

Abstract
Cone photoreceptor cyclic nucleotide-gated (CNG) channel plays a pivotal role in phototransducton. Mutations in the channel subunits are associated with achromatopsia and progressive cone dystrophy in humans. Over 50 mutations have been identified in the channel CNGA3 subunit, with 50% of them located in the carboxyl (C)-terminus. This study investigates the defects of the two frequently occurring mutations, R377W and F488L, in the C-terminus of CNGA3. Ratiometric measurement of intracellular Ca2+ concentration and electrophysiological recordings showed loss of functional activity of the mutant channels in an HEK293 heterologous expression system. Immunofluorescence labeling revealed an apparent cytosolic aggregation of the mutant channels compared to the wild-type (WT). The R377W and F488L mutants, expressed and purified from Escherichia coli as glutathione S-transferase (GST) fused to the CNGA3 C-terminal domain, showed no negative effects on interactions with the channel subunits. Circular dichroism spectrum analyses were performed to examine the structural impact of the mutations. Although the R377W and F488L C-termini mutants retained stable, folded structures, the secondary structures of both mutants differed from the WT protein. Furthermore, the WT C-terminus exhibited a significant decrease in α-helical content in response to the channel ligands, while this allosteric transition was diminished in the two mutants. This is the first study showing structural impact of the disease-causing mutations in the cone CNG channel subunit. The observed alterations in the local secondary structure and active conformational change may confer an adverse effect on the channel's activity and cellular processing.
Photoreceptor cyclic-nucleotide-gated (CNG) channels are non-selective cation channels located in the plasma membrane of the outer segments, where they function as important components of the visual transduction system (1, 2). In darkness, these channels are opened by cGMP, maintaining an inward current. Light induces hydrolysis of cGMP, resulting in closure of the channels and hyperpolarization of the cell. Structurally, CNG channels belong to the super family of voltage-gated potassium channels. The channel comprises two structurally related subunit types, the A and B subunits. The rod channel is composed of CNGA1 and CNGB1 subunits, while the cone channel comprises CNGA3 and CNGB3 subunits (3, 4). Heterologous expression studies showed that the A subunits form ion-conducting units, while the B subunits function as modulators (1, 5). Both types of subunits share a common topology characterized by cytoplasmic amino (NH2)- and carboxyl (C)-termini, six transmembrane segments (S1-S6), a pore-forming loop between S5 and S6, and a cyclic nucleotide-binding domain (CNBD) at the C-terminus. Figure 1 shows the membrane topology of the mouse CNGA3 subunit.
Figure 1.
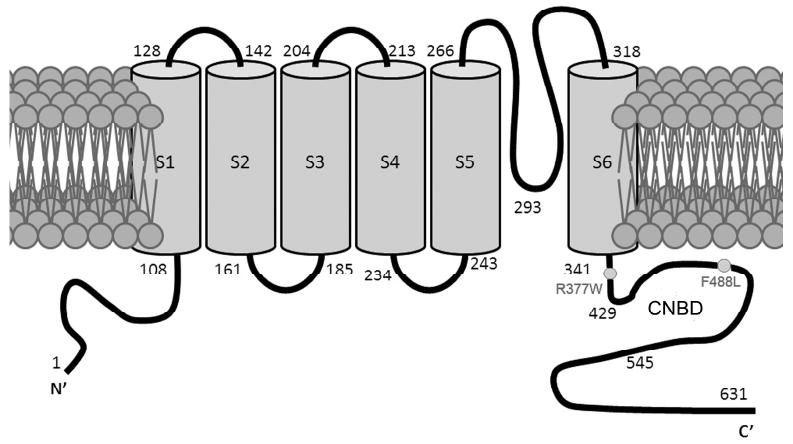
Membrane topology of the mouse CNGA3 with locations of the R377W and F488L mutations indicated. CNBD is located between residues 429 and 545.
Cone vision mediated by CNG channel activation is essential for central and color vision and visual acuity. Naturally occurring mutations in the channel subunits CNGA3 and CNGB3 are associated with achromatopsia, progressive cone dystrophy, and early-onset macular degeneration (6-10). Mutations in the CNGA3 subunit account for 25-30% of achromatopsia patients (6-8), which is an inherited disorder that affects approximately 1 in every 33,000 Americans. The condition is associated with color blindness, visual acuity loss, extreme light sensitivity, and nystagmus (8-10). Over 50 mutations have been identified in the CNGA3 subunit with nearly 50% of them located in the C-terminus. Most of these mutations are single amino acid substitutions. The R377W and F488L substitutions, located in the C-terminus, are among the four most frequently occurring mutations in the CNGA3 subunit (9). The R377W mutation is located in the C-linker while the F488L mutation is located in the CNBD (Fig. 1). The Arg377 and Phe488 residues are highly conserved among species studied (see Supplementary Figure 1 for sequence alignment of the C-terminus of CNGA3).
Many mutant CNGA3 channels have been studied in heterologous expression systems, showing loss of functional activity and impaired plasma membrane targeting (11-16). Nevertheless, little is known about the structural impact of these mutations. Understanding of the mutation effects on the channel secondary structure may provide potential mechanistic information. The purpose of this work is to identify the structural impact of the mutations in the C-terminus of CNGA3. To this end, we investigated the effects of the R377W and F488L mutations on the channel activity and cellular localization using a human embryonic kidney (HEK) 293 heterologous expression system. We also investigated the secondary structure using the channel C-termini expressed and purified from Escherichia (E.) coli. We found that both mutations abolished the channel activity and caused cytosolic aggregation of the channel. Circular dichroism (CD) spectra analysis showed that the α-helical content was decreased in the R377W and F488L C-termini compared to the wild-type (WT). Moreover, the WT C-terminus exhibited an apparent reduction of α-helical content in response to its ligands, while this effect was absent in the two mutants. Thus, this work provided the first experimental evidence showing the structural impact of the disease-causing mutations in the cone CNG channel.
Experimental Procedures
Constructs, cell culture, and transfection
The construct encoding the full-length mouse CNGA3 was generated as described previously (17). The R377W and F488L mutations in the mouse CNGA3, equivalent to the R436W and F547L mutations in the human CNGA3, respectively, were obtained by site-directed mutagenesis using the Quickchange Site-directed Mutagenesis Kit (Stratagene, La Jolla, CA). Use of the mouse sequences is to benefit future animal model studies. The glutathione-S-transferase (GST) fusion constructs encoding the NH2- and C-terminal cytoplasmic fragments of CNGA3 (amino acids 1–110 and 340–631, respectively) and CNGB3 (amino acids 1–196 and 432–694, respectively) were generated by polymerase chain reaction (PCR) amplification using the appropriate primers (see Supplementary Table 1), and conventional cloning procedures using a pGEX-4T-2 vector (Amersham Biosciences, Piscataway. NJ). The mouse CNGB3 cDNA was kindly provided by Dr. Martin Biel at the Munich Center for Integrated Protein Science, Munich, Germany.
Cell culture and transfection was performed as described previously (17). HEK293 cells were routinely cultured in Dubelcco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C in a humidified atmosphere with 5% CO2. Cells were transfected at 80-90% confluence (20 μg cDNA/per 100 mm dish) by using lipofectamine 2000® (Invitrogen, Carlsbad, CA), and used for experiments ∼48 h post-transfection.
Ratiometric measurement of intracellular Ca2+ concentration
The fluorescent indicator indo-1/AM was used to monitor Ca2+ influx through CNGA3 channels in cell suspensions. The assays were performed using a PTI QuantaMaster spectrofluorometer (Photon Technology International) as described previously (17). Briefly, cells (∼48 h post-transfection) were harvested, washed, and loaded with 2 μM Indo-1/AM (Sigma-Aldrich, St. Louis, MO) for 40 minutes at room temperature. After loading, cells were washed and resuspended in ECS buffer (140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 10 mM glucose, and 15 mM HEPES, pH 7.4). Approximately, 2 × 106 cells were used in each assay. Ca2+ entry in response to 8-pCPT-cGMP (Sigma-Aldrich, St. Louis, MO) at varying concentrations (10, 30, 100 and 300 μM) was determined by ratiometric measurement, which represents changes of free intracellular Ca2+ concentrations (expressed as a Δ340/380 ratio). Data were analyzed and graphed using GraphPad Prism software (GraphPad software, San Diego, CA).
Electrophysiological recordings
Standard whole-cell patch clamp recordings on cells grown on 35 mm culture dishes were performed as described previously (18, 19). The pipette solution consisted of (in mM): K+ aspartate 100, KCl 30, NaCl 5, MgCl2 2, Na-ATP 2, EGTA 1, HEPES 5 with pH 7.2 adjusted with KOH. Patch electrodes had resistances of 3-5 MΩ when filled with internal solution. Whole cell currents were recorded by using gap-free protocol with a holding potential at −50 mV. All experiments were performed at room temperature (23°C) and recorded using an Axopatch 200B amplifier (Axon Instrument, Foster City, CA). pCLAMP software (Axon Instruments, Foster City, CA) was used for data acquisition and analysis.
Immunofluorescence labeling and confocal microscopy
Immunofluorescence labeling was performed as described previously (17, 20). Briefly, cells were grown in DMEM medium on coverslips pre-coated with fibronectin (Sigma-Aldrich, St. Louis, MO). Cells were washed, fixed with 4% (w/v) paraformaldehyde for 10 min at room temperature, and blocked for 1 h at room temperature in 5% bovine serum albumin (BSA). Cells were then incubated with the rat monoclonal anti-CNGA3 (1:50) (kindly provided by Dr. Benjamin Kaupp at the Institute of Neurosciences and Biophysics, Jülich, Germany) overnight at 4°C, followed by incubation with Alexa-conjugated goat anti-rat secondary antibody (1:1000) for 1 h at room temperature. Vectashield™ containing DAPI stain (Vector Laboratories, Burlingame, CA) was used to mount the coverslips onto the slides.
The fluorescent signals were visualized using a 40× water immersion objective lens on an Olympus IX81-FV500 confocal laser scanning microscope (Olympus, Melville, NY) and analyzed with FluoView imaging software (Olympu, Melville, NY) as described previously (17). Multiple slices were taken at a thickness of 0.2 microns of cells. Using the intensity mapping feature of the software, cell slices with the highest localized fluorescent intensities in the intracellular area was chosen for further quantification. The cellular localization of the WT and mutant channel subunits was determined and the cytosolic aggregation was expressed in terms of percent of total cellular fluorescence intensity as described previously (17). Data were analyzed and graphed using GraphPad Prism software (GraphPad software, San Diego, CA).
Protein expression and purification
The GST fusion WT, R377W and F488L C-termini were expressed in E. coli BL21 (DE3) strain, and purified using glutathione-Sepharose resin (Amersham Biosciences, Piscataway, NJ) as described previously (21, 22). Briefly, cultures of BL21 cells possessing the pGEX-4T-2 vectors were grown at 37°C in Luria Broth (LB) medium containing ampicillin (50μg/ml). Expression was induced by adding isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 0.3 mM when the culture reached an optical density of 0.6-1.0 at 600 nm, and then the culture was incubated for an additional 20 h at 25°C prior to harvest. Bacteria were recovered by centrifugation at 6,000 × g for 15 min, and the pellets were washed in phosphate-buffered saline (PBS). Cells were then resuspended in ice-cold STE buffer [10 mM Tris, pH 8.0, 150 mM NaCl, 1 mM EDTA, 100 μg/ml of lysozyme, and 1.0 mM phenylmethylsulfonyl fluoride (PMSF)] and lysed by sonication. The lysates were then solubilized in 1.5% lauroylsarcosine for 30 min at 4°C, and the cellular debris was removed by centrifugation at 16,000×g at 4°C for 30 min. The fusion proteins were affinity-purified using glutathione-Sepharose resin and eluted with reduced glutathione (40 mM, 10 mM Tris, pH 8.0) following the manufacturer's protocol. The concentration of purified fusion protein was quantified with the Bradford assay (Bio-Rad, Hercules, CA) using BSA as the standard and was also determined by UV absorbance at 280 nm. The extinction coefficients used for the WT, R377W, and F488L fusion proteins were 64,660, 70,350, and 64,660 M-1cm-1, respectively. Expression and purification of the fusion proteins was examined by Commassie blue staining and by Western blotting using anti-GST antibody (Amersham Biosciences, Piscataway, NJ).
Preparation of retinal membrane protein extracts
Retinas from cone-dominant Nrl−/− mice, kindly provided by Dr. Anand Swaroop at NIH/NEI (Bethesda, MD), were used in the pull-down assays. Nrl−/− retinas have abundant expression of cone CNG channel and lack of rod CNG channel (20). The protein neural retina leucine zipper (Nrl) is a transcription factor that is preferentially expressed in rod photoreceptors and is essential for the normal development of rod photoreceptors. So mice lacking the Nrl gene have no rods, but have increased numbers of Scones, manifested as the loss of rod function and super-normal cone function (23). Mice were maintained under cyclic light (12-h light-dark) conditions; cage illumination was approximately 7 foot-candles during the light cycle. All experiments were approved by the local Institutional Animal Care and Use Committees (Oklahoma City, OK) and conformed to the guidelines on the care and use of animals adopted by the Society for Neuroscience and the Association for Research in Vision and Ophthalmology (Rockville, MD). Retinal membrane protein extracts were prepared as described previously (20). Briefly, after sacrifice, dissected retinas were homogenized on ice in Tris buffer (10 mM Tris-HCl, pH 7.4, 1 mM EDTA, 200 mM sucrose, 1 mM PMSF). The nuclei and cell debris were removed from the homogenate by centrifugation at 1,000 × g for 5 min at 4°C. The resulting supernatant was centrifuged at 16,000×g at 4°C for 30 min. The cell membranes from HEK293 cells were prepared similarly except sonication was used to lyse cells. The resultant membranes were solubilized in CHAPS solubilization buffer (10 mM Hepes-KOH, pH 7.4, 1 mM DTT, 10 mM CaCl2, 0.15 M KCl, 18 mM CHAPS, and 1.0 mM PMSF) at 4°C for 4 h. The supernatants were separated by centrifugation at 16,000×g at 4°C for 30 min. The solubilized membrane proteins were used in the GST pull-down experiments. Protein concentration was assessed using a Bio-Rad Protein Assay kit (Bio-Rad Laboratories, Hercules, CA).
GST pull-down assay
The assays were performed as described previously (21). Purified fusion proteins were used for the pull-down assay within 24 h after preparation. Mouse retinal membrane extracts (∼100 μg protein) were incubated with ∼5 μg of fusion protein bound to the glutathione resin in 200 μl of binding buffer (20 mM Tris-HCl, pH 7.5, 100 mM NaCl, 1 mM EDTA, and 1 mM PMSF) at 4°C for 2 h. The resin was washed three times with 0.5 ml of the binding buffer and eluted with the Laemmli sample buffer. Proteins bound to the fusion proteins were analyzed by Western blot analysis using anti-GST antibody and antibodies against CNGA3 and CNGB3.
SDS-PAGE and Western blot analysis
SDS-PAGE and Western blot analysis were performed as described previously (17). Following overnight blocking in 5% non-fat milk at 4°C, blots were incubated with primary antibodies at appropriate dilution ratios (CNGA3 and CNGB3, 1:250; actin, 1:2000; and GST, 1:1000) for 2 h at room temperature. The rabbit polyclonal anti-CNGA3 and anti-CNGB3 antibodies had been characterized and used in detection of these proteins in mouse retinas and in cells transfected with the cDNAs (17, 20). After 3×10 min washings with Tris-buffered saline with 0.1% Tween 20, the blots were incubated with HRP-conjugated secondary antibodies (1:12,500) for 1 h at room temperature. SuperSignal® West Dura Extended Duration chemiluminescent substrate (Pierce, Rockford, IL) was used to detect binding of the primary antibodies to their cognate antigens and images were captured using a Kodak Imaging Station (4000 R).
CD spectropolarimetry
The CD spectroscopy experiments were performed in the OUHSC Physical Biochemistry Core Facility using a JASCO J715 Spectropolarimeter with a PTC-348WI peltier temperature controller (Jasco, Corp., Tokyo, Japan) as described previously (24). Protein samples were dialyzed against PBS, pH 7.4, containing 2mM KH2PO4, 10mM Na2HPO47H2O, 137 mM NaCl, 2.7 mM KCl at 4°C overnight prior to CD spectropolarimetry analysis. Spectra were obtained from 6-7 μM protein with wavelength ranging from 250 to 200 nm using 0.1 cm cuvette pathlength at 25°C, and the results were expressed as the mean molar ellipticity. To examine the conformational changes of the CNGA3 C-termini in response to the ligand, 8-pCPT-cGMP was used at concentrations of 3, 10, 30, 100 μM in the same buffer. These concentrations of 8-pCPT-cGMP were shown previously to induce Ca2+ response in cells heterologously expressing CNGA3 (17). Responses to cGMP and cAMP (Sigma-Aldrich, St. Louis, MO) were also examined. Thermal denaturation curves were obtained at 220 nm from 20°C to 80°C.
Protein secondary structural contents were obtained using the CDPro software package (25) (available at http://lamar.colostate.edu/∼sreeram/CDPro). All three programs available in the CDPro package, including CONTIN, SELCON3 and CDSSTR, were executed using the same 56 protein reference library. The values of the resulting calculations were averaged to yield the percent of α-helix, β-sheet and random coil for the protein samples.
Limited tryptic digestion
Limited tryptic digestion experiments were performed as previously described (26, 27). Membranes (40 μg protein) prepared from HEK293 cells that had been transfected with cDNAs encoding the WT and mutant CNGA3 subunits were resuspended in protease inhibitor-free Tris buffer and incubated with trypsin-TPCK (Worthington Biochemicals, Lakewood, NJ) (30 μg/ml, at an approximate ratio of 1 : 6 of trypsin to membrane proteins) at 30°C for 2, 5, and 10 min. The trypsin-treated samples were then solubilized with 2× Laemmli sample buffer and resolved on 10% SDS-PAGE, followed by immunoblot analysis using anti-CNGA3 antibody.
Results
The R377W and F488L mutations cause loss of channel activity
Functional activity of the R377W and F488L mutant channels was examined by ratiometric measurement of intracellular Ca2+ concentration and by electrophysiological recordings. The concentration-dependent response of CNGA3 to 8-pCPT-cGMP was characterized in our previous studies (17, 19). HEK293 cells were transfected with cDNAs encoding the WT and mutant channels, which were then used for the functional assay 48 h post-transfection. With these assays we found that the two mutations had profound negative effects on the channel activity. Cells expressing the WT channel yielded a robust response when stimulated with 8-pCPT-cGMP, while cells expressing the R377W and F488L mutants showed no response (Fig. 2A). The deficiency of the mutant channel function was also assessed by electrophysiological recordings (Fig. 2B). The right panels of Figure 2A and B show the quantitative results of these assays. Expression and cell surface localization of the WT and mutant channels were demonstrated by Western blotting (Fig. 2C) and immunofluorescence labeling (Fig. 3). As shown in Figure 2C, expression levels of the mutant channels were comparable to the WT. Thus, the R377W and F488L mutations abolish channel activity while not effecting relative expression levels.
Figure 2.
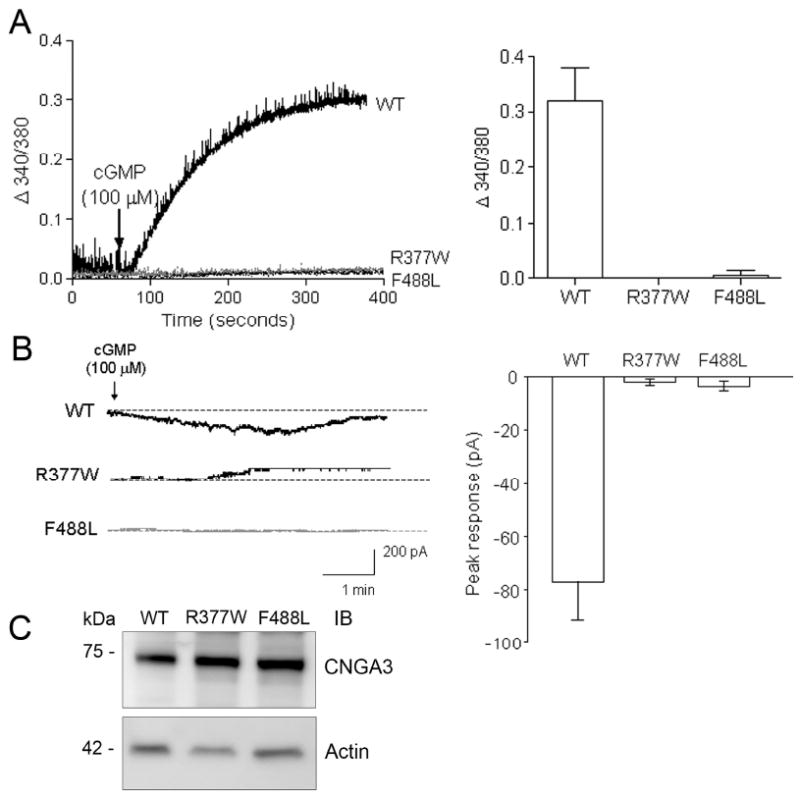
The R377W and F488L mutations cause loss of channel activity. (A). Intracellular calcium responses of HEK293 cells expressing the WT-CNGA3, R337W-CNGA3, and F488L-CNGA3 in response to 8-pCPT-cGMP (100 μM) stimulation. The left panel shows the representative response curves and the right panel is the bar graph showing the quantitative analysis of the calcium measurement (at 300 s after cGMP stimulation) from 4-5 independently performed experiments. (B). Electrophysiological recordings of HEK293 cells expressing the WT-CNGA3, R337W-CNGA3, and F488L-CNGA3 in response to 8-pCPT-cGMP (100 μM) stimulation. The recordings were performed using gap-free protocol with a holding potential at −50 mV. The left panel shows representative patch-clamp recording profiles and the right panel is the bar graph showing the quantitative analysis of peak amplitude in response to 100 μM 8-pCPT-cGMP (WT n = 18, R377W n = 13, and F488L n = 9). (C). Western blot detection of expression of the WT-CNGA3, R337W-CNGA3, and F488L-CNGA3 in HEK293 cells that had been transfected with the respective cDNAs. Cellular protein extractions were resolved on 10% SDS-PAGE, followed by Western blotting using the polyclonal anti-CNGA3 antibody.
Figure 3.
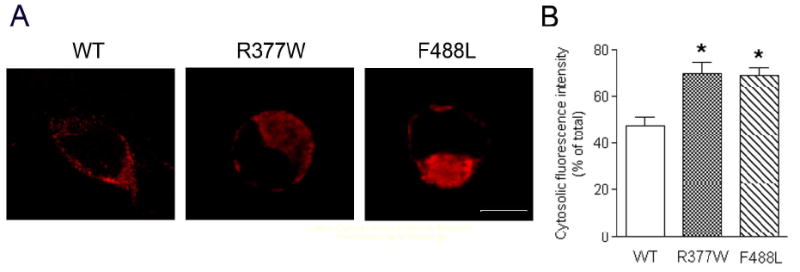
The R377W and F488L mutations cause cytosolic aggregation of the channels. Immunofluorescence labeling was performed to determine the cellular localization of the WT-CNGA3, R337W-CNGA3, and F488L-CNGA3. (A). Representative confocal images showing cellular localization of the WT-CNGA3, R337W-CNGA3, and F488L-CNGA3 in HEK293 cells that had been transfected with the respective cDNAs. Scale bar: 10 μm. (B). The bar graph shows the quantitative results of the cellular fluorescence labeling intensities. Bars represent the means ± SEM of the number of cells (12 for the WT transfected, 18 for the R377W transfected, and 16 for the F488L transfected) from 3 independently performed experiments. Unpaired Student's t test was used for determination of the significance. *, p < 0.05.
The R377W and F488L mutations lead to channel cytosolic aggregation
Immunofluorescence labeling and confocal microscopic analysis were performed to determine the cellular localization of the WT and mutant channels. Although cell surface expression of the mutant channels was detected, we observed an apparent cytosolic aggregation of the immunofluorescence signals in cells expressing the R377W and F488L mutants compared to the WT (Fig. 3A). Quantitative analysis of the confocal images shows that the fluorescence intensities of the cytosolic aggregates (expressed as percent of total cellular fluorescence intensity) in cells expressing the R377W or F488L mutants are 70% and 69%, respectively, which are significantly higher than that in cells expressing the WT channel (48%) (Fig. 3B). Hence, the R377W and F488L mutations lead to channel cellular aggregation, suggesting an impaired cellular processing and trafficking.
The R377W and F488L mutations do not affect the channel subunit interaction
Subunit interactions between CNGA3 and CNGB3 have been documented in a heterologous expression system (28) and in mouse retina (20) and the C-terminus of CNGA3 has been shown to be critical for the channel subunit interactions (28). We examined the effects of R377W and F488L mutations on the subunit interactions. The GST fusion NH2- and C-termini of CNGA3 and CNGB3 were expressed, purified, and used in the pull-down assays (Fig. 4A). The assays were performed by incubating the fusion proteins with Nrl−/− retinal extracts. Nrl−/− retinas express abundant cone CNG channel but lack expression of rod CNG channel (20). The pull-down products were resolved by 10% SDS-PAGE, followed by Western blotting using antibodies against CNGA3, CNGB3 and GST. Through these experiments, we found that the C-terminus but not the N-terminus of CNGA3 bound to CNGA3 and CNGB3 (Fig. 4B, left panel). These results are consistent with previous report by Zhong et al., using co-immunoprecipitation assays (28). We further examined the effects of the R377W and F488L mutations on the C-terminus binding to CNGA3 and CNGB3. Figure 4C shows expression of the WT and mutant fusion proteins detected by Commassie blue staining (left panel) and binding of the fusion proteins to CNGA3 and CNGB3 (right panel). Hence these results suggest that the R377W and F488L mutations do not affect the channel homo- and hetero-subunit interactions. Of note, in the pull-down assays using CNGB3 fusion proteins we detected CNGA3 but not CNGB3 (Fig. 4B, right panel), suggesting a hetero interaction with CNGA3 but not a homotypic interaction with CNGB3.
Figure 4.
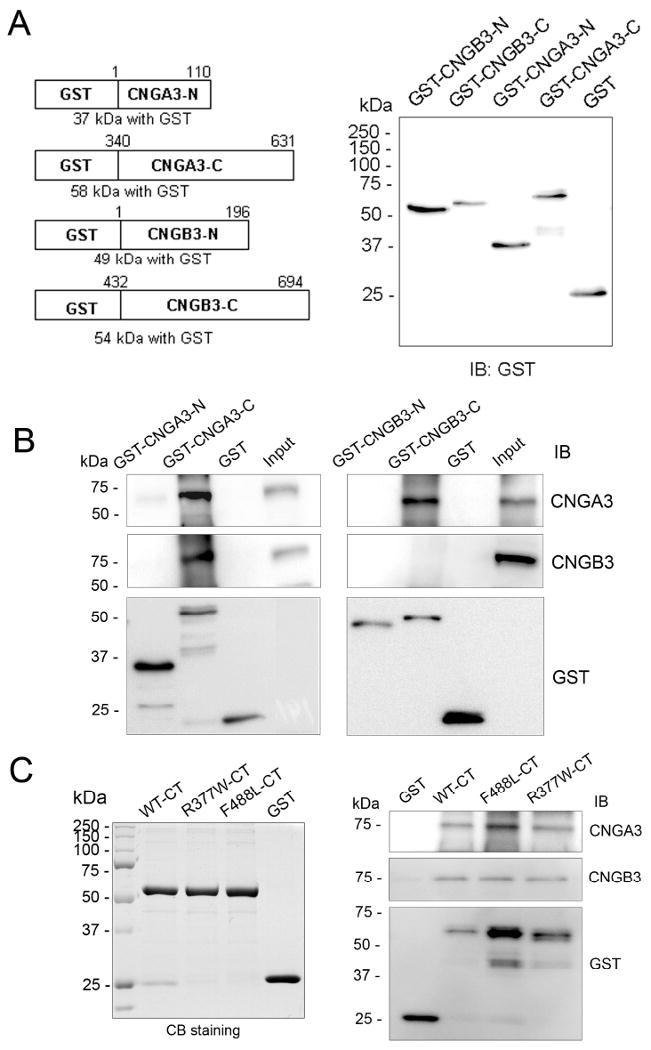
The R377W and F488L mutations do not affect the channel subunit interactions. GST pull-down assays were performed to examine the subunit interactions and the effects of mutations. The GST fusion proteins bound to glutathione resin were incubated with Nrl−/− retinal membrane extracts (∼100 μg protein) at 4°C for 2 h. The pull-down products were resolved on 10% SDS-PAGE, followed by Western blotting using antibodies as indicated. (A). Diagram of the GST-fusion proteins (left panel) and detection of their expression (right panel). (B). GST pull-down assays showing an involvement of the C-terminus of CNGA3 in the subunit interactions (left panels), and presence of a homo interaction of CNGA3 but not CNGB3 (right panels). (C). GST pull-down assays show that the R377W and F488L mutations do not affect the subunit interactions. The left panel shows expression of the GST fusion proteins of the WT, R377W, and F488L C-termini detected by Commassie blue staining, and the right panel is a representative pull-down result showing the WT and two mutant C-termini to interact with CNGA3 (upper panel) and CNGB3 (middle panel).
The R377W and F488L mutations result in decreased α-helicity in the C-terminus of CNGA3
CD spectra analyses of the WT, R377W and F488L C-termini were performed to examine the effects of the mutations on the secondary structure of CNGA3. In these experiments, the CD spectra of GST-CNGA3 C-termini were obtained first, followed by measurement of GST under identical solution conditions (CD buffer: PBS, pH 7.4, containing 2mM KH2PO4, 10mM Na2HPO47H2O, 137 mM NaCl, 2.7 mM KCl). After conversion of the spectra to units of molar ellipticity, the GST spectra were subtracted from spectra of GST-CNGA3 C-termini to yield the signals only due to CNGA3 C-termini. The assays showed that both GST and CNGA3 C-termini gave typical CD spectra of a protein with secondary structure, with the spectra of the two mutants different from the WT. Figure 5A shows typical CD spectra of the WT, R377W and F488LC-termini with subtraction of GST. We analyzed the amounts of α-helix, β-sheet and random coil using three different algorithms (Selcon, Continll, and CDSSTR). Use of these algorithms was shown to result in a high accuracy for determination of a protein secondary structural content (25, 29). From these analyses we estimated that the structure of the CNGA3 C-terminus consisted of 20±0.7 % of α-helix, 28±0.6 % of β-sheet and 49±1.1% of random coil in the buffer solution used. The α-helical contents of the two mutants were lower compared to the WT, and there were no significant differences in the amounts of β-sheet and random coil between WT and the two mutants (Fig. 5B). The results of CD profile analyzed with the three algorithms were consistent. Hence, these results suggest that the R377W and F488L mutations have a local structural impact; the mutations decrease the α-helicity in the C-terminus of CNGA3.
Figure 5.
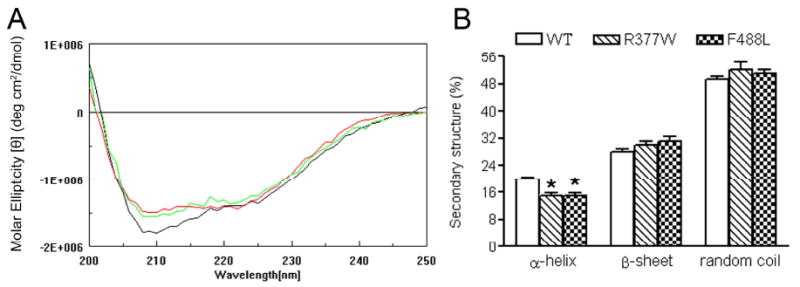
The R377W and F488L mutations result in decreased α-helicity of the C-terminus of CNGA3. CD spectra analyses were performed to examine the effects of the R377W and F488L mutation on the secondary structure of CNGA3. GST fusion proteins were expressed, purified and used in the CD analysis. (A). Representative CD spectra of the WT (black) and R377W (red) and F488L (green) mutants with subtraction of GST spectrum. The two mutants show CD spectra different from the WT. (B). Quantitative analyses of the amounts of α-helix and β-sheet were performed using three databases (Selcon, Contin, and CDSSTR). Shown are the summarized results of these analyses. The α-helical contents of the two mutants were significantly lower compared to the WT, where there were no differences in the amounts of β-sheet and random coil between WT and the two mutants. Unpaired Student's t test was used for determination of the significance (P < 0.05).
The R377W and F488L mutations abolish the conformational change of the C-terminus in response to the channel ligands
CD spectral analysis is a reliable technique for monitoring conformational change in peptide or protein upon binding of its ligand (30). Binding of cGMP to CNBD at the C-terminus of CNG channel and the subsequent conformational change is the critical step of the channel activation. The R377W mutation is located in the C-linker, while the F488L mutation is located in the CNBD (see Fig. 1). These two mutations may affect the conformational change in response to the channel ligands. Therefore, we examined the CD spectra of the WT and mutant C-termini upon addition of the ligands 8-pCPT-cGMP, cGMP and cAMP. In these experiments we found a concentration-dependent reduction of the negative ellipticity of the WT C-terminus upon addition of 8-pCPT-cGMP (Fig. 6A). As described above, contributions of GST were subtracted from the spectra of GST-CNGA3 C-termini to obtain signal due to the C-termini alone. Indeed GST showed no spectral changes with varying concentrations of 8-pCPT-cGMP (Fig. 6A). We focused primarily on changes in α-helical content, since it is the most accurately determined secondary structural element within this wavelength range (25). Quantitative analysis showed a ∼25% reduction of α-helical content in the WT C-terminus upon addition of 8-pCPT-cGMP (100 μM) (Fig. 6B, left panel). It is worth noting, however, that there was a corresponding increase in β-sheet content (Fig. 6B, right panel), suggesting a conformational shift of α-helical regions to β-sheet upon ligand binding. We attribute this relatively large reduction in CD signal to a conformational change in the WT domain upon ligand binding. It is also possible that the CD signal change may be partially due to other factors including an induced CD signal in the ligand upon binding to the WT domain or to effects on the aromatic residues in the domain upon ligand binding. Further structural studies will be necessary to elucidate the extent of the conformational change. Significantly, no such changes in CD spectral signals were observed in the two mutants upon addition of 8-pCPT-cGMP (Fig. 6). Similar to the response to 8-pCPT-cGMP, WT but not the R377W and F488L mutants responded to cGMP (Fig. 7A) and slightly to cAMP (Fig. 7B). As expected, cAMP was less potent than cGMP in eliciting a reduction of the negative ellipticity (Fig. 7B). Hence, upon addition of its ligand the WT C-terminus renders a conformational change that might be essential for activation of the channel, while this process is attenuated in the two mutants.
Figure 6.
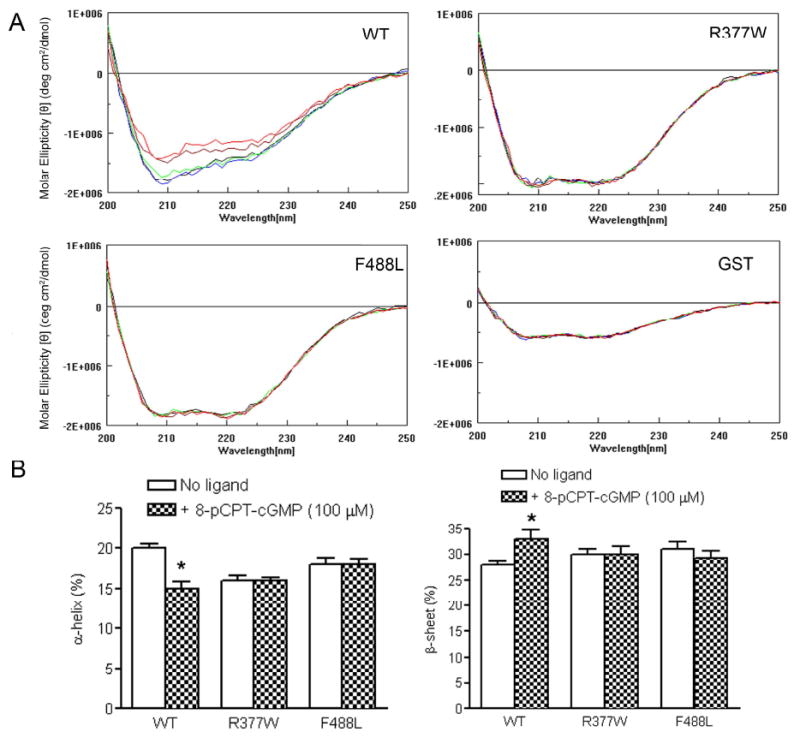
WT, but not the R377W and F488L mutants, shows a conformational change of the C-terminus in response to 8-pCPT-cGMP. (A). Representative CD profiles of the WT, R377W and F488L C-termini in response to varying concentrations (0, black; 3.0, blue; 10.0, green; 30.0, brown; and 100, red) (μM) of 8-pCPT-cGMP. A concentration-dependent reduction of negative ellipticity was seen in the WT but not the R377W and F488L mutants upon addition of 8-pCPT-cGMP. Representative CD spectral profiles of GST in response to varying concentrations of 8-pCPT-cGMP were included. (B). Quantitative analyses of α-helix and β-sheet amounts in the WT and mutant C-termini upon addition of 8-pCPT-cGMP (100 μM). Unpaired Student's t test was used for determination of the significance (P < 0.05).
Figure 7.
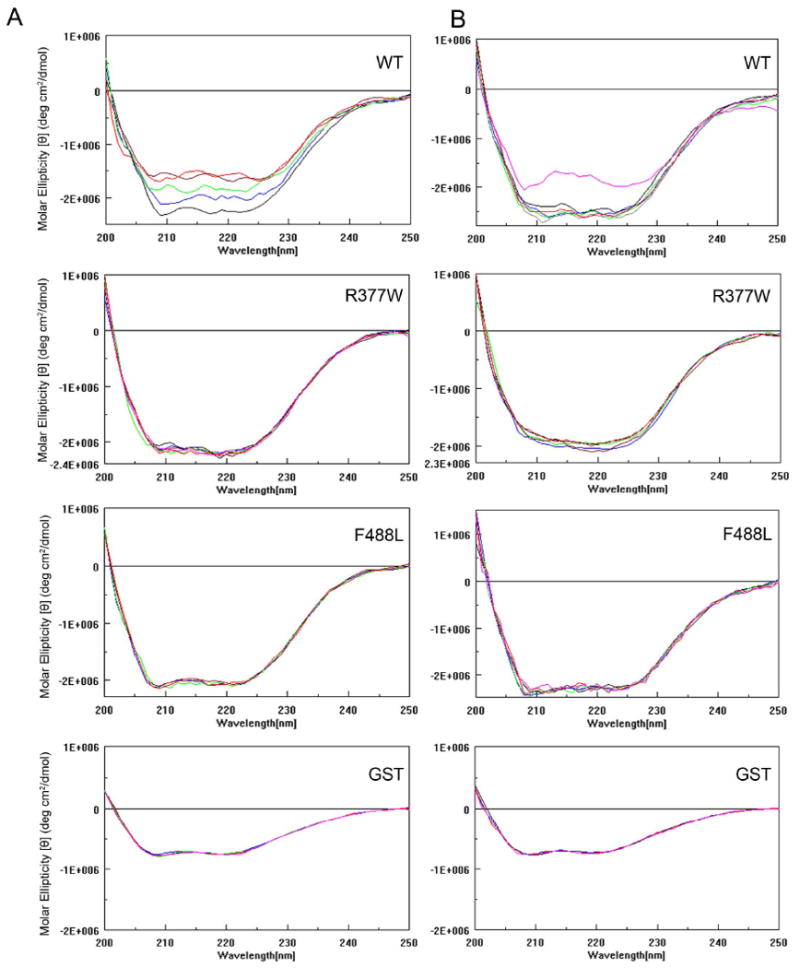
WT, but not the R377W and F488L mutants, shows a conformational change of the C-terminus in response to cGMP and cAMP. Representative CD profiles of the WT, R377W and F488L C-termini in response to varying concentrations (0, black; 3.0, blue; 10.0, green; 30.0, brown; 100, red; and 300, pink) (μM) of cGMP (A) and cAMP (B). A concentration-dependent reduction of negative ellipticity was seen in the WT but not the R377W and F488L mutants upon addition of cGMP. cAMP at the highest concentration (300 μM) used induced a CD profile change in the WT but not the mutant C-termini. Representative CD spectral profiles of GST in response to varying concentrations of cGMP and cAMP were included.
The R377W and F488L mutations do not affect the protein stability
A mutation is known quite often to decrease protein stability. We examined the effects of the R377W and F488L mutations on the stability of the channel subunit. Thermal stability was evaluated by analyzing the CD profiles of the thermal denaturation of the WT and mutant C-termini. Thermal denaturation curves were obtained at 220 nm from 20°C to 80°C. The transition temperature was at ∼ 59°C for GST unfolding and was at ∼ 68°C (a more broad transition) for the C-terminal fusion proteins (Fig. 8A). The unfolding temperature of the R377W and F488L mutants was not different from that of WT (Fig. 8A), suggesting that the mutants had a similar pattern of denaturation as WT. The protein stability was also examined by limited proteolytic trypsin digestion. The membranes prepared from cells that had been transfected with cDNAs for the WT and mutants were incubated with trypsin-TPCK (30 μg/ml) at 30°C for 2, 5, and 10 min. This method has been used to study the stability of membrane proteins including PMA1 H+-ATPase (27), cholecystokinin receptor (26) and peripherin/rds (31). From this assay we found that the R377W and F488L mutants had a similar proteolytic sensitivity as the WT. The cleavage rates of the mutants by trypsin were not different from the WT (Fig. 8B). This result is consistent with findings from the thermal denaturation analyses. Hence, the R377W and F488L mutants showed a similar thermal and proteolytic stability as the WT, indicating that the R377W and F488L mutations do not affect the overall stability of the protein. The comparable expression level of the WT and mutants (Fig. 2C) also supports an unchanged protein stability.
Figure 8.
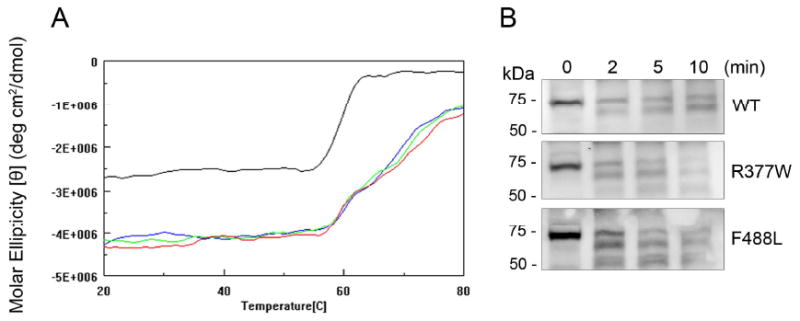
The R377W and F488L mutations do not affect protein stability. (A). Thermal denaturation study of the WT and R377W and F488L mutant C-termini. Shown are thermal denaturation curves for the WT and mutant C-termini obtained at 220 nm from 20°C to 80°C (GST, black; WT, blue; R377W, green; and F488L, red). Denaturation curves are in units of molar ellipticity vs. temperature. The transition temperature was at ∼ 59°C for GST and was at ∼ 68°C for the C-terminal fusion proteins. The unfolding temperature at the R377W and F488L mutants was not different from that in the WT. (B). Limited tryptic digestion of the WT and R377W and F488L mutant CNGA3. The membranes prepared from cells that had been transfected with cDNAs encoding the WT and R377W and F488L mutants were incubated with trypsin-TPCK (30 μg/ml) at 30°C for 2, 5, and 10 min. The digested products were resolved on 10% SDS-PAGE, followed by Western blotting using the polyclonal anti-CNGA3 antibody. Shown are representative images showing limited proteolysis of the WT and mutant CNGA3 subunits. The cleavage rates of the mutants to trypsin were about the same as the WT.
Discussion
Over twenty disease-causing mutations have been identified in the C-terminus of CNGA3 (2, 6, 7). Many of them have been examined in heterologous expression systems and shown to lack activity (12, 13, 15, 16); yet little is known about the structural impact of these mutations. This work investigated the effects of R377W and F488L mutation and revealed a functional and structural impact. Although the global structure was not significantly different between the WT and mutants, the α-helical content was decreased in the R377W and F488L C-termini. The WT C-terminus exhibited an apparent reduction of α-helical content in response to the channel ligands, while this allosteric transition was diminished in the two mutants. Hence the Arg377 and Phe488 residues have a significant allosteric role in the channel activation and the R377W and F488L mutations alter the local secondary structure of the C-terminus, implicating a structural mechanism underlying the mutation-associated channel inactivation.
The C-terminus of CNG channel subunits consists of a CNBD, the C-linker (a region that connects the CNBD to the sixth transmembrane segment in the pore region), and the distal C-terminus. Activation of the channels is promoted by ligand-induced conformational changes in their C-terminal regions and involves allosteric structural changes in several different regions of the protein (32-34). The architecture of the CNBD and C-linker regions has been revealed by the X-ray crystallographic structures of the C-terminal regions of the hyper-polarization-activated cyclic nucleotide-modulated (HCN) channels (another family of the cyclic nucleotide-modulated channels) (35, 36). This C-terminal fragment assembles as a 4-fold symmetric tetramer directly below the transmembrane portion of the channel. The primary intersubunit interactions are comprised of hydrogen bonds, hydrophobic interactions, and salt bridges. It has been shown that the salt bridges at the interface of the C-terminus are essential for the C-terminal movement during gating (37). The residue Arg377 is located in the C-linker (see Figure 1) and has been shown to participate in the salt bridge interactions (37). Via charge-reversal mutagenesis and modification of introduced cysteine residues it was found that manipulation of the charges in the residues that are involved in the salt bridge interactions dynamically changed the free energy of channel opening (38). Using X-ray crystallography, it was shown that changing the charge hindered the simultaneous formation of the salt bridges (38). Our CD data showing an alteration of the secondary structure and diminished conformational change in response to cGMP in the R377W mutant C-terminus are consistent with the mutagenesis and crystallography results (38) and demonstrate a critical allosteric role of the residue Arg377 in the channel activation. Hence the alteration of the local secondary structure caused by the R377W mutation likely abolishes the activation of the channel via affecting the salt bridge interactions.
The residue Phe488 is within the CNBD (see Figure 1). Our CD analysis demonstrated a structural impact caused by the F488L mutation. Replacing the bulky aromatic phenylalanine by a smaller leucine may cause a negative effect on the allosteric transition of the CNBD upon ligand binding, and in turn, interferes with the active conformational change. In a study by Reuter et al., culturing cells at lower temperature (27°C) rescued the response to cGMP in cells expressing the F547L mutant of human CNGA3 (equivalent to F488L mutant of mouse CNGA3) without affecting the channel surface targeting (16), suggesting an alteration of the folding states of the mutant channel. As this residue is within CNBD, it is also possible that the altered secondary structure has a negative impact on the CNBD binding ability to its ligand, leading to the observed inactive phenotype.
Our work also suggests that the structural alteration by these two mutations is local rather than global. This is supported by the overall similarity of the CD profiles between the WT and mutants, by unaffected subunit interactions, and by unchanged protein stability. Indeed the X-ray crystallography showed that changing the charge on the residues that are involved in the salt bridge interactions did not change the global protein configuration (38). The unchanged protein stability and subunit interaction suggest that the ion channel's activity seems more sensitive to a local structural change, while its stability alteration may require a more dramatic and global impact.
In addition to loss of functional activity, the R377W and F488L mutants exhibited an enhanced cytosolic aggregation. Indeed many mutations have been found to cause the channel cytosolic aggregation (12, 15, 16). Hence, an impaired channel cellular processing and trafficking appears to be a general phenomenon caused by a variety of mutations at different locations of the channel subunit. Ion channel ontogeny involves many steps, starting from subunit synthesis to the multimeric complex formation and their final targeting to the plasma membrane (39). Critical points are protein folding, subunit assembly, interacting with chaperone proteins, and targeting to the plasma membrane. It is not known why different mutations in the distinct regions cause a common phenotype. A plausible explanation is that a channel protein must maintain a certain secondary and higher order structure in order to associate with other proteins such as chaperone(s), and a mutation may cause a local alteration of the secondary structure, which alters the channel-chaperone interactions. One example is the cardiac potassium channel (40). The trafficking-deficient mutants of the cardiac potassium channel were shown to enhance the channel interaction with the chaperones Hsp90 and Hsp70; as a consequence, the mutant channels remained tightly associated with Hsp90 and Hsp70, and trapped in the endoplasmic reticulum (40). It is also likely that the mutations impair the interaction of a channel with chaperone(s), which in turn leads to a defected cellular trafficking. It would be worthwhile to explore the mutation effects on the channel-chaperone interactions. Indeed little is known about trafficking of the CNG channel and chaperones involved. The membrane adaptor protein ankyrin-G was recently shown to play a role in trafficking and membrane targeting of rod CNG channel (41). It is not known whether this protein is involved in the trafficking of cone CNG channel. Recently the anterograde molecular motor kinesin-2 was shown to play a role in transportation of cone but not rod specific proteins to the outer segments (42). It would be interesting to examine whether there is an interaction of CNGA3 with ankyrin-G and with kinesin-2, and whether the channel trafficking defective mutations have any effect on these interactions.
This work shows that the structural impact may result in two distinct consequences: the loss of channel activity and impairment of channel cellular processing. It could be difficult to distinguish whether it is more significant that the mutant channel cannot respond to cGMP properly or whether it is more significant that the mutant channel cannot be appropriately processed and transported to the plasma membrane. Our calcium and patch clamp assays showed that the mutations abolish physiological function, while our fluorescence experiments revealed that a portion of the mutant channels do, in fact, localize to the plasma membrane. Looking at these data together one could reasonably argue that loss of channel activity might be a more severe consequence and contribute more to the observed phenotype (lack of response to the channel ligands). Also, since cellular processing and trafficking of a channel is generally regulated by multiple mechanisms, a single mutation may interfere with but not completely abolish this process.
The subunit interactions of CNG channel have been studied in heterologoues expression system (4, 28) and in mouse retina (20). The C-terminus of CNGA3, specifically a C-terminal leucine zipper (CLZ) in the distal C-terminus (between the residue 566 and 613 of the mouse CNGA3), was shown to be critical for the subunit interactions (28). We examined whether the R377W and F488L mutations have any negative effect on the subunit interactions. We found that the C-terminus of CNGA3 is involved in the subunit interactions, as reported by Zhong et al. (28), and the R377W and F488L C-termini retained their ability to bind to CNGA3 and CNGB3. Hence, the local structural impact by the two mutations seems to have no negative effect on the CLZ domain's function. Of note, in the pull-down assays we detected a homo-interaction of CNGA3 but not CNGB3. This result is in line with the finding by Zhong et al. (28) showing a homotypic interaction of CNGA3, but not in agreement with the report by Peng et al. (4) showing the homo-interaction of CNGB3.
In summary, this work investigated the defects of the two most frequently occurring mutations in the C-terminus of CNGA3. The R377W and F488L mutants failed to respond to the channel ligands and aggregated in the cytosolic compartments. CD spectra analysis of the mutant C-termini revealed a decreased α-helical content and diminished conformational change in response to the channel ligands. Hence the alteration of the local secondary structure caused by these two mutations is likely responsible for the channel inactivation and impaired cellular processing. The observation that a large number of mutations at the distinct regions of CNGA3 lead to a common fate of a channel, i.e., loss of activity and impaired cellular trafficking, suggests that an integral local secondary structure is critical for these processes and an impaired local structure and folding may represent a common mechanism of channel inactivation.
Supplementary Material
Acknowledgments
We thank Drs. Benjamin Kaupp, Martin Biel, and Anand Swaroop for providing the rat monoclonal anti-CNGA3 antibody, mouse CNGB3 cDNA, and Nrl−/− mouse line, respectively. We thank Ruby Rahman, Alexander Quiambao, and Cynthia Harry for their excellent technique assistance.
This work was supported by grants from the National Center for Research Resources (P20RR017703), the National Institute of Health (P30EY12190, DK077699, and AI-054467), and the American Health Assistance Foundation.
The abbreviations used are
- WT
wild-type
- CNG
cyclic nucleotide-gated
- cGMP
cyclic guanosine monophosphate
- cAMP
cyclic adenosine monophosphate
- CNBD
cyclic nucleotide-binding domain
- CNGA1
A-subunit of the rod photoreceptor CNG channel
- CNGB1
B-subunit of the rod photoreceptor CNG channel
- CNGA3
A-subunit of the cone photoreceptor CNG channel
- CNGB3
B-subunit of the cone photoreceptor CNG channel
- GST
glutathione-S-transferase
- HEK
human embryonic kidney
- cDNA
complementary deoxyribonucleic acid
- PMSF
phenylmethylsulfonyl fluoride
- BSA
bovine serum albumin
- PBS
phosphate-buffered saline
- CD
circular dichroism
Footnotes
Supporting Information Available: Figure S1, sequence alignment of the C-terminus of CNGA3 among species studied, and Table S1, primers used to amplify the NH2- and COOH-terminal cytoplasmic domains of CNGA3 and CNGB3. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Zagotta WN, Siegelbaum SA. Structure and function of cyclic nucleotide-gated channels. Annu Rev Neurosci. 1996;19:235–263. doi: 10.1146/annurev.ne.19.030196.001315. [DOI] [PubMed] [Google Scholar]
- 2.Kaupp UB, Seifert R. Cyclic nucleotide-gated ion channels. Physiol Rev. 2002;82:769–824. doi: 10.1152/physrev.00008.2002. [DOI] [PubMed] [Google Scholar]
- 3.Weitz D, Ficek N, Kremmer E, Bauer PJ, Kaupp UB. Subunit stoichiometry of the CNG channel of rod photoreceptors. Neuron. 2002;36:881–889. doi: 10.1016/s0896-6273(02)01098-x. [DOI] [PubMed] [Google Scholar]
- 4.Peng C, Rich ED, Varnum MD. Subunit configuration of heteromeric cone cyclic nucleotide-gated channels. Neuron. 2004;42:401–410. doi: 10.1016/s0896-6273(04)00225-9. [DOI] [PubMed] [Google Scholar]
- 5.Gerstner A, Zong X, Hofmann F, Biel M. Molecular cloning and functional characterization of a new modulatory cyclic nucleotide-gated channel subunit from mouse retina. J Neurosci. 2000;20:1324–1332. doi: 10.1523/JNEUROSCI.20-04-01324.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kohl S, Marx T, Giddings I, Jagle H, Jacobson SG, Apfelstedt-Sylla E, Zrenner E, Sharpe LT, Wissinger B. Total colourblindness is caused by mutations in the gene encoding the alpha-subunit of the cone photoreceptor cGMP-gated cation channel. Nat Genet. 1998;19:257–259. doi: 10.1038/935. [DOI] [PubMed] [Google Scholar]
- 7.Nishiguchi KM, Sandberg MA, Gorji N, Berson EL, Dryja TP. Cone cGMP-gated channel mutations and clinical findings in patients with achromatopsia, macular degeneration, and other hereditary cone diseases. Hum Mutat. 2005;25:248–258. doi: 10.1002/humu.20142. [DOI] [PubMed] [Google Scholar]
- 8.Wissinger B, Gamer D, Jagle H, Giorda R, Marx T, Mayer S, Tippmann S, Broghammer M, Jurklies B, Rosenberg T, Jacobson SG, Sener EC, Tatlipinar S, Hoyng CB, Castellan C, Bitoun P, Andreasson S, Rudolph G, Kellner U, Lorenz B, Wolff G, Verellen-Dumoulin C, Schwartz M, Cremers FP, Apfelstedt-Sylla E, Zrenner E, Salati R, Sharpe LT, Kohl S. CNGA3 mutations in hereditary cone photoreceptor disorders. Am J Hum Genet. 2001;69:722–737. doi: 10.1086/323613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kohl S, Baumann B, Broghammer M, Jagle H, Sieving P, Kellner U, Spegal R, Anastasi M, Zrenner E, Sharpe LT, Wissinger B. Mutations in the CNGB3 gene encoding the beta-subunit of the cone photoreceptor cGMP-gated channel are responsible for achromatopsia (ACHM3) linked to chromosome 8q21. Hum Mol Genet. 2000;9:2107–2116. doi: 10.1093/hmg/9.14.2107. [DOI] [PubMed] [Google Scholar]
- 10.Kohl S, Baumann B, Rosenberg T, Kellner U, Lorenz B, Vadala M, Jacobson SG, Wissinger B. Mutations in the cone photoreceptor G-protein alpha-subunit gene GNAT2 in patients with achromatopsia. Am J Hum Genet. 2002;71:422–425. doi: 10.1086/341835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Faillace MP, Bernabeu RO, Korenbrot JI. Cellular processing of cone photoreceptor cyclic GMP-gated ion channels: a role for the S4 structural motif. J Biol Chem. 2004;279:22643–22653. doi: 10.1074/jbc.M400035200. [DOI] [PubMed] [Google Scholar]
- 12.Liu C, Varnum MD. Functional consequences of progressive cone dystrophy-associated mutations in the human cone photoreceptor cyclic nucleotide-gated channel CNGA3 subunit. Am J Physiol Cell Physiol. 2005;289:C187–198. doi: 10.1152/ajpcell.00490.2004. [DOI] [PubMed] [Google Scholar]
- 13.Muraki-Oda S, Toyoda F, Okada A, Tanabe S, Yamade S, Ueyama H, Matsuura H, Ohji M. Functional analysis of rod monochromacy-associated missense mutations in the CNGA3 subunit of the cone photoreceptor cGMP-gated channel. Biochem Biophys Res Commun. 2007;362:88–93. doi: 10.1016/j.bbrc.2007.07.152. [DOI] [PubMed] [Google Scholar]
- 14.Patel KA, Bartoli KM, Fandino RA, Ngatchou AN, Woch G, Carey J, Tanaka JC. Transmembrane S1 mutations in CNGA3 from achromatopsia 2 patients cause loss of function and impaired cellular trafficking of the cone CNG channel. Invest Ophthalmol Vis Sci. 2005;46:2282–2290. doi: 10.1167/iovs.05-0179. [DOI] [PubMed] [Google Scholar]
- 15.Koeppen K, Reuter P, Kohl S, Baumann B, Ladewig T, Wissinger B. Functional analysis of human CNGA3 mutations associated with colour blindness suggests impaired surface expression of channel mutants A3(R427C) and A3(R563C) Eur J Neurosci. 2008;27:2391–2401. doi: 10.1111/j.1460-9568.2008.06195.x. [DOI] [PubMed] [Google Scholar]
- 16.Reuter P, Koeppen K, Ladewig T, Kohl S, Baumann B, Wissinger B. Mutations in CNGA3 impair trafficking or function of cone cyclic nucleotide-gated channels, resulting in achromatopsia. Hum Mutat. 2008;29:1228–1236. doi: 10.1002/humu.20790. [DOI] [PubMed] [Google Scholar]
- 17.Ding XQ, Fitzgerald JB, Matveev AV, McClellan ME, Elliott MH. Functional activity of photoreceptor cyclic nucleotide-gated channels is dependent on the integrity of cholesterol- and sphingolipid-enriched membrane domains. Biochemistry. 2008;47:3677–3687. doi: 10.1021/bi7019645. [DOI] [PubMed] [Google Scholar]
- 18.Malykhina AP, Qin C, Greenwood-van Meerveld B, Foreman RD, Lupu F, Akbarali HI. Hyperexcitability of convergent colon and bladder dorsal root ganglion neurons after colonic inflammation: mechanism for pelvic organ cross-talk. Neurogastroenterol Motil. 2006;18:936–948. doi: 10.1111/j.1365-2982.2006.00807.x. [DOI] [PubMed] [Google Scholar]
- 19.Fitzgerald JB, Malykhina AP, Al-Ubaidi MR, Ding XQ. Functional expression of cone cyclic nucleotide-gated channel in cone photoreceptor-derived 661W cells. Adv Exp Med Biol. 2008;613:327–334. doi: 10.1007/978-0-387-74904-4_38. [DOI] [PubMed] [Google Scholar]
- 20.Matveev AV, Quiambao AB, Browning Fitzgerald J, Ding XQ. Native cone photoreceptor cyclic nucleotide-gated channel is a heterotetrameric complex comprising both CNGA3 and CNGB3: a study using the cone-dominant retina of Nrl-/- mice. J Neurochem. 2008;106:2042–2055. doi: 10.1111/j.1471-4159.2008.05548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ding XQ, Stricker HM, Naash MI. Role of the second intradiscal loop of peripherin/rds in homo and hetero associations. Biochemistry. 2005;44:4897–4904. doi: 10.1021/bi048414i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frangioni JV, Neel BG. Solubilization and purification of enzymatically active glutathione S-transferase (pGEX) fusion proteins. Anal Biochem. 1993;210:179–187. doi: 10.1006/abio.1993.1170. [DOI] [PubMed] [Google Scholar]
- 23.Mears AJ, Kondo M, Swain PK, Takada Y, Bush RA, Saunders TL, Sieving PA, Swaroop A. Nrl is required for rod photoreceptor development. Nat Genet. 2001;29:447–452. doi: 10.1038/ng774. [DOI] [PubMed] [Google Scholar]
- 24.Godderz LJ, Rahman NS, Risinger GM, Arbuckle JL, Rodgers KK. Self-association and conformational properties of RAG1: implications for formation of the V(D)J recombinase. Nucleic Acids Res. 2003;31:2014–2023. doi: 10.1093/nar/gkg281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sreerama N, Woody RW. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal Biochem. 2000;287:252–260. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 26.Ding XQ, Pinon DI, Furse KE, Lybrand TP, Miller LJ. Refinement of the conformation of a critical region of charge-charge interaction between cholecystokinin and its receptor. Mol Pharmacol. 2002;61:1041–1052. doi: 10.1124/mol.61.5.1041. [DOI] [PubMed] [Google Scholar]
- 27.Nakamoto RK, Verjovski-Almeida S, Allen KE, Ambesi A, Rao R, Slayman CW. Substitutions of aspartate 378 in the phosphorylation domain of the yeast PMA1 H+-ATPase disrupt protein folding and biogenesis. J Biol Chem. 1998;273:7338–7344. doi: 10.1074/jbc.273.13.7338. [DOI] [PubMed] [Google Scholar]
- 28.Zhong H, Molday LL, Molday RS, Yau KW. The heteromeric cyclic nucleotide-gated channel adopts a 3A:1B stoichiometry. Nature. 2002;420:193–198. doi: 10.1038/nature01201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pelton JT, McLean LR. Spectroscopic methods for analysis of protein secondary structure. Anal Biochem. 2000;277:167–176. doi: 10.1006/abio.1999.4320. [DOI] [PubMed] [Google Scholar]
- 30.Siligardi G, Hussain R. Biomolecules interactions and competitions by non-immobilised ligand interaction assay by circular dichroism. Enantiomer. 1998;3:77–87. [PubMed] [Google Scholar]
- 31.Ding XQ, Nour M, Ritter LM, Goldberg AF, Fliesler SJ, Naash MI. The R172W mutation in peripherin/rds causes a cone-rod dystrophy in transgenic mice. Hum Mol Genet. 2004;13:2075–2087. doi: 10.1093/hmg/ddh211. [DOI] [PubMed] [Google Scholar]
- 32.Gordon SE, Zagotta WN. Localization of regions affecting an allosteric transition in cyclic nucleotide-activated channels. Neuron. 1995;14:857–864. doi: 10.1016/0896-6273(95)90229-5. [DOI] [PubMed] [Google Scholar]
- 33.Mottig H, Kusch J, Zimmer T, Scholle A, Benndorf K. Molecular regions controlling the activity of CNG channels. J Gen Physiol. 2001;118:183–192. doi: 10.1085/jgp.118.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matulef K, Zagotta WN. Cyclic nucleotide-gated ion channels. Annu Rev Cell Dev Biol. 2003;19:23–44. doi: 10.1146/annurev.cellbio.19.110701.154854. [DOI] [PubMed] [Google Scholar]
- 35.Flynn GE, Black KD, Islas LD, Sankaran B, Zagotta WN. Structure and rearrangements in the carboxy-terminal region of SpIH channels. Structure. 2007;15:671–682. doi: 10.1016/j.str.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zagotta WN, Olivier NB, Black KD, Young EC, Olson R, Gouaux E. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature. 2003;425:200–205. doi: 10.1038/nature01922. [DOI] [PubMed] [Google Scholar]
- 37.Craven KB, Zagotta WN. Salt bridges and gating in the COOH-terminal region of HCN2 and CNGA1 channels. J Gen Physiol. 2004;124:663–677. doi: 10.1085/jgp.200409178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Craven KB, Olivier NB, Zagotta WN. C-terminal movement during gating in cyclic nucleotide-modulated channels. J Biol Chem. 2008;283:14728–14738. doi: 10.1074/jbc.M710463200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deutsch C. The birth of a channel. Neuron. 2003;40:265–276. doi: 10.1016/s0896-6273(03)00506-3. [DOI] [PubMed] [Google Scholar]
- 40.Ficker E, Dennis AT, Wang L, Brown AM. Role of the cytosolic chaperones Hsp70 and Hsp90 in maturation of the cardiac potassium channel HERG. Circ Res. 2003;92:e87–100. doi: 10.1161/01.RES.0000079028.31393.15. [DOI] [PubMed] [Google Scholar]
- 41.Kizhatil K, Baker SA, Arshavsky VY, Bennett V. Ankyrin-G promotes cyclic nucleotide-gated channel transport to rod photoreceptor sensory cilia. Science. 2009;323:1614–1617. doi: 10.1126/science.1169789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baehr W, Watt CB, Williams DS, Le YZ, Li S, Chen CK, Frederick JM, Avasthi P. Heterotrimeric Kinesin-2 is Required for Trafficking of Membrane Proteins to Cone, but Not Rod, Outer Segments. ARVO. 2009 doi: 10.1523/JNEUROSCI.3976-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.