementing the trial [11].
Although NECA is a potent A2b agonist, it is not selective and can activate other AR subtypes including A2a and A1. In the present study, NECA’s ability to increase phosphorylation and reduce infarct size at reperfusion was completely abolished by co-infusion of the highly selective A2bAR antagonist PSB1115 which indicates that NECA protected through an adenosine A2b receptor. AMP579 is closely related to NECA with a similar structure and pharmacology. Figure 6 shows the structure of the two molecules. They differ only by the side groups on the adenine moiety. It is noteworthy that AMP579 with an EC50 of about 250 nM for the A2b receptor is about 100 times more potent than NECA [4]. The highly A2b-selective BAY 60–6583 is even more potent with an EC50 of about 10 nM [14].
Fig. 6.
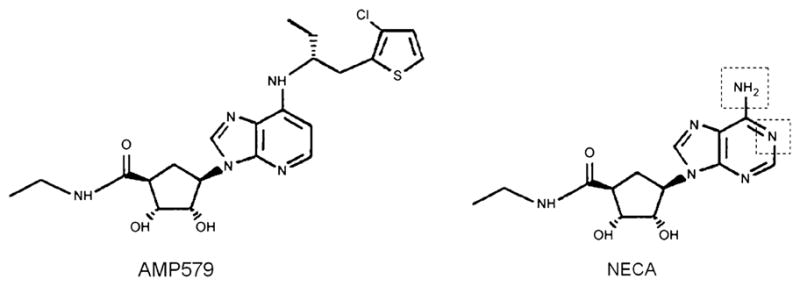
Structure of AMP579 and 5′-(N-ethylcarboxamido) adenosine (NECA). The dotted lines on the NECA structure show where the molecules differ
The cardioprotective effect of AMP579 could also be blocked by co-infusion of PSB1115, again supporting our hypothesis. We also have reported that BAY 60-6583, a highly selective A2b-receptor agonist, given at reperfusion reduced infarct size by an amount similar to that seen with NECA, and the A2b-selective antagonist MRS1754 blocked its protection as well [14].
The advantage of A2b agonists is that few tissues express these receptors so that intravenous administration of BAY 60-6583 is hemodynamically silent in rabbits (unpublished observation). AMP579 does have some bradycardic and hypotensive effects related to its A1 and A2a activity, but those side effects are minimal when AMP579 is administered at a dose that is protective. Intravenous AMP579 has already been shown to be safe in humans when it was tested in a small-scale study in patients undergoing reperfusion therapy for acute myocardial infarction [13]. Unfortunately that study did not show that AMP579 was protective, most likely because the study had a serious flaw in its design. As noted above, blood levels of drug did not reach a protective concentration until nearly 30 min after reperfusion. To inhibit lethal transition pore formation postconditioning agents must be present in the first minutes of reperfusion, and that has been specifically shown for AMP579 [29]. In conclusion, our data indicate that the anti-infarct effect of AMP579 derives from its action as a potent A2b AR agonist.
Acknowledgments
This work was supported by grant HL-20468 from the Heart, Lung, and Blood Institute of the National Institutes of Health.
References
- 1.Baxter GF, Hale SL, Miki T, Kloner RA, Cohen MV, Downey JM, Yellon DM. Adenosine A1 agonist at reperfusion trial (AART): results of a three-center, blinded, randomized, controlled experimental infarct study. Cardiovasc Drugs Ther. 2000;14:607–614. doi: 10.1023/a:1007850527878. [DOI] [PubMed] [Google Scholar]
- 2.Budde JM, Velez DA, Zhao Z-Q, Clark KL, Morris CD, Muraki S, Guyton RA, Vinten-Johansen J. Comparative study of AMP579 and adenosine in inhibition of neutrophil-mediated vascular and myocardial injury during 24 h of reperfusion. Cardiovasc Res. 2000;47:294–305. doi: 10.1016/s0008-6363(00)00115-2. [DOI] [PubMed] [Google Scholar]
- 3.Cohen MV, Downey JM. Adenosine: trigger and mediator of cardioprotection. Basic Res Cardiol. 2008;103:203–215. doi: 10.1007/s00395-007-0687-7. [DOI] [PubMed] [Google Scholar]
- 4.de Zwart M, Link R, von Frijtag Drabbe Künzel JK, Cristalli G, Jacobson KA, Townsend-Nicholson A, Ijzerman AP. A functional screening of adenosine analogues at the adenosine A2B receptor: a search for potent agonists. Nucleosides Nucleotides. 1998;17:969–985. doi: 10.1080/07328319808004215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eckle T, Krahn T, Grenz A, Köhler D, Mittelbronn M, Ledent C, Jacobson MA, Osswald H, Thompson LF, Unertl K, Eltzschig HK. Cardioprotection by ecto-5′-nucleotidase (CD73) and A2B adenosine receptors. Circulation. 2007;115:1581–1590. doi: 10.1161/CIRCULATIONAHA.106.669697. [DOI] [PubMed] [Google Scholar]
- 6.Goto M, Liu Y, Yang X-M, Ardell JL, Cohen MV, Downey JM. Role of bradykinin in protection of ischemic preconditioning in rabbit hearts. Circ Res. 1995;77:611–621. doi: 10.1161/01.res.77.3.611. [DOI] [PubMed] [Google Scholar]
- 7.Goto M, Miura T, Iliodoromitis EK, O’Leary EL, Ishimoto R, Yellon DM, Iimura O. Adenosine infusion during early reperfusion failed to limit myocardial infarct size in a collateral deficient species. Cardiovasc Res. 1991;25:943–949. doi: 10.1093/cvr/25.11.943. [DOI] [PubMed] [Google Scholar]
- 8.Hausenloy DJ, Maddock HL, Baxter GF, Yellon DM. Inhibiting mitochondrial permeability transition pore opening: a new paradigm for myocardial preconditioning? Cardiovasc Res. 2002;55:534–543. doi: 10.1016/s0008-6363(02)00455-8. [DOI] [PubMed] [Google Scholar]
- 9.Hausenloy DJ, Tsang A, Mocanu MM, Yellon DM. Ischemic preconditioning protects by activating prosurvival kinases at reperfusion. Am J Physiol. 2005;288:H971–H976. doi: 10.1152/ajpheart.00374.2004. [DOI] [PubMed] [Google Scholar]
- 10.Hausenloy DJ, Yellon DM. The mitochondrial permeability transition pore: its fundamental role in mediating cell death during ischaemia and reperfusion. J Mol Cell Cardiol. 2003;35:339–341. doi: 10.1016/s0022-2828(03)00043-9. [DOI] [PubMed] [Google Scholar]
- 11.Hausenloy DJ, Yellon DM. Clinical translation of cardioprotective strategies: report and recommendations of the Hatter Institute 5th International Workshop on Cardioprotection. Basic Res Cardiol. 2008;103:493–500. doi: 10.1007/s00395-008-0736-x. [DOI] [PubMed] [Google Scholar]
- 12.Kis A, Baxter GF, Yellon DM. Limitation of myocardial reperfusion injury by AMP579, an adenosine A1/A2A receptor agonist: role of A2A receptor and Erk1/2. Cardiovasc Drugs Ther. 2003;17:415–425. doi: 10.1023/b:card.0000015856.02691.fa. [DOI] [PubMed] [Google Scholar]
- 13.Kopecky SL, Aviles RJ, Bell MR, Lobl JK, Tipping D, Frommell G, Ramsey K, Holland AE, Midei M, Jain A, Kellett M, Gibbons RJ. A randomized, double-blinded, placebo-controlled, dose-ranging study measuring the effect of an adenosine agonist on infarct size reduction in patients undergoing primary percutaneous transluminal coronary angioplasty: The ADMIRE (AmP579 Delivery for Myocardial Infarction REduction) study. Am Heart J. 2003;146:146–152. doi: 10.1016/S0002-8703(03)00172-8. [DOI] [PubMed] [Google Scholar]
- 14.Kuno A, Critz SD, Cui L, Solodushko V, Yang X-M, Krahn T, Albrecht B, Philipp S, Cohen MV, Downey JM. Protein kinase C protects preconditioned rabbit hearts by increasing sensitivity of adenosine A2b-dependent signaling during early reperfusion. J Mol Cell Cardiol. 2007;43:262–271. doi: 10.1016/j.yjmcc.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuno A, Solenkova NV, Solodushko V, Dost T, Liu Y, Yang X-M, Cohen MV, Downey JM. Infarct limitation by a protein kinase G activator at reperfusion in rabbit hearts is dependent on sensitizing the heart to A2b agonists by protein kinase C. Am J Physiol. 2008;295:H1288–H1295. doi: 10.1152/ajpheart.00209.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Linden J, Thai T, Figler H, Jin X, Robeva AS. Characterization of human A2B adenosine receptors: radioligand binding, western blotting, and coupling to Gq in human embryonic kidney 293 cells and HMC-1 mast cells. Mol Pharmacol. 1999;56:705–713. [PubMed] [Google Scholar]
- 17.Liu GS, Richards SC, Olsson RA, Mullane K, Walsh RS, Downey JM. Evidence that the adenosine A3 receptor may mediate the protection afforded by preconditioning in the isolated rabbit heart. Cardiovasc Res. 1994;28:1057–1061. doi: 10.1093/cvr/28.7.1057. [DOI] [PubMed] [Google Scholar]
- 18.Liu GS, Thornton J, Van Winkle DM, Stanley AWH, Olsson RA, Downey JM. Protection against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit heart. Circulation. 1991;84:350–356. doi: 10.1161/01.cir.84.1.350. [DOI] [PubMed] [Google Scholar]
- 19.McVey MJ, Smits GJ, Cox BF, Kitzen JM, Clark KL, Perrone MH. Cardiovascular pharmacology of the adenosine A1/A2-receptor agonist AMP 579: coronary hemodynamic and cardioprotective effects in the canine myocardium. J Cardiovasc Pharmacol. 1999;33:703–710. doi: 10.1097/00005344-199905000-00005. [DOI] [PubMed] [Google Scholar]
- 20.Morrison RR, Talukder MAH, Ledent C, Mustafa SJ. Cardiac effects of adenosine in A2A receptor knockout hearts: uncovering A2B receptors. Am J Physiol. 2002;282:H437–H444. doi: 10.1152/ajpheart.00723.2001. [DOI] [PubMed] [Google Scholar]
- 21.National Research Council. Guide for the care and use of laboratory animals. 7. National Academy Press; Washington, DC: 1996. [Google Scholar]
- 22.Peakman M-C, Hill SJ. Adenosine A2B-receptor-mediated cyclic AMP accumulation in primary rat astrocytes. Br J Pharmacol. 1994;111:191–198. doi: 10.1111/j.1476-5381.1994.tb14043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Philipp S, Yang X-M, Cui L, Davis AM, Downey JM, Cohen MV. Postconditioning protects rabbit hearts through a protein kinase C-adenosine A2b receptor cascade. Cardiovasc Res. 2006;70:308–314. doi: 10.1016/j.cardiores.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 24.Smits GJ, McVey M, Cox BF, Perrone MH, Clark KL. Cardioprotective effects of the novel adenosine A1/A2 receptor agonist AMP 579 in a porcine model of myocardial infarction. J Pharmacol Exp Ther. 1998;286:611–618. [PubMed] [Google Scholar]
- 25.Solenkova NV, Solodushko V, Cohen MV, Downey JM. Endogenous adenosine protects preconditioned heart during early minutes of reperfusion by activating Akt. Am J Physiol. 2006;290:H441–H449. doi: 10.1152/ajpheart.00589.2005. [DOI] [PubMed] [Google Scholar]
- 26.Thornton JD, Liu GS, Olsson RA, Downey JM. Intravenous pretreatment with A1-selective adenosine analogues protects the heart against infarction. Circulation. 1992;85:659–665. doi: 10.1161/01.cir.85.2.659. [DOI] [PubMed] [Google Scholar]
- 27.Vander Heide RS, Reimer KA. Effect of adenosine therapy at reperfusion on myocardial infarct size in dogs. Cardiovasc Res. 1996;31:711–718. doi: 10.1016/0008-6363(95)00235-9. [DOI] [PubMed] [Google Scholar]
- 28.Xu Z, Downey JM, Cohen MV. AMP 579 reduces contracture and limits infarction in rabbit heart by activating adenosine A2 receptors. J Cardiovasc Pharmacol. 2001;38:474–481. doi: 10.1097/00005344-200109000-00016. [DOI] [PubMed] [Google Scholar]
- 29.Xu Z, Downey JM, Cohen MV. Timing and duration of administration are crucial for antiinfarct effect of AMP 579 infused at reperfusion in rabbit heart. Heart Dis. 2003;5:368–371. doi: 10.1097/01.hdx.0000098614.29006.a7. [DOI] [PubMed] [Google Scholar]
- 30.Xu Z, Yang X-M, Cohen MV, Neumann T, Heusch G, Downey JM. Limitation of infarct size in rabbit hearts by the novel adenosine receptor agonist AMP 579 administered at reperfusion. J Mol Cell Cardiol. 2000;32:2339–2347. doi: 10.1006/jmcc.2000.1264. [DOI] [PubMed] [Google Scholar]
- 31.Yang X-M, Krieg T, Cui L, Downey JM, Cohen MV. NECA and bradykinin at reperfusion reduce infarction in rabbit hearts by signaling through PI3 K, ERK, and NO. J Mol Cell Cardiol. 2004;36:411–421. doi: 10.1016/j.yjmcc.2003.12.008. [DOI] [PubMed] [Google Scholar]