

Abstract
Background:
Solute carriers (SLCs), in particular organic cation transporters (OCTs), have been implicated in the cellular uptake of platinum-containing anticancer compounds. The activity of these carriers may determine the pharmacokinetics and the severity of side effects, including neuro- and nephrotoxicity of platinum-based chemotherapy. As decreased drug accumulation is a key mechanism of platinum resistance, SLCs may also contribute to the development of resistance. Here, we define the role of hSLC22A2 (OCT2) in the cellular uptake of platinum compounds.
Experimental approach:
Human embryonic kidney (HEK) 293 cells stably expressing the hSLC22A2 gene (HEK293/hSLC22A2) were used in platinum accumulation studies. Following a 2 h exposure to various platinum compounds (100 µM), intracellular platinum levels were determined by flameless atomic absorption spectrometry.
Key results:
HEK293/hSLC22A2 cells, compared with HEK293/Neo control cells, displayed significant increases in oxaliplatin (28.6-fold), Pt[DACH]Cl2 (20.6-fold), ormaplatin (8.1-fold), tetraplatin (4.5-fold), transplatin (3.7-fold) and cisplatin (1.3-fold), but not carboplatin. SLC22A2-mediated transport could be inhibited by 1-methyl-4-phenylpyridinium. Furthermore, hSLC22A2-mediated oxaliplatin and cisplatin accumulation was time- and concentration-dependent, but non-saturable. Expression of hSLC22A2 in HEK293 cells resulted in enhanced sensitivity to oxaliplatin (12-fold) and cisplatin (1.8-fold). Although, hSLC22A2 mRNA expression was frequently found in ovarian cancer cell lines, its expression in clinical ovarian cancer specimens (n= 80) was low and did not correlate with the treatment outcome of platinum-based regimens.
Conclusions and implications:
The hSLC22A2 drug transporter is a critical determinant in the uptake and cytotoxicity of various platinum compounds, particularly oxaliplatin.
Keywords: platinum uptake, cisplatin, oxaliplatin, drug resistance, solute carrier (SLC), organic cation transporter (OCT), SLC22A2
Introduction
Platinum-based drugs are among the most active anticancer agents and are used as single agents or in combination with other systemic agents and/or radiation therapy in the management of many human malignancies. To date, three platinum drugs have been approved by the Food and Drug Administration (FDA), that is, cisplatin (1978), its less toxic analogue carboplatin (1989) and oxaliplatin (2002). Cisplatin has been recognized for its anti-neoplastic activity since the 1960s and is the most widely used chemotherapeutic drug in the USA (McWhinney et al., 2009). Cisplatin-based chemotherapeutic regimens are administered to a broad spectrum of solid tumours and significantly influence the treatment outcome of testicular, bladder, ovarian, endometrial, cervical, head and neck and lung cancer (Perez, 1998; Go and Adjei, 1999; Zhang et al., 2006). The second-generation platinum anticancer drug carboplatin is a more stable analogue with equivalent activity but generally has a much milder side effect profile and is part of first-line combination therapy of ovarian and lung cancer (Ardizzoni et al., 2007). Although carboplatin has the advantage of being less nephrotoxic, its cross-resistance with cisplatin limits its application in cisplatin-resistant malignancies. Oxaliplatin is a third-generation platinum agent with a different mechanism of action and a different resistance profile from that of cisplatin and carboplatin (Rixe et al., 1996). Moreover, oxaliplatin has been found to be active against cisplatin- and carboplatin-resistant tumour cells (Misset et al., 2000; Mishima et al., 2002; Raymond et al., 2002). Although oxaliplatin-based chemotherapy is regularly compromised by the development of a sensory neuropathy it has a favourable toxicity profile, being markedly less nephrotoxic and myelosuppresive (Mishima et al., 2002). It has been approved as the first-line or second-line therapy in combination with 5-fluorouracil (5-FU)/leucovorin, for locally advanced and metastatic colorectal cancer, for which cisplatin and carboplatin are essentially inactive (Rixe et al., 1996; Raymond et al., 2002).
In spite of many different efforts to circumvent platinum resistance and to reduce the toxicity profile of platinum drugs, intrinsic and/or acquired resistances together with severe side effects, such as neuro- and nephrotoxicity, are often the reason for treatment discontinuation (Yao et al., 2007; McWhinney et al., 2009). Platinum resistance is considered multifactorial and includes both mechanisms that limit the formation of platinum–DNA adducts and mechanisms that prevent cell death following drug-induced damage. With respect to cisplatin, four major cellular mechanisms of resistance have been reported, that is, (i) inactivation of cisplatin by sulphur-containing molecules like glutathione and metallothionein; (ii) increased repair of cisplatin–DNA adducts, (iii) increased cisplatin adduct tolerance and failure of apoptotic pathways; and (iv) reduced platinum accumulation by a decreased drug uptake or an increased drug efflux (Gately and Howell, 1993; Johnson et al., 1998; Perez, 1998; Kelland, 2007). Although the significance and nature of these mechanisms have not been clearly established, it is generally believed that reduced drug accumulation is a clinically important mechanism of resistance to platinum compounds (Andrews et al., 1990; Johnson et al., 1998). However, the precise mechanisms of cellular uptake of platinum drugs are largely unknown, although it is commonly supposed that both passive diffusion as well as facilitated or active transport mechanism(s) play substantial roles in the uptake of cisplatin (Gately and Howell, 1993). Putative cisplatin transporters, described in the literature and suggested to be involved in decreased platinum accumulation, are for example MRP2/ABCC2 (Taniguchi et al., 1996), the copper transporters CTR1/SLC31A1 (Kuo et al., 2007), ATP7A and ATP7B (Safaei and Howell, 2005; Huang and Sadée, 2006), and a cis-configuration specific platinum influx transporter (Helleman et al., 2006a).
A recent notion is that various anticancer drugs can enter mammalian cells by facilitated transport via solute carriers (SLCs) (Huang et al., 2004). These SLC superfamily members play a role in the physiological absorption and/or excretion of drugs and xenobiotics in intestine, liver and kidney. The SLC22 subfamily encodes organic cation, anion and zwitterion transporters and encompasses three polyspecific organic cation transporters OCT1 (SLC22A1), OCT2 (SLC22A2) and OCT3 (SLC22A3). They are involved in the translocation of endogenous and exogenous compounds across epithelial membranes and play an important role in the detoxification of a broad range of structurally diverse xenobiotics of different sizes (Koepsell and Endou, 2004). Well-documented substrates of OCTs are dopamine, choline, creatinine, monoamine neurotransmitters, a variety of xenobiotics for example tetraethylammonium (TEA; a prototypic organic cation), 1-methyl-4-phenylpyridinium (MPP+; a neurotoxin) and drugs used clinically, such as metformin, cimetidine and amantadine (Koepsell et al., 2007). Human SLC22A1 is expressed predominantly in the basolateral membrane of hepatocytes in the human liver, hSLC22A2 primarily on the basolateral membrane of the proximal tubule epithelium in the kidney and in the brain, whereas hSLC22A3 is mainly expressed in skeletal muscle, liver, placenta, kidney and heart (Koepsell et al., 2007; Terada and Inui, 2007).
Because hSLC22A2 is the most abundantly expressed OCT in the kidney that probably plays an important role in platinum-induced nephrotoxicity, in the present study we determined its potential role in the cellular uptake of cisplatin, carboplatin and oxaliplatin.
Methods
Patients
Ovarian cancer patient and tumour characteristics have been described previously (Schuyer et al., 2001; Helleman et al., 2006b). The clinical response to chemotherapy was assessed according to the standard WHO response criteria. Particular details of these primary ovarian adenocarcinoma specimens and the comprehensive response distribution have been described previously (Schuyer et al., 2001; Helleman et al., 2006b).
Cell lines and culture conditions
Human embryonic kidney (HEK) 293 cells transfected with the Neo-containing pcDNA3 vector alone (HEK293/Neo) or the hSLC22A2 gene (HEK293/hSLC22A2) were kindly provided by Dr H Koepsell (Department of Medicine, Institute of Anatomy and Cell Biology, Julius Maximilians University Würzburg, Germany). Cell lines were grown as monolayers in 75 cm2 cell culture flasks (Corning) and maintained at 37°C in a humidified incubator with 5% CO2 in HEPES-buffered RPMI 1640 medium containing Glutamax and supplemented with 10% (v/v) fetal calf serum, 100 U·mL−1 penicillin and 100 µg·mL−1 streptomycin. Frozen cell pellets of the NCI-60 panel were provided by the Developmental Therapeutics Program of the National Cancer Institute.
RNA isolation, cDNA synthesis and quantitative real-time RT-PCR
Total RNA was extracted using RNA-Bee isolation reagent (Tel-Test Temco Inc., Friendswood, TX, USA) according to the manufacturer's instructions. The details of cDNA synthesis have been described previously (Burger et al., 2003). The relative expression levels for mRNA of the human transporter genes were measured by real-time RT-PCR using Taqman Universal Master Mix and Assay-On-Demand products from Applied Biosystems (SLC22A2 assay ID: Hs00533907_m1). Reactions were run on an ABI PRISM 7700 sequence detector system (Applied Biosystems, Foster City, CA, USA) using the following cycling conditions: 50°C for 2 min, 95°C for 10 min followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. PCR data analysis was performed using the Sequence Detector Software (Applied Biosystems). Human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) assay ID: 4310884E; VIC/TAMRA (Applied Biosystems) and hypoxanthine-guanine phosphoribosyltransferase (HPRT) assay ID: 4310890E; VIC/TAMRA (Applied Biosystems) genes were used for normalization of HEK293 cells and the NCI-60 panel respectively. To compare the relative expression levels of target genes among the different tumour cell lines (e.g. between HEK293/Neo control cells and HEK293/hSLC22A2 cells), the comparative CT method was used as previously described (Burger et al., 2003).
Intracellular platinum accumulation
Cellular uptake of platinum compounds was performed in triplicate in 25 cm2 (T25) tissue culture flasks with exponentially growing cells (∼90% confluent monolayers) at 37°C. For typical uptake experiments, cells were exposed to 100 µM (unless otherwise indicated) of the platinum compounds for 2 h (unless otherwise indicated) at 37°C. Briefly, treatment was started by replacing the culture medium with fresh platinum-supplemented and fetal calf serum-containing RPMI (pre-warmed to 37°C) and immediately after 2 h of treatment the cells were washed with ice-cold phosphate-buffered saline (PBS) to remove free platinum drug. Cells were harvested by scraping the monolayer, collected by centrifugation and the resulting cell pellets were extensively washed (3 wash cycles) with ice-cold PBS. For each uptake experiment at least three separately processed aliquots (cell pellets) were prepared for intracellular platinum measurements and kept frozen (−20°C) until further processing. The protein concentration was determined in the residual cell suspension using the Bradford protein assay kit (Bio-Rad, Veenendaal, The Netherlands) and measured by an Ultrospec 3000 spectrometer (Biochrom Ltd., Cambridge, UK) at 595 nm. Total intracellular platinum content of triplicate cell pellets was determined by atomic absorption spectrometry using a flameless Perkin-Elmer (Foster City, CA, USA) 4110 ZL spectrophotometer as previously reported (Burger et al., 1997). Briefly, cell pellets were dissolved in 0.2% nitric acid and appropriately diluted in 0.2% nitric acid. Aliquots of 20 µL samples were injected in duplicate into the graphite tube of the atomic absorption spectrometry and after evaporation and ashing atomized at 2300°C. Peak area measurements were performed at a wavelength of 265.9 nm with a slit width of 0.7 nm and platinum concentrations in the cell pellets were quantified using a linear-least squares regression analysis through linear calibration curves prepared from serial diluted platinum standards in 0.2% nitric acid in the concentration range of 0.025–1.00 µg Pt·mL−1. Intracellular platinum levels were expressed as µg platinum·mg−1 protein·h−1 (µg Pt·mg−1protein·h−1).
FACScan flow cytometry
4-[4-(diethylamino)styryl]-N-methylpyridiniumiodide (ASP+) accumulation was determined by measuring the fluorescence intensity (excitation: 488 nm; emission: 590 nm) using a FACScan flow cytometer (Beckton Dickinson, Mountain View, CA, USA). A total of 2 × 106 cells in 2 mL of serum- and phenol red-free RPMI 1640 were incubated with ASP+ at 37°C for the indicated time periods in the presence or absence of a specific inhibitor/competitor (e.g. cimetidine or MPP+). Fluorescence intensity of the cells was measured as described previously (Boersma et al., 1997). At least 20.000 events were measured and the peak fluorescence signal was used as an estimate for intracellular drug concentration. Fluorescence intensity was expressed in arbitrary units (channel number), and the percentage inhibitor-mediated reduction in ASP+ fluorescence intensity was calculated by the following equation:
 |
Cytotoxicity assays
The sulphorhodamine B (SRB) colorimetric assay was used as an indicator of cell viability and to estimate drug-induced cytotoxicity (Keepers et al., 1991; Rixe et al., 1996). Each platinum compound was assayed in quadruplicate in 96-well microtitreplates. Prior to drug exposure cells were seeded (104 cells per well) and allowed to adhere for 48 h in drug-free medium at 37°C with 5% CO2. Platinum compounds were serially diluted and added to the cell cultures, which were terminated 5 days after drug addition by fixing the cells with 10% trichloroacetic acid in PBS. Cell-containing plates were kept at 4°C for 1 h and subsequently stained for at least 15 min using 0.4% SRB dissolved in 1% acetic acid to determine cellular protein content. Unbound dye was removed by washing with 1% acetic acid. Plates were air-dried and bound dye was dissolved in 10 mM Tris-base. Optical density was read at 540 nm using an automated microplate reader (Labsystems Multiskan MS 352, Labsystems, Helsinki, Finland). Dose–effect correlation curves and concordant IC50 values were generated using the Deltasoft 3 software.
Statistical analysis
Data from transport experiments were expressed as the mean ± SD, analysed statistically using Student's unpaired t-test and depicted using the graphical layout of Microsoft Excel (Microsoft).
Chemicals and reagents
Cisplatin (cis-diamminedichloroplatinum II), the clinical formulation is referred to as Platosin (Pharmachemie, Haarlem, The Netherlands), oxaliplatin or trans-L-1,2-diaminocyclohexaneoxalatoplatinum II (Eloxatin, Sanofi-Synthelabo, Maassluis, The Netherlands) and carboplatin or cis-diammine 1,1-cyclobutanedicarboxylato platinum II (Paraplatin, Bristol-Myers Squibb BV, Woerden, The Netherlands) were obtained from the Erasmus MC pharmacy as clinically formulated stock solutions of respectively 1, 5 and 10 mg·mL−1. Tetraplatin and transplatin were obtained from Sigma-Aldrich (Zwijndrecht, The Netherlands). Both ormaplatin and PtCl2[R,R-DACH] or the l,2-diaminocyclohexaneplatinum (II) compound were kindly provided by Dr J Reedijk (Leiden Institute of Chemistry, Leiden University). Chemicals: MPP+, SRB and cimetidine were obtained from Sigma-Aldrich and TEA was obtained from Merck (VWR International B.V. Amsterdam, The Netherlands); fetal calf serum was from Gibco BRL (Paisley, UK). The prototypical cationic fluorescent substrate ASP+ from Molecular Probes (Eugene, OR, USA) was dissolved as a 100 µM ASP+ stock solution in PBS, stored at 4°C and further diluted in phenol red-free RPMI 1640 for measurements on a FACScan flow cytometer (Becton Dickinson, Mountain View, CA, USA). The drug/molecular target nomenclature used (e.g. receptors, ion channels and so on) conforms to BJP's Guide to Receptors and Channels (Alexander et al., 2008).
Results
Characterization of SLC22-Transfected HEK293 cells
TaqMan real-time RT-PCR showed that the mRNA expression of hSLC22A2 in HEK293/hSLC22A2 cells was at least 6 log (106-fold) higher than that observed in the HEK293/Neo control cells (Figure 1A). Moreover, the ectopically expressed hSLC22A2 gene level was comparable to that of the endogenous GAPDH reference gene (CT values were 17.1 and 17.0 respectively). In contrast, endogenous expression of hSLC22A2 in HEK293/Neo control cells was low and barely detectable by RT-PCR (CT= 37.4). Furthermore, the expression of a variety of other SLC genes implicated in platinum transport, including hSLC22A1 (OCT1), hSLC22A3 (OCT3), hSLCO1B3 (OATP8/OATP1B3/LST-2), hSLC31A1 (CTR1), hSLC47A1/2 (MATE1/2) did not differ between HEK293/hSLC22A2 cells and HEK293/Neo control cells (Table S1).
Figure 1.
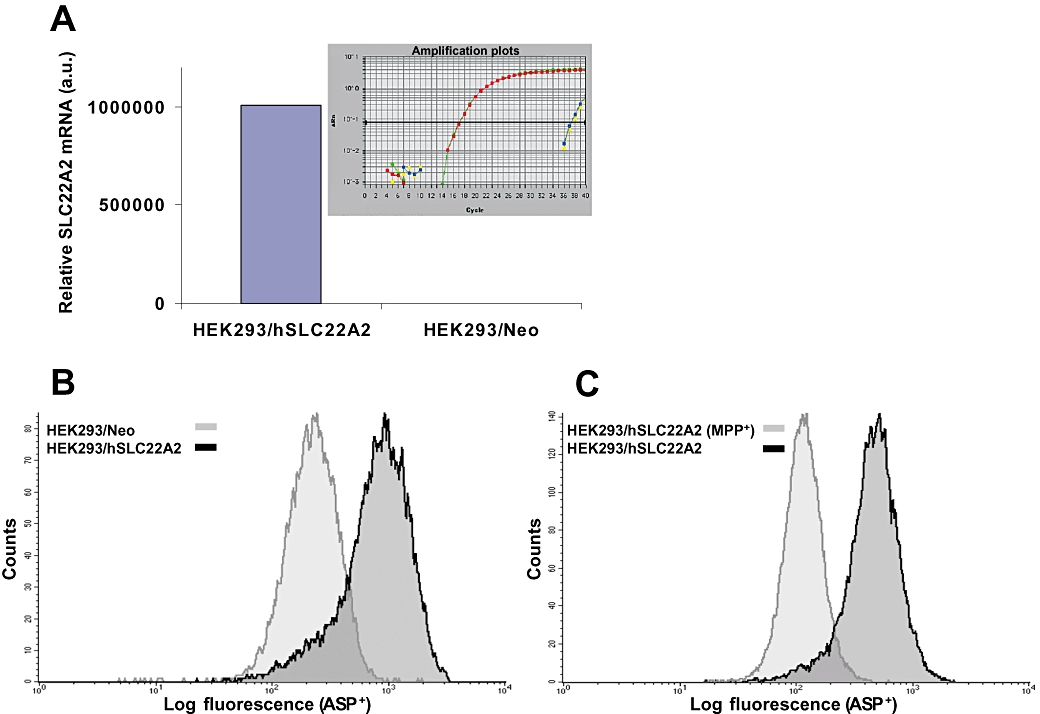
Characterization of human embryonic kidney (HEK) 293/hSLC22A2 and HEK293/Neo control cells. (A) Relative hSLC22A2 mRNA expression was determined by real-time RT-PCR using TaqMan chemistry and expressed in arbitrary units (a.u.). Insert shows the duplicate hSLC22A2 amplification plots of HEK293/hSLC22A2 and Neo control cells. (B) Analysis of 4-[4-(diethylamino)styryl]-N-methylpyridiniumiodide (ASP+) accumulation in HEK293/hSLC22A2 and HEK293/Neo control cells. Cells were incubated with 1 µM ASP+ for 20 min at 37°C and ASP+ uptake was measured by FACScan flow cytometry. ASP+ was significantly increased in the hSLC22A2-overexpressing cells compared with the Neo control cells. (C) SLC22A2-mediated accumulation of ASP+ was inhibited by addition of 1-methyl-4-phenylpyridinium (MPP+) (30 µM). ASP+ uptake in HEK293/hSLC22A2 is shown in the absence of MPP+ and in the presence of MPP+.
To confirm that the encoded hSLC22A2 protein was functional we determined the uptake of ASP+, a fluorescent prototypical substrate of hSLC22A2, in these HEK293 cells by FACScan flow cytometry. ASP+-associated fluorescence was significantly higher in the hSLC22A2-transfected cells compared with that in the HEK293/Neo control cells (Figure 1B). Next we determined whether the increased ASP+ accumulation could be reversed by inhibitors like cimetidine and MPP+ (Koepsell et al., 2007). Evidently, these specific inhibitors were able to modulate hSLC22A2-mediated ASP+ uptake (Figure 1C). Although cimetidine markedly decreased ASP+ uptake in HEK293/hSLC22A2 cells (∼40%), a larger decrease in ASP+ uptake (∼80%) was found with MPP+. Notably, these inhibitors did not affect the ASP+ uptake in HEK293/Neo control cells. Altogether, we showed that the ectopically expressed transporter in HEK293/hSLC22A2 cells is functional and can be specifically inhibited by either MPP+ or cimetidine
Increased platinum uptake in HEK293/hSLC22A2 cells
To examine whether platinum compounds are genuine substrates of hSLC22A2 we determined the intracellular platinum uptake upon drug exposure. Control and hSLC22A2-transfected cells were exposed for 2 h to the various platinum compounds (100 µM, or as otherwise stated). Notably, after 2 h continuous exposure more than 99% of both HEK293/hSLC22A2 and HEK293/Neo control cells still excluded trypan blue indicating that there were no signs of leakage and treated cells were morphologically indistinguishable from untreated cells (Table S2). The vast majority of these structurally distinct platinum compounds were found to interact with hSLC22A2. However, intracellular platinum accumulation varied extensively between each platinum compound (Figure 2; Table S3). Accordingly, the three FDA-approved platinum compounds cisplatin, oxaliplatin and carboplatin showed pronounced differences in hSLC22A2-mediated uptake. For instance, cisplatin uptake in HEK293/hSLC22A2 cells, as compared with Neo control cells, was slightly increased (1.33-fold; P < 0.05) while intracellular carboplatin accumulation was not augmented at all (Figure 2A and B, respectively). In contrast, oxaliplatin was found to be an excellent substrate as judged by the large increase (28.6-fold; P < 0.0001) in intracellular platinum accumulation in HEK293/hSLC22A2 cells (Figure 2C). Furthermore, the PtCL2[R,R-DACH] compound, ormaplatin, tetraplatin and to a lesser extent transplatin were also actively translocated by HEK293/hSLC22A2 cells resulting in 20.6-, 8.1-, 4.5- and 3.7-fold increases in uptake respectively (Figure 2D–G; Table S3).
Figure 2.
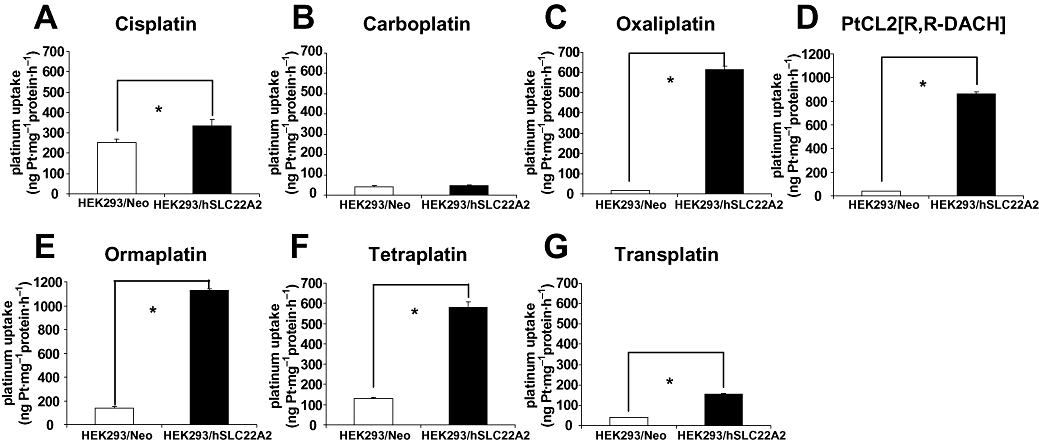
Differential intracellular accumulation of platinum compounds in human embryonic kidney (HEK) 293/hSLC22A2 cells and HEK293/Neo control cells. Cisplatin (A), carboplatin (B), oxaliplatin (C), PtCL2[R,R-DACH] (D), ormaplatin (E), tetraplatin (F) and transplatin (G). Columns and error bars represent mean values ± SD of duplicate measurements of at least three independent accumulation experiments. The asterisk (*) indicates a significant difference in accumulation (P < 0.05; Student's t-test)
Effects of specific SLC22A2 inhibitors on the platinum uptake in HEK293/hSLC22A2 cells compared with HEK293/Neo control cells
Platinum uptake experiments in the presence of specific SLC22A2 inhibitors were performed in order to explore whether the hSLC22A2-mediated platinum accumulation can be blocked. We tested a number of well-known inhibitors such as MPP+, TEA and cimetidine in combination with oxaliplatin. All three inhibitors significantly decreased intracellular oxaliplatin levels (Figure 3). Moreover, co-incubation of equimolar amounts of oxaliplatin and MPP+ (100 µM; 2 h) reduced the cellular uptake of oxaliplatin in HEK293/hSLC22A2 by almost 90% (Figure 3A). Although less pronounced, addition of TEA (Figure 3B) or cimetidine (Figure 3C) resulted in almost 70% decreased accumulation of oxaliplatin. Notably, these specific hSLC22A2 inhibitors did not affect cellular oxaliplatin accumulation in HEK293/Neo control cells. We also examined the inhibitory effects of these compounds on the hSLC22A2-mediated transport of cisplatin. Although there was a trend for both cimetidine and MPP+ (Figure 3D) to inhibit cisplatin accumulation partially in HEK293/hSLC22A2 cells, the effect was not significant and also noticeable in the HEK293/Neo control cells (Figure 3D). In addition to oxaliplatin, the hSLC22A2-mediated transport of various other platinum complexes, including the PtCl2[R,R-DACH) compound (Figure 3E) and tetraplatin (Figure 3F) were also almost completely inhibited by co-administration of MPP+.
Figure 3.
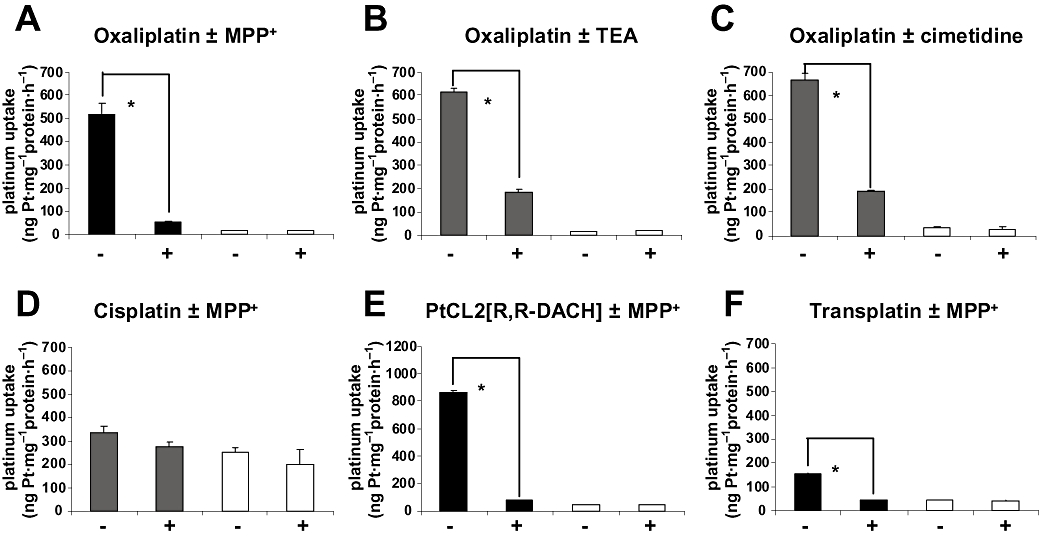
Intracellular accumulation of oxaliplatin (A–C), cisplatin (D), PtCL2[R,R-DACH] (E) and transplatin (F) in human embryonic kidney (HEK) 293/hSLC22A2 cells (solid columns) and HEK293/Neo control cells (open columns) after 2 h exposure to the different drugs (100 µM) in the presence (+) and absence (−) of 100 µM 1-methyl-4-phenylpyridinium (MPP+) (A,E,D,F), 100 µM tetraethylammonium (TEA) (B) or 300 µM cimetidine (C). Columns and error bars represent means ± SD of triplicate measurements and the asterisk (*) indicates a significant difference in accumulation (P < 0.05; Student's t-test).
SLC22A2-mediated accumulation of platinum compounds is dose- and time-dependent
An almost linear dose-dependent increase in intracellular platinum accumulation was observed in HEK293/hSLC22A2 cells upon exposure to a concentration range of oxaliplatin (Figure 4A). It should be noted that HEK293/Neo control cells hardly accumulated oxaliplatin and even a maximal exposure of 2 h to 500 µM oxaliplatin merely resulted in a platinum accumulation of 100 ng Pt·mg–1 protein·h−1, which is still more than 20-fold lower than that observed in hSLC22A2-transfected cells. Furthermore, a time-dependent increase in hSLC22A2-mediated oxaliplatin accumulation was observed, that is, accumulation was almost twice as high upon doubling the exposure time (e.g. from 30 min to 1 h or from 1 to 2 h). As judged from Figure 4A, oxaliplatin accumulation was not saturable under these conditions.
Figure 4.
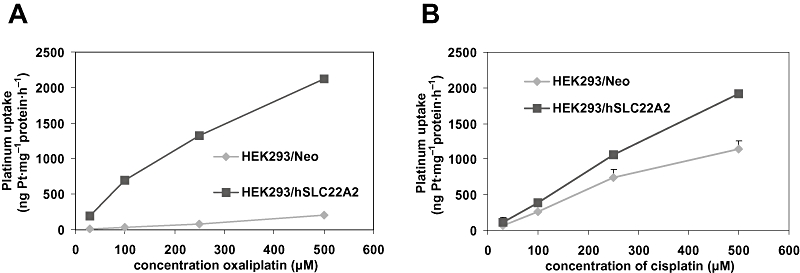
Dose-dependent intracellular accumulation of oxaliplatin (A) and cisplatin (B) in human embryonic kidney (HEK) 293/hSLC22A2 cells and HEK293/Neo control cells after 2 h exposure to these platinum drugs. Values and error bars represent means ± SD of triplicate measurements.
With respect to cisplatin accumulation, we also noted a dose-dependent and non-saturable increase in intracellular platinum accumulation in both HEK293/hSLC22A2 and the Neo control cells (Figure 4B). Surprisingly, the cisplatin accumulation in the control cells was quite high compared with that observed for oxaliplatin and reached levels up to about 1150 ng Pt·mg−1 protein·h−1 upon maximal exposure (500 µM). Nevertheless, for all the exposure concentrations investigated the uptake of cisplatin was significantly increased in hSLCA22-transfected cells compared with control cells (P < 0.05).
Cytotoxicity of oxaliplatin compared with cisplatin in HEK293/hSLC22A2 and HEK293/Neo control cells
Sulphorhodamine B assays were used to examine whether enhanced expression of hSLC22A2 renders cells more sensitive to platinum compounds. As expected from the accumulation data, compared with HEK293/Neo cells, HEK293/hSLC22A2 cells were markedly more sensitive (12-fold; IC50 values were 0.7 and 8.3 µM, respectively) to treatment with oxaliplatin (Figure 5A). Accordingly, the small increase in hSLC22A2-mediated cisplatin accumulation resulted in a comparable small increase in cisplatin sensitivity (Figure 5B). Moreover, HEK293/hSLC22A2 cells (IC50= 1.8 µM) were 1.8-fold more sensitive than Neo control cells (IC50= 3.3 µM). These results show that (over)expression of hSLC22A2 in HEK293 markedly enhanced sensitivity to oxaliplatin and although less profoundly also increased the sensitivity to cisplatin.
Figure 5.
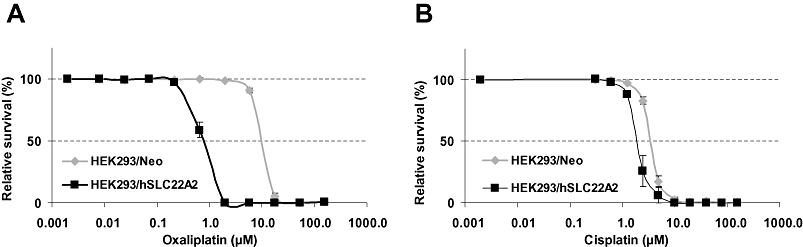
Platinum sensitivity of human embryonic kidney (HEK) 293/hSLC22A2 cells compared with HEK293/Neo cells. Cytotoxicity of oxaliplatin (A) and cisplatin (B) was measured by the sulphorhodamine B assay. The curves presented were derived from typical and representative experiments and values and error bars represent means ± SD of quadruplicate measurements.
SLC22A2 mRNA expression in cancer cell lines
We investigated the mRNA expression of hSLC22A2 in the NCI-60 panel of human cancer cell lines by real-time RT-PCR. Expression of SLC22A2 mRNA was only detected in 11 cell lines, including 4 out of 8 renal cancer cell lines, 4 out of 6 ovarian cancer cell lines, 2 out of 5 brain cancer cell lines and 1 out of 8 colon cancer cell lines (Figure 6). None of the breast, leukaemia, melanoma, lung and prostate cancer cell lines was found to express hSLC22A2. Thus, hSLC22A2 mRNA was undetectable (CT > 40) in the vast majority of cell lines, barely detectable in seven cell lines (CT between 34 and 38) and readily detectable in three ovarian cancer cell lines, that is, SKOV3 (CT= 33.4), OVCAR5 (CT= 31.2) and IGROV (CT= 30.2), having the highest expression (Figure 6).
Figure 6.
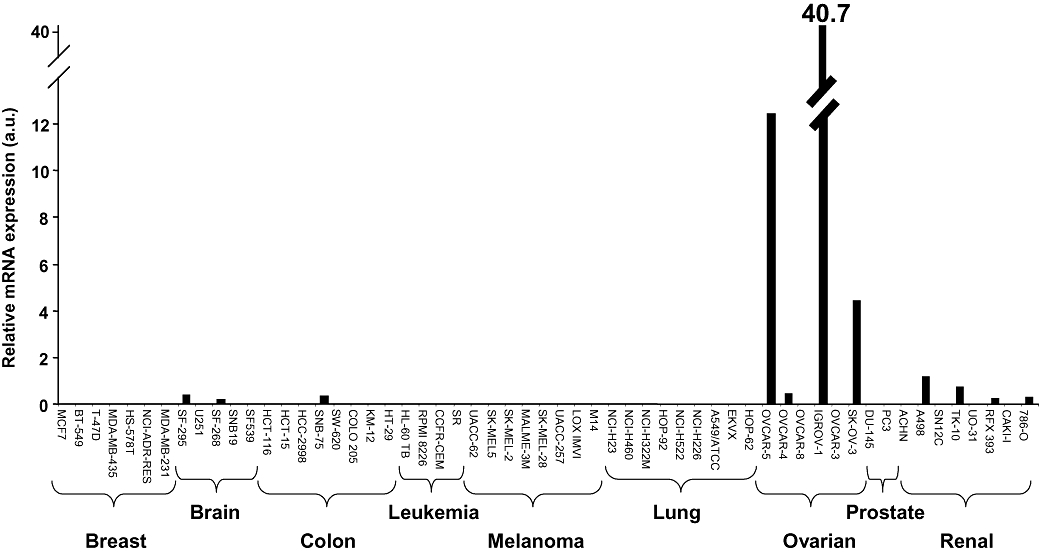
SLC22A2 expression in NCI-60 panel of human cancer cell lines. Relative hSLC22A2 mRNA expression was determined by real-time RT-PCR using TaqMan chemistry and expressed in arbitrary units (a.u.).
SLC22A2 mRNA expression in ovarian tumour samples treated with platinum-based chemotherapy
Because hSLC22A2 is frequently expressed in ovarian cancer cell lines (four out of six), (over)expression of this SLC drug transporter might affect clinical outcome of platinum-based chemotherapy in ovarian cancer. Therefore, we studied the expression of hSLC22A2 in 80 primary ovarian adenocarcinoma specimens from patients with known clinical follow-up (Table 1), including 51 patients treated with cisplatin-containing chemotherapy for which response data were available (Helleman et al., 2006b), which covered 9 non-responders and 42 responders. Quantitative RT-PCR analysis of these 80 ovarian tumour specimens revealed that the vast majority of ovarian tumours had low (n= 12; CT range between 31.1 and 38.2) or undetectable (n= 68; CT > 40) hSLC22A2 mRNA levels (Table 1). No difference, either in expression level or frequency of SLC22A2-positive tumours, was apparent between the non-responders (3 out of 9 had low but detectable expression; 33%) and the responders (6 out of 42; 14%). In fact the frequency of SLC22A2 expression was about threefold lower in responders than in non-responders. Altogether, this exploratory study strongly suggests that the role of hSLC22A2-mediated transport in ovarian cancer is limited and probably does not affect clinical outcome of platinum-based chemotherapy.
Table 1.
Expression of hSLC22A2 mRNA in ovarian cancer and correlation with clinical outcome of platinum-based chemotherapy
| Patients | hSLC22A2 expressiona | Type of chemotherapyb | ||||||
|---|---|---|---|---|---|---|---|---|
| Response | Number | Number (%) | Tax/Cis-Pt | Cyclo/Cis-Pt | Cyclo/Carbo | Cyclo | Melph | NO/Radioc |
| Total | 80 | 12 (15.0%) | 1 | 55 | 3 | 1 | 1 | 19 |
| Not informative | 29 | 3 (10.3%) | – | 5d | 3 | 1 | 1 | 19 |
| Responderse | 42 | 6 (14.3%) | 1 | 41 | – | – | – | – |
| Non-responderse | 9 | 3 (33.3%) | – | 9 | – | – | – | 9 |
The mRNA expression was determined by real-time RT-PCR, number and percentage of positive samples are shown. In all positive cases the hSLC22A2 mRNA expression was low and marginally above the detection limit (CT range: 31.1–38.2).
Tax, taxol; Cis-Pt, cisplatin; Cyclo, cyclophosphamide; Carbo, carboplatin, Melph, melphalan.
Some of the patients received no chemotherapy at all (NO) or radiotherapy (Radio).
No informative response data.
Response data for all cisplatin-treated patients. Patient and tumour characteristics and response to chemotherapy are as described previously (Schuyer et al., 2001; Helleman et al., 2006b).
Discussion
Cellular resistance to platinum-based chemotherapy is frequently observed and platinum-associated neuro- and nephrotoxicity compromise the efficacy of treatment. Altered intracellular platinum accumulation mediated by particular members of the OCT family may account for both chemotherapy resistance and the occurrence of serious side effects.
In the present study, we addressed the role of hSLC22A2 or OCT2 in intracellular accumulation and transport of platinum compounds. Uptake studies in hSLC22A2-transfected HEK293 cells revealed that hSLC22A2 is capable of transporting multiple, structurally distinct, platinum complexes including the clinically relevant anticancer agents oxaliplatin and cisplatin, but not carboplatin. Moreover, we found that in our hands the hSLC22A2-mediated intracellular accumulation of oxaliplatin was much higher than that of cisplatin. Consequently, overexpression of the hSLC22A2 drug transporter had a much more pronounced effect on the cytotoxicity of oxaliplatin than that of cisplatin. Our oxaliplatin accumulation data are in agreement with the findings of Zhang et al. (2006) who demonstrated that ectopically expressed hSLC22A2 markedly increased oxaliplatin accumulation and cytotoxicity. Similarly Inui et al. reported that both rat and hSLC22A2 efficiently transport oxaliplatin (Yonezawa et al., 2006; Yokoo et al., 2007). Only one study has shown that oxaliplatin does not interact with hSLC22A2 and that co-incubation had no effect on ASP+ uptake in HEK293-transfected cells (Ciarimboli et al., 2005). Nonetheless, using flow cytometry, we showed that the uptake of ASP+ in our HEK293/hSLC22A2 cells was clearly modulated by oxaliplatin, as co-incubation decreased ASP+-associated fluorescence by 38% in HEK293/hSLC22A2 cells, while ASP+ accumulation in HEK293/Neo control cells was not affected (Figure S1). So, hSLC22A2 was identified as a critical mediator of oxaliplatin transport and toxicity. Furthermore, our carboplatin accumulation data are in accordance with results from previous studies, using various distinct model systems, which consistently show that carboplatin is not transported by hSLC22A2 (Ciarimboli et al., 2005; Yonezawa et al., 2006; Zhang et al., 2006).
Our study revealed that hSLC22A2 has limited activity towards cisplatin. Notably, the precise role of hSLC22A2 in the cellular uptake of cisplatin and its possible impact on clinical outcome are important issues that, in fact, are still a matter of debate. For instance, initial uptake studies using Xenopus laevis oocytes revealed that cisplatin itself was not transported by hSLC22A2, although there was a role for this particular transporter in the uptake and targeting of the bile acid-cisplatin derivatives Bamet-R2 and Bamet-UD2 (Briz et al., 2002). Furthermore, Zhang et al. (2006) reported that the cellular accumulation of cisplatin in their HEK-hOCT2 cells was only slightly higher than that in HEK-MOCK cells and moreover was not affected by co-incubation with the OCT2 inhibitor cimetidine. Accordingly, the IC50 values of cisplatin were only slightly higher in HEK-MOCK cells than in HEK-hOCT2 cells indicating that hSLC22A2 (hOCT2) did not significantly enhance the cytotoxicity of cisplatin. On the other hand, there are several reports demonstrating that SLC22A2 may play a role in cisplatin-induced acute kidney injury and uraemia (Yao et al., 2007). In an early publication using a transfected cell model it was shown that cisplatin competively inhibited TEA uptake by rat Slc22a2 and the authors therefore suggested that this transporter may play a role in the renal secretion of cisplatin (Pan et al., 1999). This was further supported by the apparent association between tubular toxicity of cisplatin and expression of Slc22a2 in the rat (Yonezawa et al., 2005). Finally, there are a number of in vitro cellular accumulation studies showing that cisplatin is in fact transported by hSLC22A2 (Ciarimboli et al., 2005; Yonezawa et al., 2006; Filipski et al., 2008). The origin of the in vitro differences in hSLC22A2-mediated transport of cisplatin is unclear but may well be caused by distinct assay conditions leading to different degrees of hydration/ionization. Additional clinical studies are needed to confirm that hSLC22A2 is a determinant of cisplatin sensitivity and to elucidate its precise role in the occurrence and severity of cisplatin-induced nephrotoxicity.
In the current study we showed that a number of structurally different platinum compounds are good substrates of hSLC22A2. In particular, oxaliplatin, the PtCL2[R,R-DACH] compound and ormaplatin were efficiently translocated by hSLC22A2. Furthermore, it was recently found that cis-diammine[pyridine]chloroplatinum[II] or cDPCP, a monofunctional platinum (II) anti-tumour agent, is an outstanding substrate of hSLC22A2 (Lovejoy et al., 2008). Oxaliplatin, PtCL2[R,R-DACH], ormaplatin and this cDPCP compound all contain heterocyclic aromatic ring structures that may well be important structural features for platinum compounds to be efficiently transported by hSLC22A2. Established molecular and physicochemical descriptors of hSLC22A2-based drug interaction are limited to basicity, degree of ionization, hydrophobicity and topological polar surface area of the substrate (Zolk et al., 2008). Studies aimed at identifying generalized structural features did not result in the development of a classical pharmacophore for hSLC22A2, although typical substrates generally contain at least a hydrophobic interaction domain combined with a site that can be positively ionized (Zolk et al., 2008).
Cisplatin uptake has been shown to be non-saturable, even up to its solubility limit, and is usually not inhibited by structural analogues (Johnson et al., 1998). This non-saturable phenomenon has been used frequently as evidence to prove that diffusion is the key mechanism of cisplatin uptake. Notably, in our cell model the contribution of the diffusion component to the overall cisplatin uptake seems to be quite substantial, although the possibility that other constitutively expressed transporters (e.g. CTR1) are responsible for the relatively high accumulation rate in HEK293/Neo control cells cannot be excluded. On the other hand, we demonstrated that the cellular accumulation of oxaliplatin is also not saturable even at very high extracellular exposure concentrations, while its uptake is largely mediated by active transport (SLC22A2), apparently with a minor role for diffusion. Although the precise nature of this non-saturable accumulation of oxaliplatin in HEK293/hSLC22A2 cells is unclear, it suggests an extensive capacity of this transporter to translocate oxaliplatin. Indeed, the (diffusion-corrected) hSLC22A2-mediated oxaliplatin uptake rate is quite high (>500 ng Pt·mg−1 protein·h−1).
We clearly demonstrated that hSLC22A2 overexpression in HEK293 cells significantly affects the cytotoxicity of oxaliplatin and to a lesser extent that of cisplatin. This indicates that hSLC22A2, apart from its role in renal secretion of platinum drugs, may also play a role in platinum-based chemosensitivity of tumour cells. However, we found that the majority of ovarian tumours had no or low hSLC22A2 mRNA expression (Table 1) with no difference in expression level and/or frequency of SLC22A2-positive tumours between responders and non-responders. Although this association study was an exploratory study from which one cannot definitely conclude that SLC22A2 expression does not affect the clinical outcome, in particular the lack of substantial expression suggests that the role of hSLC22A2-mediated platinum transport in ovarian cancer is limited and probably does not affect the clinical outcome of platinum-based chemotherapy. It should, however, be noted that although we found no correlation between hSLC22A2 expression and clinical outcome in cisplatin-treated ovarian cancer patients, its role in the anti-tumour activity of cisplatin has to be further investigated. Zhang et al. (2006) speculated that hSLC22A2 expression might be an important determinant of oxaliplatin sensitivity in colorectal cancer. However, we found that of the eight colon carcinoma cell lines present in the NCI-60 panel, seven completely lacked expression and only one displayed low hSLC22A2 mRNA expression. In agreement, Zhang et al. also showed no hSLC22A2 mRNA expression in six colon cancer cell lines tested and four normal colon tissue samples. On the other hand, hSLC22A2 expression was detected in 11 of 20 colon cancer tissue samples suggesting a potential role for hSLC22A2 in oxaliplatin sensitivity in colon cancer and may explain differences in response. Altogether this warrants further (in vivo) studies designed to investigate the role of hSLC22A2 in transport and its potential correlation with oxaliplatin activity in other platinum-treated tumour types. Furthermore, replacing cisplatin/carboplatin by oxaliplatin as the drugs of choice deserves serious consideration as an alternative treatment option for tumours expressing hSLC22A2. Appropriate activation of hSLC22A2 expression to enhance the cytotoxicity of platinum drugs might be a potential therapeutic option in tumours that are intrinsically resistant to platinum-based chemotherapy. Noteworthy, hormones that bind to androgen responsive elements have been implicated in the positive regulation of SLC22A2-mediated transport in rats and this concept may be further pursued in human situations (Terada and Inui, 2007). An interesting notion is that hypermethylation of the proximal promoter region, interfering with the transactivation of hSLC22A2 by the basal transcription factor upstream stimulating factor 1, was found to be responsible for the tissue specific expression of hSLC22A2 (Aoki et al., 2008). This knowledge may also open potentially new alternative avenues for intervention.
Renal tubular secretion of platinum compounds involves uni-directed transport across the basolateral epithelial membrane of renal proximal tubular cells by OCTs and subsequent extrusion via the H+/organic cation anti-porter system at the apical or luminal brush-border membrane of tubular kidney cells. Recently it became clear that two members of the multidrug and toxin extrusion family (MATE1 and MATE2-K) are responsible for the anti-porter transport (Terada and Inui, 2007). These anti-porters are primarily expressed in the kidney and act in concert with OCTs to facilitate renal elimination of prototypical cations, drugs and xenobiotics, thereby protecting the kidney from cationic toxins. It should be noted that of the three FDA-approved platinum compounds only cisplatin induces severe nephrotoxicity. Both in vitro and in vivo cell models showed elevated renal accumulation of cisplatin, compared with oxaliplatin or carboplatin (Yokoo et al., 2007; Terada ands Inui, 2008). Interestingly, hMATE1 and in particular hMATE2-K can mediate the H+-gradient-dependent anti-port of oxaliplatin, but not that of cisplatin (Yokoo et al., 2007). This MATE-mediated renal extrusion of oxaliplatin might be the reason that oxaliplatin is not nephrotoxic, despite being a good hSLC22A2 substrate. A current working model for efficient drug clearance in the human kidney is based on the fact that hSLC22A2, mediating the cellular uptake of organic cations including oxaliplatin, metformin, cimetidine and to a lesser extent that of cisplatin, acts together with the MATE anti-porter system involved in the final renal clearance of these drugs. Oxaliplatin is transported into renal proximal tubular cells by hSLC22A2 but does not accumulate due to extrusion by the MATE anti-porter system. Thus efficient MATE-mediated efflux of oxaliplatin might protect against oxaliplatin-induced nephrotoxicity. In contrast, the inability of MATEs to translocate cisplatin that therefore may be retained in the renal proximal tubular cells probably accounts for its nephrotoxicity. Studies combining classical platinum-based chemotherapy with specific OCT and MATE inhibitors may help to understand the role of these transporters in the pharmacokinetics of platinum anticancer drugs, the occurrence of resistance and/or platinum-induced neuro-and nephrotoxicity.
With regard to the three FDA-approved platinum chemotherapeutic drugs, we conclude that the hSLC22A2 OCT is critically involved in the transport of oxaliplatin, is able to transport cisplatin, which is, however, a clearly less potent substrate compared with oxaliplatin, and is not active against carboplatin. So, tumours expressing hSLC22A2 normally treated with cisplatin or carboplatin may benefit from an alternative oxaliplatin treatment.
Acknowledgments
We gratefully express our thanks to Dr H Koepsell (Department of Medicine, Institute of Anatomy and Cell Biology, Julius Maximilians University Würzburg, Germany) for providing the HEK293/hSLC22A2 and HEK293/Neo control cells, and Dr J Reedijk (Leiden Institute of Chemistry, Leiden University, The Netherlands) for the generous gift of ormaplatin and the PtCl2[R,R-DACH] compound.
Glossary
Abbreviations:
- ASP+
4-[4-(diethylamino)styryl]-N-methylpyridiniumiodide
- DACH
diaminocyclohexane
- MPP+
1-methyl-4-phenylpyridinium
- OCT
organic cation transporter
- SLC22
solute carrier family 22
- SRB
sulphorhodamine B
Conflict of interest
The authors state no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Analysis of 4-[4-(diethylamino)styryl]-N-methylpyridiniumiodide (ASP+) accumulation in human embryonic kidney (HEK) 293/hSLC22A2 (A) and HEK293/Neo control cells (B) in the presence or absence of 30 μM oxaliplatin. Cells were incubated with 1 μM ASP+ for 20 min at 37°C and ASP+ uptake was measured by FACScan flow cytometry. In the presence of oxaliplatin, the SLC22A2-mediated ASP+ accumulation was decreased by 35% in the hSLC22A2-overexpressing cells while oxaliplatin did not affect ASP+ accumulation in the Neo control cells.
Table S1 Expression pattern of transporter genes in human embryonic kidney (HEK) 293/hSLC22A2 and HEK293/Neo control cells measured by real-time RT-PCR
Table S2 Morphological changes and trypan blue uptake in human embryonic kidney (HEK) 293/hSLC22A2 and HEK293/Neo control cells monitored upon continuous treatment with oxaliplatin or cisplatina
Table S3 Differential platinum uptake in human embryonic kidney (HEK) 293/hSLC22A2 cells compared with HEK293/Neo cells exposed to a diverse panel of distinct platinum compounds
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition (2008 revision) Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews PA, Jones JA, Varki NM, Howell SB. Rapid emergence of acquired cis-diamminedichloroplatinum(II) resistance in an in vivo model of human ovarian carcinoma. Cancer Commun. 1990;2:93–100. doi: 10.3727/095535490820874641. [DOI] [PubMed] [Google Scholar]
- Aoki M, Terada T, Kajiwara M, Ogasawara K, Ikai I, Ogawa O, et al. Kidney-specific expression of human organic cation transporter 2 (OCT2/SLC22A2) is regulated by DNA methylation. Am J Physiol Renal Physiol. 2008;295:F165–F170. doi: 10.1152/ajprenal.90257.2008. [DOI] [PubMed] [Google Scholar]
- Ardizzoni A, Boni L, Tiseo M, Fossella FV, Schiller JH, Paesmans M, et al. Cisplatin- versus carboplatin-based chemotherapy in first-line treatment of advanced non-small-cell lung cancer: an individual patient data meta-analysis. J Natl Cancer Inst. 2007;99:847–857. doi: 10.1093/jnci/djk196. [DOI] [PubMed] [Google Scholar]
- Boersma AW, Nooter K, Burger H, Kortland CJ, Stoter G. Bax upregulation is an early event in cisplatin-induced apoptosis in human testicular germ-cell tumor cell line NT2, as quantitated by flow cytometry. Cytometry. 1997;27:275–282. doi: 10.1002/(sici)1097-0320(19970301)27:3<275::aid-cyto10>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Briz O, Serrano MA, Rebollo N, Hagenbuch B, Meier PJ, Koepsell H, et al. Carriers involved in targeting the cytostatic bile acid-cisplatin derivatives cis-diammine-chloro-cholylglycinate-platinum (II) and cis-diammine-bisursodeoxycholate-platinum (II) toward liver cells. Mol Pharmacol. 2002;61:853–860. doi: 10.1124/mol.61.4.853. [DOI] [PubMed] [Google Scholar]
- Burger H, Nooter K, Boersma AWM, Kortland CJ, Stoter G. Lack of correlation between cisplatin-induced apoptosis, p53 status and expression of Bcl-2 family proteins in testicular germ cell tumour cell lines. Int J cancer. 1997;73:592–599. doi: 10.1002/(sici)1097-0215(19971114)73:4<592::aid-ijc22>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Burger H, Foekens JA, Look MP, Meijer-van Gelder ME, Klijn JGM, Wiemer EAC, et al. RNA expression of breast cancer resistance protein, lung resistance protein, multidrug resistance-associated proteins 1 and 2, and multidrug resistance gene 1 in breast cancer: correlation with chemotherapeutic response. Clin Cancer Res. 2003;9:827–836. [PubMed] [Google Scholar]
- Ciarimboli G, Ludwig T, Lang D, Pavenstädt H, Koepsell H, Piechota HJ, et al. Cisplatin nephrotoxicity is critically mediated via the human organic cation transporter 2. Am J Pathol. 2005;167:1477–1484. doi: 10.1016/S0002-9440(10)61234-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipski KK, Loos WJ, Verweij J, Sparreboom A. Interaction of cisplatin with the human cation transporter 2. Clin Cancer Res. 2008;14:3875–3880. doi: 10.1158/1078-0432.CCR-07-4793. [DOI] [PubMed] [Google Scholar]
- Gately DP, Howell SB. Cellular accumulation of the anticancer agent cisplatin: a review. Br J Cancer. 1993;67:1171–1176. doi: 10.1038/bjc.1993.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go RS, Adjei AA. Review of the comparative pharmacology and clinical activity of cisplatin and carboplatin. J Clin Oncol. 1999;17:409–422. doi: 10.1200/JCO.1999.17.1.409. [DOI] [PubMed] [Google Scholar]
- Helleman J, Burger H, Hamelers IHL, Boersma AWM, de Kroon AIPM, Stoter G, et al. Impaired cisplatin influx in an A2780 mutant cell line: evidence for a putative, cis-configuration-specific, platinum influx transporter. Cancer Biol Ther. 2006a;5:943–949. doi: 10.4161/cbt.5.8.2876. [DOI] [PubMed] [Google Scholar]
- Helleman J, Jansen MP, Span PN, van Staveren IL, Massuger LF, Meijer-van Gelder ME, et al. Molecular profiling of platinum resistant ovarian cancer. Int J Cancer. 2006b;118:1963–1971. doi: 10.1002/ijc.21599. [DOI] [PubMed] [Google Scholar]
- Huang Y, Sadée W. Membrane transporters and channels in chemoresistance and -sensitivity of tumor cells. Cancer Letters. 2006;239:168–182. doi: 10.1016/j.canlet.2005.07.032. [DOI] [PubMed] [Google Scholar]
- Huang Y, Anderle P, Bussey KJ, Barbacioru C, Shankavarm U, Dai Z, et al. Membrane transporters and channels: role of the transportome in cancer chemosensitivity and chemoresistance. Cancer Res. 2004;64:4294–4301. doi: 10.1158/0008-5472.CAN-03-3884. [DOI] [PubMed] [Google Scholar]
- Johnson SW, Ferry KV, Hamilton TC. Recent insights into platinum drug resistance in cancer. Drug Resist Updat. 1998;1:243–254. doi: 10.1016/s1368-7646(98)80005-8. [DOI] [PubMed] [Google Scholar]
- Keepers YP, Pizao PE, Peters GJ, Van Ark-Otte J, Winograd B, Pinedo HM. Comparison of the sulforhodamine B protein and tetrazolium (MTT) assays for in vitro chemosensitivity testing. Eur J Cancer. 1991;27:897–900. doi: 10.1016/0277-5379(91)90142-z. [DOI] [PubMed] [Google Scholar]
- Kelland L. The resurgence of platinum-based cancer chemotherapy. Nature Reviews Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- Koepsell H, Endou H. The SLC22 drug transporter family. Pflugers Arch. 2004;447:666–676. doi: 10.1007/s00424-003-1089-9. [DOI] [PubMed] [Google Scholar]
- Koepsell H, Lips K, Volk C. Polyspecific organic cation transporters: structure, function, physiological roles, and biopharmaceutical implications. Pharm Res. 2007;24:1227–1251. doi: 10.1007/s11095-007-9254-z. [DOI] [PubMed] [Google Scholar]
- Kuo YM, Chenn HH, Song IS, Savaraj N, Ishikawa T. The roles of copper transporters in cisplatin resistance. Cancer Metastasis Rev. 2007;26:71–83. doi: 10.1007/s10555-007-9045-3. [DOI] [PubMed] [Google Scholar]
- Lovejoy KS, Todd RC, Zhang S, McCormick MS, D'Aquino JA, Reardon JT, et al. cis-Diammine(pyridine)chloroplatinum(II), a monofunctional platinum(II) antitumor agent: uptake, structure, function, and prospects. Proc Natl Acad Sci USA. 2008;105:8902–8907. doi: 10.1073/pnas.0803441105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhinney SR, Goldberg RM, McLeod HL. Platinum neurotoxicity pharmacogenetics. Mol Cancer Ther. 2009;8:10–15. doi: 10.1158/1535-7163.MCT-08-0840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misset JL, Bleiberg H, Sutherland W, Bekradda M, Cvitkovic E. Oxaliplatin clinical activity: a review. Crit Rev Oncol Hematol. 2000;35:75–93. doi: 10.1016/s1040-8428(00)00070-6. [DOI] [PubMed] [Google Scholar]
- Mishima M, Samimi G, Kondo A, Lin X, Howell SB. The cellular pharmacology of oxaliplatin resistance. Eur J cancer. 2002;38:1405–1412. doi: 10.1016/s0959-8049(02)00096-5. [DOI] [PubMed] [Google Scholar]
- Pan BF, Sweet DH, Pritchard JB, Chen R, Nelson JA. A transfected cell model for the renal toxin transporter, rOCT2. Toxicol Sci. 1999;47:181–186. doi: 10.1093/toxsci/47.2.181. [DOI] [PubMed] [Google Scholar]
- Perez RP. Cellular and molecular determinants of cisplatin resistance. Eur J Cancer. 1998;34:1535–1542. doi: 10.1016/s0959-8049(98)00227-5. [DOI] [PubMed] [Google Scholar]
- Raymond E, Faivre S, Chaney S, Woynarowski J, Cvitkovic E. Cellular and molecular pharmacology of oxaliplatin. Mol Cancer Ther. 2002;1:227–235. [PubMed] [Google Scholar]
- Rixe O, Ortuzar W, Alvarez M, Parker R, Reed E, Paull K, et al. Oxaliplatin, tetraplatin, cisplatin, and carboplatin: spectrum of activity in drug-resistant cell lines and in the cell lines of the National Cancer Institute's Anticancer Drug Screen panel. Biochem Pharmacol. 1996;52:1855–1865. doi: 10.1016/s0006-2952(97)81490-6. [DOI] [PubMed] [Google Scholar]
- Safaei R, Howell SB. Copper transporters regulate the cellular pharmacology and sensitivity to Pt drugs. Crit Rev Oncol Hematol. 2005;53:13–23. doi: 10.1016/j.critrevonc.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Schuyer M, van der Burg MEL, Henzen-Logmans SC, Fieret JH, Klijn JGM, Look MP, et al. Reduced expression of Bax is associated with poor prognosis in patients with epithelial ovarian cancer: a multifactorial analysis of TP53, P21, Bax and Bcl-2. Brit J Cancer. 2001;85:1359–1367. doi: 10.1054/bjoc.2001.2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi K, Wada M, Kohno K, Nakamura T, Kawabe T, Kawakami M, et al. A human canalicular multispecific organic anion transporter (cMOAT) gene is overexpressed in cisplatin-resistant human cancer cell lines with decreased drug accumulation. Cancer Res. 1996;56:4124–4129. [PubMed] [Google Scholar]
- Terada T, Inui K. Gene expression and regulation of drug transporters in the intestine and kidney. Biochem Pharmacol. 2007;73:440–449. doi: 10.1016/j.bcp.2006.10.010. [DOI] [PubMed] [Google Scholar]
- Terada T, Inui K. Physiological and pharmacokinetic roles of H+/organic cation antiporters (MATE/SLC47A) Biochem Pharmacol. 2008;75:1689–1696. doi: 10.1016/j.bcp.2007.12.008. [DOI] [PubMed] [Google Scholar]
- Yao X, Panichpisal K, Kurtzman N, Nugent K. Cisplatin nephrotoxicity: a review. Am J Med Sci. 2007;334:115–124. doi: 10.1097/MAJ.0b013e31812dfe1e. [DOI] [PubMed] [Google Scholar]
- Yokoo S, Yonezawa A, Masuda S, Fukatsu A, Katsura T, Inui K. Differential contribution of organic cation transporters, OCT2 and MATE1, in platinum agent-induced nephrotoxicity. Biochem Pharmacol. 2007;74:477–487. doi: 10.1016/j.bcp.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Yonezawa A, Masuda S, Nishihara K, Yano I, Katsura T, Inui K. Association between tubular toxicity of cisplatin and expression of organic cation transporter rOCT2 (Slc22a2) in the rat. Biochem Pharmacol. 2005;70:1823–1831. doi: 10.1016/j.bcp.2005.09.020. [DOI] [PubMed] [Google Scholar]
- Yonezawa A, Masuda S, Yokoo S, Katsura T, Inui K. Cisplatin and oxaliplatin but not carboplatin and nedaplatin, are substrates for human organic cation transporters (SLC22A1-3 and multidrug and toxin extrusion family) J Pharmacol Exp Ther. 2006;319:879–886. doi: 10.1124/jpet.106.110346. [DOI] [PubMed] [Google Scholar]
- Zhang S, Lovejoy KS, Shima JE, Lagpacan LL, Shu Y, Lapuk A, et al. Organic cation transporters are determinants of oxaliplatin cytotoxicity. Cancer Res. 2006;66:8847–8857. doi: 10.1158/0008-5472.CAN-06-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolk O, Solbach TF, König J, Fromm MF. Structural determinants of inhibitor interaction with the human organic cation transporter OCT2 (SLC22A2) Naunyn Schmiedebergs Arch Pharmacol. 2008;379:337–348. doi: 10.1007/s00210-008-0369-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.