

Abstract
Objective
To localize susceptibility genes for alterations in brain structure associated with risk of stroke and dementia. We conducted genomewide linkage analyses for magnetic resonance imaging (MRI) measures of brain atrophy, ventricular, and subcortical white matter hyperintensity (leukoaraiosis) in 689 non-Hispanic white (673 sibling pairs; median age, 61 years) and 544 non-Hispanic black participants (503 sibling pairs; median age, 64 years) from sibships with at least 2 members with essential hypertension.
Design, Setting, and Patients
We determined brain, ventricular, and leukoaraiosis volumes from axial fluid-attenuated inversion recovery MRI; we calculated brain atrophy as the difference between total intracranial and brain volumes. Microsatellite markers (n=451) distributed across the 22 autosomes were genotyped, and we used variance components methods to estimate heritability and assess evidence of genetic linkage for each MRI measure.
Main Outcome Measures
Brain atrophy ventricular volume, and leukoaraiosis determined from fluid-attenuated inversion recovery MRI.
Results
In both races, the heritability of each MRI measure was statistically greater than 0 (P< .001), ranging in magnitude from 0.42 (for ventricular volume in blacks) to 0.69 (for brain atrophy in blacks). Based on multipoint logarithm of odds scores (MLS), the strongest evidence of genetic linkage was observed for brain atrophy on chromosomes 1 (MLS, 3.49 at 161 cM; P< .001) and 17 (MLS, 3.08 at 18 cM; P< .001) in whites; for ventricular volume on chromosome 12 (MLS, 3.67 at 49 cM; P< .001) in blacks and chromosome 10 (MLS, 2.47 at 110 cM; P < .001) in whites; and for leukoaraiosis on chromosome 11 (MLS, 2.21 at 118 cM; P < .001) in whites and chromosome 22 (MLS, 2.02 at 36 cM; P= .001) in blacks.
Conclusions
The MRI measures of structural brain injury are heritable in non-Hispanic black and white sibships ascertained through hypertensive sibling pairs. The susceptibility loci for brain atrophy, ventricular volume, and leukoaraiosis identified by linkage analyses differ among MRI measures and between races.
A multiplicity of vascular and neurodegenerative processes contributes to changes in brain structure with age. Despite success in identifying rare single gene mutations that cause stroke or dementia, most genes making smaller contributions to risk remain unknown.1,2 Difficulties in identifying genetic polymorphisms with small effects on clinical end points are among the motivations for studying the underlying contributory disease processes, which progress asymptomatically for decades before a clinical event. Magnetic resonance imaging (MRI) of the brain has been used as a noninvasive method to obtain accurate and reproducible quantitative measures of alterations in brain structure, including cerebral atrophy, ventricular enlargement,3 and the volume of subcortical white matter hyperintensity (leukoaraiosis).4 These MRI measures of structural brain injury are heritable, associated with hypertension and other risk factors for arteriosclerosis, and predictive of stroke and dementia.5–6
The goal of the present study was to localize regions of the genome that harbor DNA sequence variations influencing MRI measures of structural brain injury, that is, total cerebral brain atrophy, ventricular volume, and leukoaraiosis. We studied non-Hispanic white and black sibships at increased risk of structural brain injury by virtue of previously diagnosed hypertension in 2 or more members of each sibship. Our primary approach was univariate linkage analysis for each MRI measure of the brain. We also conducted bivariate linkage analyses for pairwise combinations of brain MRI measures and of each brain MRI measure with measures of blood pressure level and pulsation, attempting to leverage greater statistical power to identify loci with pleiotropic effects too small to be detected by separate univariate linkage analyses.7
METHODS
SUBJECTS
The 1233 study participants consisted of 689 non-Hispanic white adults (410 women and 279 men) and 544 non-Hispanic black adults (379 women and 165 men) from sibships enrolled in the Genetic Epidemiology Network of Arteriopathy (GENOA) of the Family Blood Pressure Program, designed to identify genetic determinants of hypertension in multiple ethnic groups.8 The Mayo Clinic diagnostic index and medical record linkage system were used to identify non-Hispanic while residents of Olmsted County, Minnesota, with a diagnosis of essential hypertension made before 60 years of age. Non-Hispanic blacks were recruited from hypertensive probands in a probability sample of black residents of that community aged 45 to 64 years.9 Sibships in which either index hypertensive sibling was known to have impaired kidney function (eg, serum creatinine level, ≥2.0 mg/dL [to convert to micromoles per liter, multiply by 88.4]) were not recruited; otheiwise, all available members of the index sibships were invited to an initial examination (1996–2000). A second phase of the Family Blood Pressure Program was designed to identify target organ complications, wherein 2721 GENOA participants (1239 white and 1482 black) underwent an examination (2000–2004) that included MRI of the brain in 1746 participants (916 white and 830 black) who also enrolled in the Genetics of Microangiopathic Brain Injury ancillary study (2001–2006). The median time between the GENOA visit and the brain MRI was 11.9 months (in white participants, 12.3 months; in black participants, 11.3 months). In 68 participants whose brain MRIs were not analyzable, the most common reason was unsuspected prior cortical/hemispheric brain infarction (n = 36 [53%]), followed by other anatomic abnormalities (n= 17 [25%]), artifacts related to subject or technical factors (n = 8 [12%]), and failure to complete the MRI (n = 7 [10%]). The analyses were conducted on a subset of 1233 participants with analyzable brain MRIs, measurements of linkage markers that were consistent with reported pedigree structure, and at least 1 sibling with these same data available.
STUDY PROTOCOL
Study protocols were approved by the Human Studies Review-Board of each institution, and written informed consent was obtained from participants. Height was measured by means of a wall stadiometer and weight by means of an electronic balance, with body mass index calculated as weight in kilograms divided by height in meters squared. Blood pressure was measured with a random zero sphygmomanometer (Hawksley & Sons Ltd, West Sussex, England) and a cuff appropriate for arm size. The second and third of 3 readings, taken from the right arm alter the participant sat for at least 5 minutes, were averaged for the analyses. Mean arterial pressure was calculated as [systolic blood pressure + (2 × diastolic blood pressure)]/3, and pulse pressure was calculated as systolic blood pressure – diastolic blood pressure. The diagnosis of hypertension was confirmed if a prior diagnosis of hypertension and use of prescription antihypertensive medication were reported, or if the systolic or diastolic blood pressures averaged at least 140 mm Hg or at least 90 mm Hg, respectively.
Magnetic resonance imaging of the brain was performed at both institutions on identically equipped 1.5-T MRI scanners (Signa; GE Medical Systems, Waukesha, Wisconsin) under the supervision of neuroradiologists. The methods for semiautomated MRI measurements of brain anatomy have been described previously.3 Total intracranial volume was measured from T1-weighted spin-echo sagittal images, each set consisting of 32 contiguous 5-mm-thick interleaved sections with no interslice gap, a field of view of 24 cm, and a matrix of 256 × 192, obtained with the following sequence: scan time, 2.5 minutes; echo time, 14 milliseconds; 2 repetitions; and repetition time, 500 milliseconds. Brain, ventricular, and leukoaraiosis volumes were determined from axial fluid-attenuated inversion recovery images, each set consisting of 48 contiguous 3-mm-thick interleaved sections with no interslice gap, a field of view of 22 cm, and a matrix of 256 × 160, obtained with the following sequence: scan time, 9 minutes; echo time, 144.8 milliseconds; inversion time, 2600 milliseconds; repetition time, 11 seconds; bandwidth, ±15.6 kHz; and an average of 1 signal. A fluid-attenuated inversion recovery image is a T2-weighted image with the signal of cerebrospinal fluid nulled, such that brain pathology appears as the brightest intracranial tissue. Interactive image-processing steps were performed by a research associate at the Mayo Clinic who had no knowledge of the subjects’ personal or medical histories or biological relationships. A fully automated algorithm was used to segment each section of the edited multislice fluid-attenuated inversion recovery sequence into voxels assigned to 1 of the following 3 categories: brain, cerebrospinal fluid, or leukoaraiosis. The, mean absolute error of this method is 1.4% for brain volume and 6.6% for leukoaraiosis volume, and the mean test-retest coefficient of variation is 0.3% for brain volume and 1.4% for leukoaraiosis volume.3 The difference between total intracranial volume and brain volume provided a measure of brain atrophy. Brain images with cortical/hemispheric infarctions were excluded from the analyses because of the distortion of the leukoaraiosis volume estimates that would be introduced in the automated segmentation algorithm. Lacunar infarctions and periventricular white matter hyperintensities were included in the leukoaraiosis intensity category and therefore in the leukoaraiosis volume estimates.
A set of 451 microsatellite markers distributed across the 22 autosomes (Cooperative Human Linkage Center/Weber screening set 9.0) was genotyped using standard polymerase chain reaction methods by the Mammalian Genotyping Center of the Marshfield Medical Research Foundation, Marshfield, Wisconsin, which provided the ordering of markers and their genetic map distances. Inconsistencies of the genotypes with pedigree structure were identified by the Lange and Goradia algorithm as implemented in the PedCheck software.10 Instances that could not be resolved as genotyping errors were considered missing data.
STATISTICAL ANALYSES
Because distributions of ventricular and leukoaraiosis volumes were positively skewed, the raw MRI trait values were transformed for the genetic analyses using the empirical normal quantile transformation,11 which was effective in normalizing the distributions.12 Adjustments for sex, age, and total intracranial volume (for brain atrophy and ventricular volume) or brain volume (for leukoaraiosis) were incorporated as co-variates in the genetic models. Genetic and environmental correlations between the adjusted (transformed) trait values were estimated by means of variance decomposition using maximum likelihood methods,13 and phenotypic correlations between traits were calculated on the basis of genetic and environmental correlations.14 Sibship structure was verified using the MERLIN software program.15 Univariate and bivariate genome-wide linkage analyses were conducted with an R/S-plus Library MULTIC routine16 that uses the variance components approach.17 We also conducted bivariate linkage analyses for pairwise combinations of brain MRI measures and of each brain MRI measure with a measure of steady-state blood pressure level and a measure of blood pressure pulsation.18 Both blood pressure level and pulsation are influenced by genetic factors,18 and each may make an additive, independent contribution to risk of structural brain injury.19 Multivariate linkage analyses provided greater statistical power to identify loci with pleiotropic effects on genetically correlated traits7 and permitted assessment of overlap of blood pressure loci with those predisposing to structural brain injury. A rationale for the bivariate analyses of pulse pressure, in addition to mean arterial pressure, was the high percentage of GENOA participants treated with anti-hypertensive medications (Table 1), which may lower both systolic and diastolic blood pressure levels but have less impact or the calculated difference between them.18
Table 1.
Descriptive Characteristics of 689 Non-Hispanic White Siblings and 544 Non-Hispanic Black Siblingsa
| Characteristic | White Siblings | Black Siblings |
|---|---|---|
| Women, No. (%) | 410 (59.5) | 379 (69.7) |
| Age, mean (range), y | 61.3 (53.5–68.2) | 63.6 (57.5–68.4) |
| BMI | 29.5 (26.5–33.3) | 29.9 (26.9–34.6)b |
| SBP, mm Hg | 130 (120–142)b | 133 (123–147) |
| DBP, nm Hg | 74 (68–80)b | 79 (73–85) |
| MAP, mm Hg | 93 (87–99)b | 98 (90–105) |
| PP, mm Hg | 55 (46–67)b | 54 (45–68) |
| Hypertension, No. (%) | 509 (73.9) | 400 (73.5) |
| Use of antihypertensive dugs, No. (%) | 491 (71.3) | 350 (64.3) |
| TIV, mL | 1452 (1354–1560) | 1359 (1272–1451) |
| Biain volume, mL | 1152 (1069–1239) | 1061 (989–1140) |
| Brain atrophy, mL | 297 (257–347) | 294 (250–344) |
| Ventricular volume, mL | 21.8 (14.8–32.2) | 17.6 (12.7–26.3) |
| Leukoaraiosis, mL | 5.92 (4.42–8.78) | 6.72 (4.79–10.35) |
Abbreviations: BMI, body mass index (calculated as weight in kilograms divided by height in meters squared); DBP, diastolic blood pressure; MAP, mean arterial pressure; PP, pulse pressure; SBP, systolic blood pressure; TIV, total intracranial volume.
White participants included 673 sibling pairs from 273 sibships in Rochester, Minnesota; black participants, 503 sibling pairs from 212 sibships in Jackson, Mississippi. Unless otherwise indicated, data are expressed as median (interquartile range) for quantitative traits.
In whites, blood pressure was not measured in 1 individual; otherwise, the number of measurements for all traits was 689; in black participants, height was not measured in 1 individual and BMI could not be calculated; otherwise, the number of measurements for all traits was 544.
The multipoint identity-by-descent sharing among pairs of relatives was calculated using SimWalk2 software20 and was based on the pedigree relationships for 1239 white and 1482 black GENOA participants in whom linkage markers were measured. A likelihood ratio test (LRT) was used to test for genetic linkage, in which the LRT is defined as −2 × ([log likelihood under the null hypothesis) − (log likelihood under the alternative hypothesis]). Under the null hypothesis, the linked genetic factors are restricted to equal 0, and the asymptotic distribution of the LRT is a mixture of χ2 statistics.21 All logarithm of odds (LOD) scores for the linkage analyses were calculated from the LRT values as LRT/(2 × log10). Conventionally, univariate multipoint LOD scores (MLSs) of at least 3.00 are considered statistically significant evidence of linkage (P ≤ 1 × 10−4). MLSs of at least 2.00 as suggestive evidence (P ≤ .001), and MLSs of at least 1.30 as tentative evidence (P ≤ .007).22,23 Because the bivariate linkage analyses have greater degrees of freedom, higher LOD score thresholds arc required for the bivariate MLSs to achieve comparable levels of statistical significance (ie, ≥4.00, ≥2.87, and ≥2.06, respectively). However, because we performed multiple univariate and bivariate linkage analyses, the MLSs and corresponding P values chiefly serve to indicate the relative strength of evidence in favor of linkage rather than to make absolute judgments regarding statistical significance. Similarly, when the bivariate MLS met the bivariate LOD score criteria for linkage, strength of the evidence in favor of pleiotropy was inferred on the basis of the magnitude of decline in the bivariate P value relative to the separate univariate P values.
RESULTS
The 689 white participants consisted of 656 full-sibling pairs from 270 sibships ranging in size from 2 individuals (191 sibships) to 9 (1 sibship) and 5 full- and 12 half-sibling pairs from 3 sibships of 2, 4, and 5 individuals. Of the 673 white full- and half-sibling pairs, 192 (28.5%) were from sibships of 2 individuals, 336 (49.9%) from sibships of 3 to 5 individuals, and 145 (21.5%) from sibships of 6 to 9 individuals. The 544 black participants consisted of 442 full-sibling pairs from 173 sibships ranging in size from 2 individuals (104 sibships) to 7 (1 sibship) and 17 full- and 61 half-sibling pairs from 39 sibships ranging in size from 2 individuals (24 sibships) to 4 (3 sibships). Of the 503 black full- and half-sibling pairs, 128 (25.4%) were from sibships of 2 individuals, 264 (52.5%) from sibships of 3 to 4 individuals, and 111 (22.1%) from sibships of 5 to 7 individuals. Most of the siblings had hypertension and were treated with antihypertensive medications (Table 1). Heritability of each MRI measure was significantly greater than 0 in whites and blacks (Table 2) (P<.001 for each estimate).
Table 2.
Heritability Estimates for Brain MRI Measures in 689 Non-Hispanic White Siblings and 544 Non-Hispanic Black Siblingsa
| Mean (SD) |
||
|---|---|---|
| Brain MRI Measure | White Siblings | Black Siblings |
| Brain atrophy | 0.61 (0.10) | 0.69 (0.12) |
| Ventricular volume | 0.66 (0.10) | 0.42 (0.10) |
| Leukoaraiosis | 0.49 (0.09) | 0.45 (0.11) |
Abbreviation: MRI, magnetic resonance imaging.
White participants included 673 sibling pairs from 273 sibships in Rochester, Minnesota; black participants, 503 sibling pairs from 212 sibships in Jackson, Mississippi. Heritability is the proportion of interindividual variance explained by additive genetic factors, measured on a scale from 0 to 1; all heritability estimates were significantly greater than 0 (P < .001). Brain MRI measures were transformed using the empirical normal quantile transformation13 and adjusted for sex, age, and total intracranial volume (brain atrophy and ventricular volume) or brain volume (leukoaraiosis).
For the MRI measure of brain atrophy, the highest level of evidence of genetic linkage (MLS ≥3.00) was achieved on chromosomes 1 and 17 in whites (Table 3 and Figure 1). In blacks, MLS values for brain atrophy achieved the intermediate level of evidence (MLS, 2.00–2.99) on chromosome 1 (Table 3). Seven other MLS values achieved the lowest level of evidence (1.30–1.99), including 2 loci in whites (on chromosomes 4 and 6) and 5 in blacks (on chromosomes 3, 9, 13, 17, and 19).
Table 3.
Univariate Maximum Multipoint LOD Scores and Nominal P Values for Brain MRI Traits Measured in 689 Non-Hispanic White Siblings and 544 Non-Hispanic Black Siblingsa
| LOD Scores (Peak Positions, cM) |
||||||
|---|---|---|---|---|---|---|
| White Siblings |
Black Siblings |
|||||
| Chromosome | Brain Atrophy | Ventricular Volume | Leukoaralosis | Brain Atrophy | Ventricular Volume | Leukoaraiosis |
| 1 | 3.49 (161)b | 1.20 (164) | 0.36 (102) | 2.00 (45) | 0.43 (268) | 1.04 (159) |
| P value | <.001 | .009 | .10 | .001 | .08 | .01 |
| 2 | 0.50 (233) | 2.05 (35) | 0.33 (260) | 0.54 (121) | 2.27 (216) | 94 (0.61) |
| P value | .06 | .001 | .11 | .06 | <.001 | .046 |
| 3 | 0.79 (188) | 2.00 (79) | 0.41 (209) | 1.90 (138) | 1.15 (12) | 1.27 (69) |
| P value | .03 | .001 | .09 | .002 | .01 | .008 |
| 4 | 1.68 (13) | 1.22 (78) | 0.36 (145) | 0.98 (144) | 1.61 (26) | 0.06 (208) |
| P value | .003 | .009 | .10 | .02 | .003 | .31 |
| 5 | 0.31 (59) | 0.27(192) | 0.97 (45) | 0.38 (139) | 0.88 (195) | 0.46 (117) |
| P value | .12 | .13 | .02 | .09 | .02 | .07 |
| 6 | 1.66 (39) | 0.99 (166) | 1.02 (128) | 0.83 (139) | 0.59 (9) | 0.53 (80) |
| P value | .003 | .02 | .02 | .03 | .05 | .06 |
| 7 | 0.70 (66) | 0.55 (108) | 0.57 (91) | 0.10 (33) | 0.28 (29) | 0.48 (177) |
| P value | .04 | .06 | .05 | .25 | .13 | .07 |
| 8 | 0.81 (64) | 0.41 (67) | 0.42(135) | 0.49 (164) | 0.58 (164) | 0.65 (8) |
| P value | .03 | .09 | .08 | .07 | .05 | .04 |
| 9 | 0.75 (143) | 0.10 (26) | 0.46 (152) | 1.52 (146) | 0.72 (152) | 0.37 (45) |
| P value | .03 | .25 | .07 | .004 | .03 | .10 |
| 10 | 0.32 (85) | 2.47 (110)c | 0.47 (63) | 0.71 (10) | 1.08 (148) | 0.34 (101) |
| P value | .11 | <.001 | .07 | .04 | .01 | .10 |
| 11 | 0.63 (9) | 0.45 (2) | 2.21 (118)d | 0.44 (59) | 1.24 (69) | 0.79 (58) |
| P value | .04 | .07 | <.001 | .08 | .009 | .03 |
| 12 | 0.93 (90) | 0.40 (67) | 0.54 (107) | 1.02 (63) | 3.67 (49)e | 0.17 (73) |
| P value | .02 | .09 | .06 | .02 | <.001 | .19 |
| 13 | 0.18 (103) | 0.56 (9) | 0.69 (51) | 1.45 (11) | 1.65 (17) | 0.25 (9) |
| P value | .18 | .054 | .04 | .005 | .003 | .14 |
| 14 | 0.27 (123) | 0.07 (126) | 0.16 (12) | 0.24 (126) | 0.36 (76) | 0.11 (26) |
| P value | .13 | .29 | .20 | .15 | .10 | .24 |
| 15 | 0.27 (90) | 0.05 (12) | 0.22 (91) | 0.57 (72) | 0.23 (78) | <0.01 (12) |
| P value | .13 | .32 | .16 | .053 | .15 | .50 |
| 16 | 1.03 (128) | 0.87 (72) | 1.11 (72) | <0.01 (51) | 0.81 (125) | 1.42 (10) |
| P value | .01 | .02 | .01 | .46 | .03 | .005 |
| 17 | 3.08 (18)f | 0.04 (45) | 1.10 (119) | 1.86 (39) | 0.85 (45) | 0.47 (86) |
| P value | <.001 | .34 | .01 | .002 | .02 | .07 |
| 18 | 0.13 (80) | 0.60 (75) | <0.01 (64) | 0.57 (116) | 0.27 (116) | 0.10 (13) |
| P value | .22 | .049 | .46 | .052 | .13 | .25 |
| 19 | 0.22 (101) | 0.16 (76) | 0.59 (33) | 1.42 (81) | 1.18 (96) | 0.65 (27) |
| P value | .16 | .20 | .050 | .005 | .01 | .04 |
| 20 | 0.79 (39) | 0.77 (80) | 0.45 (41) | 0.33 (2) | 1.79 (2) | 0.05 (2) |
| P value | .03 | .03 | .08 | .11 | .002 | .32 |
| 21 | 0.14 (58) | <0.01 (47) | 1.75 (13) | 0.05 (46) | <0.01 (3) | 1.99 (58) |
| P value | .21 | .43 | .002 | .32 | .50 | .001 |
| 22 | 0.06 (29) | 0.36 (46) | <0.01 (4) | 0.46 (11) | 0.02 (4) | 2.02 (36)g |
| P value | .30 | .10 | .50 | .07 | .38 | .001 |
Abbreviations: LOD, logarithm of odds; MRI, magnetic resonance imaging.
White participants included 673 sibling pairs from 273 sibships in Rochester, Minnesota; black participants, 503 sibling pairs from 212 sibships in Jackson, Mississippi. The brain MRI measures were transformed using the normal quantile transformation13 and adjusted for sex, age, and total intracranial volume (brain atrophy and ventricular volume) or brain size (leukoaraiosis). Positions are distances from pter in centimorgans.
Indicates between GATA12A07 (152 cM) and GATA43A04 (164 cM).
Indicates between GGAT1A4 (101 cM)and GATA115E01 (113 cM).
Indicates between GATA23E06 (113 cM) and GATA64D03 (123 cM).
Indicates between ATA27A06 (49 cM) and GATA91H06 (56 cM).
Indicates between GAAT2C03 (11 cM) and GATA8C04 (22 cM).
Indicates between GATA6F05 (32 cM) and GGAT3C10 (46 cM).
Figure 1.

Brain atrophy multipoint logarithm of odds (LOD) score plots calculated from magnetic resonance images for univariate genetic linkage analysis. Subjects included 689 non-Hispanic whites and 544 non-Hispanic blacks from sibships with at least 2 members with essential hypertension. Within the chromosome 4p 16.3-15.33 linkage region identified in whites (0–24 cM from pter) (see also Table 3). a linkage peak for white matter hyperintensity (leukoaraiosis) volume was reported and noted to harbor the gene for Huntington disease.5
For the MRI measure of ventricular volume, the highest level of evidence of genetic linkage (MLS ≥ 3.00) was achieved on chromosome 12 in blacks (Table 3 and Figure 2). The intermediate level of evidence (MLS, 2.00–2.99) was met in whites on chromosomes 2 (at 35 cM from pter), 3, and 10 and in blacks on chromosome 2 (at 216 cM from pter) (Table 3). Three other MLS values achieved the lowest level of evidence (1.30–1.99), all in blacks (on chromosomes 4, 13, and 20).
Figure 2.

Ventricular volume multipoint logarithm of odds (LOD) score plots calculated from magnetic resonance images for univariate genetic linkage analysis. Subjects included 689 non-Hispanic whites and 544 non-Hispanic blacks from sibships with at least 2 members with essential hypertension. Within the chromosome 12p12.2-12q12 linkage region identified in blacks (38.5–55.7 cM from pter) (see also Table 3), 4 single-nucleotide polymorphisms (SNPs) in the phosphodiesterase 3A gene (rs1444644, rs1444645, rs10505865, and rs1444629) were associated with total and regional cerebral volumes; 1 SNP in the contactin 1 gene (rs10506176) was associated with temporal brain volume; and 1 SNP in the leucine-rich repeat kinase 2 gene (rs10506151) was associated with frontal and temporal brain volumes.6
For the MRI measure of leukoaraiosis, 2 MLS values achieved the intermediate level of evidence of genetic linkage (MLS, 2.00–2.99), one in whites on chromosome 11 and the other in blacks on chromosome 22 (Table 3 and Figure 3). Two additional MLS values achieved the lowest level of evidence (1.30–1.99), on chromosome 21 at 13 cM from pter in white and at 58 cM from pter in blacks.
Figure 3.

Maximum multipoint logarithm of odds (LOD) score plots for univariate genetic linkage analysis of subcortical white matter hyperintensity (leukoaraiosis) volume. Subjects included 689 non-Hispanic whites and 544 non-Hispanic blacks from sibships with at least 2 members with essential hypertension. Within the chromosome 11q23.1-11q25 region identified in whites (108–139 cM from pter) (see also Table 3), single-nucleotide polymorphism in the beta-site amyloid β A4 precursor protein-cleaving enzyme 1 gene (rs1261791) was associated with parietal brain volume.6
Genetic correlations were significantly greater than 0 between MRI measures of brain atrophy and ventricular volume in both whites (0.38; P < .001) and blacks (0.45; P<.001). In whites, leukoaraiosis was also genetically correlated with brain atrophy (0.22; P< .001) and ventricular (0.40; P<.001), but neither correlation differed significantly from 0 in blacks (P = .59). The bivariate linkage analyses provided evidence of 9 loci with pleiotropic effects on pairwise combinations of brain MRI measures, based on bivariate MLS values that satisfied the intermediate or lower level of evidence of bivariate linkage (ie, bivariate MLS, 2.06–3.99) (chromosomes 2, 4, 5, 8, 10, 13, and 16 in whites and 13 and 22 in blacks ([eTable 1]). The strongest such evidence was on chromosome 13 (at 14 cM from pter), where the bivariate MLS for brain atrophy and ventricular volume rose to the intermediate level of evidence of bivariate genetic linkage (MLS, 2.95; P = .001) in blacks (Figure 4).
Figure 4.
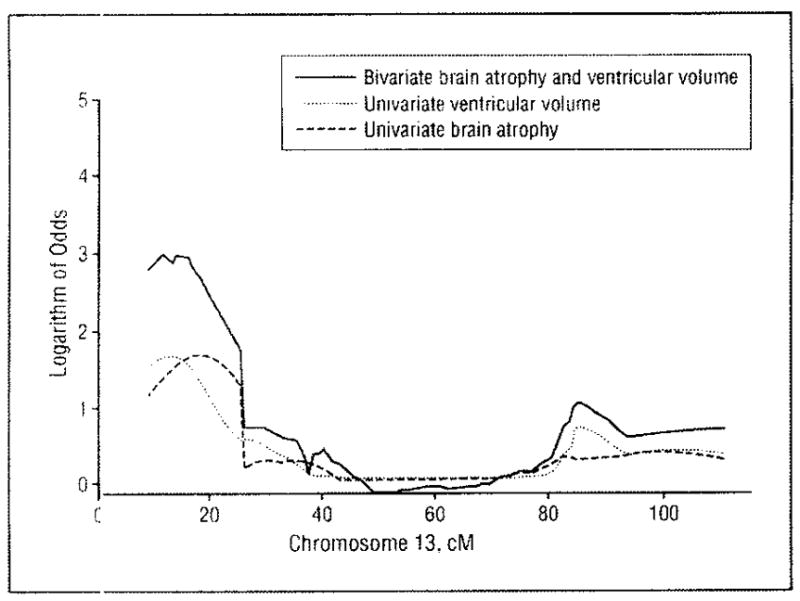
Chromosome 13 multipoint logarithm of odds score plots for genetic linkage of brain atrophy and ventricular volume. Brain atrophy and ventricular volume were analyzed as separate univariate traits and in combination as a bivariate trait in 544 non-Hispanic blacks from sibships with at least 2 members with essential hypertension.
Genetic correlations were also significantly greater than 0 between mean arterial pressure and leukoaraiosis in whites (0.61); P < .001) and blacks (0.40; P < .001) and with brain atrophy in whites (0.27; P < .001). Likewise, for pulse pressure, genetic correlations were significantly greater than 0 with brain atrophy in whites (0.45; P < .001) and with leukoaraiosis in blacks (0.29; P < .001). The bivariate linkage analyses with measures of blood pressure provided only the lowest level of evidence of bivariate genetic linkage (ie, bivariate MLS, 2.06–2.86) and for only 2 loci, one on chromosome 19 that may have pleiotropic effects on brain atrophy and mean arterial pressure in blacks (bivariate MLS, 2.72 at 26 cM from pter; P = .002) and the other on chromosome 7 that may have pleiotropic effects on ventricular volume and pulse pressure in whites (bivariate MLS, 2.32 at 181 cM from pter; P = .004) (eTable 2).
COMMENT
Results of this study provide evidence of genetic influences on MRI measures of structural brain injury in non-Hispanic black and white sibships in which at least 2 siblings had essential hypertension. The estimated heritabilities for MRI measures of brain atrophy, ventricular volume, and leukoaraiosis were similar in magnitude to those reported initially for a cohort of elderly male twins24,25 and subsequently for the community-based Framingham Heart Study cohort.26 The present study provides estimates of heritability in hypertensive sibships and in non-Hispanic black sibships, which were not included in previous samples. Consistency of the heritability estimates with samples that were not ascertained through hypertension suggests independence of the larger genetic effects detectable by linkage analyses from those with major effects on blood pressure. This latter inference is also consistent with previous genetic analyses that adjusted brain MRI measures for blood pressure and other risk factors for arteriosclerosis27 and with results of our bivariate linkage analyses that detected only 2 loci (and at only the lowest level of statistical support) with pleiotropic effects on brain MRI and blood pressure measures.
The chromosomal regions defined by 1-LOD score-down intervals around the 2 highest MLS peaks for each MRI measure contain numerous plausible candidate genes for structural brain entry; potentially relevant metabolic, vascular, and neurodegenerative disorders contributing to structural brain injury have been mapped to these regions (eTable 3).28–68 For example, within the intervals identified by the highest level of evidence of genetic linkage (ie, MLSs > 3.00), the chromosome 1p12-1q23.3 region for brain atrophy and the chromosome 12p12.2-12q12 region for ventricular volume include genes implicated in Parkinson disease (PARK10 and LRRK2, respectively), and the chromosome 17p13.3-17p12 region for brain atrophy includes genes implicated in type 2 diabetes mellitus (SLC2A4) and the metabolic insulin-resistance, syndrome (AOMS2). The chromosome 1 and 12 regions also harbor genes implicated in hypertension (eg, SELE, HYT4, and HTNB) and atherosclerosis (eg, SELP, APOA2, and CRP), which, like diabetes and insulin resistance, are risk factors for structural brain injury.69 Moreover, diabetes, hypertension, and other risk factors for arteriosclerosis have been implicated in Alzheimer disease,70 in which brain atrophy and ventricular enlargement are prominent features. Because biologically plausible candidate genes may be located under linkage peaks by chance alone, comparisons with results from other genome scans is indicated.
The Framingham Heart Study has reported results of genome scans for brain MRI measures.5,6 An initial scan for white matter hyperintensity volume used 387 highly polymorphic linkage markers in 2259 family members.5 The significant linkage criterion was satisfied by a single locus on chromosome 4p16.2 (LOD, 3.69) that was noted to harbor genes contributing to brain injury, including the gene for Huntington disease (HTT). This region corresponds to one of the linkage regions for brain atrophy in our white sample (0–24 cM from 4 pter) (Table 3 and Figure 1). Subsequently, results of genomewide association analyses of 1 00 000 single-nucleotide polymorphisms (SNPs) have been reported for 705 members of the largest Framingham pedigrees.6 Comparison with the 2 highest MLS peaks that we observed for each brain MRI measure validates association of candidate genes within 2 of the 6 regions. Within the chromosome 12p12.2-12q12 linkage region identified for ventricular volume in blacks (38.5–55.7 cM from pter) (Table 3), the Framingham investigators6 noted that 4 SNPs in the phosphodiesterase 3A gene (PDE3A; rs1444644, rs1444645, rs10505865, and rs1444629) were associated with total and regional cerebral volumes, 1 SNP in the contactin 1 gene (CNTN1; rs10506176) was associated with temporal brain volume, and 1 SNP in the leucine-rich repeat kinase 2 gene (LRRK2; rs10506151) was associated with frontal and temporal brain volumes. The PDE3A expressed in human platelets plays a role in activation,71 CNTN1 has been associated with cerebellar ataxia in an animal model,72 and LRRK2 has been associated with Parkinson disease73 and tau pathology.74 Within the chromosome 11q23.1–11q25 region for leukoaraiosis in whites (108–139 cM from pter) (Table 3), the Framingham investigators noted that 1 SNP in the beta-site amyloid β precursor protein-cleaving enzyme 1 gene (BACE1; rs1261791) was associated with parietal brain volume. Variation in BACE1 has been previously associated with Alzheimer disease.75
We attempted to leverage the genetic correlations among traits by performing bivariate linkage analyses with greater statistical power76 to localize additional regions with effects on brain MRI measures too small to be detected in univariate linkage analyses and to assess overlap with regions influencing blood pressure. Although results of the genetic correlation analyses supported the concept of pleiotropy, those regions implicated by the bivariate linkage analyses to harbor genes influencing more than 1 MRI measure were also detected in the univariate linkage analyses for the separate MRI measures (as illustrated in Figure 4). Moreover, evidence of overlap between linkage regions influencing blood pressure and brain MRI measures was relatively sparse and statistically weak but consistent with findings in an animal model in which stroke genes detected by linkage analysis were separate from genes determining blood pressure.77,78 Hence, the theoretical advantages of multivariate linkage analyses were not realized in the present study.
Results of this and previous studies have several implications for the genetic architecture of MRI measures of structural brain injury. First, high heritabilities of the MRI measures are likely, for the most part, to be the consequence of many genes with small effects (ie, polygenes), in as much as relatively few regions had large enough effects to satisfy conventional criteria for significant evidence of genetic linkage or association.6 Second, because the regions identified by genetic linkage or association analyses differ among MRI measures,6 the genetic correlations among MRI measures are also likely to reflect, for the most part, polygenes. Third, most of the genetic variation in MRI measures of structural brain injury appears to be separate from genetic variation in measures of blood pressure or other established risk factors for arteriosclerosis.27 Fourth, because most regions reported to harbor variants influencing a given MRI measure also differ between racial groups and between different samples from the same race,5 the larger genetic effects detected by this and previous linkage or association analyses may be context dependent, possibly reflecting gene-gene or gene-environment interactions that are sample specific or study specific.
Our study has the strength of relatively large, well-characterized biracial cohorts; however, it also has limitations. Use of genetic markers to scan the genome is a hypothesis-generating first step toward localizing particular DNA sequence variants that have functional effects contributing to structural brain injury. Replication of linkage or association findings in independent samples will be necessary before undertaking fine mapping of the regions to prioritize which positional candidate genes may merit further investigation for functional variants. In addition, the traditional linkage approach we applied may have insufficient power to detect many additional loci hypothesized to have smaller effects on brain MRI measures.79 Despite apparent independence of the genes influencing brain MRI measures from those influencing blood pressure, because our results are based on sibships with at least 2 members diagnosed as having essential hypertension before 60 years of age in whom the full range of untreated blood pressure levels was not observed, extension of inferences to groups of different risk factor profiles must be cautious.
The present study confirms a large genetic component of interindividual variation in MRI measures of structural brain injury and makes a first step toward localizing regions of the genome that may harbor DNA sequence variants meriting further investigation as potential risk factors for stroke and dementia.
Acknowledgments
Funding/Support: This study was supported by US Public Health Service gr ants U01 HL 54464, U01 HL 54457, U01 HL 54481, R01 HL 71917, and M01 RR 00585 from the National Institutes of Health.
Footnotes
Financial Disclosure: None reported.
Additional Information: The eTables are available at http://www.archneurol.com.
Author Contributions: Dr Turner had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: Turner, Jack, Mosley, Boerwinkle, and de Andrade. Acquisition of data: Turner, Fornage, Jack, Mosley, Kardia, and Boerwinkle. Analysis and interpretation of data: Turner, Mosley, Knopman, Kardia, Boerwinkle, and de Andrade. Drafting of the manuscript: Turner. Critical revision of the manuscript for important intellectual content: Turner, Fornage, Jack, Mosley, Knopman, Kardia, Boerwinkle, and de Andrade. Statistical analysis: Kardia, Boerwinkle, and de Andrade. Obtained funding: Turner, Mosley, Kardia, and Boerwinkle. Administrative, technical, and material support: Turner, Jack, and Mosley. Study supervision: Turner and de Andrade.
Additional Contributions: Jodie Van De Rostyne, BS, Debra Gearhart, Maria Shiung, BA, and Bill Li, MS, provided expert technical support.
References
- 1.Joutel A, Corpechot C, Ducros A, et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383(6602):707–710. doi: 10.1038/383707a0. [DOI] [PubMed] [Google Scholar]
- 2.Campion D, Dumanchin C, Hannequin D, et al. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet. 1999;65(3):664–670. doi: 10.1086/302553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jack CR, Jr, O’Brien PC, Rettman DW, et al. FLAIR histogram segmentation for measurement of leukoaraiosis volume. J Magn Reson Imaging. 2001;14(6):668–676. doi: 10.1002/jmri.10011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pantoni L, Garcia JH. Pathogenesis of leukoaraiosis: a review. Stroke. 1997;28(3):652–659. doi: 10.1161/01.str.28.3.652. [DOI] [PubMed] [Google Scholar]
- 5.DeStefano AL, Atwood LD, Massaro JM, et al. Genome-wide scan for white matter hyperintensity: the Framingham Heart Study. Stroke. 2006;37(1):77–81. doi: 10.1161/01.STR.0000196987.68770.b3. [DOI] [PubMed] [Google Scholar]
- 6.Seshadri S, DeStefano AL, Au R, et al. Genetic correlates of brain aging on MRI and cognitive test measures: a genome-wide association and linkage analysis in the Framingham Study. BMC Med Genet. 2007;8(suppl 1):S15. doi: 10.1186/1471-2350-8-S1-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amos C, de Andrade M, Zhu D. Comparison of multivariate tests for genetic linkage. Hum Hered. 2001;51(3):133–144. doi: 10.1159/000053334. [DOI] [PubMed] [Google Scholar]
- 8.FBPP Investigators. Multi-center genetic study of hypertension: the Family Blood Pressure Program (FBPP) Hypertension. 2002;39(1):3–9. doi: 10.1161/hy1201.100415. [DOI] [PubMed] [Google Scholar]
- 9.ARIC Investigators. The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. Am J Epidemiol. 1989;129(4):687–702. [PubMed] [Google Scholar]
- 10.O’Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998;63(1):259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lehmann EL, D’Abrera HJM. Nonparametrics: Statistical Methods Based on Ranks. San Francisco, CA: Holden-Day; 1975. [Google Scholar]
- 12.Peng B, Yu RK, Dehoff KL, Amos CI. Normalizing a large number of quantitative traits using empirical normal quantile transformation. BMC Proc. 2007;1(suppl 1):S156. doi: 10.1186/1753-6561-1-s1-s156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lange K, Boehnke M. Extensions to pedigree analysis, IV: covariance components models for multivariate traits. Am J Med Genet. 1983;14(3):513–524. doi: 10.1002/ajmg.1320140315. [DOI] [PubMed] [Google Scholar]
- 14.Jaquish CE, Blangero J, Haffner SM, Stern MP, Maccluer JW. Quantitative genetics of dehydroepiandrosterone sulfate and its relation to possible cardiovascular disease risk factors in Mexican Americans. Hum Hered. 1996;46(6):301–309. doi: 10.1159/000154368. [DOI] [PubMed] [Google Scholar]
- 15.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. MERLIN: rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30(1):97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 16.de Andrade M, Atkinson EJ, Lunde EM, Amos CI, Chen J. Estimating Genetic Components of Variance in Family Studies Using the MULTIC Routines. Rochester, MN: Dept of Health Science Research, Mayo Clinic; 2006. Technical Report Series No. 78. [Google Scholar]
- 17.de Andrade M, Thiel TJ, Yu L, Amos CI. Assessing linkage on chromosome 5 using components of variance approach: univariate versus multivariate. Genet Epidemiol. 1997;14(6):773–778. doi: 10.1002/(SICI)1098-2272(1997)14:6<773::AID-GEPI35>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 18.Bielinski SJ, Lynch Al, Miller MB, et al. Genome-wide linkage analysis for loci affecting pulse pressure: the Family Blood Pressure Program. Hypertension. 2005;46(6):1286–1293. doi: 10.1161/01.HYP.0000191706.41980.29. [DOI] [PubMed] [Google Scholar]
- 19.Domanski M, Norman J, Wolz M, Mitchell G, Pfeffer M. Cardiovascular risk assessment using pulse pressure in the first national health and nutrition examination survey (NHANES I) Hypertension. 2001;38(4):793–797. doi: 10.1161/hy1001.092966. [DOI] [PubMed] [Google Scholar]
- 20.Sobel E, Sengul H, Weeks DE. Multipoint estimation of identity-by-descent probabilities at arbitrary positions among marker loci on general pedigrees. Hum Hered. 2001;52(3):121–131. doi: 10.1159/000053366. [DOI] [PubMed] [Google Scholar]
- 21.Self S, Liang K-Y. Asymptotic properties of maximum likelihood estimators and likelihood ratio tests under nonstandard conditions. J Am Stat Assoc. 1987;82:605–610. [Google Scholar]
- 22.Morton NE. Significance levels in complex inheritance. Am J Hum Genet. 1998;62(3):690–697. doi: 10.1086/301741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11(3):241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- 24.Carmelli D, DeCarli C, Swan GE, et al. Evidence for genetic variance in white matter hyperintensity volume in normal elderly male twins. Stroke. 1998;29(6):1177–1181. doi: 10.1161/01.str.29.6.1177. [DOI] [PubMed] [Google Scholar]
- 25.Pfefferbaum A, Sullivan EV, Swan GE, Carmelli D. Brain structure in men remains highly heritable in the seventh and eighth decades of life. Neurobiol Aging. 2000;21(1):63–74. doi: 10.1016/s0197-4580(00)00086-5. [DOI] [PubMed] [Google Scholar]
- 26.Atwood LD, Wolf PA, Heard-Costa NL, et al. Genetic variation in white matter hyperintensity volume in the Framingham Study. Stroke. 2004;35(7):1609–1613. doi: 10.1161/01.STR.0000129643.77045.10. [DOI] [PubMed] [Google Scholar]
- 27.Carmelli D, Swan GE, Reed T, Wolf PA, Miller BL, DeCarli C. Midlife cardiovascular risk factors and brain morphology in identical older male twins. Neurology. 1999;52(6):1119–1124. doi: 10.1212/wnl.52.6.1119. [DOI] [PubMed] [Google Scholar]
- 28.Li YJ, Scott WK, Hedges DJ, et al. Age at onset in two common neurodegenerative diseases is genetically controlled. Am J Hum Genet. 2002;70(4):985–993. doi: 10.1086/339815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hicks AA, Petursson H, Jonsson T, et al. A susceptibility gene for late-onset idiopathic Parkinson’s disease. Ann Neurol. 2002;52(5):549–555. doi: 10.1002/ana.10324. [DOI] [PubMed] [Google Scholar]
- 30.Niemann S, Muller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. 2000;26(3):268–270. doi: 10.1038/81551. [DOI] [PubMed] [Google Scholar]
- 31.Niemann S, Muller U, Engelhardt D, Lohse P. Autosomal dominant malignant and catecholamine-producing paraganglioma caused by a splice donor site mutation in SDHC. Hum Genet. 2003;113(1):92–94. doi: 10.1007/s00439-003-0938-0. [DOI] [PubMed] [Google Scholar]
- 32.Cohn DH, Shohat T, Yahav M, et al. A locus for an autosomal dominant form of progressive renal failure and hypertension at chromosome 1q21. Am J Hum Genet. 2000;67(3):647–651. doi: 10.1086/303051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takada D, Emi M, Ezura Y, et al. Interaction between the LDL-receptor gene bearing a novel mutation and a variant in the apolipoprotein A-II promoter: molecular study in a 1135-member familial hypercholesterolemia kindred. J Hum Genet. 2002;47(12):656–664. doi: 10.1007/s100380200101. [DOI] [PubMed] [Google Scholar]
- 34.Pajukanta P, Lilja HE, Sinsheimer JS, et al. Familial combined hyperlipidemia is associated with upstream transcription factor 1 (USF1) Nat Genet. 2004;36(4):371–376. doi: 10.1038/ng1320. [DOI] [PubMed] [Google Scholar]
- 35.Pajukanta P, Terwilliger JD, Perola M, et al. Genomewide scan for familial combined hyperlipidemia genes in Finnish families, suggesting multiple susceptibility loci influencing triglyceride, cholesterol, and apolipoprotein B levels. Am J Hum Genet. 1999;64(5):1453–1463. doi: 10.1086/302365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wenzel K, Felix S, Kleber FX, et al. E-selectin polymorphism and atherosclerosis: an association study. Hum Mol Genet. 1994;3(11):1935–1937. doi: 10.1093/hmg/3.11.1935. [DOI] [PubMed] [Google Scholar]
- 37.Takei T, Iida A, Nitta K, et al. Association between single-nucleotide polymorphisms in selectin genes and immunoglobulin A nephropathy. Am J Hum Genet. 2002;70(3):781–786. doi: 10.1086/339077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang YP, Liu X, Kim JD, et al. Multiple genes for essential-hypertension susceptibility on chromosome 1q. Am J Hum Genet. 2007;80(2):253–264. doi: 10.1086/510918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Herrmann SM, Ricard S, Nicaud V, et al. The P-selectin gene is highly polymorphic: reduced frequency of the Pro715 allele carriers in patients with myocardial infarction. Hum Mol Genet. 1998;7(8):1277–1284. doi: 10.1093/hmg/7.8.1277. [DOI] [PubMed] [Google Scholar]
- 40.Carlson CS, Aldred SF, Lee PK, et al. Polymorphisms within the C-reactive protein (CRP) promoter region are associated with plasma CRP levels. Am J Hum Genet. 2005;77(1):64–77. doi: 10.1086/431366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kusari J, Verma US, Buse JB, Henry RR, Olefsky JM. Analysis of the gene sequences of the insulin receptor and the insulin-sensitive glucose transporter (GLUT-4) in patients with common-type non–insulin-dependent diabetes mellitus. J Clin Invest. 1991;88(4):1323–1330. doi: 10.1172/JCI115437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kissebah AH, Sonnenberg GE, Myklebust J, et al. Quantitative trait loci on chromosomes 3 and 17 influence phenotypes of the metabolic syndrome. Proc Natl Acad Sci U S A. 2000;97(26):14478–14483. doi: 10.1073/pnas.97.26.14478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tatsuguchi M, Furutani M, Hinagata J, et al. Oxidized LDL receptor gene (OLR1) is associated with the risk of myocardial infarction. Biochem Biophys Res Commun. 2003;303(1):247–250. doi: 10.1016/s0006-291x(03)00326-7. [DOI] [PubMed] [Google Scholar]
- 44.Lambert JC, Luedecking-Zimmer E, Merrot S, et al. Association of 3′-UTR polymorphisms of the oxidised LDL receptor 1 (OLR1) gene with Alzheimer’s disease. J Med Genet. 2003;40(6):424–430. doi: 10.1136/jmg.40.6.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fogli A, Schiffmann R, Hugendubler L, et al. Decreased guanine nucleotide exchange factor activity in elF2B-mutated patients. Eur J Hum Genet. 2004;12(7):561–566. doi: 10.1038/sj.ejhg.5201189. [DOI] [PubMed] [Google Scholar]
- 46.Gong M, Zhang H, Schulz H, et al. Genome-wide linkage reveals a locus for human essential (primary) hypertension on chromosome 12p. Hum Mol Genet. 2003;12(11):1273–1277. doi: 10.1093/hmg/ddg135. [DOI] [PubMed] [Google Scholar]
- 47.Schuster H, Wienker TF, Toka HR, et al. Autosomal dominant hypertension and brachydactyly in aTurkish kindred resembles essential hypertension. Hypertension. 1996;28(6):1085–1092. doi: 10.1161/01.hyp.28.6.1085. [DOI] [PubMed] [Google Scholar]
- 48.Pericak-Vance MA, Bass MP, Yamaoka LH, et al. Complete genomic screen in late-onset familial Alzheimer disease: evidence for a new locus on chromosome 12. JAMA. 1997;278(15):1237–1241. [PubMed] [Google Scholar]
- 49.Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 50.Dong C, Wang S, Li WD, Li D, Zhao H, Price RA. Interacting genetic loci on chromosomes 20 and 10 influence extreme human obesity. Am J Hum Genet. 2003;72(1):115–124. doi: 10.1086/345648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Souto JC, Blanco-Vaca F, Soria JM, et al. A genomewide exploration suggests a new candidate gene at chromosome 11q23 as the major determinant of plasma homocysteine levels: results from the GAIT project. Am J Hum Genet. 2005;76(6):925–933. doi: 10.1086/430409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Neumann HP, Bausch B, McWhinney SR, et al. Freiburg-Warsaw-Columbus Pheochromocytoma Study Group. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346(19):1459–1466. doi: 10.1056/NEJMoa020152. [DOI] [PubMed] [Google Scholar]
- 53.von Eckardstein A, Holz H, Sandkamp M, Weng W, Funke H, Assmann G. Apolipoprotein C-III(Lys58—Glu): identification of an apolipoprotein C-III variant in a family with hyperalphalipoproteinemia. J Clin Invest. 1991;87(5):1724–1731. doi: 10.1172/JCI115190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ye S, Eriksson P, Hamsten A, Kurkinen M, Humphries SE, Henney AM. Progression of coronary atherosclerosis is associated with a common genetic variant of the human stromelysin-1 promoter which results in reduced gene expression. J Biol Chem. 1996;271(22):13055–13060. doi: 10.1074/jbc.271.22.13055. [DOI] [PubMed] [Google Scholar]
- 55.Franceschini G, Sirtori CR, Capurso A, II, Weisgraber KH, Mahley RW. A-IMilano apoprotein: decreased high density lipoprotein cholesterol levels with significant lipoprotein modifications and without clinical atherosclerosis in an Italian family. J Clin Invest. 1980;66(5):892–900. doi: 10.1172/JCI109956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dallinga-Thie GM, van Linde-Sibenius Trip M, Rotter JI, et al. Complex genetic contribution of the Apo AI-CIII-AIV gene cluster to familial combined hyperlipidemia: identification of different susceptibility haplotypes. J Clin Invest. 1997;99(5):953–961. doi: 10.1172/JCI119260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pennacchio LA, Olivier M, Hubacek JA, et al. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science. 2001;294(5540):169–173. doi: 10.1126/science.1064852. [DOI] [PubMed] [Google Scholar]
- 58.Endo K, Yanagi H, Araki J, Hirano C, Yamakawa-Kobayashi K, Tomura S. Association found between the promoter region polymorphism in the apolipoprotein A-V gene and the serum triglyceride level in Japanese schoolchildren. Hum Genet. 2002;111(6):570–572. doi: 10.1007/s00439-002-0825-0. [DOI] [PubMed] [Google Scholar]
- 59.Kort EN, Ballinger DG, Ding W, et al. Evidence of linkage of familial hypoalphalipoproteinemia to a novel locus on chromosome 11q23. Am J Hum Genet. 2000;66(6):1845–1856. doi: 10.1086/302945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vaughan CJ, Casey M, He J, et al. Identification of a chromosome 11q23.2-q24 locus for familial aortic aneurysm disease, a genetically heterogeneous disorder. Circulation. 2001;103(20):2469–2475. doi: 10.1161/01.cir.103.20.2469. [DOI] [PubMed] [Google Scholar]
- 61.Prakash S, Chung KW, Sinha S, et al. Autosomal dominant progressive nephropathy with deafness: linkage to a new locus on chromosome 11q24. J Am Soc Nephrol. 2003;14(7):1794–1803. doi: 10.1097/01.asn.0000071513.73427.97. [DOI] [PubMed] [Google Scholar]
- 62.Atwond LD, Heard-Costa NL, Cupples LA, Jaquish CE, Wilson PW, D’Agostino RB. Genomewide linkage analysis of body mass index across 28 years of the Framingham Heart Study. Am J Hum Genet. 2002;71(5):1044–1050. doi: 10.1086/343822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Feitosa MF, Borecki IB, Rich SS, et al. Quantitative-trait loci influencing body-mass index reside on chromosomes 7 and 13: the National Heart, Lung, and Blood Institute Family Heart Study. Am J Hum Genet. 2002;70(1):72–82. doi: 10.1086/338144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hanson RL, Ehm MG, Pettitt DJ, et al. An autosomal genomic scan for loci linked to type II diabetes mellitus and body-mass index in Pima Indians. Am J Hum Genet. 1998;63(4):1130–1138. doi: 10.1086/302061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen YH, Lin SJ, Lin MW, et al. Microsatellite polymorphism in promoter of heme oxygenase-1 gene is associated with susceptibility to coronary artery disease in type 2 diabetic patients. Hum Genet. 2002;111(1):1–8. doi: 10.1007/s00439-002-0769-4. [DOI] [PubMed] [Google Scholar]
- 66.Tai ES, Demissie S, Cupples LA, et al. Association between the PPARA L162V polymorphism and plasma lipid levels: the Framingham Offspring Study. Arterioscler Thromb Vasc Biol. 2002;22(5):805–810. doi: 10.1161/01.atv.0000012302.11991.42. [DOI] [PubMed] [Google Scholar]
- 67.Ozaki K, Inoue K, Sato H, et al. Functional variation in LGALS2 confers risk of myocardial infarction and regulates lymphotoxin-alpha secretion in vitro. Nature. 2004;429(6987):72–75. doi: 10.1038/nature02502. [DOI] [PubMed] [Google Scholar]
- 68.Elbaz A, Levecque C, Clavel J, et al. CYP2D6 polymorphism, pesticide exposure, and Parkinson’s disease. Ann Neurol. 2004;55(3):430–434. doi: 10.1002/ana.20051. [DOI] [PubMed] [Google Scholar]
- 69.Manolio TA, Burke GL, O’Leary DH, et al. CHS Collaborative Research Group. Relationships of cerebral MRI findings to ultrasonographic carotid atherosclerosis in older adults: the Cardiovascular Health Study. Arterioscler Thromb Vasc Biol. 1999;19(2):356–365. doi: 10.1161/01.atv.19.2.356. [DOI] [PubMed] [Google Scholar]
- 70.Staessen JA, Richart T, Birkenhager WH. Less atherosclerosis and lower blood pressure for a meaningful life perspective with more brain. Hypertension. 2007;49(3):389–400. doi: 10.1161/01.HYP.0000258151.00728.d8. [DOI] [PubMed] [Google Scholar]
- 71.Feijge MA, Ansink K, Vanschoonbeek K, Heemskerk JW. Control of platelet activation by cyclic AMP turnover and cyclic nucleotide phosphodiesterase type-3. Biochem Pharmacol. 2004;67(8):1559–1567. doi: 10.1016/j.bcp.2003.12.028. [DOI] [PubMed] [Google Scholar]
- 72.Berglund EO, Murai KK, Fredette B, et al. Ataxia and abnormal cerebellar micro-organization in mice with ablated contactin gene expression. Neuron. 1999;24(3):739–750. doi: 10.1016/s0896-6273(00)81126-5. [DOI] [PubMed] [Google Scholar]
- 73.Clark LN, Wang Y, Karlins E, et al. Frequency of LRRK2 mutations in early- and late-onset Parkinson disease. Neurology. 2006;67(10):1786–1791. doi: 10.1212/01.wnl.0000244345.49809.36. [DOI] [PubMed] [Google Scholar]
- 74.Rajput A, Dickson DW, Robinson CA, et al. Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology. 2006;67(8):1506–1508. doi: 10.1212/01.wnl.0000240220.33950.0c. [DOI] [PubMed] [Google Scholar]
- 75.Rogaeva E, Meng Y, Lee JH, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39(2):168–177. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Amos CI, Laing AE. A comparison of univariate and multivariate tests for genetic linkage. Genet Epidemiol. 1993;10(6):671–676. doi: 10.1002/gepi.1370100657. [DOI] [PubMed] [Google Scholar]
- 77.Jacob HJ, Lindpaintner K, Lincoln SE, et al. Genetic mapping of a gene causing hypertension in the stroke-prone spontaneously hypertensive rat. Cell. 1991;67(1):213–224. doi: 10.1016/0092-8674(91)90584-l. [DOI] [PubMed] [Google Scholar]
- 78.Ikeda K, Yamori Y. Gene related to stroke in stroke-prone spontaneously hypertensive rats. Clin Exp Pharmacol Physiol. 1999;26(7):566–567. doi: 10.1046/j.1440-1681.1999.03087.x. [DOI] [PubMed] [Google Scholar]
- 79.Bourgain C, Genin E, Cox N, Clerget-Darpoux F. Are genome-wide association studies all that we need to dissect the genetic component of complex human diseases? Eur J Hum Genet. 2007;15(3):260–263. doi: 10.1038/sj.ejhg.5201753. [DOI] [PubMed] [Google Scholar]