

Abstract
Seizures are relatively common in Alzheimer disease (AD) and other neurodegenerative disorders. To our knowledge, however, there have been no reports of seizures associated with corticobasal degeneration (CBD). We describe a patient with brain biopsy features suggestive of CBD whose course was complicated by complex partial seizures with secondary generalization. Thus, the occurrence of seizures in a patient with dementia should not exclude the diagnosis of CBD.
CASE HISTORY
A 65-year-old right-handed woman presented to our clinic with a 2-year history of difficulty in driving and a 1-year history of declining speech, reading, and writing. At her initial visit she also complained of gait imbalance, mild short-term memory deficits, and difficulty in performing household chores. Her medical history was significant for hyperlipidemia, hypertension, osteoporosis, mild left hearing loss, and cervical radiculopathy. Her daily medications consisted of atorvastatin, alendronate, aspirin, calcium, a multivitamin, and vitamins C and D. She was college educated. She had never used tobacco or illicit drugs, and she drank alcohol only rarely. There was no family history of early-onset dementia.
Her neurological examination showed slow, spastic speech with phonemic paraphasias, mild naming difficulty, mildly impaired memory, perseveration, frequent (and at times inappropriate) laughter, a partial Balint syndrome with asimultanagnosia and optic apraxia, visual extinction to double simultaneous stimulation on the left, bilateral blepharospasm, and square-wave jerks. There was no supranuclear gaze palsy. She had asymmetric limb ideomotor apraxia, rigidity, and impaired rapid alternating movements, all worse on the left. Her gait was slow and slightly wide-based. Sensation, reflex, and coordination testing were unremarkable, and her plantar responses were flexor.
Her mini-mental state examination (MMSE) score was 26/30, losing one point each for loss of orientation, failing to recall 1/3 words, failing to correctly spell “world” backwards, and poor copy of intersecting pentagons. Neuropsychological testing showed deficits in visual spatial skills, confrontation naming and language, calculations, abstraction, working memory, and set-shifting, with relative sparing of episodic memory. Her initial brain MRI demonstrated moderate bilateral parietal, frontal and caudate atrophy. There were no white matter abnormalities, post-contrast enhancement or restricted diffusion.
Shortly after her clinic visit the patient was found unconscious by a family member. She was admitted to a local hospital where she was observed to have two left-sided focal motor seizures. A repeat brain MRI was unchanged. She was prescribed levetiracetam and discharged home. One week later, after missing two doses of her antiepileptic medication, she had a precipitous change and was noted to be neglectful of her left side. Examination revealed a stable MMSE score of 26, left visual, auditory, and tactile neglect and a right gaze preference. A complete blood count, electrolytes, liver and renal function studies were normal. An MRI with fluid-attenuated inversion recovery (FLAIR) sequence showed hyperintensity in right parietal and posterior temporal lobes without restricted diffusion (Fig 1). Electroencephalogram (EEG) at the time showed a semirhythmic, medium amplitude background with a well-developed 9-10 Hz alpha rhythm. On the right, the background was of higher amplitude, disordered and slowed to the theta range, with spikes and sharp waves arising predominantly from the right parietal region in 3-4 second bursts every 8-12 seconds. Sharp waves from the right hemisphere occasionally spread to the left anterior quadrant (Fig 1). There was no clear clinical correlate to these discharges, although the degree of neglect and right gaze preference fluctuated while the recording was being obtained. Repeat MRI one week later showed cortical T1 shortening suggestive of laminar necrosis in the region of the FLAIR abnormality seen previously. The FLAIR changes resolved after several months and were attributed to postictal factors (Fig 1).
Figure 1. MRI and Electroencephalogram after onset of complex partial seizures.
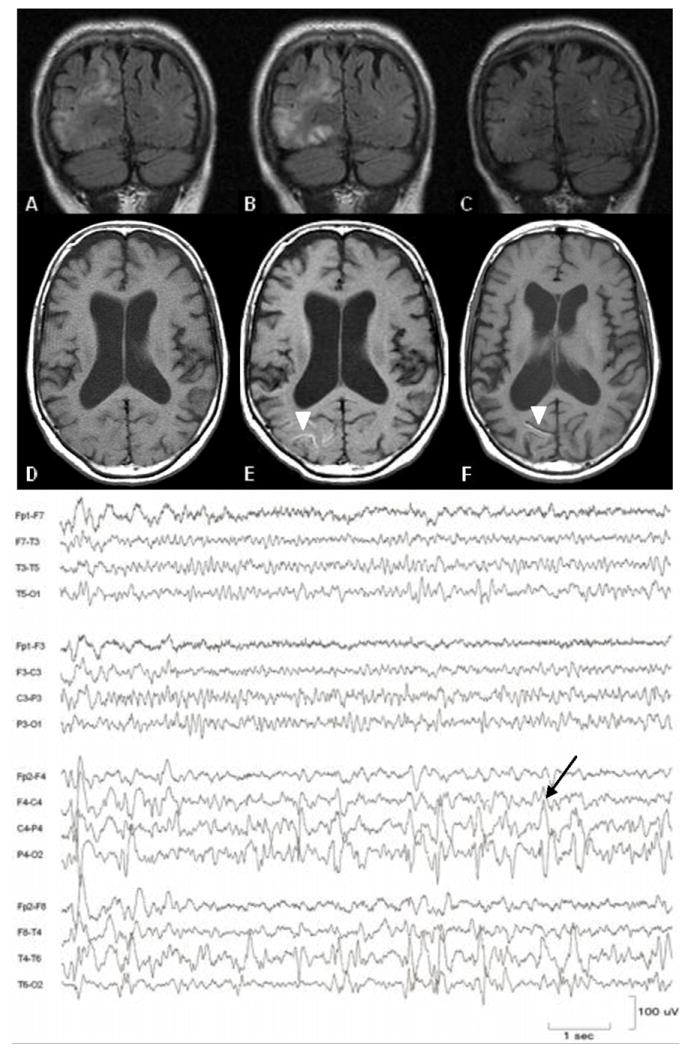
Evolution of FLAIR abnormalities (upper panel) and cortical T1 shortening (lower panel, arrowheads) one week (A, D), two weeks (B, E), and four months (C, F) after sudden onset of left hemineglect. EEG demonstrates epileptiform discharges in the right posterior quadrant, arising predominantly from the parietal region (arrow).
On further evaluation, no embolic source for recurrent vascular events was found on computed tomography (CT) angiogram of the neck and brain, electrocardiogram or echocardiogram. A rheumatologic panel was negative, and thyroid studies, rapid plasma reagin, vitamin B12, human immunodeficiency virus, and Lyme titers were unremarkable. Her spinal fluid had a normal white cell count, protein content and IgG index, negative cytology and was negative on Whipple’s polymerase chain reaction. Spinal fluid and serum testing for the paraneoplastic antibodies Hu, Yo, Ri, Ma1, Ma2, CV2/CRMP5, amphiphysin, Tr, and voltage-gated potassium channels was unrevealing. CT of the chest, abdomen and pelvis did not show a primary neoplasm. Twenty-four hour urine levels of tin, lead, mercury and arsenic were normal. Copper and manganese were slightly above the upper limits of normal. Genetic testing for mitochondrial myopathy, encephalopathy, lactic acidosis and stroke; myoclonic epilepsy with ragged-red fibers; and neuropathy, ataxia and retinitis pigmentosa mutations were negative. Positron-emission tomography with 18fluoro-labeled fluorodeoxyglucose revealed decreased glucose metabolism in the right parietal, lateral and medial temporal and posterior frontal lobes (Fig 2). A nuclear scan with 11carbon-labeled Pittsburgh-compound B (PIB) (Klunk, 2004) did not show evidence of beta-amyloid deposition. A brain biopsy of the right parietal cortex and periventricular white matter showed findings consistent with corticobasal degeneration, including neuropil threads in the cortex and white matter, with occasional coiled bodies and Pick-like bodies on CP-13 and Gallyas silver staining (Dickson, 2002). Hematoxylin and eosin staining showed ballooned neurons. Bielschowsky staining revealed no plaques or neurofibrillary tangles to suggest AD, and staining for alpha-synuclein and beta-amyloid was negative (Fig 3). The specimen was processed using previously reported standardized techniques (Forman, 2006).
Figure 2. Positron-emission tomography with 18fluoro-labeled fluorodeoxyglucose .
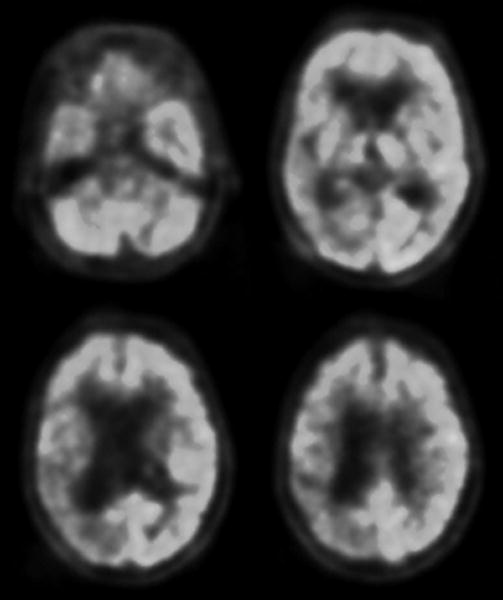
Axial sections are shown in radiological orientation. Darker areas correspond to decreased glucose metabolism, as observed in the right parietal, lateral and medial temporal and posterior frontal lobes.
Figure 3. Right parietal lobe brain biopsy (40x) showing characteristic features of corticobasal degeneration.
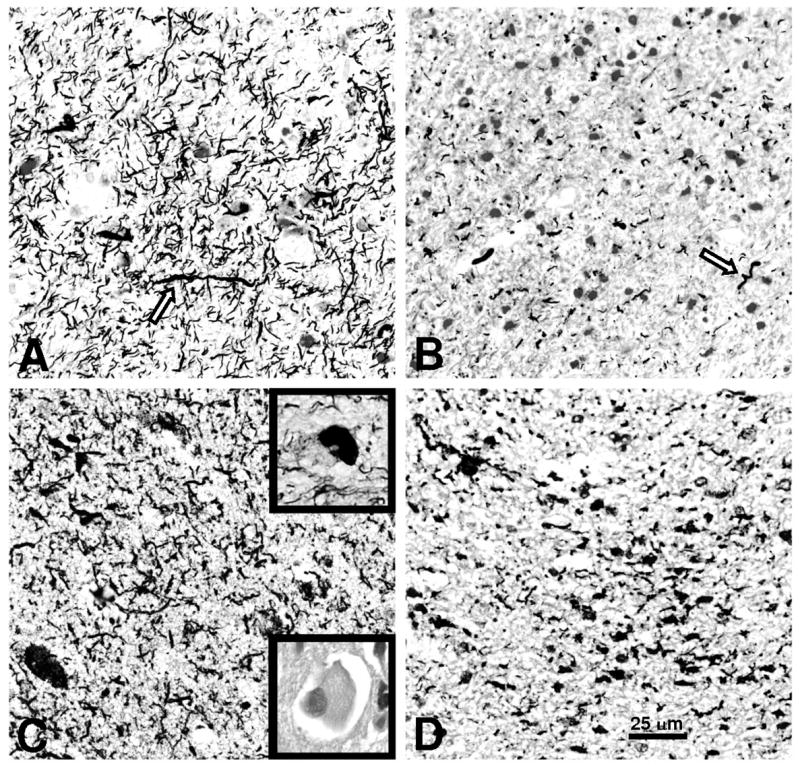
Gallyas silver stain of (A) parietal cortex and (B) periventricular white matter showed abundant neuropil threads with occasional coiled bodies (arrows). CP-13, an antibody against hyperphosphorylated tau, stained (C) parietal cortex and (D) periventricular white matter similarly. Occasional Pick-like bodies were present (upper inset, C). Ballooned neurons were present on Hematoxylin and Eosin staining (lower inset, C).
Treatment with levetiracetam was continued. Despite good control of seizures, the patient continued to exhibit symptoms of left hemineglect. She developed Balint syndrome and her right gaze preference worsened. Her left arm developed dystonia and would at times levitate involuntarily or antagonistically grab at clothes she was trying to remove, consistent with an alien limb phenomenon. Axial rigidity progressed and her gait became slow and shuffling and she became wheelchair bound one year following presentation. Episodic memory was relatively well-preserved until late in her course. Eventually she was transferred to hospice care where she passed away less than two years following presentation. Permission for an autopsy was not granted.
DISCUSSION
Seizures occur at a higher frequency in patients with dementia than in age-matched controls (Brodie, 2005). The association between neurodegenerative disease and seizures was first demonstrated in patients with Alzheimer disease who were noted to develop convulsions after the onset of dementia without a clear symptomatic seizure focus (Hauser, 1986). It has since been noted that other types of degenerative diseases including dementia with Lewy bodies (DLB), frontotemporal dementia and progressive supranuclear palsy (PSP) also are associated with seizures (Forsgren, 1996; Hesdorffer, 1996; Nygaard, 1989; Sperfeld, 1999; Weiner, 2003). Many seizure types have been reported in patients with dementia, including generalized tonic-clonic seizures, simple partial seizures with or without secondary generalization, and complex partial seizures (Lozsadi, 2006; Nygaard, 1989; Rao, 2009). Although seizures are typically a late complication of dementia, they may also manifest within the first few years of the illness (Lozsadi, 2006).
The patient described in this report had a clinical condition consistent with corticobasal syndrome (CBS), with progressive aphasia, parkinsonism, and asymmetric apraxia, rigidity, dystonia, and alien limb phenomenon. CBS has been associated with a variety of underlying histopathologies, including CBD, AD, PSP, Pick’s disease, and prion disease (Boeve, 1999). In our patient, testing for other possible causes of dementia was unrevealing, and brain biopsy showed findings typical of CBD. PSP, a tauopathy closely related to CBD, can present clinically as CBS (Josephs, 2006) and is known to be associated with seizures. However, our patient never developed a supranuclear gaze palsy and the presence of ballooned neurons, Pick-like bodies and abundant tau-positive thread-like processes in both the cortex and white matter on biopsy is more consistent with a pathologic diagnosis of CBD (Dickson, 2002). Similarly, AD can mimic CBS clinically (Alladi, 2007; Jagust, 1990), but the absence of PIB uptake or AD pathology on biopsy make underlying AD unlikely in this patient.
Prior to the onset of her seizures and immediately after the first seizure, the patient’s MRI showed only atrophy without enhancing lesions or evidence of acute ischemia. After the sudden onset of fluctuating left hemineglect, she developed T2 signal abnormalities and laminar necrosis without restricted diffusion. The timing of these changes, their evolution, the fact they did not conform to a vascular territory, and the lack of restricted diffusion is consistent with change due to prolonged seizures (Yaffe, 1995). Therefore we are confident there was no seizure focus other than that due to her underlying neurodegenerative disease.
Despite the relative frequency of seizures in the setting of neurodegenerative disease, we could not find any reports of seizures occurring as a complication of CBD. In the largest case series of clinically diagnosed CBS, no seizures were reported among the 147 patients (Kompoliti, 1998). Similarly, seizures were not listed as a characteristic of 14 pathology-proven cases in another large series (Wenning, 1998). The reason that CBD does not lead to seizures as frequently as other types of dementia is not clear. It is possible that seizures have been underreported in CBD. Another possibility is that CBD tends to spare the medial temporal lobe and hippocampus (Boxer, 2006; Josephs, 2008), areas of cortex that are thought to be especially epileptogenic. These areas are commonly affected in AD and DLB, diseases in which seizures appear to be more frequent (Rezaie, 1996).
We report the first case, to our knowledge, of unprovoked seizures occurring in the setting of CBD. Physicians caring for patients with this illness should be aware that it can be complicated by seizures, and elderly patients presenting with seizures should be evaluated for neurodegenerative diseases, including CBD.
Acknowledgments
Dr. Rabinovici is supported by a National Institute on Aging grant K23-AG031861 (GDR), an Alzheimer’s Association grant NIRG-07-59422, and the John Douglas French Alzheimer’s Association. Drs. Miller, DeArmond, and Rabinovici are supported by an Alzheimer’s Disease Research Center grant NIA P50-AG023501. The funding sources did not have any role in the design, writing, or review of this manuscript.
References
- Alladi S, Xuereb J, Bak T, Nestor P, Knibb J, Patterson K, et al. Focal cortical presentations of Alzheimer’s disease. Brain. 2007;130(Pt 10):2636–2645. doi: 10.1093/brain/awm213. [DOI] [PubMed] [Google Scholar]
- Boeve BF, Maraganore DM, Parisi JE, Ahlskog JE, Graff-Radford N, Caselli RJ, et al. Pathologic heterogeneity in clinically diagnosed corticobasal degeneration. Neurology. 1999;53(4):795–800. doi: 10.1212/wnl.53.4.795. [DOI] [PubMed] [Google Scholar]
- Boxer AL, Geschwind MD, Belfor N, Gorno-Tempini ML, Schauer GF, Miller BL, et al. Patterns of brain atrophy that differentiate corticobasal degeneration syndrome from progressive supranuclear palsy. Arch Neurol. 2006;63(1):81–86. doi: 10.1001/archneur.63.1.81. [DOI] [PubMed] [Google Scholar]
- Brodie MJ, Kwan P. Epilepsy in elderly people. Bmj. 2005;331(7528):1317–1322. doi: 10.1136/bmj.331.7528.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K, et al. Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol. 2002;61(11):935–946. doi: 10.1093/jnen/61.11.935. [DOI] [PubMed] [Google Scholar]
- Forman MS, Farmer J, Johnson JK, Clark CM, Arnold SE, Coslett HB, et al. Frontotemporal dementia: clinicopathological correlations. Ann Neurol. 2006;59(6):952–962. doi: 10.1002/ana.20873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsgren L, Bucht G, Eriksson S, Bergmark L. Incidence and clinical characterization of unprovoked seizures in adults: a prospective population-based study. Epilepsia. 1996;37(3):224–229. doi: 10.1111/j.1528-1157.1996.tb00017.x. [DOI] [PubMed] [Google Scholar]
- Hauser WA, Morris ML, Heston LL, Anderson VE. Seizures and myoclonus in patients with Alzheimer’s disease. Neurology. 1986;36(9):1226–1230. doi: 10.1212/wnl.36.9.1226. [DOI] [PubMed] [Google Scholar]
- Hesdorffer DC, Hauser WA, Annegers JF, Kokmen E, Rocca WA. Dementia and adult-onset unprovoked seizures. Neurology. 1996;46(3):727–730. doi: 10.1212/wnl.46.3.727. [DOI] [PubMed] [Google Scholar]
- Jagust WJ, Davies P, Tiller-Borcich JK, Reed BR. Focal Alzheimer’s disease. Neurology. 1990;40(1):14–19. doi: 10.1212/wnl.40.1.14. [DOI] [PubMed] [Google Scholar]
- Josephs KA, Petersen RC, Knopman DS, Boeve BF, Whitwell JL, Duffy JR, et al. Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology. 2006;66(1):41–48. doi: 10.1212/01.wnl.0000191307.69661.c3. [DOI] [PubMed] [Google Scholar]
- Josephs KA, Whitwell JL, Dickson DW, Boeve BF, Knopman DS, Petersen RC, et al. Voxel-based morphometry in autopsy proven PSP and CBD. Neurobiol Aging. 2008;29(2):280–289. doi: 10.1016/j.neurobiolaging.2006.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55(3):306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Kompoliti K, Goetz CG, Boeve BF, Maraganore DM, Ahlskog JE, Marsden CD, et al. Clinical presentation and pharmacological therapy in corticobasal degeneration. Arch Neurol. 1998;55(7):957–961. doi: 10.1001/archneur.55.7.957. [DOI] [PubMed] [Google Scholar]
- Lozsadi DA, Larner AJ. Prevalence and causes of seizures at the time of diagnosis of probable Alzheimer’s disease. Dement Geriatr Cogn Disord. 2006;22(2):121–124. doi: 10.1159/000093664. [DOI] [PubMed] [Google Scholar]
- Nygaard TG, Duvoisin RC, Manocha M, Chokroverty S. Seizures in progressive supranuclear palsy. Neurology. 1989;39(1):138–140. doi: 10.1212/wnl.39.1.138. [DOI] [PubMed] [Google Scholar]
- Rao SC, Dove G, Cascino GD, Petersen RC. Recurrent seizures in patients with dementia: frequency, seizure types, and treatment outcome. Epilepsy Behav. 2009;14(1):118–120. doi: 10.1016/j.yebeh.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezaie P, Cairns NJ, Chadwick A, Lantos PL. Lewy bodies are located preferentially in limbic areas in diffuse Lewy body disease. Neurosci Lett. 1996;212(2):111–114. doi: 10.1016/0304-3940(96)12775-0. [DOI] [PubMed] [Google Scholar]
- Sperfeld AD, Collatz MB, Baier H, Palmbach M, Storch A, Schwarz J, et al. FTDP-17: an early-onset phenotype with parkinsonism and epileptic seizures caused by a novel mutation. Ann Neurol. 1999;46(5):708–715. doi: 10.1002/1531-8249(199911)46:5<708::aid-ana5>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Weiner MF, Hynan LS, Parikh B, Zaki N, White CL, 3rd, Bigio EH, et al. Can alzheimer’s disease and dementias with Lewy bodies be distinguished clinically? J Geriatr Psychiatry Neurol. 2003;16(4):245–250. doi: 10.1177/0891988703258671. [DOI] [PubMed] [Google Scholar]
- Wenning GK, Litvan I, Jankovic J, Granata R, Mangone CA, McKee A, et al. Natural history and survival of 14 patients with corticobasal degeneration confirmed at postmortem examination. J Neurol Neurosurg Psychiatry. 1998;64(2):184–189. doi: 10.1136/jnnp.64.2.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe K, Ferriero D, Barkovich AJ, Rowley H. Reversible MRI abnormalities following seizures. Neurology. 1995;45(1):104–108. doi: 10.1212/wnl.45.1.104. [DOI] [PubMed] [Google Scholar]