

Abstract
The frequencies of antigen-specific CD4+ T cells in samples of human tissue has been difficult to determine accurately ex vivo, particularly for autoimmune diseases such as multiple sclerosis or Type 1 diabetes. Conventional approaches involve the expansion of primary T cells in vitro to increase the numbers of cells, and a subsequent assessment of the frequencies of antigen-specific T cells in the expanded population by limiting dilution or by using fluorescently labeled tetramers of peptide-loaded major histocompatibility complex (MHC) receptors. Here we describe an alternative approach that uses arrays of subnanoliter wells coated with recombinant peptide-loaded MHC Class II monomers to isolate and stimulate individual CD4+ T cells in an antigen-specific manner. In these experiments, activation was monitored using microengraving to capture two cytokines (IFNγ and IL-17) released from single cells. This new method should enable direct enumeration of antigen-specific CD4+ T cells ex vivo from clinical samples.
Keywords: Single-cell assays, immunology, soft lithography, microengraving, antigen-specific T cells
During immune responses, only a small fraction of lymphocytes within the adaptive immune system of the host recognize a specific antigen. One subset of these cells—CD4+ T helper (T) cells—play a critical role in coordinating the adaptive immune response. Clonotypic variants of this lineage bear unique T cell receptors (TCRs) that can recognize antigenic peptides of approximately 15 amino acids bound in heterodimeric, surface-expressed receptors—major histocompatibility complex (MHC) Class II—displayed by antigen-presenting cells (APCs) such as dendritic cells and macrophages.1 This event induces activation and proliferation of the interacting T cells, who then coordinate both local and systemic events in the evolving immune response by releasing cytokines such as IFNγ, IL-4, or IL-17.2 To understand how CD4+ T cells influence the development of an immune response during the course of an infection or an autoimmune disease,3 it is important to identify, quantify, and characterize them. The T cells that respond to a specific antigen, however, are infrequent: in peripheral blood, the number is reported to range from 1:1,000 to 1:1,000,000 depending on the quality of the host's immune system and the progression of the response.4
The low frequency of antigen-specific CD4+ T cells necessitates sensitive assays to detect and recover them from a clinical sample (e.g., blood, tissue biopsy, cerebrospinal fluid). The presence of antigen-specific T cells traditionally has been inferred by assessing the extent of proliferation in response to the antigen of interest, or by measuring cytokine production using either enzyme-linked immunosorbant assays (ELISA) or the related ELISpot method to detect single cells releasing cytokines.5 Recombinant monomers of MHC Class II reconstituted with specific peptides of interest, or tetrameric constructs formed from biotinylated monomers and streptavidin, have been used to label and activate CD4+ T cells for detection and isolation by fluorescence-assisted cell sorting (FACS).6 Although these reagents can facilitate the enumeration of T cells ex vivo following an infection,7 the numbers of cells typically present in samples of small volumes from patients with autoimmune diseases such as multiple sclerosis or Type 1 diabetes are low,8 and the avidities of the tetramers for the TCR can be poor. These factors hinder the detection of autoantigen-reactive T cells.9 To assess the frequencies of these cells, the current practice involves in vitro expansion of the T cells by exposure to the antigen of interest, followed by labeling with MHC Class II tetramers. Expansion of cells in vitro for long periods (1-2 weeks), however, can introduce selective bias in the populations of cells analyzed.10 New methods sensitive to low-frequency antigen-specific CD4+ T cells ex vivo would improve the study of human diseases, especially ones where clinical samples are limited.
The maturation of microfabricated systems compatible with living cells has enabled new approaches to study individual cells, and to characterize the heterogeneity within populations of cells. Many reported microsystems for determining the identities and functional responses of single cells rely on microfluidics or arrays of microwells to position them.11 In most cases, rare cells have been identified by differentially labeled surface markers and imaging cytometry.12 Other demonstrations have used array-based variations on intracellular staining or ELISpot to assess functional responses of T cells exposed to broadly activating, exogenously applied stimuli.13 For B cells, detection of antigen-specific cells in arrays of microwells has been accomplished by applying the antigen to the cells themselves,14 or by detecting antibodies produced by the cells that bind to the antigen of interest.15 One strategy for detecting antigen-induced calcium release from T cells in microfluidic channels has been reported,16 but there remains a significant need for methods to identify and classify antigen-specific T cells on the basis of the cytokines they release.
Microengraving is a soft lithographic method that uses an elastomeric array of subnanoliter wells loaded with cells to print microarrays of proteins in which each element maps directly to a particular well with a cell.15b, c The microarray of captured proteins can be configured to detect antibodies or cytokines from primary human lymphocytes.15a Here we report a simple adaptation of the technique to activate T cells directly in the wells in an antigen-specific manner and to detect them by the capture of released cytokines using microengraving (Figure 1). We demonstrate that this approach enables the specific activation and enumeration of T cell clones reactive to two different peptides derived from haemagglutenin (HA p306-318) and myelin oligodendrocyte glycoprotein (MOG p97-109). This method should enable detailed analyses of the frequencies and functions of antigen-specific T cells with sensitivity sufficient to assess clinical samples ex vivo.
Figure 1.
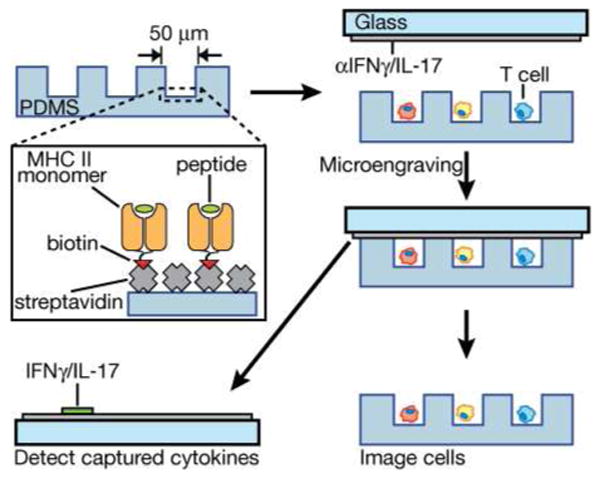
Schematic illustration of surface modification for on-chip stimulation of antigen-specific T cells and subsequent detection of released cytokines by microengraving. Streptavidin is applied to oxidized PDMS, then exposed to biotinylated monomers of recombinant, peptide-loaded MHC Class II and biotinylated anti-CD28 (not shown for clarity). The modified surface is blocked with bovine serum albumin immediately prior to loading cells. Cells are then deposited into the microwells and incubated to allow activation. After a period of time (6-18 h), the array of cells is sealed against a glass slide bearing anti-cytokine antibodies and incubated for 2 h. After this process (microengraving), the array of cells is imaged to determine the number of cells per well, and the array of captured cytokines is labeled and imaged.
The material used to form the arrays of wells used for microengraving—poly(dimethylsiloxane) (PDMS)—readily adsorbs proteins from solution if not treated to prevent this modification.17 Given the ubiquity of biotinylated reagents—particularly MHC Class I and Class II monomers used for labeling in flow cytometry, we chose to adsorb streptavidin directly onto the surface of plasma-oxidized PDMS to provide a surface on which recombinant monomers of MHC Class II would bind with a predominantly favorable orientation to engage the TCRs on T cells. We treated an array of wells with an oxygen plasma, and then applied a solution of streptavidin to the surface of the array for 2 h at 37°C. To verify that the physisorbed coating remained functionally active, we applied a series of defined spots of a biotinylated antibody (OKT3) at different concentrations onto a streptavidin-coated slab of PDMS. After washing and staining with a fluorescently labeled streptavidin, we measured the background-corrected relative fluorescence intensities of each spot (Figure S-1). These experiments suggested that the streptavidin remained capable of binding the poly-biotinylated antibody, and that the maximum amount of antibody captured increased with the concentration of streptavidin applied (with saturation of the available binding sites occurring at concentrations greater than 3-5 μg/mL of the poly-biotinylated antibody).
The measurements on the modified PDMS surfaces indicated that the streptavidin-coated surfaces would bind biotinylated antibodies, but our primary purpose for this modification was to enable activation of T cells deposited in the treated wells. To test the ability of the functionalized array to activate T cells, we prepared a surface coated with a combination of biotinylated anti-CD3 (OKT3) and anti-CD28 (28.2)—a combined stimulus commonly employed for a polyclonal activation of T cells through engagement of the TCR/CD3 complex and the co-stimulatory receptor CD28.18 We deposited peripheral blood mononuclear cells (PBMCs) into the array of wells supporting the combination of antibodies, and in parallel, deposited a portion of the same cells into a 96-well microtiter plate coated with streptavidin and both biotinylated antibodies as a control. After defined periods of stimulation, we transferred the cells stimulated in the 96-well plate onto an untreated array of wells, and conducted a microengraving experiment with both arrays to measure the frequencies of cells producing IFNγ (Figure 2a,b). These experiments indicated that the two approaches for polyclonal activation of primary T cells (in an array of wells or in a microtiter plate) induced cytokine responses of similar magnitudes at each time point evaluated.
Figure 2. Optimization and analysis of on-chip stimulation.
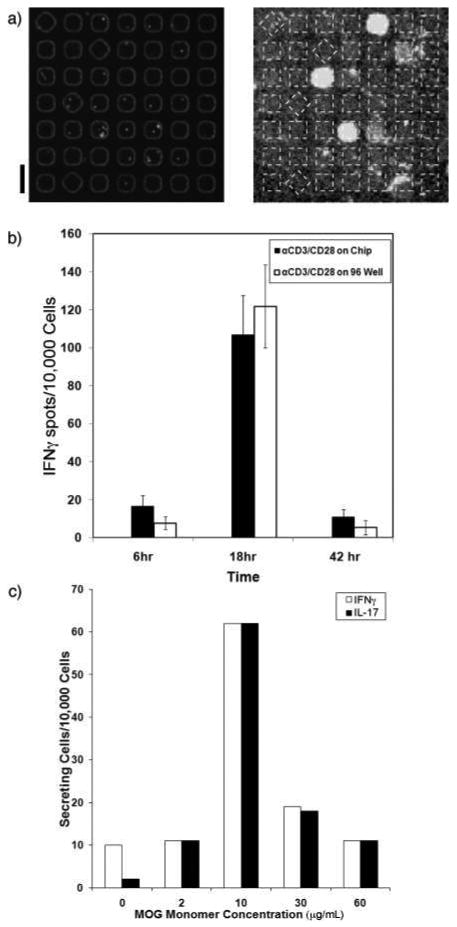
a) Fluorescent micrographs of a block of microwells containing PBMCs (labeled with Hoescht dye) and the matched region of the printed microarray of captured IFNγ. The array of microwells was coated with streptavidin (200 μg/mL), followed by biotinylated anti-CD3 (10 μg/mL) and anti-CD28 (1 μg/mL). The cells were incubated on the modified wells for 6 h at 37 °C prior to microengraving. The boxes (dashed lines) on the right hand image have been overlaid to clarify the regions of interest. The scale bar is 100 μm. b) A plot of the measured frequencies of IFNγ-secreting PBMCs after stimulation either directly on-chip for 6, 18, or 42 h (black) or in bulk in a 96-well microtiter plate (white) for 6, 18, or 42 h. After bulk stimulation, the cells were deposited into microwells for microengraving. The error bars represent the range in responses measured in three independent experiments. c) A plot of the frequencies of IFNγ-secreting (white) and IL-17-secreting (black) MOG-specific clones following on-chip stimulation for 6 h as a function of applied concentration of MOGp97-109-loaded HLA DRB1*0401 monomer (0, 2, 10, 30, 60 μg/mL).
To test whether the scheme for modifying the surfaces of the microwells would then allow antigen-specific activation of T cells, we conducted an experiment using a MOG-reactive CD4+ T cell clone expanded from a multiple sclerosis patient, and selected using MHC Class II tetramers of human leukocyte antigen (HLA) DRB1*0401 loaded with MOG97-109 peptide. The clone was distributed into microwells modified with streptavidin, biotinylated anti-CD28, and biotinylated MOG-loaded MHC Class II monomer. We found that the maximum response for this clone after on-chip stimulation occurred 6 h after deposition into the wells (Figure S-2). This time was faster than the maximum activation observed for the primary T cells from whole PBMCs, but is consistent with the known sensitivity of T cell clones to activating stimuli after expansion in vitro.8
We then measured the response induced in the clone as a function of the concentration of monomer applied to the streptavidin-coated microwells (Figure 2c). This particular clone could produce either IFNγ or IL-17 upon activation. The maximum number of cytokine-secreting cells occurred when a solution of 10 μg/mL of biotinylated MHC Class II monomer was applied. A low concentration of monomer (2 μg/mL) failed to induce a response distinct from the negative control (0 μg/mL). This observation indicates that there was insufficient stimulation to exceed the threshold of signal required for activation. Concentrations above 10 μg/mL reduced the number of activated cells detected. This result suggests that, like polyclonal activation, there is an optimal concentration for the surface-bound activating molecules. Overstimulation may induce cell death or non-responsiveness. Alternatively, saturation of the available binding sites on the immobilized streptavidin by the MHC Class II monomers at high concentrations may limit the subsequent immobilization of the requisite costimulatory factor (biotinylated anti-CD28), and therefore, reduces the quality of activation.
Using the optimal determined conditions for activating the MOG-specific clone with MOG-loaded MHC Class II monomers, we then evaluated the specificity of the response. We measured the responses of both a MOG-specific clone and a HA-specific clone when stimulated with either MOG-loaded or HA-loaded MHC Class II monomers (Figure 3). The frequencies of IFNγ+ and IL-17+ cells were greatest when the clones were matched to the appropriate peptide-MHC Class II complex. For both clones evaluated, antigen-specific stimulation induced cytokine secretion from a similar number of cells as the polyclonal stimulation (anti-CD3/anti-CD28). This result suggests that the fraction of clonal cells in these populations that can be activated under these conditions is independent of the nature of the activating signal (antigen-specific or polyclonal stimulus).
Figure 3. Specific activation of T cell clones on-chip.
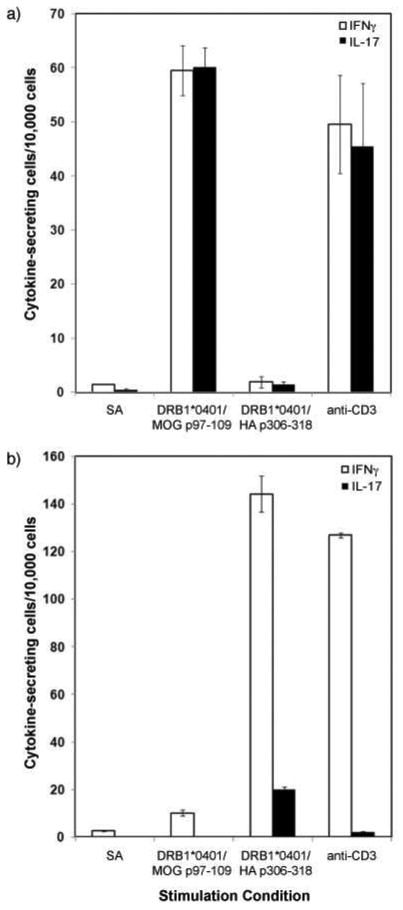
Plots of the frequencies of IFNγ-secreting (white) and IL-17 secreting (black) (a) MOG-specific and (b) HA-specific T cell clones following on-chip stimulation for 6 h. The stimulation conditions included streptavidin only (SA), MOGp97-109-loaded HLA DRB1*0401 monomer (10 μg/mL), HAp306-318-loaded HLA DRB1*0401 monomer (10 μg/mL), and anti-CD3 (OKT3; 10 μg/mL). All conditions, except streptavidin only, also included anti-CD28 (1 μg/mL) for co-stimulation. Both IFNγ (white bars) and IL-17 (black bars) were captured by microengraving after stimulation. The error bars indicate the range in frequencies measured from three independent experiments.
The HA-reactive clones were consistently more responsive than the MOG-reactive clones. The MOG-reactive and HA-reactive clones evaluated here were selected by their affinity for MHC Class II tetramer loaded with their respective peptides from two different individuals, not by which cytokines they secreted. The data on their functional responses suggest that the MOG-reactive clone derived from a Th17 cell (IL-17 and IFNγ secretion; 96% dual secretion with monomeric stimulation) while the HA-reactive clone derived from a Th1 cell (IFNγ; 14% dual secretion). It is interesting that only a fraction of the total number of clones loaded on to the array responded. This outcome was reproducible, and consistent with the frequencies of individual activated, antigen-specific clones measured by a traditional ELISpot assay (Figure S-3). The small frequency of IL-17+ cells detected among the activated HA-reactive clones further indicates there is functional heterogeneity among these cells. These observations underscore the variability intrinsic to the dynamic functional responses among isogenic cells.19
There was no significant difference between the number of cells activated by stimulation on the streptavidin alone, or on the irrelevant peptide-loaded monomer. The detected false-positive events, therefore, are a result of non-specific activation, rather than cross-reactivity of the clones to an alternate antigen. The percentage of cells activated non-specifically was comparable to that observed by a traditional ELISpot assay (Figure S-3), and indicates an approximate false discovery rate of 3-6% for each clone. This rate suggests that the minimum number of antigen-specific events scored should be ∼10-20 per assay. Given that our current arrays of microwells can hold up to ∼250,000 cells, these estimates suggest a lower limit of detection of ∼1 in 10,000 cells—an order of magnitude more sensitive than assessing frequencies with flow cytometry and labeled tetramers and comparable to the estimated frequencies of circulating antigen-specific CD4+ T cells.6
Taken together, these experiments demonstrate that the simple method described here for modifying the surface of arrays of microwells with peptide-loaded recombinant MHC Class II monomers allows antigen-specific activation of CD4+ T cells. A similar approach using monomers of peptide-loaded MHC Class I monomers should also allow functional assessments of CD8+ T cells. Combined with the microengraving method, on-chip stimulation of either primary T cells, or clonal T cells with known reactivities, can be assessed by the capture of secreted cytokines—a functional measure of activation. Using either polyclonal or antigen-specific stimuli, isolated single T cells were induced to secrete cytokines as effectively as traditional methods to activate T cells in bulk cultures. One advantage of stimulating T cells in isolation is that it should minimize non-specific activation mediated by other ‘by-stander’ cells in a mixed culture. A specific advantage for using microengraving to detect activation is that it will make it possible to evaluate the functional responses for individual T cells over time by serial microengraving.15d Although physical adsorption of activating biomolecules onto PDMS is sufficient, long-term studies of on-chip activation (>24-48 h) may benefit from further refinements that allow covalent attachment of the proteins. Nevertheless, we believe that the combined approach described here should now enable the detection and characterization of primary autoantigen-reactive CD4+ T cells ex vivo from patients with autoimmune diseases—a technical challenge that currently available analytical tools have been unable to address.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health (5U19AI050864-07). JCL is a Texaco-Mangelsdorf Career Development Professor.
Footnotes
Supporting Information Available. Materials and methods, three supporting figures.
References
- 1.Huppa JB, Davis MM. Nat Rev Immunol. 2003;3:973–83. doi: 10.1038/nri1245. [DOI] [PubMed] [Google Scholar]
- 2.Reinhardt RL, Kang SJ, Liang HE, Locksley RM. Curr Opin Immunol. 2006;18:271–7. doi: 10.1016/j.coi.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 3.Ohashi PS. Nat Rev Immunol. 2002;2:427–38. doi: 10.1038/nri822. [DOI] [PubMed] [Google Scholar]
- 4.(a) Bieganowska KD, Ausubel LJ, Modabber Y, Slovik E, Messersmith W, Hafler DA. J Exp Med. 1997;185:1585–94. doi: 10.1084/jem.185.9.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Martin R, Jaraquemada D, Flerlage M, Richert J, Whitaker J, Long EO, McFarlin DE, McFarland HF. J Immunol. 1990;145:540–8. [PubMed] [Google Scholar]; (c) Ota K, Matsui M, Milford EL, Mackin GA, Weiner HL, Hafler DA. Nature. 1990;346:183–7. doi: 10.1038/346183a0. [DOI] [PubMed] [Google Scholar]; (d) Pette M, Fujita K, Wilkinson D, Altmann DM, Trowsdale J, Giegerich G, Hinkkanen A, Epplen JT, Kappos L, Wekerle H. Proc Natl Acad Sci USA. 1990;87:7968–72. doi: 10.1073/pnas.87.20.7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pelfrey CM, Rudick RA, Cotleur AC, Lee JC, Tary-Lehmann M, Lehmann PV. J Immunol. 2000;165:1641–51. doi: 10.4049/jimmunol.165.3.1641. [DOI] [PubMed] [Google Scholar]
- 6.Mallone R, Nepom GT. Clin Immunol. 2004;110:232–42. doi: 10.1016/j.clim.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 7.Meyer AL, Trollmo C, Crawford F, Marrack P, Steere AC, Huber BT, Kappler J, Hafler DA. Proc Natl Acad Sci USA. 2000;97:11433–8. doi: 10.1073/pnas.190335897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mallone R, Kochik SA, Reijonen H, Carson B, Ziegler SF, Kwok WW, Nepom GT. Blood. 2005;106:2798–805. doi: 10.1182/blood-2004-12-4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Gebe JA, Falk BA, Rock KA, Kochik SA, Heninger AK, Reijonen H, Kwok WW, Nepom GT. Eur J Immunol. 2003;33:1409–17. doi: 10.1002/eji.200323871. [DOI] [PubMed] [Google Scholar]; (b) Novak EJ, Liu AW, Nepom GT, Kwok WW. J Clin Invest. 1999;104:R63–7. doi: 10.1172/JCI8476. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Standifer NE, Burwell EA, Gersuk VH, Greenbaum CJ, Nepom GT. Clin Immunol. 2009;132:312–20. doi: 10.1016/j.clim.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geginat J, Sallusto F, Lanzavecchia A. J Exp Med. 2001;194:1711–9. doi: 10.1084/jem.194.12.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) El-Ali J, Sorger P, Jensen KF. Nature. 2006;442:403–11. doi: 10.1038/nature05063. [DOI] [PubMed] [Google Scholar]; (b) Di Carlo D, Lee L. Anal Chem. 2006;78:7918–25. doi: 10.1021/ac069490p. [DOI] [PubMed] [Google Scholar]
- 12.(a) Whitaker R, Walt D. Anal Chem. 2007 doi: 10.1021/ac701744x. [DOI] [PubMed] [Google Scholar]; (b) Rettig J, Folch A. Anal Chem. 2005 doi: 10.1021/ac0505977. [DOI] [PubMed] [Google Scholar]; (c) Revzin A, Sekine K, Sin A, Tompkins R, Toner M. Lab Chip. 2005;5:30. doi: 10.1039/b405557h. [DOI] [PubMed] [Google Scholar]; (d) Hosokawa M, Arakaki A, Takahashi M, Mori T, Takeyama H, Matsunaga T. Anal Chem. 2009;81:5308–13. doi: 10.1021/ac900535h. [DOI] [PubMed] [Google Scholar]
- 13.(a) Tajiri K, Kishi H, Ozawa T, Sugiyama T, Muraguchi A. Cytometry A. 2009;75:282–8. doi: 10.1002/cyto.a.20675. [DOI] [PubMed] [Google Scholar]; (b) Zhu H, Stybayeva G, Silangcruz J, Yan J, Ramanculov E, Dandekar S, George MD, Revzin A. Anal Chem. 2009;81:8150–6. doi: 10.1021/ac901390j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Tajiri K, Kishi H, Tokimitsu Y, Kondo S, Ozawa T, Kinoshita K, Jin A, Kadowaki S, Sugiyama T, Muraguchi A. Cytometry A. 2007;71:961–7. doi: 10.1002/cyto.a.20471. [DOI] [PubMed] [Google Scholar]; (b) Tokimitsu Y, Kishi H, Kondo S, Honda R, Tajiri K, Motoki K, Ozawa T, Kadowaki S, Obata T, Fujiki S, Tateno C, Takaishi H, Chayama K, Yoshizato K, Tamiya E, Sugiyama T, Muraguchi A. Cytometry. 2007;71:1003–10. doi: 10.1002/cyto.a.20478. [DOI] [PubMed] [Google Scholar]
- 15.(a) Bradshaw EM, Kent SC, Tripuraneni V, Orban T, Ploegh HL, Hafler DA, Love JC. Clin Immunol. 2008;129:10–18. doi: 10.1016/j.clim.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Love JC, Ronan JL, Grotenbreg GM, van der Veen AG, Ploegh HL. Nat Biotech. 2006;24:703–707. doi: 10.1038/nbt1210. [DOI] [PubMed] [Google Scholar]; (c) Ogunniyi AO, Story CM, Papa E, Guillen E, Love JC. Nature Protocols. 2009;4:767–782. doi: 10.1038/nprot.2009.40. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Story CM, Papa E, Hu CC, Ronan JL, Herlihy K, Ploegh HL, Love JC. Proc Natl Acad Sci USA. 2008;105:17902–17907. doi: 10.1073/pnas.0805470105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Jin A, Ozawa T, Tajiri K, Obata T, Kondo S, Kinoshita K, Kadowaki S, Takahashi K, Sugiyama T, Kishi H, Muraguchi A. Nat Med. 2009;15:1088–1092. doi: 10.1038/nm.1966. [DOI] [PubMed] [Google Scholar]
- 16.Faley S, Seale K, Hughey J, Schaffer DK, VanCompernolle S, McKinney B, Baudenbacher F, Unutmaz D, Wikswo JP. Lab Chip. 2008;8:1700–12. doi: 10.1039/b719799c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ostuni E, Chen C, Ingber D, Whitesides G. Langmuir. 2001;17:2828–2834. [Google Scholar]
- 18.Thompson CB, Lindsten T, Ledbetter JA, Kunkel SL, Young HA, Emerson SG, Leiden JM, June CH. Proc Natl Acad Sci USA. 1989;86:1333–7. doi: 10.1073/pnas.86.4.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spencer SL, Gaudet S, Albeck J, Burke JM, Sorger P. Nature. 2009;459:428–32. doi: 10.1038/nature08012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.