

Abstract
Background and purpose:
Apremilast is an orally administered phosphodiesterase-4 inhibitor, currently in phase 2 clinical studies of psoriasis and other chronic inflammatory diseases. The inhibitory effects of apremilast on pro-inflammatory responses of human primary peripheral blood mononuclear cells (PBMC), polymorphonuclear cells, natural killer (NK) cells and epidermal keratinocytes were explored in vitro, and in a preclinical model of psoriasis.
Experimental approach:
Apremilast was tested in vitro against endotoxin- and superantigen-stimulated PBMC, bacterial peptide and zymosan-stimulated polymorphonuclear cells, immunonoglobulin and cytokine-stimulated NK cells, and ultraviolet B light-activated keratinocytes. Apremilast was orally administered to beige-severe combined immunodeficient mice, xenotransplanted with normal human skin and triggered with human psoriatic NK cells. Epidermal skin thickness, proliferation index and inflammation markers were analysed.
Key results:
Apremilast inhibited PBMC production of the chemokines CXCL9 and CXCL10, cytokines interferon-γ and tumour necrosis factor (TNF)-α, and interleukins (IL)-2, IL-12 and IL-23. Production of TNF-α by NK cells and keratinocytes was also inhibited. In vivo, apremilast significantly reduced epidermal thickness and proliferation, decreased the general histopathological appearance of psoriasiform features and reduced expression of TNF-α, human leukocyte antigen-DR and intercellular adhesion molecule-1 in the lesioned skin.
Conclusions and implications:
Apremilast displayed a broad pattern of anti-inflammatory activity in a variety of cell types and decreased the incidence and severity of a psoriasiform response in vivo. Inhibition of TNF-α, IL-12 and IL-23 production, as well as NK and keratinocyte responses by this phosphodiesterase-4 inhibitor suggests a novel approach to the treatment of psoriasis.
Keywords: immunopharmacology, skin pharmacology, chemokines, anti-inflammatory drugs, PDE inhibitor, inflammation
Introduction
cAMP is a pivotal second messenger that is well known to regulate inflammatory responses (Tasken and Aandahl, 2004). The sole means of degrading cAMP is through the activity of the large superfamily of phosphodiesterases (PDE) (Conti and Beavo, 2007). Within this group, enzymes of the four-gene PDE4 family play a key role in degrading cAMP in inflammatory cells, as well as endothelial cells, smooth muscle cells and keratinocytes (Houslay et al., 2005). Indeed, it is now well appreciated that PDE4 enzymes play a key role in the compartmentalization of cAMP signalling in various cell types (Terrin et al., 2006; Willoughby et al., 2007), due to isoforms being targeted to specific signalling complexes (Baillie and Houslay, 2005; Huston et al., 2006; Houslay et al., 2007), including those associated with activation of inflammatory cells. This makes PDE4 potentially a very important therapeutic target.
The role of PDE4 in regulating inflammation has been well documented, due in large part to the study of small molecular weight PDE4 inhibitors, such as rolipram (Giembycz, 2008; Spina, 2008; Press and Banner, 2009). PDE4 inhibitors are a well-characterized class of pharmaceutical agents, with a broad range of anti-inflammatory activities in vitro, and in in vivo preclinical models of asthma, lung neutrophilia, arthritis, inflammatory bowel disease, and multiple sclerosis, osteoporosis and other conditions (Houslay et al., 2005; Videla et al., 2006; Keshavarzian et al., 2007; Yao et al., 2007; Spina, 2008; Press and Banner, 2009). During the past decade, PDE4 inhibitors have been studied in clinical trials of chronic obstructive pulmonary disease (arofylline, cilomilast and roflumilast), asthma (roflumilast) and ulcerative colitis (tetomilast) (Lipworth, 2005; Fan Chung, 2006; Martina et al., 2006; Schreiber et al., 2007; Giembycz, 2008).
Phosphodiesterase-4 inhibitors have also been studied as topical agents for the treatment of psoriasis and atopic dermatitis with positive results. Ro 20-1724 1% cream was efficacious in psoriasis, although similar to occlusive treatment with 0.025% triamcinolone acetonide cream (Stawiski et al., 1979). Another PDE4 inhibitor, CP-80633 (0.5% ointment) significantly improved clinical scores (erythema, induration and excoriation) in atopic dermatitis (Hanifin et al., 1996). However, the efficacy of PDE4 inhibitors may be greater as systemic rather than topical agents (Teixeira et al., 1994). When taken together, available data provide a strong rationale for further exploring the utility of systemic PDE4 inhibitors in the clinical treatment of psoriasis and atopic dermatitis.
Psoriasis is considered to be a Th1 autoimmune skin disease because of the involvement of pro-inflammatory cytokines, interferon (IFN)-γ and tumour necrosis factor (TNF)-α. While psoriasis can be either triggered or worsened by various environmental factors, some of which are bacterial or viral, the disease is also associated with specific genetic markers (Gottlieb, 2005). The psoriatic immune response involves monocytes, dendritic cells, neutrophils and T cells, all of which contribute to aberrant keratinocyte proliferation (Lowes et al., 2007). PDE4 inhibitors have been shown to inhibit production of pro-inflammatory cytokines, such as TNF-α, IFN-γ and interleukin (IL)-2 from peripheral blood monocytes and T cells (Claveau et al., 2004). In neutrophils, PDE4 inhibitors are known to inhibit production of leukotriene B4 (LTB4) and IL-8, both autocrine chemotactic factors, which promote further neutrophilia in inflamed tissues (Schudt et al., 1991). However, little is known about the effect of PDE4 inhibition in other cells involved in psoriasis, namely natural killer (NK) cells and keratinocytes. The importance of cells bearing NK markers in the pathophysiology of psoriasis has become better understood in recent years (Bos et al., 2005). Gilhar et al. demonstrated that injection of NK cells from psoriatic donors into non-lesioned skin, engrafted onto beige-severe combined immunodeficient (SCID) mice, could induce classic psoriasis histology (Gilhar et al., 2002). Despite extensive study of the role of PDE4 in T lymphocytes (Prehn et al., 2001; Claveau et al., 2004), the function of PDE4 in the phenotypically related NK cells has not been previously examined. PDE4 function in keratinocyte biology has only been superficially explored in keratinocyte cell lines (Tenor et al., 1995; Mammone et al., 1998). Taken together, results from these studies suggest that elevation of cAMP in human keratinocytes, via PDE4 inhibition, might mitigate some of the pathophysiological responses of keratinocytes in psoriasis.
Apremilast is a novel PDE4 inhibitor with TNF-α inhibitory activity (Man et al., 2009), currently under clinical investigation for the treatment of psoriasis and other inflammatory conditions (Khobzaoui et al., 2005; Gottlieb et al., 2008;Papp et al., 2008). The current study demonstrates the broad anti-inflammatory effects of apremilast in vitro, namely the inhibition of production of multiple mediators including TNF-α, IFN-γ, CXCL9 (monokine induced by IFN-γ, or MIG), CXCL10 (IFN-γ-induced protein of 10 kDa, or IP-10), IL-2, IL-12, IL-23, macrophage inflammatory protein (MIP)-1α, monocyte chemoattractant protein (MCP)-1 and granulocyte macrophage-colony stimulating factor (GM-CSF) from PBMC. The responses of polymorphonuclear cells (PMN), including IL-8 and LTB4 production, were also inhibited by apremilast. TNF-α production by NK cells and keratinocytes was inhibited by apremilast in vitro, demonstrating for the first time that these two cell types, involved in psoriasis pathophysiology, are directly affected by a PDE4 inhibitor. These in vitro anti-inflammatory activities, in particular the inhibition of TNF-α, IL-12 and IL-23 production, and the ability of apremilast to suppress psoriasiform lesions in vivo suggests that this compound may be a useful agent in the treatment of psoriasis via a multifaceted mechanism.
Methods
PDE enzyme assays
Crude PDE4 enzyme was purified from U937 human monocytic cells by gel filtration chromatography, and PDE reactions were carried as previously described at 1 µM cAMP (Muller et al., 1998). The binding constant, Ki, for apremilast was calculated from a Lineweaver-Burke analysis, where the y-intercept = 1/Vmax, and the control plot x-intercept = −1/KM. These data yielded an average PDE4 Vmax of 1.16 ± 0.15 pmol·min−1 mg−1 protein, and an average PDE4 KM of 2.82 ± 0.25 μM (mean ± SD from three experiments). Because the mode of inhibition by apremilast was partially competitive in this enzyme preparation, the affinity constant Ki of the inhibitor for the free enzyme could not be obtained. However, the Ki value, or the affinity constant of the inhibitor for the enzyme-substrate complex, was calculated from a secondary replot of 1/delta slope versus 1/[Inhibitor]. From this secondary slope replot, the x-intercept = −1/Ki. The Ki value is the mean from three experiments.
Plasmids encoding recombinant human PDE4A4, PDE4B2, PDE4C2 and PDE4D3 have been described previously (Huston et al., 1996; 1997; Bolger et al., 1997; Owens et al., 1997). These were expressed in DEAE-Dextran transfected Cos-7 cells with confirmation of expression performed, as previously described, by immunoblotting using antisera specific for each PDE4 subfamily (MacKenzie and Houslay, 2000; MacKenzie et al., 2002; Lynch et al., 2005) and for cAMP PDE activity at 1 µM cAMP (Marchmont et al., 1981; Lobban et al., 1994). As before (Bolger et al., 1996; MacKenzie et al., 2002), >98% of the cAMP PDE activity in transfected cells was due to expression of the appropriate recombinant PDE4 enzyme. Cos-7 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 10 000 U·mL−1 penicillin/streptomycin and 2 mM glutamine. Cell lines were transfected using Lipofectamine™ 2000 reagent according to recommended protocols.
The specificity of apremilast for PDE4 versus enzymes from other PDE families was assessed at a single concentration of 10 µM against PDE1A, PDE1C, PDE2A, PDE3A, PDE3B, PDE5A1, PDE7A, PDE7B, PDE8A1, PDE9A2, PDE10A1 and PDE11A4 (BPS Bioscience, San Diego, CA, USA) using IMAP TR-FRET Screening Express with Progressive Binding kit, 100 nM FAM-AMP or 100 nM FAM-cGMP (AXXORA, San Diego, CA, USA).
PBMC purification and stimulation
Human leukocytes obtained from healthy blood donors (Blood Center of New Jersey, East Orange, NJ, USA) were diluted 1:1 with sterile Hank's Balanced Salt Solution and centrifuged over room temperature Ficoll-Paque Plus to yield peripheral blood mononuclear cells (PBMC). PBMC were washed in Hank's Balanced Salt Solution and resuspended in Roswell Park Memorial Institute (RPMI) complete medium (RPMI 1640), 5% human serum, 100 U·mL−1 penicillin, 100 mg mL−1 streptomycin, 2 mM L-glutamine) and counted. One hundred microlitres (2 × 106 L−1) of PBMC were added to each well of a 96-well flat-bottom plate (final cell count = 2 × 105 per well) and incubated at 37°C for 1 h. Fifty microlitres of (4×) compound was added to each test well, and 50 µL medium containing 1% dimethylsulphoxide (DMSO) was added to each control well ([DMSO]final = 0.25%) and the plate was incubated for 1 h at 37°C. Cells were then stimulated with 50 µL, 4 ng·mL−1 lipopolysaccharide (LPS) from Salmonella abortus equii ([LPS]final = 1 ng mL−1) and incubated for 18 h at 37°C. For stimulation with superantigen, PBMC were plated in 96-well tissue culture plates at 3 × 105 cells per well in complete medium, pretreated with compounds at 37°C for 1 h, then stimulated with 100 ng mL−1 Staphylococcal enterotoxin B (SEB) for 18 h.
Quantitative reverse transcription-polymerase chain reaction
Cells were harvested and RNA isolation was performed using RNeasy according to the manufacturer's instructions. Reverse transcription was performed, converting 1 µg RNA to cDNA for each sample according to manufacturer's protocol (RT Kit). Quantitative real-time polymerase chain reaction (PCR) was performed for gene expression analysis using 50 ng cDNA per sample. Gene expression assays for target genes and endogenous glyceraldehyde 3-phosphate dehydrogenase control were from Applied Biosystems. Expression was measured on a real-time PCR System 7500 (Applied Biosystems). Relative quantifications were calculated with SDS v.1.3.1 software.
Cytokine and chemokine protein analysis
A 50 µL sample of supernatant from each well was transferred into new round-bottomed 96-well plates and stored at −20°C for cytokine analysis by cytometric bead array using a Luminex IS100 instrument (Luminex Corporation, Austin, TX, USA). LincoPlex kits with antibody bound beads for Luminex xMAP Technology (Millipore) were combined into multiplex format prior to assay. Data analysis was performed using Upstate Beadview software. IL-2 and IFN-γ levels from SEB-stimulated PBMC were measured by enzyme-linked immunoabsorbant assay (elisa) (R&D Systems).
PMN isolation and stimulation
Polymorphonuclear cells were isolated from human leukocytes by separation from PBMC using Ficoll gradient centrifugation to remove PBMC. Erythrocytes were removed by sedimentation in 3% dextran followed by hypotonic lysis in 0.2% saline. Finally, any contaminating monocytes were depleted using human leukocyte antigen (HLA) class II magnetic beads. The resulting PMN were determined to be approximately 74% CD16+ and Mac-1+ by flow cytometry. Eosinophil contamination was determined to be approximately 3% by CD9 staining. No CD14+ monocytes were detected. PMN (3 × 105 cells per well) were pretreated with titrated apremilast for 1 h, and then stimulated with zymosan A particles (heat-killed Saccharomyces cerevisiae) at various doses. Polymyxin B sulphate (40 nM final) was added to all samples to neutralize any contaminating LPS. After overnight incubation, supernatants were harvested and assayed for IL-8 by elisa.
For LTB4 production, PMN were resuspended in phosphate-buffered saline without calcium or magnesium (BioWhittaker) containing 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulphonic acid (HEPES) (pH 7.2) and plated in 96-well tissue culture plates at a concentration of 1.7 × 106 cells per well. Cells were treated with 50 μM thimerosal/1 mM CaCl2/1 mM MgCl2 for 15 min at 37°C 5% CO2, then treated with apremilast in a final DMSO concentration of 0.01% in duplicate for 10 min. PMN were stimulated with 1 μM N-formyl-Met-Leu-Phe (fMLF) for 30 min, then lysed by the addition of methanol (20% final concentration) and frozen in a dry ice/isopropanol bath for 10 min. Lysates were stored at −70°C until the LTB4 content was measured by competitive LTB4elisa (R&D Systems).
For CD18/CD11b (Mac-1) expression, PMN were pretreated with apremilast for 10 min and stimulated with fMLF for 30 min, then placed on ice, stained with anti-CD18-FITC and anti-CD11b-PE and analysed by flow cytometry using a fluorescence-activated cell sorter Calibur flow cytometer (BD Biosciences, San Jose, CA, USA).
To measure adhesion of neutrophils to endothelial cells, human umbilical vein endothelial cells (HUVEC) were plated out onto 96-well plates (5 × 103 cells per well) in medium containing 2% fetal bovine serum 4 days prior to the experiment to ensure adhesion of HUVEC to the plate. On the day of the experiment, neutrophils were isolated from human leukocytes and labelled with the fluorescent dye Calcein-AM for 1 h. Labelled neutrophils (2 × 105 per well) were added to the adhered HUVECs and pretreated with apremilast for 10 min at 37°C in a humidified incubator at 5% CO2. fMLF was added to trigger neutrophil adhesion to HUVECs for 30 min. The cells were washed with phosphate-buffered saline containing 2% glucose to remove non-adherent neutrophils, and the number of adherent neutrophils was measured on a fluorimeter.
For IL-8 production assays, PMN were plated in 96-well tissue culture plates at 3 × 105 cells per well in complete medium, treated with apremilast in duplicate in a final DMSO concentration of 0.1% for 1 h at 37°C 5% CO2. PMN were then stimulated with unopsonized, boiled zymosan A at 2.5 × 105 particles per well for 18 h. Supernatants were harvested and tested for IL-8 by elisa (R&D Systems).
NK cell purification and stimulation
Natural killer cells were isolated from leukocyte units from healthy blood donors by a 30 min incubation with RossetteSep cocktail for NK cell enrichment by negative selection (StemCell Technologies, Inc., Vancouver, BC, Canada) followed by Ficoll-Hypaque density gradient centrifugation. CD56+ NK cells were isolated to ∼85% purity as determined by flow cytometry. Flat-bottom plates were coated with 100 µg·mL−1 of human IgG overnight at 4°C. The unbound IgG was washed away. NK cells were plated at 2 × 105 cells per well into the 96-well plates, and 10 ng·mL−1 of IL-2 was added. Apremilast was then added to the plate wells. After a 48 h incubation, the supernatants were harvested and analysed for levels of TNF-α, IFN-γ, GM-CSF and MIP-1α by elisa (R&D Systems).
Keratinocyte proliferation, TNF-α production and viability
For proliferation studies, human neonatal foreskin epidermal keratinocytes (HEKn cells) were plated at 3000 cells per well in 96-well flat bottom tissue culture plates for 2 days. Cell proliferation was measured using the Cell Counting Kit. For TNF-α production and viability studies, HEKn cells were obtained from Cascade Biologics (Portland, OR, USA) and were grown in serum-free medium supplemented with growth factors. When cells reached 80% confluency, cells were trypsinized and plated at 1 × 105 cells per well in 6-well dishes. Plates were incubated for 24 h to allow cell adhesion. HEKn cells were treated with apremilast or 0.1% DMSO as the vehicle control for 1 h before ultraviolet B (UVB) irradiation with 50 mJ·cm−2 in a UV Stratalinker 2400 (Stratagene, La Jolla, CA, USA) calibrated with 312 nm UVB bulbs. Media and compounds were replaced, and cells were incubated for 18 h. Supernatants were removed for testing in a TNF-αelisa before 100 µL of ATP-lite reagent was added to each well to assay for viability. Lysates were transferred to plates and shaken for 2 min before chemiluminescence was read on a TopCount NXT Luminescence Counter (PerkinElmer Life and Analytical Sciences).
Psoriasis mouse model
This study was conducted after receiving approval of the institutional ethics committee of the Technion-Istael Institute of Technology. Six psoriatic patients were included in this study (mean age = 42 years, range = 29–58 years). All patients had classical plaque psoriasis, and none were being treated. Skin from seven normal volunteers was also obtained for grafting. Healthy human skin pieces having a width of 0.4 mm and surface area of 3 × 3 cm were provided from residual skin of routine plastic surgery procedures from the Plastic Surgery Department of the Rambam Medical Center, Israel. In addition, blood samples from psoriatic patients were taken at a volume of 25 mL.
Beige-SCID (weight ∼20 g; each, 7 per group final) were included in this study. Normal human skin was transplanted onto the beige-SCID mice as previously described (Nickoloff et al., 1995; Wrone-Smith and Nickoloff, 1996). A sample from each donor was transplanted onto four mice so that each treatment group was similar.
Peripheral blood mononuclear cells from the psoriatic patient blood were isolated and cultured in the presence of IL-2 (100 U·mL−1 of media) for 14 days to activate the NK cells, as previously described (Gilhar et al., 2002). Four weeks following the engraftment, each mouse was injected with 1 × 107 activated allogeneic NK cells from the psoriatic patients. Two weeks following the injections, the mice were divided and treated, twice a day for 14 days. Apremilast and cyclosporine A were each dosed at (5 mg·kg−1·day−1, divided into b.i.d. doses). A volume of 0.05 mL of a 1 mg·mL−1 aqueous solution of the compounds was administered b.i.d. with a syringe through a blunt-ended curved feeding tube. The vehicle (negative) control groups received 0.05 mL (b.i.d.) of a 0.5% carboxymethylcellulose and 0.25% Tween 80. Two weeks after the start of the treatments (4 weeks following the injections), the skins were harvested. Grafts were analysed by histology and immunohistochemistry.
Determination of epidermal thickness
Skin graft histological assessment was performed by light microscopy both before and after transplantation, with two blinded observers performing the evaluations. Epidermal thickness was determined with an ocular micrometer, at a minimum of 50 points along the epidermis selected to represent points of maximal and minimal thickness. Thickness of the suprapapillary plate was similarly measured at 50 points for each sample.
Immunohistochemical staining
For frozen sections, monoclonal antibodies to human antigens used were as follows for immunohistochemistry on frozen sections: anti-HLA-DR (Becton Dickenson, San Jose, CA, USA) and anti-CD54 [intercellular adhesion molecule (ICAM)-1] (Biodesign, Saco, Maine). Purified murine IgG was used as a control for the above antibodies. Immunohistochemistry was performed on OCT-embedded specimens with a biotin-avidin system (Vectostain, Vector Laboratories, Burlingame, CA, USA). For paraffin sections, goat anti-human TNF-α (R&D Systems, Minneapolis, MN, USA) was used on deparaffinized and peroxidase blocked slides. Sections were treated with citrate buffer (pH 6) in the microwave oven for 20 min. The sections were then cooled for 30 min at room temperature and blocked for non-specific binding as well as avidin-biotin. All washes were performed with phosphate-buffered saline-saponin. Anti-TNF-α was applied overnight at 4°C. Slides were then incubated with biotinylated rabbit anti goat-IgG (DAKO, Carpenteria, CA, USA), followed by streptavidin horseradish peroxidase (Jackson Immunoresearch, West Grove, PA, USA). The colour was developed with 3-amino-9-ethylcarbazole. The epidermal proliferation index was determined as a percentage of keratinocytes expressing Ki-67 as detected by the monoclonal anti-human Ki-67 antibody (Zymed Laboratories, San Francisco, CA, USA) using the above procedure, except that antigen retrieval was achieved with EDTA buffer (pH 8).
Scoring of immunohistochemical staining
Diffuse staining was defined as positive and intense expression of more than 50% of the epidermis versus focal staining, which was defined as <50% of the epidermis. Focal staining may represent positive expression of very small areas.
IC50 calculation and statistics
IC50 (EC50) calculations were made using non-linear regression, sigmoidal dose–response, constraining the top to 100% and bottom to 0%, allowing variable slope using GraphPad Prism v4.00. Statistical analyses for multiple groups comparisons were by repeated measured analysis of variance (anova) followed by Bonferroni's post-test, or by one-way anova followed by Dunnett's post-test using GraphPad Prism v4.00.
Materials
Apremilast (CC-10004), or (S)-N-{2-[1-(3-ethoxy-4-methoxyphenyl)-2-methanesulphonylethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}, was synthesized by Celgene Corporation (Summit, NJ, USA) (Man et al., 2009). Lipofectamine™ 2000 reagent was obtained from Invitrogen (Paisley, UK); IMAP TR-FRET Screening Express with Progressive Binding Kit from Molecular Devices (Sunnyvale, CA, USA); RPMI 1640 from BioWhittaker (Walkersville, MD, USA). LPS from Salmonella abortus equii was obtained from Sigma; Ficoll-Paque Plus from GE Healthcare; SEB from Toxin Technology (Sarasota, FL, USA); RNeasy from Qiagen (Valencia, CA, USA); the RT Kit from Applied Biosystems (Foster City, CA, USA); thimerosal, unopsonized, boiled zymosan A, human IgG and fMLP were all from Sigma.
Calcein-AM was obtained from Molecular Probes (Eugene, OR, USA); the HUVEC from Anthrogenesis Corporation (Cedar Knolls, NJ, USA); IL-2 from R&D Systems (Minneapolis, MN, USA). HEKn cells for proliferation studies were obtained from Cell Applications, Inc. (San Diego, CA, USA); the Cell Counting Kit was from Dojindo Molecular Technologies, Inc. (Gaithersburg, MD, USA); ATP-lite reagent from PerkinElmer Life and Analytical Sciences (Shelton, CT, USA).
Results
PDE4 inhibition
Apremilast, or (S)-N-{2-[1-(3-Ethoxy-4-methoxyphenyl)-2-methanesulphonylethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide (Figure 1), was synthesized as previously described (Man et al., 2009). Apremilast was initially screened for PDE4 inhibition using a partially purified enzyme preparation from U937 human monocytic cells and which has been shown previously to contain predominantly PDE4B and PDE4D activities (Shepherd et al., 2004). Using this preparation, apremilast was found to exhibit an IC50 of around 74 nM using 1 µM cAMP as substrate (Table 1). When PDE4 activity in lysates from U937 human monocytic cells was evaluated at various concentrations (0.03–5 μM) of apremilast using a range of substrate concentrations (0.625–10 μM cAMP) then Lineweaver-Burke analysis indicated a competitive binding mode (data not shown) as has been noted before for active-site directed inhibitors such as the archetypal PDE4 selective inhibitor, rolipram (Huston et al., 1996; Wilkinson et al., 1997). The affinity constant of the inhibitor for the enzyme-substrate complex, Ki value, was determined as 68 nM (Table 1). By comparison, the affinity constant for rolipram was slightly higher (Ki = 159 nM). Again, as with rolipram inhibition was fully reversible by dilution (>98%).
Figure 1.
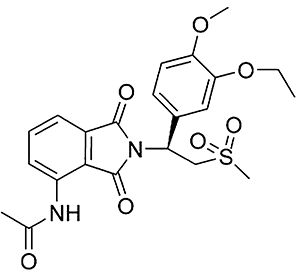
Chemical structure of apremilast, (S)-N-{2-[1-(3-ethoxy-4-methoxyphenyl)-2-methanesulphonylethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide.
Table 1.
Selectivity of apremilast for phosphodiesterase-4 (PDE4) inhibition
| Apremilast (nM) | |
|---|---|
| PDE4 (U937 cell) IC50 | 74 ± 34 |
| PDE4 (U937 cell) Ki | 68 ± 26 |
| PDE4A4 (recombinant) IC50 | 20 ± 3 |
| PDE4B2 (recombinant) IC50 | 49 ± 5 |
| PDE4C2 (recombinant) IC50 | 50 ± 3 |
| PDE4D3 (recombinant) IC50 | 30 ± 4 |
PDE4 isoforms were assayed in the presence of 1 µM cAMP as substrate. IC50 values are given as means ± SD for three separate experiments.
Apremilast was also tested for any PDE4 subfamily selectivity in cAMP PDE assays using recombinant human PDE4A4, PDE4B2, PDE4C2 and PDE4D3 isoforms representative of key species, including PDE4A4, a form that is up-regulated in COPD (Barber et al., 2004). Apremilast showed no marked selectivity among these individual PDE4 isozymes (Table 1). However, apremilast, while not selective for PDE4 isotypes A-D, was more potent for inhibition of PDE4 compared with cAMP or cGMP hydrolysing enzymes from other PDE families, all of which showed no significant inhibition at 10 μM apremilast (data not shown).
Inhibition of pro-inflammatory gene expression
TNF-α, IFN-γ, IL-12 and IL-23 are pro-inflammatory cytokines produced by mononuclear cells and are thought to play a role in inflammatory diseases such as psoriasis. Although PDE4 inhibitors are known to suppress the transcription of TNF-α mRNA, and reductions in IFN-γ and IL-12 production have been reported (Claveau et al., 2004), the effects of a PDE4 inhibitor on IL-23 mRNA levels have not been studied. Therefore the effect of apremilast on mRNA levels for these cytokines was studied using PBMC stimulated with LPS, an endotoxin produced by gram-negative bacteria such as Escherichia coli. LPS induces the production of many pro-inflammatory cytokines, including TNF-α, IFN-γ, IL-12 and IL-23. A time course was conducted to find the optimal stimulation time of each cytokine mRNA. The results below show that the maximum expression time varied from 2 h for TNF-α, to 4 h for IFN-γ, to 8 h for IL-12A and IL-23A (Figure 2A). Apremilast significantly inhibited expression of all four of these genes at their peak time points (Figure 2B)
Figure 2.
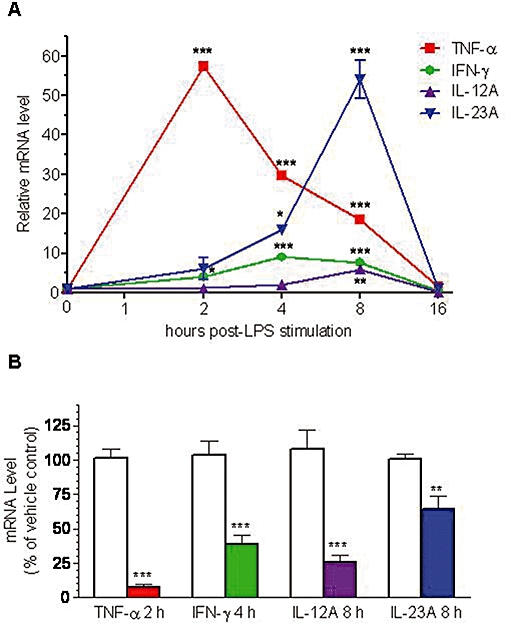
(A) Time course of pro-inflammatory cytokine mRNA expression in LPS-stimulated PBMC. PBMC were either not stimulated (time = 0) or stimulated with LPS for 2, 4, 8 and 16 h. Cytokine mRNA levels were measured by RT-PCR and normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA levels. All cytokine mRNAs at time zero were assigned a value of one. Data represent the mean ± SEM from two to three experiments. *P < 0.05, **P < 0.01, ***P < 0.001, as determined by one-way anova followed by Dunnett's multiple comparison test, comparing each mRNA level with its level at time 0. (B) Reduction of pro-inflammatory cytokine mRNA levels by apremilast. PBMC were treated with vehicle (open columns) or 10 μM apremilast (coloured columns) for 1 h prior to stimulation with LPS for the indicated duration to achieve the maximum mRNA expression for each particular cytokine. mRNA levels were measured by RT-PCR and normalized to GAPDH mRNA levels. Data represent the mean ± SEM from three experiments. **P < 0.01, ***P < 0.001, as determined by a repeated measures anova followed by a Bonferroni's post-test comparing each pair of groups. IFN-γ, interferon-γ; IL, interleukin; LPS, lipopolysaccharide; PBMC, peripheral blood mononuclear cells; TNF-α, tumour necrosis factor-α.
Inhibition of cytokine and chemokine protein expression
Cytokine (Figure 3A) and chemokine (Figure 3B) profiling using LPS-stimulated healthy human donor PBMC demonstrated varied potencies for inhibition by apremilast, with the rank order of inhibition being IP-10 > IFN-γ, MIG > TNF-α > IL-12p70, MIP-1α, MCP-1 and GM-CSF. IL-10 and IL-6 production were enhanced by apremilast at 1 µM and 10 µM, respectively, while production of IL-1β, IL-8 and RANTES (regulated upon activation, normal T cell expressed and secreted) were unaffected (Figure 3C). No changes in PBMC viability were observed upon apremilast treatment, as measured by MTT assay (data not shown).
Figure 3.
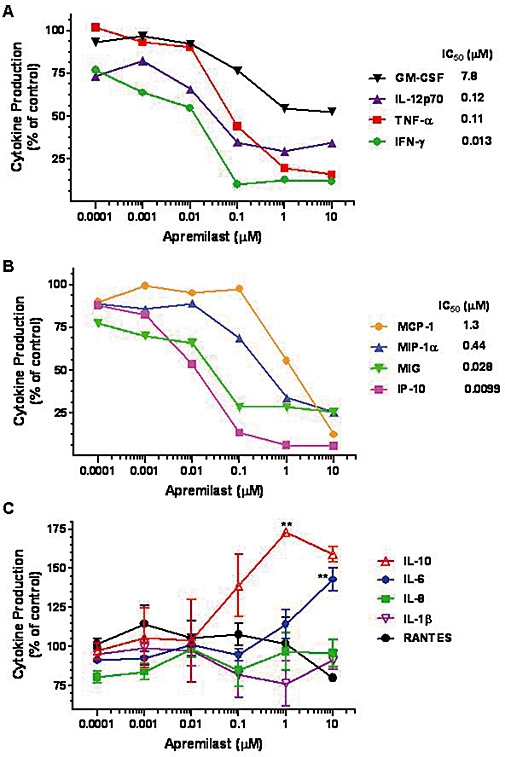
Effect of apremilast on cytokine and chemokine production by LPS-stimulated PBMC. (A) Inhibition of cytokines by apremilast. (B) Inhibition of chemokines by apremilast. Results are means from three experiments, except for IL-12p70 (n = 4). SEMs are not shown to improve graph readability, but were similar to the SEMs shown in (C). Inhibition of all cytokines and chemokines shown in (A) and (B) was statistically significant by one-way anova (P < 0.05). (C) Apremilast elevated IL-10 and IL-6 production and had no effect on IL-8, IL-1β and RANTES production. Data are mean ± SEM from three experiments, except for IL-10 and RANTES (n = 2). **P < 0.01, by one-way anova followed by Dunnett's multiple comparisons post-test, as compared with vehicle control cultures. GM-CSF, granulocyte macrophage-colony stimulating factor; IFN-γ, interferon-γ; IL, interleukin; LPS, lipopolysaccharide; MCP, monocyte chemoattractant protein; MIG, monokine induced by IFN-γ (CXCL9); MIP, macrophage inflammatory protein; PBMC, peripheral blood mononuclear cells; RANTES, regulated on activation, normal T cell expressed and secreted; TNF-α, tumour necrosis factor-α.
Inhibition of neutrophil responses
A previous study has shown that cilomilast blocks zymosan (yeast particle)-induced IL-8 production by human neutrophils with an IC50 of approximately 700 nM (Au et al., 1998). Although many cell types produce IL-8, its activity is restricted primarily to inducing neutrophil chemotaxis to inflamed tissues. Apremilast was found here to inhibit the zymosan-induced PMN production of IL-8 with an IC50 value of 94 nM (Table 2).
Table 2.
Inhibition of neutrophil (PMN) and T cell (superantigen-stimulated PBMC) responses by apremilast
| Stimulus | Response | Apremilast IC50 (95% CI), nM |
|---|---|---|
| Polymorphonuclear cells | ||
| Zymosan A | IL-8 production | 94 (56–110) |
| fMLF | CD18 expression | 390 (270–550) |
| fMLF | CD11b expression | 74 (13–430) |
| fMLF | Adhesion to HUVEC | 150 (80–280) |
| fMLF | LTB4 production | 2.5 (1.9–3.2) |
| Peripheral blood mononuclear cells | ||
| SEB | IL-2 production | 290 (170–500) |
| SEB | IFN-γ production | 46 (31–67) |
IC50 values were calculated using pooled data from three separate experiments. The 95% confidence interval (95% CI) for the IC50 is shown in parentheses. IC50 values for fMLP-induced CD18 and CD11b expression were calculated from a single representative experiment from three similar experiments.
fMLF, N-formyl-Met-Leu-Phe; HUVEC, human umbilical vein endothelial cells; IFN-γ, interferon-γ; IL, interleukin; LTB4, leukotriene B4; PBMC, peripheral blood mononuclear cells; PMN, polymorphonuclear cells; SEB, Staphylococcal enterotoxin B.
During inflammatory responses, neutrophils infiltrate into tissue by first adhering to the vascular wall endothelium. This adhesion is mediated by induced neutrophil expression of the β2-integrin Mac-1, which consists of a CD18/CD11b heterodimer, and lymphocyte function-associated antigen-1, a response that can be inhibited by rolipram (Derian et al., 1995). Certain chemotactic factors such as the bacterial peptide fMLF induce increased expression of adhesion molecules. Apremilast inhibited fMLF-induced PMN CD18 and CD11b expression with IC50 values of 390 nM and 74 nM, respectively, and inhibited fMLF-induced adhesion of PMN to HUVECs with an IC50 value of 150 nM (Table 2)
Leukotriene B4 is a product of arachidonic acid metabolism of the 5-lipoxygenase pathway, released by neutrophils and other myeloid cells. It is a pro-inflammatory neutrophil chemoattractant and activator of lymphocytes. There are reports in the literature stating that neutrophil LTB4 release can be blocked by PDE4 inhibitors. Apremilast inhibited LTB4 production by PMN with an IC50 of 2.5 nM. This is a very potent effect considering that the IC50 of apremilast for inhibition of PDE4 is at least an order of magnitude higher.
Because neutrophils are known to produce adenosine, which can inhibit LTB4 production via agonism of adenosine receptors, an experiment was conducted to determine if the adenosine receptor antagonist 8-(3-chlorostyryl) (CSC) caffeine or adenosine deaminase could raise the LTB4 IC50 of apremilast. CSC and adenosine deaminase reversed the inhibition of LTB4 by apremilast, indicating that endogenously produced adenosine was having a synergistic effect with apremilast to inhibit LTB4 production in this assay (data not shown).
Inhibition of T cell responses to superantigen
One of the proposed pro-inflammatory triggers of psoriasis is SEB (Bour et al., 1995). Apremilast inhibited SEB-induced PBMC production of IL-2 and IFN-γ production (Table 2). This indicates that T cell responses to a known physiological psoriasis trigger are inhibited by apremilast.
Inhibition of NK cell responses
A growing body of evidence also points to the involvement of NK and NK T cells in the pathogenesis of psoriasis (Bos et al., 2005). Psoriatic skin lesions contain an increased number of NK and NKT cells compared with healthy control skin biopsies (Cameron et al., 2002). Although NK cells are derived from lymphoid cells, they are considered part of the innate immune system because their activation is not clonally restricted by specific antigen receptors, but rather by pro-inflammatory cytokines, immunoglobulin and lack of MHC class I expression. In purified human NK cells stimulated with IL-2 and IgG, apremilast was found to significantly inhibit TNF-α and GM-CSF production, but not that of MIP-1α. A trend for increased IFN-γ production was observed but did not reach statistical significance (Figure 4).
Figure 4.
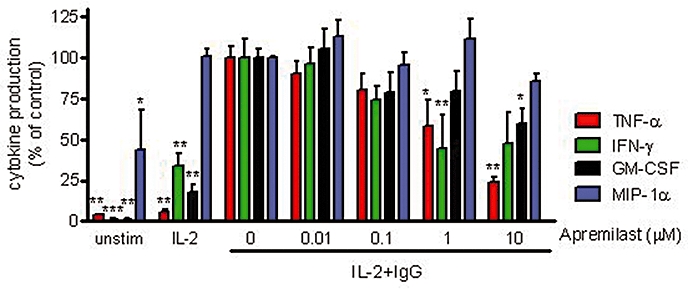
Inhibition of cytokine production by NK cells. Human NK cells from peripheral blood were stimulated with IL-2 only or with IL-2 plus IgG. Data shown are mean ± SEM from two experiments. *P < 0.05, **P < 0.01, ***P < 0.001 by one-way anova followed by Dunnett's multiple comparisons post-test, as compared with the IL-2 + IgG stimulated control samples. GM-CSF, granulocyte macrophage-colony stimulating factor; IFN-γ, interferon-γ; IL, interleukin; MIP, macrophage inflammatory protein; NK, natural killer; TNF-α, tumour necrosis factor-α.
Keratinocyte TNF-α production
The effects of PDE4 inhibition on keratinocyte responses have not been well described in the literature. We therefore examined the effect of apremilast on proliferation, TNF-α production and viability of primary normal human epidermal keratinocytes. Cyclosporin A, a psoriasis medication, was included as a positive control. A series of three experiments indicated that apremilast alone has no significant effect on normal keratinocyte proliferation. Cyclosporin A inhibited proliferation, as expected (Figure 5A). To verify that apremilast actually activates the cAMP/protein kinase A (PKA)/cyclic AMP-responsive element binding protein (CREB) signalling pathway in keratinocytes, cell cultures were first treated with 10 μM apremilast plus 10 μM isoprenaline for 30 min. They were then lysed to examine the phosphorylation status of the transcriptional regulator, CREB, which is activated upon phosphorylation by cAMP-activated PKA (Tasken and Aandahl, 2004) and can become phosphorylated by PKA subsequent to PDE4 inhibition by rolipram (MacKenzie and Houslay, 2000). Our results indicate that CREB becomes PKA phosphorylated by the combination of these agents (data not shown). Therefore, we conclude that activation of the cAMP pathway in normal primary human epidermal keratinocytes with apremilast does not inhibit proliferation.
Figure 5.
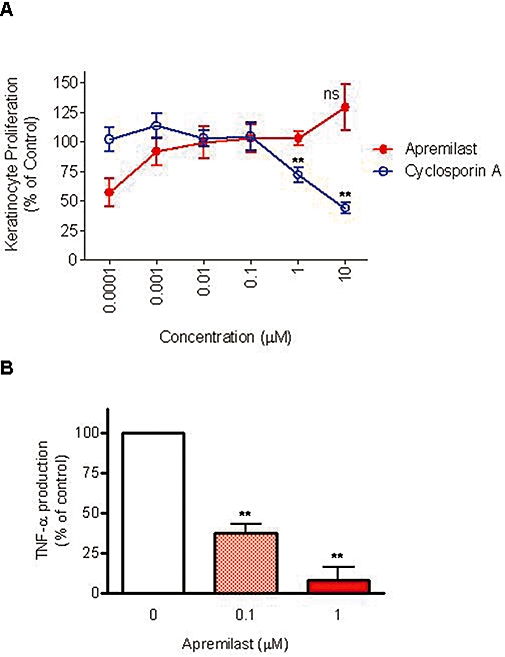
Apremilast inhibited keratinocyte tumour necrosis factor (TNF)-α production but not proliferation or cell viability. (A) Normal human epidermal keratinocytes were treated with apremilast or cyclosporine A for 2 days and assayed for cell proliferation. Data are mean ± SEM from three experiments. **P < 0.01, ns, not significant, by one-way anova followed by Dunnett's multiple comparison test versus vehicle control. (B) Normal human epidermal keratinocytes were treated with apremilast or 0.1% dimethylsulphoxide as a control, prior to ultraviolet B irradiation to induce TNF-α production. Supernatants were analysed for TNF-α protein production by elisa. TNF data are mean ± SEM from three experiments. **P < 0.01 by one-way anova followed by Dunnett's multiple comparisons post-test, as compared with vehicle control group.
To stimulate TNF-α production and apoptosis of keratinocytes, cells were irradiated with 50 mJ·cm−2 of UVB light. Apremilast inhibited keratinocyte TNF-α production at both 0.1 and 1 µM (P < 0.01) (Figure 5B), but did not affect keratinocyte cell viability as measured by intracellular ATP levels (0% inhibition at 0.1 µM, and 25% inhibition at 1 µM, which was not statistically significant, n = 4).
Treatment of psoriasiform features in beige-SCID mouse human skin/psoriatic NK cell xenograft model
The pharmacological activity of apremilast (5 mg·kg−1·day−1 total divided into 2 daily doses) was tested in comparison with cyclosporine (5 mg·kg−1·day−1 total divided into 2 daily doses) and vehicle control (0.1 mL·day−1 divided into twice daily doses) in a mouse xenograft model of psoriasis. This human skin transplantation model was originally developed using human psoriatic skin engrafted onto SCID mice, resulting in psoriasiform lesions involving T lymhocytes, monocytes, macrophages and dendritic cells (Nickoloff et al., 1995). It was later adapted to use skin grafts from symptomless/uninvolved skin from psoriasis patients and immunocytes stimulated with IL-2 and Staphylococcal enterotoxins SEB and SEC2, to activate T cell populations (Wrone-Smith and Nickoloff, 1996). This model produced plaques that were characterized by visible presence of flaking and thickened skin, loss of the granular cell layer, prominent elongation of rete ridges with a dermal angiogenic tissue reaction involving murine neutrophils, infiltration within the epidermis by human T cells, and displayed twenty different antigenic determinants of the psoriatic phenotype. Later, the model was further refined to use normal human skin engrafted onto Beige-SCID mice (which lack murine NK cells) and injection of allogeneic psoriatic NK cells that had been stimulated in culture with IL-2 but in the absence of superantigens, to avoid T cell activation via T cell receptor signalling (Gilhar et al., 2002). In the current version of this model, therefore, psoriasiform lesions are triggered by psoriatic NK cells and thus provide an ideal model for testing whether suppression of NK cells can affect skin lesion morphology. Furthermore, the interaction of NK cells and the surrounding myeloid cell populations (e.g. monocytes and neutrophils) with human keratinocytes allows the assessment of TNF-α production, as well as keratinocyte expression of activation markers ICAM-1 and MHC class II (HLA-DR).
The normal donor skin sections xenografted onto beige-SCID mice and treated with psoriatic patient NK cells and vehicle developed psoriasiform features. These features included significant epidermal thickening (acanthosis), hyperkeratosis and parakeratosis, along with a dermal lymphocytic infiltrate, some areas with retention, and others areas with lack of the granular layer (Figure 6A and B). These skin grafts exhibited significant increases in epidermal thickness and the keratinocyte proliferative index. Additionally, elongation of rete ridges was observed in most grafts. Vascular dilatation associated with a perivascular lymphocytic infiltrate was noted in the papillary dermis. Overall, these grafts showed histological features similar to psoriasis but combined with some signs of dermatitis.
Figure 6.
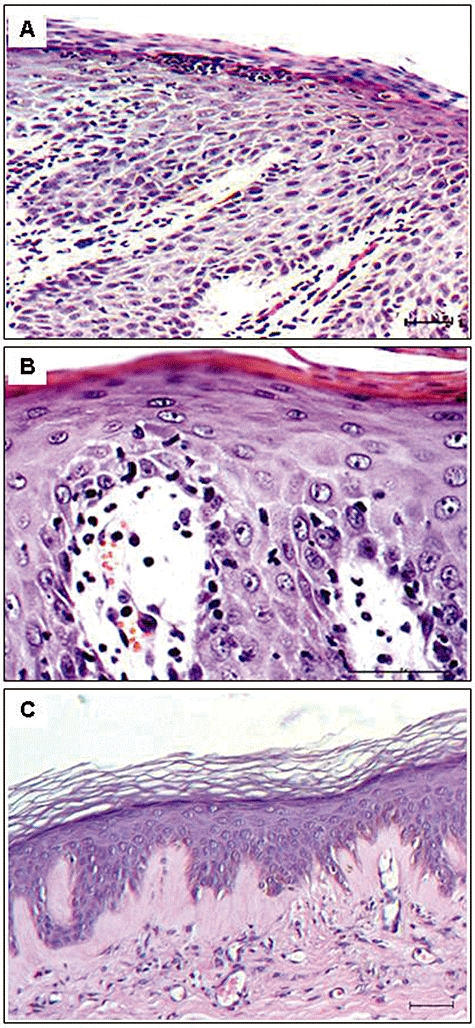
(A) Histological section of human skin graft on severe combined immunodeficient (SCID) mouse, injected with natural killer (NK)-like cells of psoriatic patient, exhibits histological parameters of psoriasis, as follows: parakeratosis, hyperkeratosis, collection of neutrophils (Munro micro-abscess formation), absence of the granular layer and a focal area of spongiosis with a lymphocytic infiltrate. Additionally, epidermal thickening (acanthosis) and elongation of the rete ridges are detected. Vascular dilatation associated with a perivascular inflammatory cell infiltrates is observed in the dermis. (B) Histological section of another human skin graft on SCID mouse, injected with NK-like cells of psoriatic patient, demonstrates parakeratosis, hyperkeratosis, absence of granular layer, hyperplasia and suprapapillary epidermal thinning. Oedema of the dermal papilla is seen combined with dilated, tortuous dermal blood vessel, surrounded by several lymphocytes and neutrophils. (C) Histological section of human skin graft on SCID mouse, injected with NK-like cells of psoriatic patient, shows a complete recovery of the psoriatic phenotype, following treatment with apremilast (5 mg·kg−1·day−1 divided into two daily oral doses). Bar length = 50 µm.
Cyclosporine treatment resulted in the complete/partial recovery of psoriasis features in three of seven mice. Similarly, apremilast treatment resulted in the complete/partial recovery of psoriasis features in four of seven mice (Table 3). The recovery included a morphological normalization of the epidermis and the absence of the lymphocyte infiltration (Figure 6C).
Table 3.
Histological evaluation of human skin grafts in beige-severe combined immunodeficient mice following treatment
| Histological features | Vehicle | Apremilast | Cyclosporine |
|---|---|---|---|
| Psoriasiform | 7/7 | 3/7 | 4/7 |
| Complete recovery | 0/7 | 3/7 | 2/7 |
| Partial recovery | 0/7 | 1/7 | 1/7 |
Epidermal thickness and proliferation index correlated with histological findings and demonstrated significant differences between treatment groups. Notably, apremilast caused statistically significant reductions in epidermal thickness (P < 0.001) (Figure 7A) and proliferation index (P < 0.001) (Figure 7B), compared with the vehicle-treated groups. Apremilast was as effective as cyclosporine for reducing epidermal thickness and reducing the proliferation index.
Figure 7.
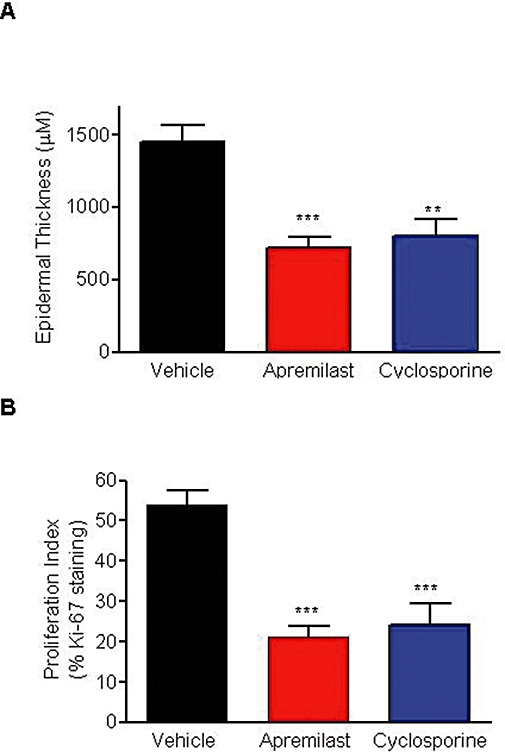
Apremilast reduced epidermal thickness and proliferation index in the psoriasiform xenograft model. (A) Epidermal thickness and (B) proliferation index in normal human skin xenotransplanted along with psoriatic patient NK cells into beige-severe combined immunodeficient (SCID) mice. Columns represent the mean ± SEM of seven beige-SCID mice. **P < 0.01, ***P < 0.001, one-way anova followed by Dunnett's multiple comparison test versus vehicle control.
Qualitative reductions in the expression of TNF-α, ICAM-1 and HLA-DR on the skin grafts were also observed in both the apremilast- and cyclosporine-treated groups (Table 4). Specifically, the immunohistochemical TNF-α expression was also 100% in the vehicle-treated mice grafts and showed multiple TNF-α-positive cells. HLA-DR and ICAM-1 markers were diffusely expressed in 6/7 (86%) grafts from the vehicle-treated mice. These vehicle treatment findings demonstrated the presence of active psoriasis symptoms in control mice, validating the effectiveness of the human skin xenotransplant/psoriatic patient NK cell-injected SCID mouse model of psoriasis. Moreover, psoriasiform histology was reduced in four of seven (57%) apremilast-treated mice and three of seven (42.9%) in the cyclosporine group. Comparatively, TNF-α expression was down-regulated in 57% (4/7) of the grafts treated with apremilast and 86% (6/7) of those in the cyclosporine treatment group. HLA-DR expression was decreased in 42.9% (3/7) of graphs treated with either apremilast or cyclosporine. However, the distinguishing feature of the HLA-DR data was that apremilast decreased HLA-DR expression to undetectable levels in the three of seven (42.9%) responding graphs. Moreover, apremilast treatment reduced ICAM-1 expression in 5/7 (71%) grafts, while cyclosporine treatment was effective in four of seven (57%) grafts. This study, therefore, demonstrated some similarities between apremilast and cyclosporine with regards to their therapeutic effects.
Table 4.
Summary of immunohistochemical staining of inflammation markers in human skin grafts in beige-SCID mice
| Inflammation marker | Vehicle | Apremilast | Cyclosporine |
|---|---|---|---|
| TNF-α | 7 Multiple | 3 Multiple | 1 Multiple |
| 0 Few | 1 Few | 4 Few | |
| 0 Negative | 3 Negative | 2 Negative | |
| HLA-DR | 6 Diffuse | 4 Diffuse | 4 Diffuse |
| 0 Focal | 0 Focal | 1 Focal | |
| 1 Negative | 3 Negative | 2 Negative | |
| ICAM-1 | 6 Diffuse | 2 Diffuse | 3 Diffuse |
| 1 Focal | 4 Focal | 2 Focal | |
| 0 Negative | 1 Negative | 2 Negative |
Numbers shown are the total number of grafts expressing each marker (n = 7 per group).
Diffuse, diffuse pattern throughout the epidermis; Few, few TNF-α-positive cells; Focal, focal pattern of expression; HLA-DR, human leukocyte antigen-DR; ICAM-1, intercellular adhesion molecule-1; Multiple, multiple TNF-α-positive cells; Negative, negative expression (0%); SCID, severe combined immunodeficient; TNF-a, tumour necrosis factor-α.
Discussion and conclusions
Apremilast is a novel inhibitor of the important PDE4 family of enzymes responsible for hydrolysing the key second messenger cAMP; and these enzymes are a recognized therapeutic target for treating inflammatory diseases (Houslay et al., 2005). Results from this study show that in human PBMCs stimulated with LPS, apremilast potently inhibited the production of multiple cytokines and chemokines, with selectivity for IP-10, IFN-γ, MIG, TNF-α, IL-12p70, MIP-1α, MCP-1 and GM-CSF, but not IL-8, IL-1β, RANTES, IL-10 and IL-6 production. The greatest potency was observed against the interferon-dependent chemokines IP-10 and MIG, and IFN-γ itself, suggesting a general selectivity for the interferon responsive genes. The mechanisms by which PDE4 inhibitors and cAMP-elevating agents block transcription of genes such as TNF-α and IL-12, but elevate other genes such as IL-10 and IL-6, have been described previously (Seldon et al., 1998; Liu et al., 2000; Persson et al., 2005). The cAMP elevation caused by PDE4 inhibition triggers activation of PKA, which phosphorylates the transcription factor CREB, thereby inducing transcription of genes such as IL-10 and IL-6 that bear CRE sites within their promoters. However, the expression of genes such as TNF-α, which are driven primarily by the transcription factor nuclear factor κ B (NF-κB), tends to be inhibited by cAMP elevation because of competition between CREB and the NF-κB p65 subunit for binding the co-activator CREB binding protein, which binds directly to the TATA box and initiates transcription (Parry and Mackman, 1997). The relative roles for NF-κB, CREB and other transcription factors in the regulation of pro-inflammatory mediator expression have been reviewed in detail (Barnes and Adcock, 1998). Thus, in general, NF-κB-dependent gene transcription tends to be inhibited by cAMP-elevating agents, while CREB-dependent gene expression tends to be augmented. The interplay between CREB-dependent and non-CREB-dependent gene transcription thus defines the relative sensitivity of individual promoters to the effects of PDE4 inhibition.
Inhibition of TNF-α, IFN-γ, IL-12A and IL-23A by apremilast was found to occur through suppression of mRNA expression (Figure 2B), consistent with the evidence in the literature described above that cAMP-elevating agents regulate expression of genes at the transcriptional level. Expression of the common IL-12 and IL-23 p40 subunit, encoded by the IL-12B gene, is known to be NF-κB-dependent (Ma et al., 2004) and inhibited by the cAMP analogue 8-Br-cAMP (Feng et al., 2002). However, suppression of the IL-12A (p35) and IL-23A (p19) subunits by a PDE4 inhibitor has not been reported previously. The inhibition of IL-12 gene expression is reflected in the decreased IL-12 p70 protein levels (Figure 3A). Inhibition of IL-12 and IL-23 may be an important aspect of psoriasis therapy, as supported by the significant efficacy of ustekinumab, an anti-IL-12/IL-23 p40 monoclonal antibody in development for the treatment of patients with chronic moderate to severe plaque psoriasis (Chien et al., 2009).
In contrast to inhibition of IP-10, IFN-γ, MIG, TNF-α, IL-12p70, MIP-1α, MCP-1 and GM-CSF, apremilast enhanced LPS-stimulated IL-10 production (with significant elevation at 1 µM), and to a lesser extent, IL-6 production (with significant elevation only at 10 µM) (Figure 3C). The pharmacokinetics of apremilast in psoriasis patients have been described previously, with a mean steady-state apremilast Cmax of 207 ng·mL−1 (450 nM) (Gottlieb et al., 2008). At concentrations in vitro of up to 1 µM, IL-6 elevation by apremilast was not significant (Figure 3C).
Unlike the biological TNF inhibitors, which bind directly to soluble TNF-α, apremilast inhibits TNF production at the level of gene expression. The potency of apremilast against TNF-α production in PBMC (IC50 = 110 nM) was similar to its potency for PDE4 enzymatic inhibition (IC50 = 74 nM) and its affinity constant (Ki = 68 nM), illustrating that regulation of TNF-α expression closely parallels both PDE4 enzymatic activity and inhibitor binding. The affinity of apremilast for PDE4 is at least 7900-fold lower than the binding avidities of the biological TNF-α inhibitors for soluble TNF, (etanercept KD = 0.4 pM; adalimumab KD = 8.6 pM; infliximab KD = 4.2 pM) (Kaymakcalan et al., 2009), all of which are approved for use against moderate to severe plaque psoriasis. In the first phase 2 clinical study of apremilast in patients with severe plaque-type psoriasis, the day 29 mean steady-state apremilast Cmax was 207 ng·mL−1 (450 nM) (Gottlieb et al., 2008). At this concentration, for example, apremilast inhibits approximately 70% of the TNF-α produced by LPS-stimulated PBMC (Figure 3A). Therefore, unlike the biological TNF neutralizing agents, apremilast is not expected to completely suppress TNF-α production in the clinical setting. Rather, its mode of action causes broad, but not complete, inhibition of multiple pro-inflammatory mediators. By comparison, etanercept was also reported to inhibit expression of IP-10, MIG and IFN-γ, as well as other pro-inflammatory mediators, in skin biopsies of psoriasis patients (Gottlieb et al., 2005). Thus, compared with etanercept, apremilast may show an overlapping but non-identical pattern of gene expression changes.
Psoriasis is characterized by distinct histological features of the skin including hyperproliferation of kertinocytes, and thickened epidermis with infiltration of mononuclear cells including monocytes, neutrophils, T cells and dendritic cells (Lowes et al., 2007). In addition to suppression of LPS-stimulated monocyte responses (Figures 2 and 3), apremilast also inhibited the response of T cells to the known psoriasis superantigen SEB, as shown by reduction in IL-2 and IFN-γ production (Table 2). The majority of CD8 and CD4 T cells from the epidermis or peripheral blood of psoriasis patients display a bias towards producing the Th1 cytokines IFN-γ and IL-2, illustrating the importance of these T cell-derived cytokines in psoriasis (Austin et al., 1999).
Several responses of PMN, mainly attributable to neutrophils, were blocked in vitro by apremilast, including the production of IL-8 and LTB4 (Table 2), as previously reported for other PDE4 inhibitors (Schudt et al., 1991). IL-8 is an autocrine chemotactic factor, which attracts additional neutrophils to sites of inflammation. Neutrophil infiltration is apparent in acute psoriatic lesions, which are characterized by epidermal hyperplasia and concomitant infiltration of CD11c+ dendritic cells and T cells (Bowcock and Krueger, 2005). In the mouse experiments described in this current study, apremilast inhibited the infiltration of neutrophils and lymphocytes into the dermis (Figure 6C), thereby confirming the relevance of the in vitro findings. Both neutrophils and dendritic cells are dependent upon the growth and differentiation factor GM-CSF, production of which was modestly inhibited by apremilast in both PBMC (Figure 3A) and NK cells (Figure 4). This degree of inhibition is consistent with the known regulation of GM-CSF expression by the transcription factors CREB, AP-1 and NFAT (Cockerill et al., 1993). In dendritic cells cultured from the dermis of psoriatic lesions, anti-GM-CSF antibody reduces both CD86 expression and antigen presenting cell function as measured by T cell stimulating capacity (Mitra et al., 1995). Similarly, exogenous IL-10 inhibited CD86 expression and antigen presenting cell function in these studies. This is intriguing in light of the fact that apremilast both inhibits GM-CSF production and enhances IL-10 elevation (Figure 3). This combination of effects would therefore be expected to act in concert to inhibit psoriatic dermal dendritic cell costimulatory marker expression and T cell stimulation.
In purified human NK cells, apremilast inhibited production of TNF-α and GM-CSF, and to a lesser extent, IFN-γ, induced by IL-2 and Fc receptor (FcRγ) cross-linking. This in vitro inhibition of NK-derived TNF-α is consistent with the reduced TNF-α expression observed in vivo in the psoriatic mice treated with apremilast (Table 4). Prior to this study, the effect of PDE4 inhibitors on NK cell responses had not been reported. In contrast to inhibition of NK cell production TNF-α, IFN-γ and GM-CSF, apremilast did not inhibit NK cell MIP-1α expression (Figure 4). This selectivity is dependent upon the context of the various signalling pathways that control transcription factor activity and cytokine gene expression. MIP-1α was the only cytokine or chemokine that was not induced by FcRγ receptor cross-linking via IgG, as the production of MIP-1α was the same in the presence of IL-2 and IL-2+IgG (Figure 4), suggesting that signalling through the Fc receptor is sensitive to PDE4 inhibition. The MIP-1α promoter also contains a CRE (Matsumoto et al., 2008), which would tend to enhance transcription in response to cAMP elevation, and thus make inhibition of this chemokine by a PDE4 inhibitor more difficult. Supernatants from psoriatic NK cells have been shown to produce large amounts of IFN-γ, and to induce expression of ICAM-1 and MHC class II in psoriatic keratinocytes (Ottaviani et al., 2006). Therefore, inhibition of NK cell inflammatory cytokine production by apremilast in vitro is consistent with the observed decrease in ICAM-1 and HLA-DR expression in vivo in the psoriatic mice treated with apremilast (Table 4).
Although PDE4 activity had been identified in the human keratinocyte cell line HaCaT, and its activity increased in response to the β2 adrenoceptor agonist salbutamol (Tenor et al., 1995), prior to this report, the role of selective PDE4 inhibitors in keratinocyte biology has not been explored. Elevation of cAMP in HaCaT cells by treatment with the β2 adrenoceptor agonist, isoprenaline, and the non-specific PDE inhibitor, IBMX, suppressed cell growth and induced expression of terminal differentiation markers K1, K10, involucrin and transglutaminase (Mammone et al., 1998). The new data presented in this study, namely that the PDE4 inhibitor apremilast blocks UVB-induced production of TNF-α by primary human keratinocytes without affecting proliferation or viability, are a significant finding that increases our understanding of how therapeutic PDE4 inhibitors might contribute to the treatment of inflammatory dermatological conditions.
In a model of psoriasis utilizing normal human skin xenotransplanted onto beige-SCID mice and triggered with human psoriatic NK cells, orally administered apremilast (5 mg·kg−1·day−1) significantly reduced epidermal thickness and proliferation, and decreased the general histopathological appearance of psoriasiform features in a manner similar to cyclosporine A. Notably, four of seven (57%) grafts from the apremilast-treated mice underwent a partial or complete recovery of histological features; whereas, three of seven grafts from cyclosporine-treated group displayed a histological recovery. The recovery included a morphological normalization of the epidermis and the absence of the lymphocytic infiltrations. Staining for TNF-α, HLA-DR and ICAM-1 in the lesioned skin was also qualitatively reduced by apremilast treatment. All of these results are consistent with the potent in vitro anti-inflammatory activities of apremilast. The present results also provide the first demonstration that a PDE4 inhibitor can directly suppress NK cell and keratinocyte pro-inflammatory cytokine production in vitro, and reduce the severity of a psoriasiform response in vivo.
Collectively, these data suggest a correlation between the histological evaluations (Table 3) and the inflammatory markers expression (TNF-α, HLA-DR and ICAM-1; Table 4).
Recently, the first open-label phase 2 pilot study to evaluate the effects of apremilast in patients with severe plaque-type psoriasis, over half (53.3%) demonstrated a ≥20% reduction from baseline in epidermal thickness at day 29. In addition, the majority (73.7%) of patients demonstrated improvement in their psoriasis symptoms, with 15.8% of these patients showing a >50% reduction from baseline in their total PASI score at day 29. Over half (52.6%) of all patients had a reduction from baseline in their psoriasis body surface area. Based on the sPGA, 52.9% of patients demonstrated improvement in plaque elevation, erythema and scaling. Reductions from baseline in epidermal and dermal T cells, CD83+ and CD11c cells as well as key inflammatory markers were observed at day 29, suggesting an improvement in the underlying inflammatory condition of the skin with apremilast treatment (Gottlieb et al., 2008). In a subsequent double-blinded placebo-controlled phase 2 study, a significantly higher proportion of subjects treated with apremilast (20 mg, b.i.d.) achieved a 75% reduction in the psoriasis area and severity index (PASI-75) compared with the placebo group after 12 weeks of treatment (24% vs. 10%) (P = 0.023). At week 12/last treatment, subjects achieved a mean decrease of 52% versus 17% in PASI from baseline in the apremilast (20 mg, b.i.d.) versus placebo groups respectively (Papp et al., 2008).
In summary, the epidermal thickness and proliferation index data in the mouse model yielded statistically significant results for apremilast suggesting favourable outcomes as a psoriasis treatment. The immunohistochemical staining data partially illustrated the positive mechanistic effects of the PDE4 inhibitors in psoriasis. Together, these data suggest that the human skin xenotransplant SCID mouse model may serve as a tool for investigating potential agents directed against the pathophysiological mechanisms of psoriasis. The anti-inflammatory activity of apremilast, as demonstrated in vitro and in a preclinical psoriasis model, has translated into clinical efficacy in this disease.
Acknowledgments
This work was supported by Celgene Corporation.
Glossary
Abbreviations:
- CRE
cyclic AMP-responsive element
- CREB
CRE binding protein
- CSC
8-(3-chlorostyryl) caffeine
- fMLF
N-formyl-Met-Leu-Phe
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GM-CSF
granulocyte macrophage-colony stimulating factor
- HEKn
human neonatal foreskin epidermal keratinocytes
- HLA-DR
human leukocyte antigen-DR
- HUVEC
human umbilical vein endothelial cells
- ICAM-1
intercellular adhesion molecule-1
- IFN-γ
interferon-γ
- IL
interleukin
- LFA-1
lymphocyte function-associated antigen 1
- LPS
lipopolysaccharide
- LTB4
leukotriene B4
- Mac-1
CD11b/CD18
- MCP
monocyte chemoattractant protein
- MIG
monokine induced by IFN-γ (CXCL9)
- MIP
macrophage inflammatory protein
- NK
natural killer
- PASI-75
75% reduction in the psoriasis area and severity index
- PBMC
peripheral blood mononuclear cells
- PDE4
phosphodiesterase-4
- PKA
protein kinase A
- PMN
polymorphonuclear cells
- RANTES
regulated on activation, normal T cell expressed and secreted
- RPMI
Roswell Park Memorial Institute
- SCID
severe combined immunodeficient
- SEB
Staphylococcal enterotoxin B
- TNF-α
tumour necrosis factor-α
Conflicts of interest
PHS, AP, AKG, LC, MA, LW, JBB, MAL, H-WM, GWM and DIS are employees of Celgene Corporation. This research was supported by Celgene Corporation.
References
- Au BT, Teixeira MM, Collins PD, Williams TJ. Effect of PDE4 inhibitors on zymosan-induced IL-8 release from human neutrophils: synergism with prostanoids and salbutamol. Br J Pharmacol. 1998;123(6):1260–1266. doi: 10.1038/sj.bjp.0701723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin LM, Ozawa M, Kikuchi T, Walters IB, Krueger JG. The majority of epidermal T cells in Psoriasis vulgaris lesions can produce type 1 cytokines, interferon-gamma, interleukin-2, and tumor necrosis factor-alpha, defining TC1 (cytotoxic T lymphocyte) and TH1 effector populations: a type 1 differentiation bias is also measured in circulating blood T cells in psoriatic patients. J Invest Dermatol. 1999;113(5):752–759. doi: 10.1046/j.1523-1747.1999.00749.x. [DOI] [PubMed] [Google Scholar]
- Baillie GS, Houslay MD. Arrestin times for compartmentalised cAMP signalling and phosphodiesterase-4 enzymes. Curr Opin Cell Biol. 2005;17(2):129–134. doi: 10.1016/j.ceb.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Barber R, Baillie GS, Bergmann R, Shepherd MC, Sepper R, Houslay MD, et al. Differential expression of PDE4 cAMP phosphodiesterase isoforms in inflammatory cells of smokers with COPD, smokers without COPD, and nonsmokers. Am J Physiol Lung Cell Mol Physiol. 2004;287(2):L332–L343. doi: 10.1152/ajplung.00384.2003. [DOI] [PubMed] [Google Scholar]
- Barnes PJ, Adcock IM. Transcription factors and asthma. Eur Respir J. 1998;12(1):221–234. doi: 10.1183/09031936.98.12010221. [DOI] [PubMed] [Google Scholar]
- Bolger GB, McPhee I, Houslay MD. Alternative splicing of cAMP-specific phosphodiesterase mRNA transcripts. Characterization of a novel tissue-specific isoform, RNPDE4A8. J Biol Chem. 1996;271(2):1065–1071. doi: 10.1074/jbc.271.2.1065. [DOI] [PubMed] [Google Scholar]
- Bolger GB, Erdogan S, Jones RE, Loughney K, Scotland G, Hoffmann R, et al. Characterization of five different proteins produced by alternatively spliced mRNAs from the human cAMP-specific phosphodiesterase PDE4D gene. Biochem J. 1997;328(2):539–548. doi: 10.1042/bj3280539. Pt. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos JD, de Rie MA, Teunissen MB, Piskin G. Psoriasis: dysregulation of innate immunity. Br J Dermatol. 2005;152(6):1098–1107. doi: 10.1111/j.1365-2133.2005.06645.x. [DOI] [PubMed] [Google Scholar]
- Bour H, Demidem A, Garrigue JL, Krasteva M, Schmitt D, Claudy A, et al. In vitro T cell response to staphylococcal enterotoxin B superantigen in chronic plaque type psoriasis. Acta Derm Venereol. 1995;75(3):218–221. doi: 10.2340/0001555575218221. [DOI] [PubMed] [Google Scholar]
- Bowcock AM, Krueger JG. Getting under the skin: the immunogenetics of psoriasis. Nat Rev Immunol. 2005;5:699–711. doi: 10.1038/nri1689. [DOI] [PubMed] [Google Scholar]
- Cameron AL, Kirby B, Fei W, Griffiths CE. Natural killer and natural killer-T cells in psoriasis. Arch Dermatol Res. 2002;294(8):363–369. doi: 10.1007/s00403-002-0349-4. [DOI] [PubMed] [Google Scholar]
- Chien AL, Elder JT, Ellis CN. Ustekinumab: a new option in psoriasis therapy. Drugs. 2009;69(9):1141–1152. doi: 10.2165/00003495-200969090-00001. [DOI] [PubMed] [Google Scholar]
- Claveau D, Chen SL, O'Keefe S, Zaller DM, Styhler A, Liu S, et al. Preferential inhibition of T helper 1, but not T helper 2, cytokines in vitro by L-826,141 [4-[2-(3,4-Bisdifluromethoxyphenyl)-2-[4-(1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl)-phenyl]-ethyl]3-methylpyridine-1-oxide], a potent and selective phosphodiesterase 4 inhibitor. J Pharmacol Exp Ther. 2004;310(2):752–760. doi: 10.1124/jpet.103.064691. [DOI] [PubMed] [Google Scholar]
- Cockerill PN, Shannon MF, Bert AG, Ryan GR, Vadas MA. The granulocyte-macrophage colony-stimulating factor/interleukin 3 locus is regulated by an inducible cyclosporin A-sensitive enhancer. Proc Natl Acad Sci USA. 1993;90(6):2466–2470. doi: 10.1073/pnas.90.6.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76:481–511. doi: 10.1146/annurev.biochem.76.060305.150444. [DOI] [PubMed] [Google Scholar]
- Derian CK, Santulli RJ, Rao PE, Solomon HF, Barrett JA. Inhibition of chemotactic peptide-induced neutrophil adhesion to vascular endothelium by cAMP modulators. J Immunol. 1995;154(1):308–317. [PubMed] [Google Scholar]
- Fan Chung K. Phosphodiesterase inhibitors in airways disease. Eur J Pharmacol. 2006;533(1–3):110–117. doi: 10.1016/j.ejphar.2005.12.059. [DOI] [PubMed] [Google Scholar]
- Feng WG, Wang YB, Zhang JS, Wang XY, Li CL, Chang ZL. cAMP elevators inhibit LPS-induced IL-12 p40 expression by interfering with phosphorylation of p38 MAPK in murine peritoneal macrophages. Cell Res. 2002;12(5–6):331–337. doi: 10.1038/sj.cr.7290135. [DOI] [PubMed] [Google Scholar]
- Giembycz MA. Can the anti-inflammatory potential of PDE4 inhibitors be realized: guarded optimism or wishful thinking? Br J Pharmacol. 2008;155(3):288–290. doi: 10.1038/bjp.2008.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilhar A, Ullmann Y, Kerner H, Assy B, Shalaginov R, Serafimovich S, et al. Psoriasis is mediated by a cutaneous defect triggered by activated immunocytes: induction of psoriasis by cells with natural killer receptors. J Invest Dermatol. 2002;119(2):384–391. doi: 10.1046/j.1523-1747.2002.01812.x. [DOI] [PubMed] [Google Scholar]
- Gottlieb AB. Psoriasis: emerging therapeutic strategies. Nat Rev Drug Discov. 2005;4(1):19–34. doi: 10.1038/nrd1607. [DOI] [PubMed] [Google Scholar]
- Gottlieb AB, Chamian F, Masud S, Cardinale I, Abello MV, Lowes MA, et al. TNF inhibition rapidly down-regulates multiple proinflammatory pathways in psoriasis plaques. J Immunol. 2005;175(4):2721–2729. doi: 10.4049/jimmunol.175.4.2721. [DOI] [PubMed] [Google Scholar]
- Gottlieb AB, Strober B, Krueger JG, Rohane P, Jones M, Sutherland D, et al. An open-label, single-arm pilot study in patients with severe plaque-type psoriasis treated with an oral anti-inflammatory agent, apremilast. Curr Med Res Opin. 2008;24(5):1529–1538. doi: 10.1185/030079908x301866. [DOI] [PubMed] [Google Scholar]
- Hanifin JM, Chan SC, Cheng JB, Tofte SJ, Henderson WR, Jr, Kirby DS, et al. Type 4 phosphodiesterase inhibitors have clinical and in vitro anti-inflammatory effects in atopic dermatitis. J Invest Dermatol. 1996;107(1):51–56. doi: 10.1111/1523-1747.ep12297888. [DOI] [PubMed] [Google Scholar]
- Houslay MD, Schafer P, Zhang KY. Keynote review: phosphodiesterase-4 as a therapeutic target. Drug Discov Today. 2005;10(22):1503–1519. doi: 10.1016/S1359-6446(05)03622-6. [DOI] [PubMed] [Google Scholar]
- Houslay MD, Baillie GS, Maurice DH. cAMP specific phosphodiesterase-4 enzymes in the cardiovascular system: a molecular toolbox for generating compartmentalised cAMP signalling. Circ Res. 2007;100(7):950–966. doi: 10.1161/01.RES.0000261934.56938.38. [DOI] [PubMed] [Google Scholar]
- Huston E, Pooley L, Julien P, Scotland G, McPhee I, Sullivan M, et al. The human cyclic AMP-specific phosphodiesterase PDE-46 (HSPDE4A4B) expressed in transfected COS7 cells occurs as both particulate and cytosolic species that exhibit distinct kinetics of inhibition by the antidepressant rolipram. J Biol Chem. 1996;271(49):31334–31344. doi: 10.1074/jbc.271.49.31334. [DOI] [PubMed] [Google Scholar]
- Huston E, Lumb S, Russell A, Catterall C, Ross AH, Steele MR, et al. Molecular cloning and transient expression in COS7 cells of a novel human PDE4B cAMP-specific phosphodiesterase, HSPDE4B3. Biochem J. 1997;328(2):549–558. doi: 10.1042/bj3280549. Pt. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huston E, Houslay TM, Baillie GS, Houslay MD. cAMP phosphodiesterase-4A1 (PDE4A1) has provided the paradigm for the intracellular targeting of phosphodiesterases, a process that underpins compartmentalized cAMP signalling. Biochem Soc Trans. 2006;34(4):504–509. doi: 10.1042/BST0340504. Pt. [DOI] [PubMed] [Google Scholar]
- Kaymakcalan Z, Sakorafas P, Bose S, Scesney S, Xiong L, Hanzatian DK, et al. Comparisons of affinities, avidities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clin Immunol. 2009;131(2):308–316. doi: 10.1016/j.clim.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Keshavarzian A, Mutlu E, Guzman JP, Forsyth C, Banan A. Phosphodiesterase 4 inhibitors and inflammatory bowel disease: emerging therapies in inflammatory bowel disease. Expert Opin Investig Drugs. 2007;16(9):1489–1506. doi: 10.1517/13543784.16.9.1489. [DOI] [PubMed] [Google Scholar]
- Khobzaoui M, Gutke HJ, Burnet M. Apremilast. Curr Opin Investig Drugs. 2005;6(5):518–525. [PubMed] [Google Scholar]
- Lipworth BJ. Phosphodiesterase-4 inhibitors for asthma and chronic obstructive pulmonary disease. Lancet. 2005;365(9461):167–175. doi: 10.1016/S0140-6736(05)17708-3. [DOI] [PubMed] [Google Scholar]
- Liu J, Chen M, Wang X. Calcitonin gene-related peptide inhibits lipopolysaccharide-induced interleukin-12 release from mouse peritoneal macrophages, mediated by the cAMP pathway. Immunology. 2000;101(1):61–67. doi: 10.1046/j.1365-2567.2000.00082.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobban M, Shakur Y, Beattie J, Houslay MD. Identification of two splice variant forms of type-IVB cyclic AMP phosphodiesterase, DPD (rPDE-IVB1) and PDE-4 (rPDE-IVB2) in brain: selective localization in membrane and cytosolic compartments and differential expression in various brain regions. Biochem J. 1994;304(2):399–406. doi: 10.1042/bj3040399. Pt. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowes MA, Bowcock AM, Krueger JG. Pathogenesis and therapy of psoriasis. Nature. 2007;445(7130):866–873. doi: 10.1038/nature05663. [DOI] [PubMed] [Google Scholar]
- Lynch MJ, Baillie GS, Mohamed A, Li X, Maisonneuve C, Klussmann E, et al. RNA silencing identifies PDE4D5 as the functionally relevant cAMP phosphodiesterase interacting with beta arrestin to control the protein kinase A/AKAP79-mediated switching of the beta2-adrenergic receptor to activation of ERK in HEK293B2 cells. J Biol Chem. 2005;280(39):33178–33189. doi: 10.1074/jbc.M414316200. [DOI] [PubMed] [Google Scholar]
- Ma W, Gee K, Lim W, Chambers K, Angel JB, Kozlowski M, et al. Dexamethasone inhibits IL-12p40 production in lipopolysaccharide-stimulated human monocytic cells by down-regulating the activity of c-Jun N-terminal kinase, the activation protein-1, and NF-kappa B transcription factors. J Immunol. 2004;172(1):318–330. doi: 10.4049/jimmunol.172.1.318. [DOI] [PubMed] [Google Scholar]
- MacKenzie SJ, Houslay MD. Action of rolipram on specific PDE4 cAMP phosphodiesterase isoforms and on the phosphorylation of cAMP-response-element-binding protein (CREB) and p38 mitogen-activated protein (MAP) kinase in U937 monocytic cells. Biochem J. 2000;347(2):571–578. doi: 10.1042/0264-6021:3470571. Pt. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie SJ, Baillie GS, McPhee I, MacKenzie C, Seamons R, McSorley T, et al. Long PDE4 cAMP specific phosphodiesterases are activated by protein kinase A-mediated phosphorylation of a single serine residue in upstream conserved region 1 (UCR1) Br J Pharmacol. 2002;136(3):421–433. doi: 10.1038/sj.bjp.0704743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammone T, Marenus K, Maes D, Lockshin RA. The induction of terminal differentiation markers by the cAMP pathway in human HaCaT keratinocytes. Skin Pharmacol Appl Skin Physiol. 1998;11(3):152–160. doi: 10.1159/000029821. [DOI] [PubMed] [Google Scholar]
- Man HW, Schafer P, Wong LM, Patterson RT, Corral LG, Raymon H, et al. Discovery of (S)-N-[2-[1-(3-ethoxy-4-methoxyphenyl)-2-methanesulfonylethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl] acetamide (apremilast), a potent and orally active phosphodiesterase 4 and tumor necrosis factor-alpha inhibitor. J Med Chem. 2009;52(6):1522–1524. doi: 10.1021/jm900210d. [DOI] [PubMed] [Google Scholar]
- Marchmont RJ, Ayad SR, Houslay MD. Purification and properties of the insulin-stimulated cyclic AMP phosphodiesterase from rat liver plasma membranes. Biochem J. 1981;195(3):645–652. doi: 10.1042/bj1950645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina SD, Ismail MS, Vesta KS. Cilomilast: orally active selective phosphodiesterase-4 inhibitor for treatment of chronic obstructive pulmonary disease. Ann Pharmacother. 2006;40(10):1822–1828. doi: 10.1345/aph.1H049. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Murao K, Imachi H, Nishiuchi T, Cao W, Yu X, et al. The role of calcium/calmodulin-dependent protein kinase cascade on MIP-1alpha gene expression of ATL cells. Exp Hematol. 2008;36(4):390–400. doi: 10.1016/j.exphem.2007.11.013. [DOI] [PubMed] [Google Scholar]
- Mitra RS, Judge TA, Nestle FO, Turka LA, Nickoloff BJ. Psoriatic skin-derived dendritic cell function is inhibited by exogenous IL-10. Differential modulation of B7-1 (CD80) and B7-2 (CD86) expression. J Immunol. 1995;154(6):2668–2677. [PubMed] [Google Scholar]
- Muller GW, Shire MG, Wong LM, Corral LG, Patterson RT, Chen Y, et al. Thalidomide analogs and PDE4 inhibition. Bioorg Med Chem Lett. 1998;8(19):2669–2674. doi: 10.1016/s0960-894x(98)00475-2. [DOI] [PubMed] [Google Scholar]
- Nickoloff BJ, Kunkel SL, Burdick M, Strieter RM. Severe combined immunodeficiency mouse and human psoriatic skin chimeras. Validation of a new animal model. Am J Pathol. 1995;146(3):580–588. [PMC free article] [PubMed] [Google Scholar]
- Ottaviani C, Nasorri F, Bedini C, de Pità O, Girolomoni G, Cavani A. CD56brightCD16(-) NK cells accumulate in psoriatic skin in response to CXCL10 and CCL5 and exacerbate skin inflammation. Eur J Immunol. 2006;36(1):118–128. doi: 10.1002/eji.200535243. Links. [DOI] [PubMed] [Google Scholar]
- Owens RJ, Lumb S, Rees-Milton K, Russell A, Baldock D, Lang V, et al. Molecular cloning and expression of a human phosphodiesterase 4C. Cell Signal. 1997;9(8):575–585. doi: 10.1016/s0898-6568(97)00072-7. [DOI] [PubMed] [Google Scholar]
- Papp K, Bolduc C, Rohane P, Sutherland D, Jones M, Hu A, et al. A Phase 2 Study Demonstrating the Efficacy and Safety of the Oral Therapy CC-10004 in Subjects with Moderate-to-Severe Psoriasis. 2008. 66th Annual AAD Meeting, San Antonio, Texas, Feb 1–5.
- Parry GC, Mackman N. Role of cyclic AMP response element-binding protein in cyclic AMP inhibition of NF-kappaB-mediated transcription. J Immunol. 1997;159(11):5450–5456. [PubMed] [Google Scholar]
- Persson E, Voznesensky OS, Huang YF, Lerner UH. Increased expression of interleukin-6 by vasoactive intestinal peptide is associated with regulation of CREB, AP-1 and C/EBP, but not NF-kappaB, in mouse calvarial osteoblasts. Bone. 2005;37(4):513–529. doi: 10.1016/j.bone.2005.04.043. [DOI] [PubMed] [Google Scholar]
- Prehn JL, Landers C, Muller GW, Man HW, Stirling DI, Targan SR. Potent inhibition of cytokine production from intestinal lamina propria T cells by phosphodiesterase-4 inhibitory thalidomide analogues. J Clin Immunol. 2001;21(5):357–364. doi: 10.1023/a:1012292703871. [DOI] [PubMed] [Google Scholar]
- Press NJ, Banner KH. 2 PDE4 inhibitors – a review of the current field. Prog Med Chem. 2009;47:37–74. doi: 10.1016/S0079-6468(08)00202-6. [DOI] [PubMed] [Google Scholar]
- Schudt C, Winder S, Forderkunz S, Hatzelmann A, Ullrich V. Influence of selective phosphodiesterase inhibitors on human neutrophil functions and levels of cAMP and Cai. Naunyn Schmiedebergs Arch Pharmacol. 1991;344(6):682–690. doi: 10.1007/BF00174752. [DOI] [PubMed] [Google Scholar]
- Schreiber S, Keshavarzian A, Isaacs KL, Schollenberger J, Guzman JP, Orlandi C, et al. A randomized, placebo-controlled, phase II study of tetomilast in active ulcerative colitis. Gastroenterology. 2007;132(1):76–86. doi: 10.1053/j.gastro.2006.11.029. [DOI] [PubMed] [Google Scholar]
- Seldon PM, Barnes PJ, Giembycz MA. Interleukin-10 does not mediate the inhibitory effect of PDE-4 inhibitors and other cAMP-elevating drugs on lipopolysaccharide-induced tumors necrosis factor-alpha generation from human peripheral blood monocytes. Cell Biochem Biophys. 1998;29(1–2):179–201. doi: 10.1007/BF02737835. [DOI] [PubMed] [Google Scholar]
- Shepherd MC, Baillie GS, Stirling DI, Houslay MD. Remodelling of the PDE4 cAMP phosphodiesterase isoform profile upon monocyte-macrophage differentiation of human U937 cells. Br J Pharmacol. 2004;142(2):339–311. doi: 10.1038/sj.bjp.0705770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spina D. PDE4 inhibitors: current status. Br J Pharmacol. 2008;155(3):308–315. doi: 10.1038/bjp.2008.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stawiski MA, Rusin LJ, Burns TL, Weinstein GD, Voorhees JJ. Ro 20-1724: an agent that significantly improves psoriatic lesions in double-blind clinical trials. J Invest Dermatol. 1979;73(4):261–263. doi: 10.1111/1523-1747.ep12531617. [DOI] [PubMed] [Google Scholar]
- Tasken K, Aandahl EM. Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol Rev. 2004;84(1):137–167. doi: 10.1152/physrev.00021.2003. [DOI] [PubMed] [Google Scholar]
- Teixeira MM, Rossi AG, Williams TJ, Hellewell PG. Effects of phosphodiesterase isoenzyme inhibitors on cutaneous inflammation in the guinea-pig. Br J Pharmacol. 1994;112(1):332–340. doi: 10.1111/j.1476-5381.1994.tb13073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenor H, Hatzelmann A, Wendel A, Schudt C. Identification of phosphodiesterase IV activity and its cyclic adenosine monophosphate-dependent up-regulation in a human keratinocyte cell line (HaCaT) J Invest Dermatol. 1995;105(1):70–74. doi: 10.1111/1523-1747.ep12313330. [DOI] [PubMed] [Google Scholar]
- Terrin A, Di Benedetto G, Pertegato V, Cheung YF, Baillie G, Lynch MJ, et al. PGE(1) stimulation of HEK293 cells generates multiple contiguous domains with different [cAMP]: role of compartmentalized phosphodiesterases. J Cell Biol. 2006;175(3):441–451. doi: 10.1083/jcb.200605050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Videla S, Vilaseca J, Medina C, Mourelle M, Guarner F, Salas A, et al. Selective inhibition of phosphodiesterase-4 ameliorates chronic colitis and prevents intestinal fibrosis. J Pharmacol Exp Ther. 2006;316(2):940–945. doi: 10.1124/jpet.105.090837. [DOI] [PubMed] [Google Scholar]
- Wilkinson I, Engels P, Houslay MD. Rolipram inhibition of the human cyclic AMP specific phosphodiesterase splice variant PDE4D3 expressed in yeast. Pharmacol Revs Commun. 1997;9:215–226. [Google Scholar]
- Willoughby D, Baillie GS, Lynch MJ, Ciruela A, Houslay MD, Cooper DM. Dynamic regulation, desensitization, and cross-talk in discrete subcellular microdomains during beta2-adrenoceptor and prostanoid receptor cAMP signaling. J Biol Chem. 2007;282(47):34235–34249. doi: 10.1074/jbc.M706765200. [DOI] [PubMed] [Google Scholar]
- Wrone-Smith T, Nickoloff BJ. Dermal injection of immunocytes induces psoriasis. J Clin Invest. 1996;98(8):1878–1887. doi: 10.1172/JCI118989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao W, Tian XY, Chen J, Setterberg RB, Lundy MW, Chmielzwski P, et al. Rolipram, a phosphodiesterase 4 inhibitor, prevented cancellous and cortical bone loss by inhibiting endosteal bone resorption and maintaining the elevated periosteal bone formation in adult ovariectomized rats. J Musculoskelet Neuronal Interact. 2007;7(2):119–130. [PubMed] [Google Scholar]