

Abstract
The membrane bound receptor tyrosine kinase Her2 is overexpressed in approximately 30% of human breast cancers which correlates with poor prognosis. Her2-induced signaling pathways include MAPK and PI3K/Akt, of which the latter has been shown to be critical for Her2+ breast cancer cell growth and survival. Additionally, the NF-κB pathway has been shown to be activated downstream of Her2 overexpression, however the mechanisms leading to this activation are not currently clear. Using Her2+/ER- breast cancer cells, we show that Her2 activates NF-κB through the canonical pathway which, surprisingly, involves IKKα. Knockdown of IKKα led to a significant decrease in transcription levels of multiple NF-κB-regulated cytokine and chemokine genes. siRNA-mediated knockdown of IKKα resulted in a decrease in cancer cell invasion, but had no effect on cell proliferation. Inhibition of the PI3K/Akt pathway had no effect on NF-κB activation, but significantly inhibited cell proliferation. Our study suggests different roles for the NF-κB and PI3K pathways downstream of Her2, leading to changes in invasion and proliferation of breast cancer cells. Additionally this work indicates the importance of IKKα as a mediator of Her2-induced tumor progression.
Keywords: Her2, IKKalpha, NF-KappaB
Introduction
The epidermal growth factor receptor Her2 is amplified in 20-30% of breast cancers, which typically do not express estrogen receptor, and are often correlated with poor prognosis and/or chemoresistance, making Her2 an important therapeutic target (Hynes & Stern, 1994; Klapper et al., 2000; Slamon et al., 1987; Slamon et al., 1989). The Her2-specific antibody trastuzumab and the dual EGFR/Her2 inhibitor lapatinib have been shown to decrease growth of Her2-overexpressing tumors (Baselga et al., 1999; Pegram et al., 1998), however a majority of patients treated with trastuzamab develop resistance (Slamon et al., 2001), indicating the importance of elucidating alternative therapeutic targets in this disease. Her2-overexpression was first shown to activate NF-κB over a decade ago (Galang et al., 1996), however, the role NF-κB plays in development and progression of Her2-overexpressing breast cancer is still poorly understood. Additionally, the pathway leading to NF-κB activation downstream of Her2 is not well characterized.
NF-κB is an important transcription factor that has been shown to be involved in expression of genes involved in key cellular processes including innate and adaptive immunity (Bonizzi & Karin, 2004), cell proliferation and survival (Papa et al., 2006), lymphoid organ development (Weih & Caamano, 2003), as well as being activated in a variety different cancers, including breast cancer (Basseres & Baldwin, 2006; Belguise & Sonenshein, 2007; Cogswell et al., 2000). The NF-κB family of transcription factors consists of five subunits: RelA (p65), RelB, c-Rel, p105/p50 and p100/p52. These subunits are evolutionarily conserved and exist as hetero- or homodimers (Hayden & Ghosh, 2004). The p65/p50 heterodimer is the most abundant NF-κB complex in the cell and is regulated by the so-called canonical pathway. Following stimulation with activators such as TNF, IκB is phosphorylated by the Inhibitor of KappaB Kinase (IKK) complex. The IKK complex consists of two catalytic subunits IKKα and IKKβ, and a regulatory subunit IKKγ (NEMO), which binds both catalytic subunits at their NEMO-binding domains (NBD)(Gilmore, 2006). In the canonical pathway, IKKβ phosphorylates IκBα leading to its degradation and NF-κB nuclear accumulation (Ghosh & Karin, 2002). Furthermore, the p65 subunit of NF-κB can be phosphorylated on multiple residues, including serine 536, which is important for transactivation potential (Sakurai et al., 1999). NF-κB activation can also occur via the alternative, or non-canonical pathway. Activation of NF-κB in the non-canonical pathway, most common in B cells, involves Inhibitor of KappaB Kinase α (IKKα) and is IκBα-independent (Solt & May, 2008). Thus most current models place IKKβ as the dominant IKK subunit in the canonical pathway with IKKα functioning in the non-canonical system. Few studies have addressed the individual roles of IKKα and IKKβ downstream of oncoprotein-dependent signaling.
Using an siRNA approach, we set out to determine how NF-κB is activated downstream of Her2, and what role the IKK complex plays in this signaling cascade, as well as how the activation of the IKK kinases may lead to a malignant state. While the classical pathway has long been thought to require IKKβ, here we show that IKKα plays a larger role than IKKβ in the activation of NF-κB in Her2+ breast cancer cells, including the phosphorylation of the p65 subunit at serine 536. Using siRNA to the IKK kinases, we show that knockdown of IKKα leads to a change in the gene expression profile in Her2+ cells, including a notable cytokine and chemokine gene expression signature. Furthermore, knockdown of IKKα by siRNA led to a marked decrease in invasive ability in SKBr3 cells, yet had no effect on cell proliferation. Taken together, our data suggests that Her2 can activate NF-κB through the canonical pathway. Surprisingly, this activation occurs primarily through IKKα, a subunit typically not thought to be involved in the canonical pathway. Interestingly, we have discovered differential roles for the IKK kinases with IKKα specifically involved in an invasive oncogenic phenotype in Her2+ breast cancer cells.
Materials and Methods
Cell culture and reagents
The tumor-derived SKBr3 cell lines were maintained in McCoy’s 5A medium (Mediatech) supplemented with 10% fetal bovine serum (FBS) and 100 units/mL penicillin/streptomycin. The tumor-derived MCF7, MDA-MB-453 and MDA-MB-231 cell lines, as well as Mouse Embryonic Fibroblast (MEF) cell lines, were maintained in Dulbecco’s Modified Eagle Medium (Gibco) supplemented with 10% FBS and 100 units/mL penicillin/streptomycin. The human mammary epithelial cell lines (H16N2-pTP and H16N2-Her2) were maintained as previously described (Ethier et al., 1993). The stable 3x-κB luciferase SKBr3 cell line was established by transfection of a luciferase reporter construct containing tandem NF-κB binding sites from the MHC class I promoter region into SKBr3 cells with Fugene (Roche) and maintained under selection with G418 (Geneticin, Live Technologies). The Her2 wild-type and mutant (V654E) plasmids were constructed previously (Li et al., 2004) (Addgene plasmid 16257 and Addgene plasmid 16259). The Her2 coding sequences were subcloned into retroviral pLHCX vector (Stratagene) and virus was produced in 293T cells with cotransfection of AmphoPAK. MEFs were transduced with virus with polybrene and lysed 48 hours later. The following antibodies were purchased from commercial sources: antibodies against phospho-p65 (Ser536), phospho-Akt (Ser473), Akt, phospho-IκBα (Ser32/36) and IκBα from Cell Signaling Technology; antibodies against Her2, IKKα clone 14A231 and IKKβ clone10AG2 and p100/p52 from Millipore, antibodies against p65 and p50 (supershift), β-tubulin and IKKγ from Santa Cruz Biotechnology, antibody against total p65 from Rockland (PA, USA). LY294002 and Wortmannin were purchased from Cell Signaling Technology. Lapatinib (GW572016; Tykerb) was a gift from Dr. H. Shelton Earp (University of North Carolina at Chapel Hill).
Immunoblots
Whole cell extracts were prepared on ice with Mammalian Protein Extraction Reagent (Thermo Scientific) according to manafacturer’s instructions supplemented with protease inhibitor mix (Roche, IN, USA) and phosphotase inhibitor mix (Sigma, MO, USA). Nuclear and cytoplasmic extracts were prepared as previously described (Mayo et al., 1997). Protein concentrations were determined by Bradford assay (Biorad Laboratories) and SDS-PAGE analysis was performed as previously described (Steinbrecher et al., 2005).
Small RNA interference
The following small interfering RNAs (siRNA; siGenome SMARTpool) were obtained from Dharmacon as a pool of four annealed double-stranded RNA oligonucleotides: IKKα (M-003473-02), IKKβ (M-003503-03), NEMO (M-003767-02), RelA (p65) (M-003533-02) and nontargeting control #3 (D001201-03). Cells were grown to approximately 50% confluency and transfected with 100 nmol/L siRNA with Dharmafect 1 reagent according to manafacturer’s instructions.
Quantitative Real-time PCR
Total RNA extracts were obtained from cells approximately 72 hours post-transfection by Trizol (Invitrogen) extraction. Two micrograms of RNA was reverse transcribed using random primers and MMLV-reverse transcriptase (Invitrogen). Real-time PCR was performed and analyzed as previously described (Steinbrecher et al., 2005) using Taqman Gene Expression Assay primer-probe sets IL-6 (Hs00174131_m1), IL-8 (Hs001741103_m1), CCL2 (Hs00234140_m1), TNF (Hs99999043_m1), and uPA (Hs00170182_m1).
Electorphoretic Mobility Shift Assay
Electrophoretic mobility shift assay (EMSA) and NF-κB supershift analysis were done on nuclear extracts as previously described (Steinbrecher et al., 2005) using 32P-labeled oligonucleotide probe corresponding to an NF-κB site within the MHC class I promoter region.
IKK Kinase Assay
Whole cell lysates were prepared on ice for 45 minutes in lysis buffer containing 20 mmol/L Tris (pH 8.0), 500 mmol/L NaCl, 0.25% Triton X-100, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L DTT, 1x protease inhibitor (Roche Applied Science), and 1x phosphatase inhibitor cocktail (Sigma-Aldrich). IKK complexes were immunoprecipitated from 500-μg total protein extract using IKKγ antibody (Santa Cruz Biotechnology). An in vitro kinase assay was done and analyzed as previously described (Steinbrecher et al., 2005) using GST-IκBα as a substrate.
Luciferase Assay
SKBr3 cells stably expressing the 3x-κB plasmid were plated in equal number in triplicate in 24-well plates and transfected with siRNA for 72 hours or treated overnight with LY294002. Cells were lysed in MPER and luciferase activity was measured with Promega Luciferase Assay System (Promega). Luciferase levels were normalized by protein concentration using a Bradford assay. H16N2-Her2 and MDA-MB-453 cells were transfected with siRNA 72 hours before lysates were obtained, and were transfected with 3x-κB reporter plasmid and pRL-CMV (Promega) renilla plasmid 24 hours prior to lysate collection. Lysates were collected as mentioned above and luciferase levels were normalized to renilla.
Cell invasion assay
Innocyte™ Cell Invasion Assay Kit was purchased from Calbiochem (San Diego, California). Cells were transfected with siRNA for 48 hours before seeding. Invasion assay was performed as per manafacturer’s protocol for 48 hours. The number of invading cells was measured fluorometrically with Calcein AM.
Cell Proliferation Assay
Cell proliferation assay was performed as previously described (Wilson & Baldwin, 2008). Cells were cultured in the presence or absence of inhibitors, or transiently transfected with siRNA to IKK subunits and measured at the indicated timepoints post-transfection.
Results
Lapatinib inhibits Her2 activation of NF-κB and Akt
It has previously been shown that Her2-overexpression leads to activation of NF-κB family members involved in the canonical pathway, specifically the p65/p50 heterodimeric complex (Biswas et al., 2004; Galang et al., 1996). Given this result, we investigated whether the dual EGFR/Her2 inhibitor Lapatinib (Tykerb, GW572016) could block Her2-induced p65 phosphorylation at serine 536, a marker of increased NF-κB transcriptional activity (Sakurai et al., 1999). Five breast cancer cell lines were treated with 1 μM of lapatinib for 12 hours and whole cell extracts were analyzed for expression of phosphorylated p65. A marked decrease in p65 phosphorylation was observed in Her2-ovexpressing tumor cell lines (SKBr3 and MDA-MB-453) upon treatment with lapatinib, while non Her2-overexpresing tumor cell lines (MCF7 and MDA-MB-231) showed no change (Fig. 1A). The H16N2-Her2 cell line also showed a decrease in p65 phosphorylation upon lapatinib treatment. Overexpression of Her2 in this cell line results in NF-κB activation, as the parental cell line, H16N2-pTP, has very little basal p65 phosphorylation (Supplemental Figure 1). In order to further investigate how Her2 signals to NF-κB, we chose to use the tumor-derived SKBr3 cell line, as it has previously proven to be an excellent in vitro model for Her2+/ER- breast cancer (Singh et al., 2007). SKBr3 cells were treated with 1 μM lapatinib or vehicle control over a course of 24 hours and whole cell extracts were analyzed for levels of phosphorylated IκBα. Phosphorylation of IκBα at serines 32 and 36 was inhibited within 3 hours of lapatinib treatment (Fig. 1B). Stabilization of IκBα was also observed, consistent with loss of phosphorylated IκBα. It has previously been shown that Her2-overexpression activates the PI3K/Akt pathway and that lapatinib can inhibit Akt phosphorylation in lapatinib-sensitive Her2 overexpressing breast cancer cell lines (Hegde et al., 2007). Similarly, we observe a decrease in phosphorylation of Akt at serine 473 in the lapatinib-sensitive SKBr3 cell line upon treatment with lapatinib (Fig. 1C). This indicates that Her2 can activate both the NF-κB and the PI3K/Akt pathways, and that pharmacological inhibition of Her2 leads to subsequent inhibition of these survival pathways.
Figure 1. Lapatinib treatment inhibits the NF-κB and PI3K pathways in Her2-overexpressing cells.
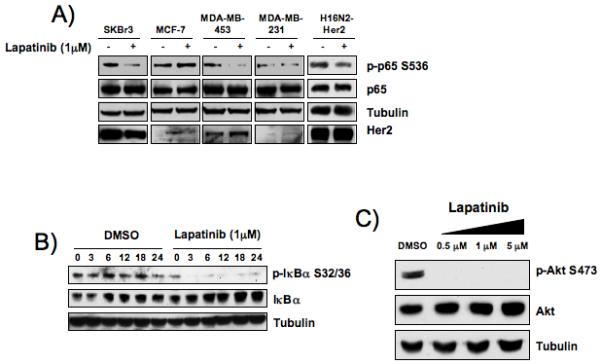
A) Western blot of phospho-p65S536 in multiple breast cancer cell lines treated with lapatinib. Breast cancer cell lines were treated with 1 μM dual EGFR/Her2 inhibitor lapatinib or DMSO vehicle control for 12 hours. Western blots were performed with 25 μg protein from whole cell extracts. B) Western blot of phospho-IκBαS32/36 in SKBr3 cells treated with lapatinib. SKBr3 cells were treated with lapatinib (1 μM) or DMSO control over a course of 24 hours and levels of phospho-IκBαS32/36 were measured by western blot of 25 μg total protein from whole cell extracts. C) Western blot of phospho-AktS473 in SKBr3 cells treated with lapatinib. SKBr3 cells were treated for 12 hours with dual EGFR/Her2 inhibitor lapatinib and levels of phospho-AktS473 were measured by western blot of 25 μg protein from whole cell extracts.
Her2 activates the NF-κB canonical pathway through IKKα and IKKβ
We next examined the role of the IKK complex in the activation of NF-κB downstream of Her2. siRNA targeting the catalytic subunits of the IKK complex (IKKα and IKKβ) was transfected into Her2-overexpressing breast cancer cells and whole cell extracts were analyzed for markers of NF-κB activation. In the Her2-ovexpressing SKBr3, H16N2-Her2 and MDA-MB-453 cells, knockdown of IKKα led to a greater decrease in p65 phosphorylation than knockdown of IKKβ (Fig. 2A). Mouse embryonic fibroblasts (MEFs) lacking IKKα, as well as wild-type cells, were transduced with Her2 wild-type and constitutively active constructs. Transduction of these constructs resulted in increased p65 phosphorylation in wild-type MEFs, however, no increase in phosphorylation was seen in IKKα -/- cells (Supplemental Figure 2). A similar result was obtained using IKKβ -/- cells (data not shown), indicating that both IKKα and IKKβ are important for Her2 to activate NF-κB in murine fibroblasts. In order to further investigate the role IKKα may play in the activation of classical NF-κB complexes downstream of Her2, siRNA was again used to target IKK in SKBr3 cells stably expressing a 3x-κB luciferase reporter construct, as well as in H16N2-Her2 and MDA-MB-453 transiently transfected with the 3x-κB reporter plasmid. Knockdown of IKKα or a combination of IKKα and IKKβ led to a significant decrease in luciferase reporter activity (student’s T-test *<0.05 and **<0.001 respectively), while knockdown of IKKβ did not show a significant decrease in luciferase reporter activity in two of the three cell lines (Fig. 2B). An Electrophoretic Mobility Shift Assay (EMSA) was performed to further investigate the role of IKK in Her2 activation of NF-κB in SKBr3 cells. Knockdown of IKKα led to a greater decrease in NF-κB DNA binding activity than IKKβ knockdown (Fig. 2C). Supershift analysis indicated that loss of IKKα leads to a decrease in DNA binding of classical-pathway NF-κB heterodimers p65/p50. Phosphorylation of IκBα by the catalytic subunits of the IKK complex is a hallmark of activation of the canonical NF-κB pathway, therefore we measured this kinase activity upon knockdown of IKKα or IKKβ. The IKK complex was immunoprecipitated with IKKγ, the scaffolding subunit of the IKK complex. Knockdown of IKKα led to a greater decrease of in vitro phosphorylation of IκBα than knockdown of IKKβ (Fig. 2D), further indicating IKKα plays a prominent role in the canonical pathway in Her2-overexpressing cells. Taken together, these results demonstrate that IKKα plays a more significant role than IKKβ in activation of the NF-κB canonical pathway in Her2-overexpressing breast cancer cells.
Figure 2. Her2 activation of NF-κB via IKKα and IKKβ involves the canonical pathway.
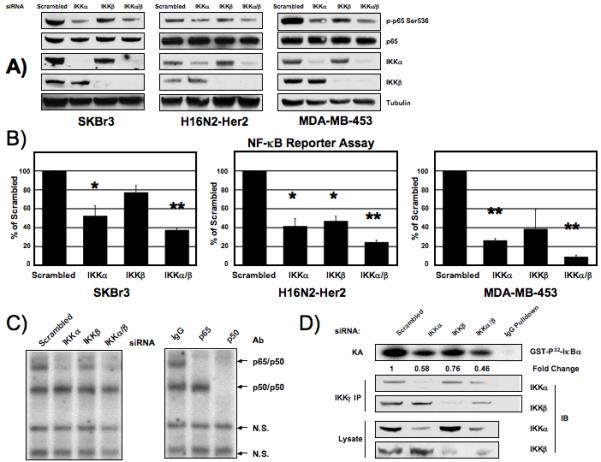
A) Western blot of phospho-p65S536 in Her2-overexpressing breast cancer cells transfected with siRNA to IKK catalytic subunits. SKBr3 (left), H16N2-Her2 (center) and MDA-MB-453 (right) cells were were transfected with 100 nM siRNA to IKKα and IKKβ and whole cell extracts were prepared after 72 hours and western blot analysis performed. B) NF-κB luciferase reporter assay of SKBr3, H16N2-Her2 and MDA-MB-453 cells transfected with IKK siRNA. Whole cell extracts were prepared 72 hours post-siRNA transfection and luciferase levels were measured. Statistically significant differences were determined by students t-test (*<0.05 **<0.001). Fold change of reporter activity with IKK knockdown is shown relative to scrambled siRNA treated cells. Values are the average of at least 3 experiments. Error bars are ± 1 S.E. Samples are normalized by protein concentration (SKBr3) or renilla (H16N2-Her2 and MDA-MB-453). C) Electrophoretic mobility shift assay (EMSA) of SKBr3 cells transfected with IKK siRNA. Nuclear extracts were prepared after 72 hours. Identities of the bound complexes were determined by super-shift with antibodies to p65 and p50. Non-specific binding complexes are noted with as N.S. D) Kinase assay measuring IKK in vitro phosphorylation of IκBα. SKBr3 cells were transfected with IKK siRNA for 72 hours and IKKγ was immunoprecipitated from 500 μg whole cell extracts. Ability of immunoprecipitated complex to phosphorylate purified GST-IκBα was measured (KA). Amount of IKKα and IKKβ in immunoprecipitated complex (IP) and whole cell extracts (lysate) were measured. Fold change in kinase activity was calculated using pixel densitometry and compared to scrambled siRNA transfected cells.
Knockdown of IKKα and IKKβ leads to distinct gene expression profiles
We next determined if knockdown of the two IKK catalytic subunits leads to differential changes in gene expression in Her2-overexpressing cells. A chemiluminescent oligo-based array was used to measure expression of 219 genes. Upon knockdown of IKKα or IKKβ, significant decrease in expression was seen in 14 genes (Supplementary Table 1). Genes that showed significant changes in expression upon siRNA transfection were validated by quantitative real-time PCR. Decrease in expression of pro-inflammatory cytokines and chemokines IL-6, IL-8, CCL-2, TNF, and the serine-protease uPA, was greater upon siRNA knockdown of IKKα than IKKβ in both SKBr3 and H16N2-Her2 breast cancer cell lines (Fig. 3A). In order to demonstrate that IKK dependent changes in gene expression were occurring through modulation of NF-κB transcriptional activity, we performed RNAi against the classic subunit p65 in SKBr3 and H16N2-Her2 cells and assayed expression of mRNA by quantitative real-time PCR. Gene expression analysis showed that knockdown of p65 by siRNA led to a significant decrease in gene transcription levels of IL-8, IL-6, TNF and uPA (Fig. 3B). This transcriptional profile mirrors that seen upon knockdown of IKK, specifically IKKα, suggesting that induction of chemokines and cytokines in Her2 breast cancer cells occurs through IKK activation of p65. We next measured changes in expression of these genes in SKBr3 cells following treatment with lapatinib to confirm this activation of NF-κB regulated genes was induced downstream of overexpression of Her2. Treatment of SKBr3 cells with 1 μM of lapatinib led to a significant decrease in gene expression of IL-6, IL-8, CCL-2, TNF and uPA at both 8 and 16 hours post treatment (Fig. 3C). Taken together, this suggests that Her2 activates NF-κB through the canonical pathway involving IKKα and leading to an increase in multiple NF-κB regulated genes involved in tumor progression.
Figure 3. Her2 induces NF-κB-regulated gene expression through IKKα and IKKβ.
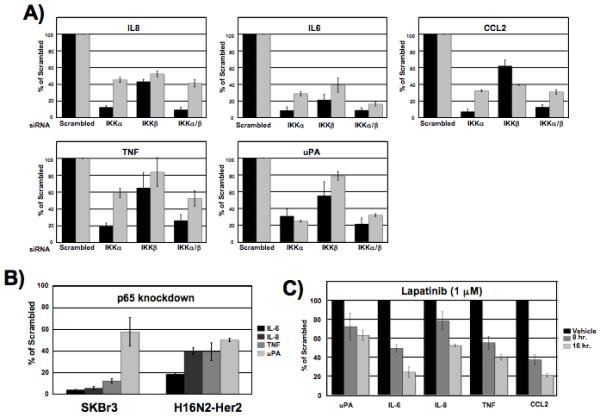
(A) Quantitative real-time PCR of multiple genes shows different gene expression profiles upon IKKα or IKKβ knockdown. qRT-PCR was performed on extracts from SKBr3 (black bars) and H16N2-Her2 (gray bars) cells transfected with 100 nM IKKα or IKKβ siRNA for 72 hours. Gene expression levels were normalized to Gus or GAPDH and presented as fold change versus cells transfected with scrambled control siRNA. Values are the average of at least 3 experiments. Error bars are ± 1 S.E. (B) Quantitative real-time PCR of multiple genes upon knockdown of p65 by siRNA. SKBr3 and H16N2-Her2 cells were transfected with 100 nM siRNA for 72 hours and gene expression levels were measured. Fold change of transcript levels is shown relative to scrambled siRNA treated cells. Values are the average of at least 3 experiments. Error bars are ± 1 S.E. C) Quantitative real-time PCR shows inhibition of Her2 by lapatinib blocks NF-κB regulated gene expression. SKBr3 cells were treated with 1 μM lapatinib for 8 or 16 hours and gene expression levels of uPA, IL-6, IL-8, TNF and CCL2 were compared to DMSO treated cells. Fold change of transcript levels is shown relative to scrambled siRNA treated cells. Error bars are ± 1 S.E.
Activation of NF-κB in Her2-overexpressing cells requires NEMO
The scaffolding subunit of the IKK complex, IKKγ (NEMO), is required for activation of NF-κB canonical pathway involving IKKβ (Gilmore, 2006), and inhibition of the IKK signalsome with the Nemo Binding Domain (NBD) peptide can block NF-κB activation (Biswas et al., 2004). We used an siRNA approach to determine the importance of NEMO in NF-κB activation in Her2-overexpressing cell lines. siRNA knockdown of NEMO led to a marked decrease in p65 phosphorylation in all three Her2+ cell lines (Fig. 4A). NF-κB luciferase reporter activity was also significantly decreased in these cell lines upon siRNA knockdown of NEMO (Fig. 4B). We performed quantitative real-time PCR analysis in the SKBr3 cell line upon NEMO knockdown to determine if this resulted in a similar gene expression profile as IKKα knockdown. Consequently, IL-6, IL-8, TNF and CCL2 all showed a significant decrease in expression upon NEMO knockdown, though uPA expression levels did not change (Fig. 4C). In order to rule out any effect loss of IKKα could have on non-classical activation of NF-κB, we analyzed processing of the p100 subunit. Cleavage of the precursor NF-κB protein p100 to p52 is a hallmark of activation of the non-canonical pathway. No significant effect was seen on p100 processing to p52 upon knockdown of either of the IKK subunits in Her2+ cells (Fig. 4D). These results suggest that NF-κB activation in Her2+ cells occurs through IKKα and this requires the NEMO subunit. Additionally, these results indicate that the non-canonical NF-κB signaling pathway is not activated in Her2+ breast cancer cells.
Figure 4. Knockdown of NEMO blocks NF-κB activation through the canonical pathway.
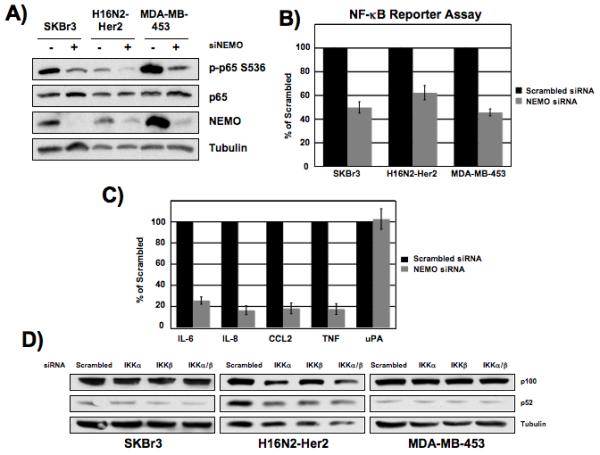
A) Her2+ breast cancer cells were transfected with 100 nM NEMO siRNA and whole cell lysates were collected 72 hours post transfection and western blot analysis of phosphorylated p65 was performed using 25 μg total protein. B) Her2+ cell lines were transfected with 100 nM NEMO siRNA and whole cell extracts were prepared 72 hours post-siRNA transfection and luciferase levels were measured. Fold change of reporter activity with IKK knockdown is shown relative to scrambled siRNA treated cells. Values are the average of at least 3 experiments. Error bars are ± 1 S.E. Samples are normalized by protein concentration (SKBr3) or renilla (H16N2-Her2 and MDA-MB-453). C) SKBr3 cells were transfected with 100 nM NEMO siRNA and extracts were isolated after 72 hours and qRT-PCR was performed. Fold change of transcript levels is shown relative to scrambled siRNA treated cells. Error bars are ± 1 S.E. D) Her2-overexpressing breast cancer cells were transfected with 100 nM siRNA to IKKα or IKKβ and whole cell extracts were collected 72 hours post transfection. Levels of p100 and p52 were measured by western blot analysis using 25 μg total protein.
Activation of the NF-κB canonical pathway is independent of the PI3K pathway
It has previously been reported that expression of dominant-negative PI3K and Akt plasmids can block NF-κB DNA binding (Pianetti et al., 2001). Therefore, we investigated if NF-κB activation downstream of Her2 is dependent on the PI3K/Akt pathway. Upon treatment of SKBr3 cells with lapatinib, phosphorylation of Akt at Serine 473 decreases dramatically (Fig. 1A). Treatment with the PI3K inhibitor LY294002 also blocked phosphorylation of Akt at serine 473, however LY294002 had no effect on the phosphorylation status of p65 at serine 536 in SKBr3, H16N2-Her2, or MDA-MB-453 cells (Fig. 5A-C). Furthermore, treatment of SKBr3 cells stably expressing the 3x-κB luciferase reporter with LY294002 had no effect on NF-κB transcriptional activity (Fig. 5D). These results demonstrate that Her2 activates Akt through PI3K, and that the Her2-induced activation of NF-κB is independent of this pathway.
Figure 5. Inhibition of the PI3K-pathway does not block NF-κB activation.
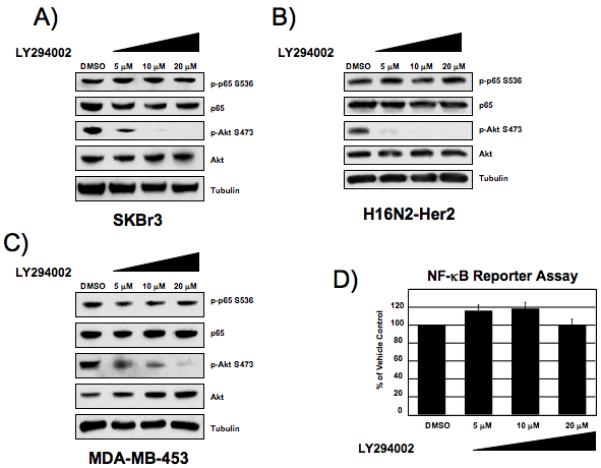
Western blot of phospho-p65 serine 536 from SKBr3 (A), H16N2-Her2 (B) and MDA-MB-453 (C) cells treated with PI3K-inhibitor inhibitor LY294002 for 2 hours. Western blot analysis was performed with 25 μg whole cell extracts. D) Luciferase reporter assay of SKBr3 cells treated with LY294002 overnight. Fold change of reporter activity with PI3K-inhibitor treatment is shown relative to vehicle treated cells. Values are the average of at least 3 experiments. Error bars are ± 1 S.E. Samples are normalized by protein concentration.
IKKα induces cell invasion but not cell proliferation
Having determined that overexpression of Her2 leads to IKKα-dependent activation of the NF-κB classical pathway, we next sought to determine how this signaling may promote oncogenic phenotypes. We investigated the effect IKK activation may have on proliferation of Her2-overexpressing breast cancer cells. SKBr3 cells were transfected with siRNA to the IKK catalytic subunits and cell proliferation was measured by MTS assay. Knockdown of IKKα or IKKβ had no effect on cell proliferation (Fig. 6A). As a control, SKBr3 cells were treated with the PI3K-inhibitor LY294002, as well as lapatinib. Inhibition of PI3K/Akt or Her2 led to a dramatic decrease in cell growth (Fig. 6B), consistent with what has been previously reported, suggesting that Her2 drives cell proliferation through the PI3K/Akt pathway. Our previous results have shown IKK/NF-κB dependent increases in proinflammatory cytokines downstream of Her2, and these genes have been shown to promote increased motility and invasiveness. Furthermore, overexpression of Her2 has been shown to lead to increase in invasiveness of breast cancer cells (Arora et al., 2008). We reasoned that NF-κB activity downstream of Her2 may contribute to increased invasiveness of Her2 breast cancer. To address this question, SKBr3 cells were transfected with siRNA to IKKα and IKKβ and the ability of the cells to invade through a basement membrane was measured. Knockdown of IKKα led to a significant decrease in invasiveness of SKBr3 cells while knockdown of IKKβ had no effect (Figure 6C). This suggests that Her2 overexpression results in activation of at least two independent oncogenic signaling pathways, one involving PI3K/Akt and another involving NF-κB, which have two different but important roles in promoting tumorigenesis (Fig. 6D).
Figure 6. Inhibition of PI3K blocks cell proliferation, knockdown of IKKα blocks cell invasion.
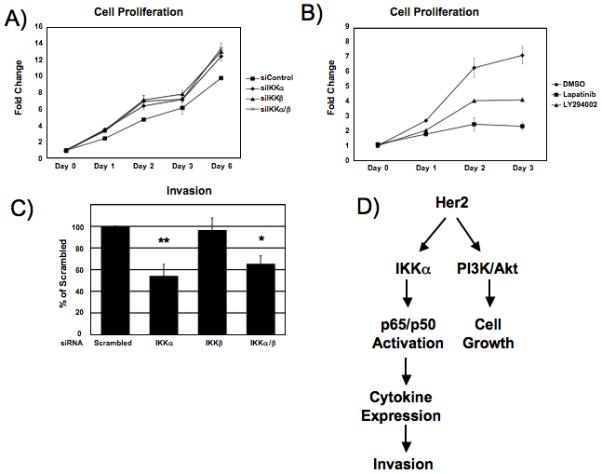
A) Cell proliferation of SKBr3 cells transfected with siRNA to IKKα or IKKβ was measured to for 6 days post-transfection compared to scrambled siRNA treated cells using CellTiter cell viability reagent. Knockdown of IKK by siRNA led to a slight increase in cell proliferation. Error bars represent ± 1 S.D. (B) Cell proliferation of SKBr3 cells treated with PI3K inhibitors LY294002 (10 μM) or EGFR/Her2 inhibitor lapatinib (1 μM) was measured over 3 days. Both inhibitors showed a significant decrease in cell proliferation over a course of 3 days. Error bars represent ± 1 S.D. C) SKBr3 cells were transfected with 100 nM siRNA to IKKα or IKKβ and cell invasion was measured after 48 hours fluorometrically. Statistical significance was measured by student’s T-test (*<0.01, **<0.001). Error bars represent ± 1 S.D.
Discussion
While Her2-positive breast cancer is known to activate both NF-κB and PI3K/Akt pathways, (Biswas et al., 2004; Knuefermann et al., 2003; Pianetti et al., 2001; She et al., 2008), it has been unclear how Her2 induces NF-κB and whether PI3K is involved with this pathway. Additionally, potential roles for IKKα and IKKβ in controlling Her2-induced NF-κB have not been addressed. The latter point is of interest since IKKα and IKKβ have previously been associated with controlling distinct NF-κB pathways, with IKKβ controlling the so-called canonical pathway and IKKα controlling the non-canonical pathway. These issues are potentially quite important in the therapeutic setting. Our data indicate the following: (i) IKKα plays an important role in controlling the ability of Her2 to activate NF-κB through the canonical pathway (including phosphorylation of IκBα, phosphorylation of RelA/p65, activation of IKK, and regulation of gene expression), (ii) IKKα controls invasion of Her2+ cells, with apparent little contribution of IKKβ in this process, and (iii) PI3K-dependent pathways do not contribute to the direct activation of NF-κB in these cells.
Previous experiments from several groups have shown that IKKβ plays a major role in controlling canonical NF-κB activation downstream of inflammatory cytokines such as TNF (Verma et al., 1995). The potential contribution of IKKα to NF-κB activation downstream of Her2-dependent signaling or to that induced by other oncoproteins has not been fully elucidated. Lapatinib has been shown to be effective in its inhibition of the Akt and Erk pathways in Her2 overexpressing breast cancer cell lines and human tumor xenografts, but there are no reports of it having an effect on the NF-κB pathway (Xia et al., 2002; Zhou et al., 2004), although Herceptin has been shown to inhibit NF-κB activation in SKBr3 cells (Biswas et al., 2004). In our studies, treatment of Her2-overexpressing cell lines with 1 μM lapatinib led to a marked decrease in phosphorylation of NF-κB subunit p65 at serine 536 and of IκBα at serines 32 and 36 (Fig. 1A and B). Lapatinib also blocked NF-κB-induced gene transcription (Fig. 3C). Treatment of SKBr3 cells with lapatinib led to complete loss of phosphorylation of Akt at serine 473 (Fig. 1C), a marker for Akt activation.
To address potential contributions of IKKα and IKKβ to NF-κB activation in Her2+ cells and to the oncogenic phenotype, we used an IKK knockdown approach in Her2-overexpressing cells. Knockdown of IKKα led to a more dramatic reduction in p65 phosphorylation at Ser536 than did knockdown of IKKβ (Fig. 2A). Furthermore, knockdown of IKKα strongly reduced NF-κB activation as measured through EMSA and NF-κB-dependent reporter assays while IKKβ knockdown had less of an effect (Fig. 2B and 2C). Similarly, knockdown of IKKα was more effective at blocking IKK activity than knockdown of IKKβ (Fig. 2D). SKBr3 cells exhibit low levels of p52/NF-κB2, which is derived from IKKα-dependent processing of the p100/NF-κB2 precursor. Knockdown of IKKα had little effect on p52 levels in these cells, indicating that non-canonical pathway does not appear to be active in SKBr3 cells at a measurable level. Consistent with this, very low to undetectable levels of p52 or RelB are detected in the nuclei of SKBr3 cells (data not shown). It is important to note that inhibition of IKKβ can lead to a compensatory response whereby IKKα controls canonical NF-κB activation in some cell types (Lam et al., 2008). Our studies clearly indicate that loss of IKKα leads to reduced NF-κB activation downstream of Her2-induced signaling. A study showing that IKKα is necessary for self-renewal of Her2-transformed mammary initiating tumor cells (Cao et al., 2007) is consistent with our results demonstrating the importance of IKKα in controlling NF-κB downstream of Her2. The way in which Her2 may selectively activate IKKα in breast cancer remains to be investigated. One possibility is selective activation of IKKα by the kinase NIK, as NIK has been shown to associate with ErbB2 family member EGFR (Habib et al., 2001), and has been shown to be recruited to EGF/heregulin receptor signaling complexes (Chen et al., 2003).
The knockdown studies were extended to analysis of NF-κB-dependent target gene expression (Fig. 3A). Knockdown of IKKα lead to a more dramatic reduction in gene expression of IL-6, IL-8, CCL2, TNF and uPA than did knockdown of IKKβ. Decreased expression of these genes upon knockdown of the p65 subunit of NF-κB indicates that this activation is occurring through the canonical pathway. (Fig. 3B). To demonstrate that these genes are controlled through Her2, and not through Her2-independent pathways, lapatinib was shown to block target gene expression (Fig. 3C). This increase in chemokine and cytokine gene expression by Her2, as well as the increase in the expression of the serine protease uPA, shows a large similarity to Her2 induced gene expression signatures which have been previously reported, and this increase has been implicated in progression of multiple different cancers, including breast cancer (Arihiro et al., 2000; Chavey et al., 2007; Vazquez-Martin et al., 2008; Wang et al., 1999). Therefore, our gene expression data suggests that IKKα plays in important role in regulating genes involved in breast cancer progression, and this requires the scaffolding subunit NEMO (Fig. 4).
Some studies indicate that NF-κB can be activated downstream of PI3K/Akt (Dan et al., 2008; Makino et al., 2004). However, experiments using the PI3K inhibitor LY294002 indicate that NF-κB is not activated in Her2+ cells downstream of PI3K (Fig. 5). Thus, this pathway is not a link between Her2, IKKα and NF-κB activation. We cannot rule out a PI3K-independent Akt-controlled pathway in NF-κB activation. Additionally, we cannot rule out that PI3K and/or Akt have effects on NF-κB-target gene expression that function separately from the induction of NF-κB activation as assayed through experiments described above. Future studies will address Her2-regulated pathways that lead to activation of IKK. Other studies (Dillon et al., 2007) as well as our own (Fig. 6B) show that activation of the PI3K pathway plays an important role in cell proliferation/viability. Interestingly, knockdown of IKKα or IKKβ subunits (individually or together) by siRNA has no measurable effect on cell proliferation (Fig. 6A).
In order to determine if IKKα or IKKβ controls other oncogenic phenotypes, we used siRNA treatment and measured cell invasion of SKBr3 cells. Her2 overexpression has been shown to induce cell invasion, consistent with its ability to promote upregulation of genes such as IL-8 and uPA (Gum et al., 1995; Vazquez-Martin et al., 2008). Knockdown of IKKα, but not knockdown of IKKβ, significantly blocks the invasive phenotype of SKBr3 cells (Fig. 6C). This result is consistent with the regulation of target genes by IKKα that are associated with invasive phenotype. Interestingly, other factors have linked breast cancer invasion and NF-κB, including microRNAs (Ma et al., 2007). MicroRNAs have been shown to negatively regulate NF-κB activity and gene expression, such as microRNA-146, which can suppress expression of IL-6 and IL-8 through a reduction in levels of IRAK1 and TRAF6 in MDA-MB-231 cells, leading to the metastatic phenotype (Bhaumik et al., 2008).
This study shows that Her2 activation of NF-κB requires IKKα, and this PI3K-independent activation leads to an increase in cytokine and chemokine expression, as well as an increase in invasive phenotype (Fig. 6D). This data suggests that targeting multiple pathways in Her2+ breast cancer may be advantageous for effective therapy, and development of inhibitors of IKKα or the use of dual IKKα/IKKβ inhibitors may prove therapeutic in Her2+ cancer cells.
Supplementary Material
Acknowledgements
We’d like to thank William Comb for his assistance in the preparation of this manuscript, as well as Dr. H. Shelton Earp and Dr. Carolyn Sartor for generously providing reagents. This work is funded by NIH RO1CA73756 and RO1CA75080, and Department of Defense grant BC074027. Support is also provided by the Samuel Waxman Cancer Research Foundation.
References
- Arihiro K, Oda H, Kaneko M, Inai K. Cytokines facilitate chemotactic motility of breast carcinoma cells. Breast Cancer. 2000;7:221–30. doi: 10.1007/BF02967464. [DOI] [PubMed] [Google Scholar]
- Arora P, Cuevas BD, Russo A, Johnson GL, Trejo J. Persistent transactivation of EGFR and ErbB2/HER2 by protease-activated receptor-1 promotes breast carcinoma cell invasion. Oncogene. 2008;27:4434–45. doi: 10.1038/onc.2008.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga J, Tripathy D, Mendelsohn J, Baughman S, Benz CC, Dantis L, Sklarin NT, Seidman AD, Hudis CA, Moore J, Rosen PP, Twaddell T, Henderson IC, Norton L. Phase II study of weekly intravenous trastuzumab (Herceptin) in patients with HER2/neu-overexpressing metastatic breast cancer. Semin Oncol. 1999;26:78–83. [PubMed] [Google Scholar]
- Basseres DS, Baldwin AS. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25:6817–30. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- Belguise K, Sonenshein GE. PKCtheta promotes c-Rel-driven mammary tumorigenesis in mice and humans by repressing estrogen receptor alpha synthesis. J Clin Invest. 2007;117:4009–21. doi: 10.1172/JCI32424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaumik D, Scott GK, Schokrpur S, Patil CK, Campisi J, Benz CC. Expression of microRNA-146 suppresses NF-kappaB activity with reduction of metastatic potential in breast cancer cells. Oncogene. 2008;27:5643–5647. doi: 10.1038/onc.2008.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas DK, Shi Q, Baily S, Strickland I, Ghosh S, Pardee AB, Iglehart JD. NF-kappa B activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc Natl Acad Sci U S A. 2004;101:10137–42. doi: 10.1073/pnas.0403621101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–8. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Cao Y, Luo JL, Karin M. I{kappa}B kinase {alpha} kinase activity is required for self-renewal of ErbB2/Her2-transformed mammary tumor-initiating cells. Proc Natl Acad Sci U S A. 2007;104:15852–7. doi: 10.1073/pnas.0706728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavey C, Bibeau F, Gourgou-Bourgade S, Burlinchon S, Boissiere F, Laune D, Roques S, Lazennec G. Oestrogen receptor negative breast cancers exhibit high cytokine content. Breast Cancer Res. 2007;9:R15. doi: 10.1186/bcr1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Xu LG, Chen L, Li L, Zhai Z, Shu HB. NIK is a component of the EGF/heregulin receptor signaling complexes. Oncogene. 2003;22:4348–55. doi: 10.1038/sj.onc.1206532. [DOI] [PubMed] [Google Scholar]
- Cogswell PC, Guttridge DC, Funkhouser WK, Baldwin AS., Jr. Selective activation of NF-kappa B subunits in human breast cancer: potential roles for NF-kappa B2/p52 and for Bcl-3. Oncogene. 2000;19:1123–31. doi: 10.1038/sj.onc.1203412. [DOI] [PubMed] [Google Scholar]
- Dan HC, Cooper MJ, Cogswell PC, Duncan JA, Ting JP, Baldwin AS. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008;22:1490–500. doi: 10.1101/gad.1662308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon RL, White DE, Muller WJ. The phosphatidyl inositol 3-kinase signaling network: implications for human breast cancer. Oncogene. 2007;26:1338–45. doi: 10.1038/sj.onc.1210202. [DOI] [PubMed] [Google Scholar]
- Ethier SP, Mahacek ML, Gullick WJ, Frank TS, Weber BL. Differential isolation of normal luminal mammary epithelial cells and breast cancer cells from primary and metastatic sites using selective media. Cancer Res. 1993;53:627–35. [PubMed] [Google Scholar]
- Galang CK, Garcia-Ramirez J, Solski PA, Westwick JK, Der CJ, Neznanov NN, Oshima RG, Hauser CA. Oncogenic Neu/ErbB-2 increases ets, AP-1, and NF-kappaB-dependent gene expression, and inhibiting ets activation blocks Neu-mediated cellular transformation. J Biol Chem. 1996;271:7992–8. doi: 10.1074/jbc.271.14.7992. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109(Suppl):S81–96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680–4. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- Gum R, Wang SW, Lengyel E, Yu D, Hung MC, Juarez J, Boyd D. Up-regulation of urokinase-type plasminogen activator expression by the HER2/neu proto-oncogene. Anticancer Res. 1995;15:1167–72. [PubMed] [Google Scholar]
- Habib AA, Chatterjee S, Park SK, Ratan RR, Lefebvre S, Vartanian T. The epidermal growth factor receptor engages receptor interacting protein and nuclear factor-kappa B (NF-kappa B)-inducing kinase to activate NF-kappa B. Identification of a novel receptor-tyrosine kinase signalosome. J Biol Chem. 2001;276:8865–74. doi: 10.1074/jbc.M008458200. [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- Hegde PS, Rusnak D, Bertiaux M, Alligood K, Strum J, Gagnon R, Gilmer TM. Delineation of molecular mechanisms of sensitivity to lapatinib in breast cancer cell lines using global gene expression profiles. Mol Cancer Ther. 2007;6:1629–40. doi: 10.1158/1535-7163.MCT-05-0399. [DOI] [PubMed] [Google Scholar]
- Hynes NE, Stern DF. The biology of erbB-2/neu/HER-2 and its role in cancer. Biochim Biophys Acta. 1994;1198:165–84. doi: 10.1016/0304-419x(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Klapper LN, Kirschbaum MH, Sela M, Yarden Y. Biochemical and clinical implications of the ErbB/HER signaling network of growth factor receptors. Adv Cancer Res. 2000;77:25–79. [PubMed] [Google Scholar]
- Knuefermann C, Lu Y, Liu B, Jin W, Liang K, Wu L, Schmidt M, Mills GB, Mendelsohn J, Fan Z. HER2/PI-3K/Akt activation leads to a multidrug resistance in human breast adenocarcinoma cells. Oncogene. 2003;22:3205–12. doi: 10.1038/sj.onc.1206394. [DOI] [PubMed] [Google Scholar]
- Lam LT, Davis RE, Ngo VN, Lenz G, Wright G, Xu W, Zhao H, Yu X, Dang L, Staudt LM. Compensatory IKKalpha activation of classical NF-kappaB signaling during IKKbeta inhibition identified by an RNA interference sensitization screen. Proc Natl Acad Sci U S A. 2008;105:20798–803. doi: 10.1073/pnas.0806491106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YM, Pan Y, Wei Y, Cheng X, Zhou BP, Tan M, Zhou X, Xia W, Hortobagyi GN, Yu D, Hung MC. Upregulation of CXCR4 is essential for HER2-mediated tumor metastasis. Cancer Cell. 2004;6:459–69. doi: 10.1016/j.ccr.2004.09.027. [DOI] [PubMed] [Google Scholar]
- Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449:682–8. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- Makino K, Day CP, Wang SC, Li YM, Hung MC. Upregulation of IKKalpha/IKKbeta by integrin-linked kinase is required for HER2/neu-induced NF-kappaB antiapoptotic pathway. Oncogene. 2004;23:3883–7. doi: 10.1038/sj.onc.1207485. [DOI] [PubMed] [Google Scholar]
- Mayo MW, Wang CY, Cogswell PC, Rogers-Graham KS, Lowe SW, Der CJ, Baldwin AS., Jr. Requirement of NF-kappaB activation to suppress p53-independent apoptosis induced by oncogenic Ras. Science. 1997;278:1812–5. doi: 10.1126/science.278.5344.1812. [DOI] [PubMed] [Google Scholar]
- Papa S, Bubici C, Zazzeroni F, Pham CG, Kuntzen C, Knabb JR, Dean K, Franzoso G. The NF-kappaB-mediated control of the JNK cascade in the antagonism of programmed cell death in health and disease. Cell Death Differ. 2006;13:712–29. doi: 10.1038/sj.cdd.4401865. [DOI] [PubMed] [Google Scholar]
- Pegram MD, Lipton A, Hayes DF, Weber BL, Baselga JM, Tripathy D, Baly D, Baughman SA, Twaddell T, Glaspy JA, Slamon DJ. Phase II study of receptor-enhanced chemosensitivity using recombinant humanized anti-p185HER2/neu monoclonal antibody plus cisplatin in patients with HER2/neu-overexpressing metastatic breast cancer refractory to chemotherapy treatment. J Clin Oncol. 1998;16:2659–71. doi: 10.1200/JCO.1998.16.8.2659. [DOI] [PubMed] [Google Scholar]
- Pianetti S, Arsura M, Romieu-Mourez R, Coffey RJ, Sonenshein GE. Her-2/neu overexpression induces NF-kappaB via a PI3-kinase/Akt pathway involving calpain-mediated degradation of IkappaB-alpha that can be inhibited by the tumor suppressor PTEN. Oncogene. 2001;20:1287–99. doi: 10.1038/sj.onc.1204257. [DOI] [PubMed] [Google Scholar]
- Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W. IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J Biol Chem. 1999;274:30353–6. doi: 10.1074/jbc.274.43.30353. [DOI] [PubMed] [Google Scholar]
- She QB, Chandarlapaty S, Ye Q, Lobo J, Haskell KM, Leander KR, DeFeo-Jones D, Huber HE, Rosen N. Breast tumor cells with PI3K mutation or HER2 amplification are selectively addicted to Akt signaling. PLoS ONE. 2008;3:e3065. doi: 10.1371/journal.pone.0003065. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Singh S, Shi Q, Bailey ST, Palczewski MJ, Pardee AB, Iglehart JD, Biswas DK. Nuclear factor-kappaB activation: a molecular therapeutic target for estrogen receptor-negative and epidermal growth factor receptor family receptor-positive human breast cancer. Mol Cancer Ther. 2007;6:1973–82. doi: 10.1158/1535-7163.MCT-07-0063. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–82. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J, Ullrich A, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–12. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–92. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- Solt LA, May MJ. The IkappaB kinase complex: master regulator of NF-kappaB signaling. Immunol Res. 2008;42:3–18. doi: 10.1007/s12026-008-8025-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbrecher KA, Wilson W, 3rd, Cogswell PC, Baldwin AS. Glycogen synthase kinase 3beta functions to specify gene-specific, NF-kappaB-dependent transcription. Mol Cell Biol. 2005;25:8444–55. doi: 10.1128/MCB.25.19.8444-8455.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Martin A, Colomer R, Menendez JA. Her-2/neu-induced “cytokine signature” in breast cancer. Adv Exp Med Biol. 2008;617:311–9. doi: 10.1007/978-0-387-69080-3_29. [DOI] [PubMed] [Google Scholar]
- Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S. Rel/NF-kappa B/I kappa B family: intimate tales of association and dissociation. Genes Dev. 1995;9:2723–35. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- Wang W, Abbruzzese JL, Evans DB, Chiao PJ. Overexpression of urokinase-type plasminogen activator in pancreatic adenocarcinoma is regulated by constitutively activated RelA. Oncogene. 1999;18:4554–63. doi: 10.1038/sj.onc.1202833. [DOI] [PubMed] [Google Scholar]
- Weih F, Caamano J. Regulation of secondary lymphoid organ development by the nuclear factor-kappaB signal transduction pathway. Immunol Rev. 2003;195:91–105. doi: 10.1034/j.1600-065x.2003.00064.x. [DOI] [PubMed] [Google Scholar]
- Wilson W, 3rd, Baldwin AS. Maintenance of constitutive IkappaB kinase activity by glycogen synthase kinase-3alpha/beta in pancreatic cancer. Cancer Res. 2008;68:8156–63. doi: 10.1158/0008-5472.CAN-08-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W, Mullin RJ, Keith BR, Liu LH, Ma H, Rusnak DW, Owens G, Alligood KJ, Spector NL. Anti-tumor activity of GW572016: a dual tyrosine kinase inhibitor blocks EGF activation of EGFR/erbB2 and downstream Erk1/2 and AKT pathways. Oncogene. 2002;21:6255–63. doi: 10.1038/sj.onc.1205794. [DOI] [PubMed] [Google Scholar]
- Zhou H, Kim YS, Peletier A, McCall W, Earp HS, Sartor CI. Effects of the EGFR/HER2 kinase inhibitor GW572016 on EGFR- and HER2-overexpressing breast cancer cell line proliferation, radiosensitization, and resistance. Int J Radiat Oncol Biol Phys. 2004;58:344–52. doi: 10.1016/j.ijrobp.2003.09.046. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.