

Abstract
Background and purpose:
Tetrandrine, a well-known naturally occurring calcium antagonist with anti-inflammatory, antioxidant and anti-fibrogenetic activities, has long been used clinically for treatment of cardiovascular diseases such as hypertension and arrhythmia. However, little is known about the effect of tetrandrine on cardiac hypertrophy. The aims of the present study were to determine whether tetrandrine could attenuate cardiac hypertrophy and to clarify the underlying molecular mechanisms.
Experimental approach:
Tetrandrine (50 mg·kg−1·day−1) was administered by oral gavage three times a day for one week and then the mice were subjected to either chronic pressure overload generated by aortic banding (AB) or sham surgery (control group). Cardiac function was determined by echocardiography.
Key results:
Tetrandrine attenuated the cardiac hypertrophy induced by AB, as assessed by heart weight/body weight and lung weight/body weight ratios, cardiac dilatation and the expression of genes of hypertrophic markers. Tetrandrine also inhibited fibrosis and attenuated the inflammatory response. The cardioprotective effects of tetrandrine were mediated by blocking the increased production of reactive oxygen species and the activation of ERK1/2-dependent nuclear factor-κB and nuclear factor of activated T cells that occur in response to hypertrophic stimuli.
Conclusions and implications:
Taken together, our results suggest that tetrandrine can improve cardiac function and prevent the development of cardiac hypertrophy by suppressing the reactive oxygen species-dependent ERK1/2 signalling pathway.
Keywords: tetrandrine, cardiac hypertrophy, reactive oxygen species, mitogen-activated protein kinase, transcription factor, nuclear factor-κB
Introduction
Cardiovascular disease, such as that caused by myocardial infarction, obesity or drug abuse promotes cardiac hypertrophy and subsequent heart failure, which is one of the leading causes of mortality in the world and encompasses a wide spectrum of cardiac pathologies (Barry et al., 2008). When the heart experiences extended periods of elevated workload, it undergoes hypertrophic enlargement in response to the increased demand. In the case of pathological hypertrophy, although the increased heart size is initially a compensatory mechanism, sustained hypertrophy can ultimately lead to a decline in left ventricular function and thus represents an independent risk factor for heart failure (Dhalla et al., 2009). Therefore, it is important to clarify the specific molecular mechanism and signalling pathways that lead to hypertrophy. A number of signalling modulators in the vasculature milieu are known to regulate heart mass including those that influence gene expression, apoptosis, cytokine release and growth factor signalling (Olson, 2004; Jenkins et al., 2009). Recent evidence, obtained using genetic and cellular models of cardiac hypertrophy, suggests that pathological hypertrophy can be prevented or reversed and has promoted an enormous drive in drug discovery research aiming to identify novel and specific regulators of hypertrophy (Li et al., 2005; 2006; Cai et al., 2009a).
Tetrandrine is a bisbenzylisoquinoline alkaloid isolated from the Chinese medicinal herb-root of Stephania tetrandra S Moore, which was traditionally used as an anti-inflammatory, antipyretic and analgesic herb in Chinese medicine (Kwan and Achike, 2002; Yao and Jiang, 2002). The empirical formula of tetrandrine is C38H42O8N2 with molecular weight 622 Da. The first pharmacological and toxicological study of tetrandrine was published in 1937; this demonstrated its hypotensive and cardiodepressant effects. Since the 1950s, tetrandrine has been used in China as an antihypertensive agent (Yao and Jiang, 2002). Subsequently, tetrandrine has been shown to act as a calcium antagonist, inhibiting the ICa-L and reducing Ca2+ flows into the cytosol in myocyte and vascular smooth muscle cells (Rao, 2002; Wang et al., 2004). Recently, it was reported that tetrandrine improved haemodynamic changes during remodelling, especially diastolic function such as LV compliance and stiffness, increased cardiac myosin ATPase activity and Na+-K+, Ca2+ ATPase activity, and normalized vascular reactivity (Chen et al., 2000). Tetrandrine is also known for its anti-inflammatory and anti-fibrogenic actions, which make tetrandrine and related compounds potentially useful in the treatment of lung silicosis, liver cirrhosis and rheumatoid arthritis (Rao, 2002; Wang et al., 2004). However, the effects of tetrandrine on cardiac hypertrophy and the related signalling mechanisms have not been elucidated. The aims of this study were, therefore, to determine whether tetrandrine, which is recognized to possess anti-inflammatory, antioxidant, as well as anti-fibrogenetic activities, can attenuate pressure overload-induced cardiac hypertrophy in mice, and to identify the molecular mechanisms responsible for these putative effects.
Methods
Materials and preparation of tetrandrine
The antibodies used to recognize total and phosphorylation of ERK1/2, p38, JNK1/2, phospho-I κ B kinase β (IKKβ), phospho-IκBα and IκBα were purchased from Cell Signaling Technology. The BCA protein assay kit was purchased from Pierce and IKK activity kit was obtained from B&D Bioscience. The antibodies used to recognize tropomyosin and α-sarcomeric actin were obtained from Abacm. All other antibodies were purchased from Santa Cruz Biotechnology. Fetal calf serum (FCS) was obtained from Hyclone. Nuclear factor-κB (NF-κB)- and nuclear factor of activated T cells (NFAT)-luc report constructs were as described previously (Cai et al., 2009a,b;) Cell culture reagents and all other reagents were from Sigma. Tetrandrine was obtained from MP Biomedicals and dissolved in 0.1 N HCl at a concentration of 25 mg·mL−1 and it was passed through a 0.22 µm filter for bacteriological sterilization, as described previously (Ho et al., 2004). The pH was adjusted to 6.8–7 before use.
The LipofectAMINE reagent and TRIzol were from Invitrogen; the Advantage RT-for-PCR kit was from BD Biosciences; SYBR Green PCR Master Mix was from Applied Biosystems; BCA Protein Assay Reagents were from PIERCE (Rockford, IL, USA). The electrophoretic mobility shift assays (EMSA) (Gel Shift Assay System E3300) was obtained from Promega (Madison, WI, USA) and the CardiaoTACS in situ Apoptosis Detection Kit from R&D Systems.
Neonatal rat cultured cardiac myocytes
Primary cultures of cardiac myocytes were prepared as described previously (Li et al., 2005; 2006;). Cells from the hearts of 1- to 2-day-old Sprague-Dawley rats were seeded at a density of 1 × 106 per well onto six-well culture plates in plating medium consisting of F10 medium supplemented with 10% FCS and penicillin/streptomycin. After 48 h, the culture medium was replaced with F10 medium containing 0.1% FCS and bromodeoxyuridine (BrdU, 100 µM). After 24 h of serum deprivation, tetrandrine alone or tetrandrine followed by angiogensin II (Ang II, 1 µM) were added to the medium and the cultures were incubated for the indicated time. Viability was determined by the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay. For cell infection, cardiac myocytes or cardiac fibroblasts (1 × 106 per well) were cultured in six-well plates and exposed to 2 × 108 pfu of each virus in 1 mL of serum-free medium for 24 h. The cells were then washed and incubated in serum-containing media for 24 h.
Reporter assays, Western blotting and quantitative real-time PCR
Cardiac myocytes were seeded in triplicate in six-well plates. Cells were transfected with 0.5 µg of luciferase reporter constructs NF-κB and NFAT, and internal control plasmid DNA using 10 µL of LipofectAMINE reagent, according to the manufacturer's instructions. After 6 h of exposure to the DNA-LipofectAMINE complex, cells were cultured in medium containing 10% serum for 24 h and then incubated with serum-free medium for 12 h. Cells were pretreated with tetrandrine for 60 min and then treated with Ang II and harvested using passive lysis buffer (Promega) according to the manufacturer's protocol. The luciferase activity was normalized by control plasmid. All experiments were done in triplicate and repeated at least three times. For Western blot, cardiac tissue and cultured cardiac myocytes or fibroblasts were lysed in RIPA lysis buffer. Fifty micrograms of cell lysate was used for SDS-PAGE, and proteins were then transferred to an Immobilon-P membrane (Millipore, Beijing, China). Specific protein expression levels were normalized to either the glyceraldehydes-3-phosphate dehydrogenase (GAPDH) protein for total cell lysate and cytosolic protein, or the Lamin-B1 protein for nuclear protein signal on the same PVDF membrane. For real-time PCR, total RNA was extracted from frozen, pulverized mouse tissues using TRIzol and synthesized cDNA using oligo (dT) primers with the Advantage RT-for-PCR kit. We quantified PCR amplifications using SYBR Green PCR Master Mix and normalized the results against GAPDH gene expression.
Electrophoretic mobility shift assay and IKK assay
To examine the DNA-binding activities of NF-κB, EMSA were performed according to the manufacturer's instructions. Nuclear proteins were isolated as described previously (Li et al., 2007a). Protein concentrations were measured by BCA Protein Assay Reagents using bovine serum albumin as a standard.
Animal models, echocardiography and blood pressure
The protocols for the animal experiments were approved by our hospital guidelines. All surgery and subsequent analyses were performed in a blinded fashion for all groups. Adult male C57/B6 mice (8–10 weeks old) were used in the current study, which were purchased from Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences, Beijing, China and acclimatized for more than one week prior to experimental use. Aortic banding (AB) was performed as described previously (Bian et al., 2008; Tang et al., 2009). Tetrandrine suspension was prepared using 0.5% carboxy methylcellulose solution for animal experiments. Suspensions were freshly prepared and administered at a constant volume of 1 mL·100 g−1 body weight (BW) by oral gavage three times a day. The control group for these animal experiments was given the same volume of liquid but consisted solely of the vehicle solution (0.5% carboxy methylcellulose). We administered tetrandrine (50 mg·kg−1·day−1) by oral gavage three times a day for one week and then subjected the mice to either chronic pressure overload generated by AB or sham surgery as the control group. Mice were randomly assigned into four groups as tetrandrine + sham, tetrandrine + AB, vehicle + sham and vehicle + AB. Pretreatment with vehicle or 50 mg·kg−1·day−1 tetrandrine for one week prior to AB surgery or sham operation allowed for critical evaluation. Doppler analysis was performed to ensure that physiological constriction of the aorta was induced. The internal diameter and wall thickness of LV were assessed by echocardiography at the indicated time after surgery or infusion. After the mice had been killed, the hearts and lungs were dissected out and weighed to compare heart weight/body weight (HW/BW, mg·g−1) and lung weight/body weight (LW/BW, mg·g−1) ratios in tetrandrine-treated and vehicle-treated mice. Echocardiography was performed by SONOS 5500 ultrasound (Philips Electronics, Amsterdam, the Netherlands) with a 15 MHz linear array ultrasound transducer. The LV was assessed in both parasternal long-axis and short-axis views at a frame rate of 120 Hz. End-systole or end-diastole was defined as the phase in which the smallest or largest area of LV, respectively, was obtained. Left ventricular end-diastolic diameter (LVEDD) and left ventricular end-systolic diameter (LVESD) were measured from the LV M-mode tracing with a sweep speed of 50 mm·s−1 at the mid-papillary muscle level. Blood pressure was recorded by a microtip catheter transducer (SPR-839, Millar Instruments, Houston, TX, USA) inserted into the right carotid artery and advanced into the left ventricle for haemodynamic measurements.
Histological and apoptosis analysis
Hearts were excised, washed with saline solution and placed in 10% formalin. Hearts were cut transversely close to the apex to visualize the left and right ventricles. Several sections of heart (4–5 µm thick) were prepared and stained with haematoxylin and eosin (HE) for histopathology or picrosirius red for collagen deposition and then visualized by light microscopy. For myocyte cross-sectional area, sections were stained HE. A single myocyte was measured with an image quantitative digital analysis system (Image Pro-Plus 4.5). The outline of 100 myocytes was traced in each group. Apoptosis was assessed by the TdT-mediated dUTP nick end labelling (TUNEL) assay that was performed in sections using the CardiaoTACS in situ Apoptosis Detection Kit according to manufacturer's recommendations and was confirmed by detection of the activation of caspase-3/8/9.
Measurements of reactive oxygen species (ROS) and myofibrillar protein oxidation
Cardiac myocytes were cultured on coverslips in 35 mm dishes and then pretreated with tetrandrine and subsequently stimulated with 1 µM Ang II for the indicated times. Intracellular generation of ROS was quantified using 2′,7′-dichlorofluorescein dilacerate (DCFH-DA). The cells were incubated with 5 µM DCFH-DA in the dark for 60 min and immunofluorescence was visualized using laser scanning confocal microscope (488 nm, 200 mW). ROS in the heart tissue were quantified using electron spin resonance (ESR) spectroscopy with 4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl (hydroxy-TEMPO) as described previously (Li et al., 2006). The procedure for myofibrillar protein extraction was carried out according to the method described by Canton et al. (2004; 2006;) and Barbato et al. (1996). Myofibrillar proteins were extracted, in the presence of protease and phosphatases inhibitors, from frozen cardiac tissue of the indicated groups. After the extraction of the myofilament-enriched fraction had finished, oxidation of actin and tropomyosin were measured as protein carbonyl groups by using OxyBlot Protein Oxidation Detection Kit (Millipore, S7150). The detailed procedure for measuring myofibrillar protein oxidation was following the method described by Canton et al. (2006) and Duncan et al. (2005).
Statistical analysis
All the experiments were repeated at least three times with a similar pattern of results. Results are expressed as mean ± SEM of multiple experiments for in vitro assays and mean ± SEM of n number of animals for in vivo experiments. Student's t-test was used to compare two groups and anova (two-tailed P-value) was used with the Dunnett post hoc test for multiple groups. A P-value <0.05 was considered significant.
Results
Tetrandrine attenuates cardiac hypertrophy in vivo
To investigate the effects of tetrandrine on cardiac hypertrophy in vivo, we used a chronic pressure overload model to induce hypertrophy. As expected, all vehicle-treated AB mice 8 weeks post-surgery demonstrated the classical increase in heart size and dilatation of ventricular chambers as compared with the sham control group. Tetrandrine treatment of the AB mice, on the other hand, resulted in a marked reduction in hypertrophic growth as measured by HW/BW, LW/BW and cardiomyocyte cross-sectional area as well as systolic blood pressure. No significant changes were observed in the sham-operated mice treated with tetrandrine or vehicle (Table 1). Subsequent assessment of chamber size and wall thickness using M-mode echocardiography confirmed these findings. In vehicle-AB mice, decreased fractional shortening (FS) with dilated posterior wall thickness (PWT), LVEDD, LVESD, left ventricular septum, diastolic (LVSD), left ventricular posterior wall thickness, diastolic (LVPWD) were seen compared with the sham group; this corresponded to cardiac hypertrophy. However, tetrandrine treatment prevented the development of the adverse effects due to cardiac remodelling and ventricular dysfunction, as evidenced by decreased PWT, LVEDD, LVSD, LVPWD and a higher percent of fractional shortening (%FS) compared with AB mice treated with vehicle. Sham-operated animals exhibited no signs of myocardial dysfunction as assessed by echocardiography (Table 1). Gross hearts, whole hearts and HE staining of histological sections further confirmed the inhibitory effect of tetrandrine on cardiac remodelling in AB hearts (Figure 1A). To determine whether tetrandrine affected the mRNA expression levels of markers of cardiac hypertrophy, real-time PCR analysis of atrial natriuretic peptide, B-type natriuretic peptide, myosin heavy chain 6 (Myh6) and myosin heavy chain 7 (Myh7) was performed. The results revealed a significant attenuation of the observed increase in expression level from the vehicle-treated AB group when these animals were treated with tetrandrine (Figure 1B). In addition, similar results were obtained with the protein expression levels of markers of cardiac hypertrophy, β-MHC, atrial natriuretic peptide and B-type natriuretic peptide (Figure 1C,D). These results indicate that tetrandrine inhibits the expression of cardiac hypertrophy markers in the heart and this results in an attenuated cardiac hypertrophic response induced by pressure overload. Taken together, our findings indicate that tetrandrine prevents the development of cardiac hypertrophy in vivo.
Table 1.
Echocardiographic data showed the effects of tetrandrine on cardiac hypertrophy induced by aortic banding
| Parameter | Vehicle-sham | Tetrandrine-sham | Vehicle-AB | Tetrandrine-AB |
|---|---|---|---|---|
| Number (n) | 10 | 10 | 8 | 9 |
| BW (g) | 27.5 ± 1.5 | 26.8 ± 2.1 | 26.6 ± 1.5 | 27.5 ± 1.2 |
| HW/BW (mg·g−1) | 4.66 ± 0.14 | 4.72 ± 0.13 | 8.11 ± 0.25* | 5.43 ± 0.31† |
| LW/BW (mg·g−1) | 4.54 ± 0.22 | 4.63 ± 0.12 | 7.69 ± 0.31* | 5.34 ± 0.15† |
| CSA (µm2) | 272 ± 30 | 269 ± 21 | 447 ± 28* | 325 ± 18† |
| SBP (mmHg) | 110.6 ± 1.8 | 114.5 ± 1.4 | 153.5 ± 4.2* | 143.2 ± 3.1* |
| HR (beats·min−1) | 471 ± 25 | 488 ± 25 | 456 ± 34 | 466 ± 30 |
| PWT (mm) | 1.23 ± 0.01 | 1.22 ± 0.02 | 2.42 ± 0.04* | 1.53 ± 0.03† |
| LVEDD (mm) | 3.55 ± 0.02 | 3.58 ± 0.02 | 5.54 ± 0.03* | 4.10 ± 0.01† |
| LVESD (mm) | 2.35 ± 0.03 | 2.37 ± 0.04 | 3.47 ± 0.02* | 2.66 ± 0.02† |
| LVSD (mm) | 0.61 ± 0.02 | 0.64 ± 0.03 | 1.51 ± 0.04* | 0.96 ± 0.03† |
| LVPWD (mm) | 0.65 ± 0.01 | 0.64 ± 0.06 | 1.32 ± 0.01* | 0.87 ± 0.05† |
| FS (%) | 52.7 ± 2.4 | 54.5 ± 3.1 | 32.4 ± 2.6* | 47.5 ± 1.5*† |
All values are mean ± SEM.
P < 0.01 was obtained for the vehicle-sham values.
P < 0.05 was obtained for the vehicle-AB values after AB.
AB, aortic banding; BW, body weight; CSA, cardiomyocyte cross-sectional area; FS, fractional shortening; HR, heart rate; HW, heart weight; LVEDD, left ventricular end-diastolic diameter; LVESD, left ventricular end-systolic diameter; LVPWD, left ventricular posterior wall, diastolic; LVSD, left ventricular septum, diastolic; LW, lung weight; PWT, posterior wall thickness; SBP, systolic blood pressure.
Figure 1.
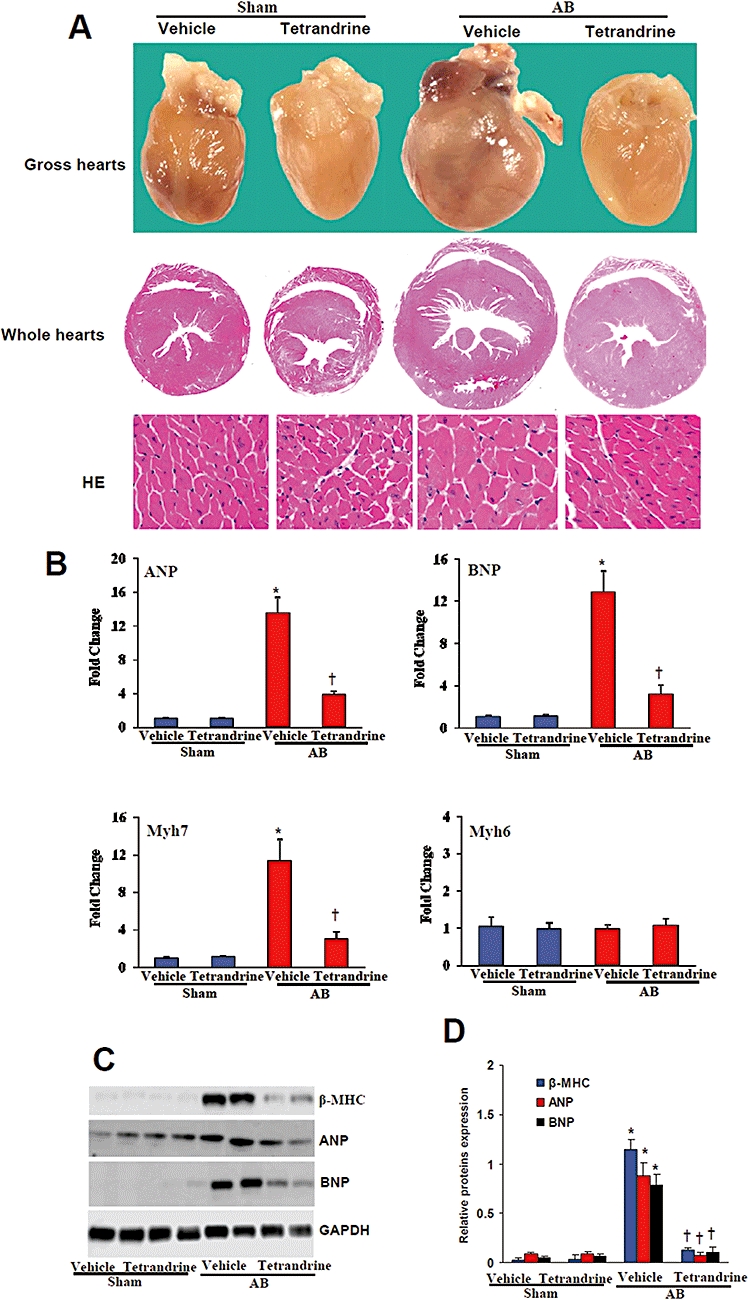
Tetrandrine attenuated cardiac hypertrophy in vivo. (A) Gross hearts (top), representative whole hearts section (middle) and HE staining (bottom) at 8 weeks after aortic banding or sham operation(n= 6). (B–D) mRNA and protein expression levels of hypertrophic markers, ANP, BNP and Myh7, which were evaluated by real-time PCR (n= 3) and western blot (n= 4). *P < 0.01, compared with the vehicle-sham values. †P < 0.01, compared with the vehicle-AB values after AB. AB, aortic banding; ANP, atrial natriuretic peptide; BNP, B-type natriuretic peptide; GAPDH, glyceraldehydes-3-phosphate dehydrogenase; HE, haematoxylin and eosin; Myh7, myosin heavy chain 7.
Tetrandrine blocks fibrosis, but not apoptosis
To investigate the mechanism by which tetrandrine inhibits cardiac hypertrophy further, we examined the ability of tetrandrine to inhibit fibrosis. In order to determine the extent of fibrosis in the heart 8 weeks post-AB, paraffin-embedded slides were stained with picrosirius red and the LV collagen volume was scored under a light microscope. Tetrandrine treatment significantly reduced the LV collagen volume in contrast to the vehicle-treated AB mice (Figure 2A,B). Subsequent analysis of protein and mRNA expression levels of known mediators of fibrosis including connective tissue growth factor, TGF-β1, collagen I and III demonstrated a blunted response following tetrandrine administration compared with the vehicle-treated group (Figure 2C,D). In addition, tetrandrine has been shown to induce apoptosis in different cancer cell lines (Chen et al., 2007; McCubrey et al., 2008). Therefore, we performed TUNEL assay on sections of cardiac tissues from indicated groups. Our results showed that the number of apoptotic cells versus total cells was comparable with or without tetrandrine treatment (Figure 2E). Further experiments demonstrated that the amounts of cleaved caspase-3/8/9 as well as the protein expression of Bax, Bad and Bcl-2 were not different in tetrandrine- or vehicle-treated mice (Figure 2F). These data suggest that the decreased cardiac hypertrophy and fibrosis induced by tetrandrine were not caused by decreased cardiomyocyte apoptosis.
Figure 2.
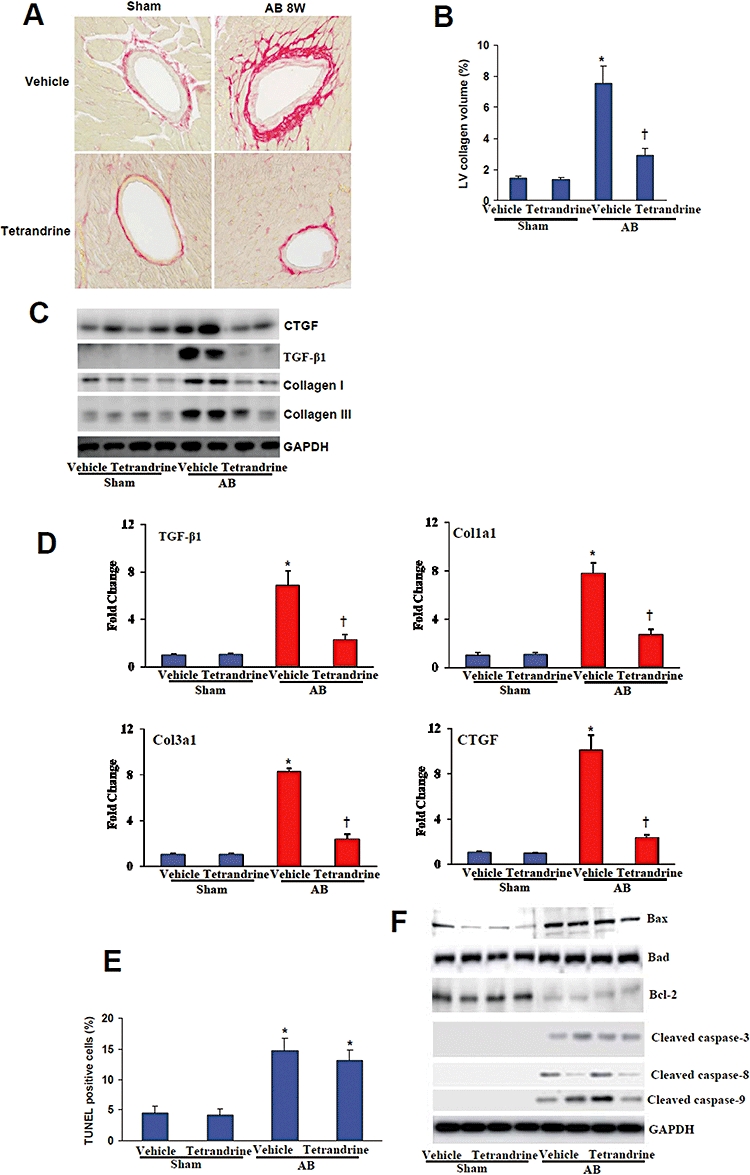
Tetrandrine blocked fibrosis, but not apoptosis. (A,B) Representative images of PSR staining form indicated groups and quantitative analysis of left ventricle interstitial collagen volume fraction in indicated groups (n= 6). *P < 0.01, compared with the vehicle-sham values. †P < 0.01, compared with the vehicle-AB values after AB. (C) Protein expression of CTGF, collagen I and III were tested by western blot analysis. GAPDH was used as internal control (n= 4). (D) Real-time PCR analysis of TGF-β1, Col1a1, Col1a3 and CTGF mRNA level (n= 4). *P < 0.01, compared with the vehicle-sham values. †P < 0.01, compared with the vehicle-AB values after AB. (E) TUNEL positive cells from histological sections of indicated groups were quantified (n= 5). *P < 0.01, compared with the vehicle-sham values. (F) Western blot analysis of cleaved caspase-3, caspase-8 and caspase-9 as well as the expression of Bcl-2, Bad and Bax proteins in the indicated groups (n= 5). AB, aortic banding; BW, body weight; CTGF, connective tissue growth factor; GAPDH, glyceraldehydes-3-phosphate dehydrogenase; PSR, picrosirius red; TUNEL, TdT-mediated dUTP nick end labelling.
Tetrandrine attenuates the excess production of ROS in vitro and in vivo
Reactive oxygen species play a critical role in the progression of cardiac hypertrophy and heart failure (Li et al., 2005; 2006;). In order to measure the level of ROS, we used DCFH-DA. Cardiac myocytes were pretreated with 10 µM tetrandrine for 30 min and subsequently incubated with 1 µM Ang II for 120 min. The results show that Ang II markedly increased the levels of ROS and this effect was significantly blocked by tetrandrine treatment (Figure 3A). To confirm these in vitro findings, we evaluated the levels of ROS in mouse with or without tetrandrine treatment. The production of ROS in the murine hearts was evaluated by ESR spectroscopy with hydroxy-TEMPO as a spin probe. The intensity of ESR signals declined more rapidly in aortic-banded mice than controls, and a linear relation was observed in the semi-logarithmic plot of peak signal intensity versus time (data not shown). The rate of signal decay has been shown to reflect the concentration of ROS in the reaction mixture, which was significantly higher in AB mice than sham groups and this increase was markedly attenuated by tetrandrine (Figure 3B). Recent studies have indicated that myofibrillar protein oxidation has an important impact on ventricular remodelling (Dalla Libera et al., 2005; Duncan et al., 2005; Machackova et al., 2006). Therefore, we examined the effects of tetrandrine on the oxidation of the myofibrillar proteins tropomyosin and α-sarcomeric actin. The data show that the oxidation of tropomyosin and α-sarcomeric actin were significantly increased in vehicle-treated AB mice, and this increase was markedly attenuated by tetrandrine treatment (Figure 3C). These findings indicate that tetrandrine inhibits the excess production of ROS and the oxidation of myofibrillar proteins.
Figure 3.
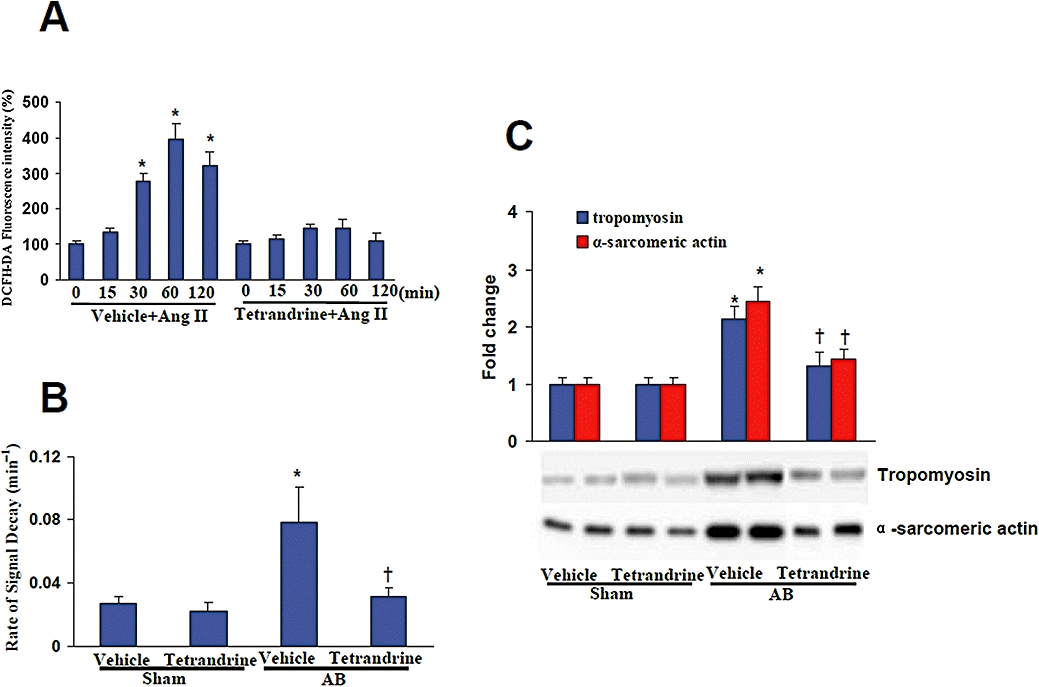
Tetrandrine attenuated the excess production of reactive oxygen species (ROS) in vitro and in vivo. (A) The time courses for the effect of tetrandrine on the generation of ROS induced by angiogensin II (Ang II). Cardiac myocytes were pretreated with 10 µM tetrandrine for 30 min and subsequently incubated with 1 µM Ang II for 120 min. Four parallel experiments were indicated. *P < 0.05, compared with control (0 Ang II). (B) Tetrandrine inhibited the pressure overload-induced increase of signal decay rate (n= 5). (C) Representative blots for the oxidation of myofibrillar proteins tropomyosin and α-sarcomeric actin and their quantitative analysis for each indicated group of mice. *P < 0.01, compared with the vehicle-sham values. †P < 0.01, compared with the vehicle-aortic banding (AB) values after AB.
Tetrandrine blunts the inflammatory response by blocking NF-κB signalling
An increasing number of studies suggest that inflammation plays an important role in the development of cardiac and vascular diseases (Bian et al., 2008; Dorn, 2009). To determine whether tetrandrine can suppress the inflammatory responses in the heart, we evaluated NF-κB signalling. Treatment with tetrandrine abolished the increased activation of NF-κB observed in the myocardium of vehicle-treated mice 8 weeks post-AB and myocytes treated with Ang II (Figure 4A,B). To determine the molecular mechanisms through which tetrandrine blocks NF-κB activation, we analysed IκBα phosphorylation and the process of IKK activation. Heart lysates from samples obtained from mice 8 weeks post-AB were prepared and Western blot analysis was performed. The detection of IκBα phosphorylation and IκBα degradation in vehicle-treated AB mice was significantly impaired after treatment with tetrandrine. Because the phosphorylation of IκBα is mediated through IKKβ, these findings suggested to us that tetrandrine might have an inhibitory role in IKKβ activation. Indeed, we observed that the activation of IKKβ was almost completely blocked by the administration of tetrandrine (Figure 4C). Furthermore, we examined the expression of inflammatory mediators IL-6, tumour necrosis factor α (TNF-α) and monocyte chemoattractant protein-1 (MCP-1) in cardiac tissue. Our results showed that tetrandrine significantly decreased the mRNA levels of IL-6, TNF-α and MCP-1 compared with vehicle-treated AB mice (Figure 4D). These results indicate that tetrandrine inhibits inflammation by blocking NF-κB signalling in response to chronic pressure overload.
Figure 4.
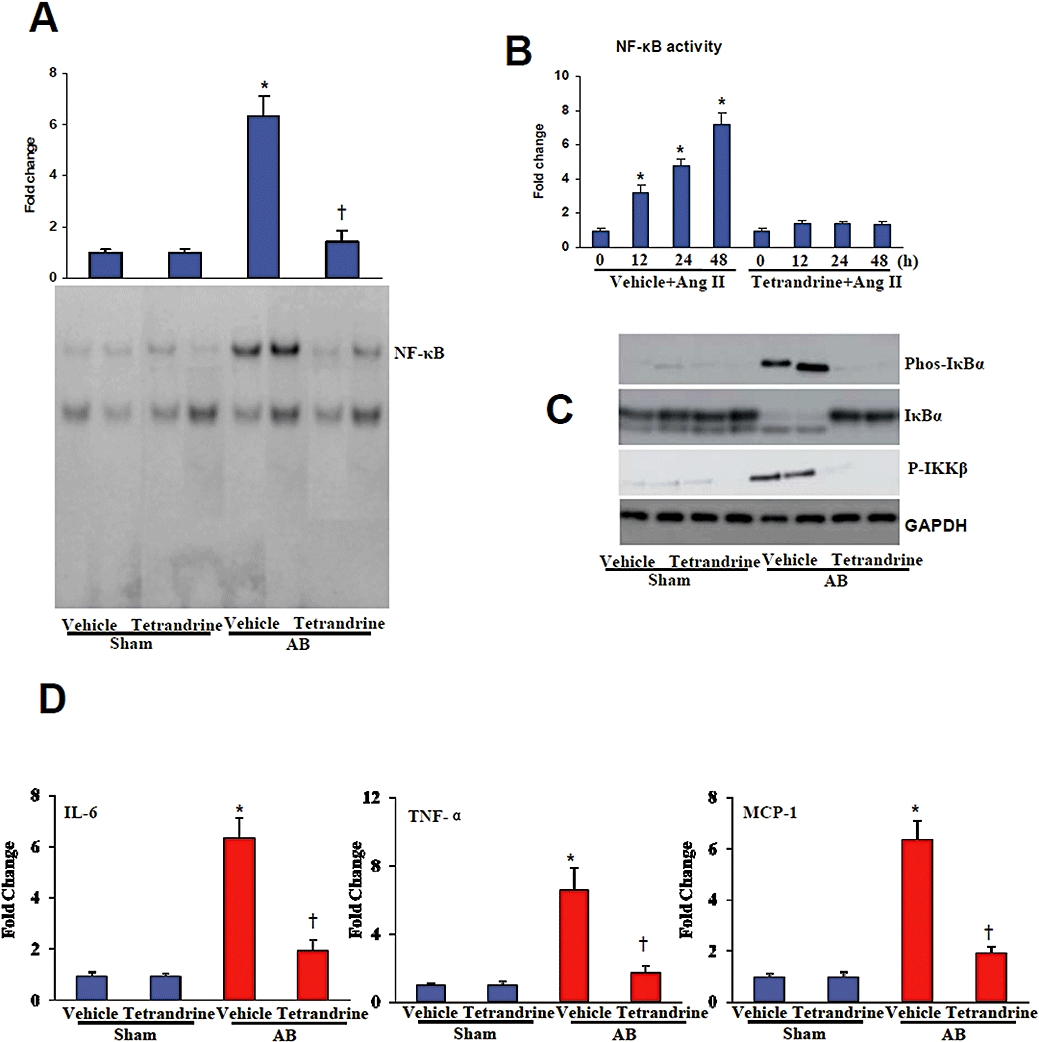
Tetrandrine inhibited the inflammatory response induced by pressure overload. (A) The DNA binding activity of NF-κB evaluated by EMSA in the hearts from the indicated groups (n= 6). (B) NF-κB activity was determined in cardiomyocytes treated with Ang II for the indicated times (n= 6). The concentration of tetrandrine was 10 µM. (C) Representative blots of IκBα degradation, and IκBα and IKKβ phosphorylation in the myocardium obtained from the indicated groups (n= 4). (D) Quantification of IL-6, TNF-α and MCP-1 mRNA levels by use of real-time PCR (n= 4). *P < 0.01, compared with the vehicle-sham values. †P < 0.01, compared with the vehicle-AB values after AB. AB, aortic banding; Ang II, angiogensin II; EMSA, electrophoretic mobility shift assays; GAPDH, glyceraldehydes-3-phosphate dehydrogenase; IKK, IκB kinase; MCP-1, monocyte chemoattractant protein-1; NF-κB, nuclear factor-κB; TNF-α, tumour necrosis factor α.
Tetrandrine inhibits calcineurin/NFAT signalling in response to hypertrophic stimuli
In the heart, the calcineurin/NFAT pathway has been shown to be a key signalling cascade that promotes cardiac hypertrophy (Vega et al., 2003; Clerk et al., 2007). Thus, we measured the activity and nuclear translocation of NFAT to investigate whether tetrandrine can regulate the calcineurin/NFAT signalling. The activity of NFAT in Ang II-treated myocytes was significantly impaired after treatment with 10 µM tetrandrine (Figure 5A). Also the increased nuclear levels of NFATc4 in the myocytes induced by Ang II in vitro or AB in vivo were obviously reduced after treatment with tetrandrine (Figure 5B,C).
Figure 5.
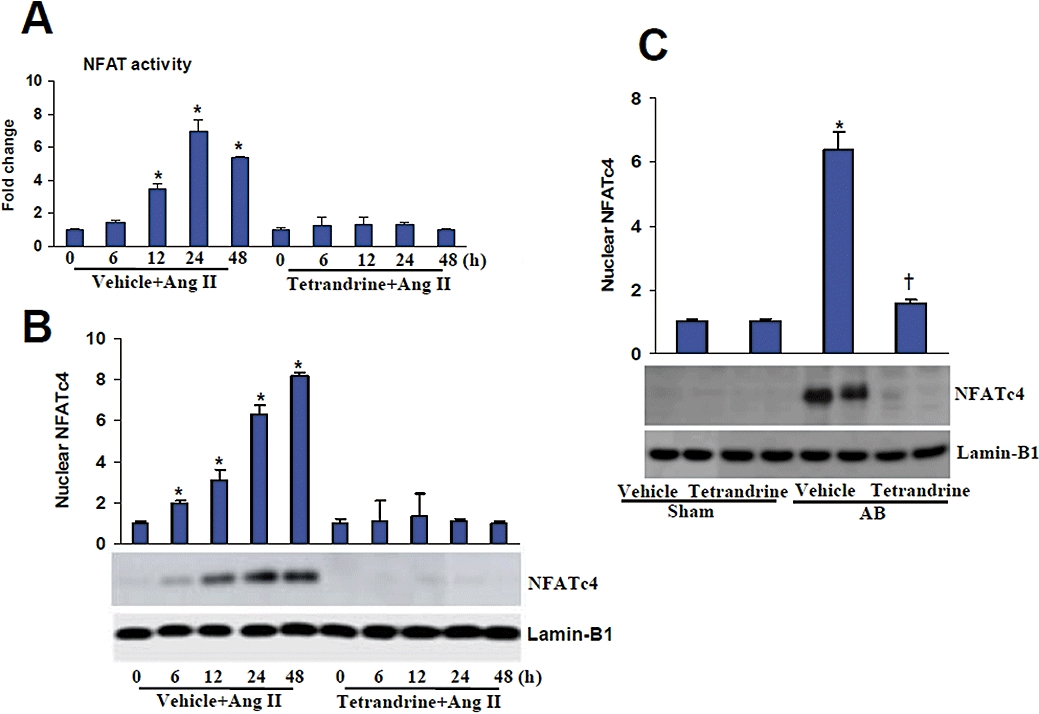
Tetrandrine inhibited the calcineurin/NFAT signalling in response to hypertrophic stimuli. (A) NFAT activity was determined in cardiomyocytes treated with Ang II for the indicated times (n= 6). (B,C) Expression of the nuclear protein NFATc4 in the myocytes induced by Ang II in vitro and AB in vivo (n= 4). The concentration of tetrandrine used for in vitro experiments was 10 µM. *P < 0.01, compared with the vehicle-sham values. †P < 0.01, compared with the vehicle-AB values after AB. AB, aortic banding; Ang II, angiogensin II; NFAT, nuclear factor of activated T cells.
Tetrandrine inhibits ROS-dependent ERK1/2 signalling
To explore the molecular mechanisms through which tetrandrine impairs the cardiac hypertrophic response further, we examined the effects of tetrandrine on the mitogen-activated protein kinase (MAPK) signalling pathway. We exposed neonatal rat cultured cardiac myocytes to 1 µM Ang II with or without 10 µM tetrandrine. Ang II induced a significant increase in the phosphorylated levels of ERK1/2, p38 and JNK1/2; however, tetrandrine treatment markedly blocked the activation of ERK1/2 and this effect was sustained for all the time points tested, whereas p38 and JNK1/2 were not significantly affected (Figure 6A). In line with the in vitro findings, we found that ERK1/2, p38 and JNK1/2 were also significantly phosphorylated in AB mice. However, only the phosphorylation of ERK1/2 was almost completely blocked by tetrandrine, not that of p38 or JNK1/2 (Figure 6B). Further experiments demonstrated that N-acetylcysteine (NAC, 10 mM), a typical antioxidant, markedly inhibited the activation of ERK1/2, NF-κB and NFAT mediated by Ang II treatment (Figure 6C,D), suggesting that the activation of ERK1/2 signalling is dependent on the generation of ROS. Blocking the ERK1/2 signalling with the MEK inhibitor U0126 significantly attenuated the Ang II-induced increase in NF-κB and NFAT activity (Figure 6D), indicating that the activation of NF-κB and NFAT are dependent on ERK1/2 signalling. These findings suggest that tetrandrine inhibits the ROS-dependent ERK1/2 signalling that occurs in response to hypertrophic stimuli.
Figure 6.
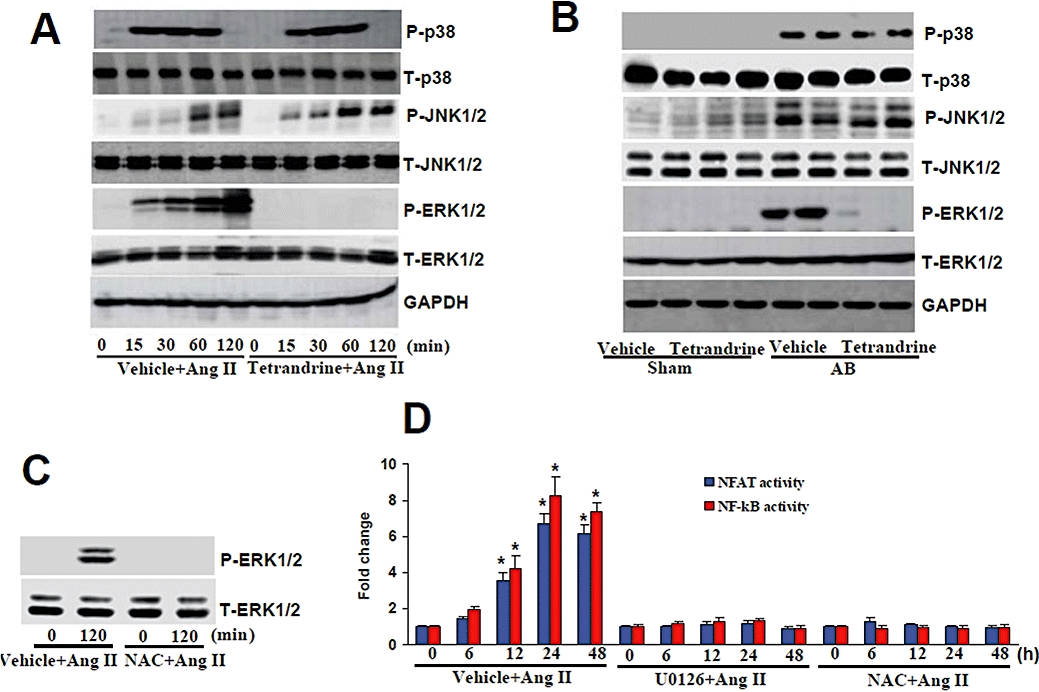
The effects of tetrandrine on MAPKs signalling in response to hypertrophic stimuli. (A) Time course of phosphorylated and total ERK1/2, p38, JNK1/2, and the effects of tetrandrine (10 µM) on them, in cardiomyocytes treated with Ang II (n= 4). (B) Representative blots of ERK1/2, p38 and JNK1/2 phosphorylation and their total protein expression in the indicated groups of mice (n= 4). (C) The effect of NAC on ERK1/2 activation induced by Ang II in cultured myocytes. Cardiac myocytes were pretreated with 10 mM NAC for 30 min and incubated with Ang II for 120 min. (D) Luciferase assay of the effects of U0126 and NAC on the activities of NF-κB and NFAT (n= 6). Cells were incubated with 1 µM Ang II for up to 48 h. The luciferase assay was performed as described in the Methods. The results were reproducible in three separate experiments. *P < 0.05 versus corresponding control. Ang II, angiogensin II; GAPDH, glyceraldehydes-3-phosphate dehydrogenase; MAPK, mitogen-activated protein kinase; NAC, N-acetylcysteine; NFAT, nuclear factor of activated T cells; NF-κB, nuclear factor-κB.
Discussion
The results from our current study demonstrate that tetrandrine protects against hypertrophic growth of cardiomyocytes in vitro and in vivo. The protective role of tetrandrine on cardiac hypertrophy is mediated by interruption of ROS-dependent ERK1/2 signalling. This results in the protection of the host from the combined deleterious effects of cardiac hypertrophy, inflammation and fibrosis. These findings support the concept that tetrandrine could be a preventive and therapeutic candidate for use in cardiac hypertrophy and heart failure.
In the current study, we have demonstrated that tetrandrine not only attenuated cardiac hypertrophy in vivo in response to hypertrophic stimuli, but also improved cardiac performance and reduced chamber dimensions. These findings are consistent with those from previous studies showing that tetrandrine decreases the severity of ventricular hypertrophy in spontaneously hypertensive rats and deoxycorticosterone-acetate-salt hypertensive rats (Chen et al., 1993; Xu and Rao, 1995). Although tetrandrine has been shown to reduce blood pressure in these studies, we did not observe a hypotensive effect of tetrandrine in aortic-banded mice. The reason for this discrepancy is not clear at this point. However, several factors may account for this apparent contradiction, including animal species, animal models and treatment time points. We used C57 mice and the AB model to induce cardiac hypertrophy, whereas Xu and Rao (1995) used deoxycorticosterone-acetate-salt hypertensive rats and Chen et al. (1993) used spontaneously hypertensive rats subjected to acute coronary artery occlusion in their studies.
Fibrosis is another classical feature of pathological hypertrophy; it is characterized by the expansion of the extracellular matrix by the accumulation of collagen. Tetrandrine has been shown to possess anti-fibrotic effects in hepatic cirrhosis, silicotic fibrosis and renal interstitial fibrosis (Hsu et al., 2006; Yin et al., 2007). Therefore, we investigated whether tetrandrine exerts an anti-fibrotic property in the hypertrophic hearts. Our study demonstrated that tetrandrine attenuates cardiac fibrosis and inhibits the expression of several fibrotic mediators induced by pressure overload. This study is the first to report inhibition of cardiac fibrosis by tetrandrine.
The mechanism by which tetrandrine mediates its anti-hypertrophic and anti-fibrotic effects remains largely unclear. Mounting evidence has suggested that several DNA-binding transcription factors are implicated in cardiac hypertrophy, including NF-κB and nuclear factors of activated T cells (NFATs) (Molkentin, 2004; Schulz and Yutzey, 2004; Hall et al., 2006; Li et al., 2007b). Therefore, the inhibitory mechanisms of tetrandrine on cardiac hypertrophy were examined by investigating its effect on NF-κB and NFAT activation. Our results show that tetrandrine significantly abrogated NF-κB activation by disrupting DNA-binding and transcriptional activity. It did this by blocking the phosphorylation and degradation of IκBα, IKKβ activation as well as inhibiting the expression of inflammatory mediators (IL-6, TNF-α and MCP-1). In accordance with our findings, three recent reports also found that tetrandrine suppressed NF-κB signalling (Ho et al., 2004; Feng et al., 2008; Liou et al., 2008). Ho et al. (2004) demonstrated that tetrandrine blocked IκBα degradation by inhibiting IKKα and IKKβ activities and then inhibited NF-κB activation. Inflammation plays a key role in the development of cardiac hypertrophy and heart failure. By blocking NF-κB signalling, tetrandrine inhibited the early steps of inflammation and modulated the amplification of multiple cytokine signalling cascades, which partly contributes to the cardioprotective effects of tetrandrine. In addition, a similar profile was observed with respect to NFAT signalling. Our data clearly revealed that tetrandrine blocked NFATc4 activation induced by Ang II or pressure overload. Because NFATs are probably the best-known of the DNA-binding transcription factors in relation to cardiac hypertrophy, our results implicate tetrandrine as an inhibitor of NFATc4 and suggest that such an effect may be related to the cardioprotective effect of tetrandrine against cardiac hypertrophy. The exact mechanisms for the inhibitory effect of tetrandrine on NFAT activation remain unclear. Because tetrandrine is a calcium antagonist in cardiac myocytes and vascular smooth cells, we speculate that tetrandrine blocks NFAT signalling by affecting the calcium concentration.
Increasing evidence shows that MAPKs signalling participates in the activation of transcription factors, including NF-κB and NFAT (Molkentin, 2004; Li et al., 2006). Therefore, we examined the effects of tetrandrine on MAPKs signalling and further explored the role of MAPKs in hypertrophic stimuli-induced NF-κB and NFAT activation. In line with results from previous studies (Li et al., 2005; 2006;), we observed a significant increase in the phosphorylated levels of ERK1/2, JNK1/2 and p38 both in Ang II-treated cardiac myocytes and in the hypertrophic hearts of AB mice. When tetrandrine was administered, the phosphorylation of ERK1/2 was markedly blocked. However, the activation of JNK1/2 and p38 was not significantly affected by tetrandrine treatment. Consistent with our findings, Fang et al. (2004) reported that tetrandrine inhibits the proliferation of aortic smooth muscle cells by inhibiting the activation of ERK1/2. More importantly, we found that U0126, an ERK1/2 inhibitor, significantly attenuated Ang II-induced NF-κB and NFAT activation, indicating that NF-κB and NFAT activation are dependent on ERK1/2 signalling. Recent studies showed that ROS play an important role in the cardiac hypertrophy induced by Ang II or pressure overload, and increased ROS generation leads to the activation of the downstream signalling pathways and transcription factors including NF-κB and NFAT (Luo et al., 2001; Yoshioka et al., 2007; Ago et al., 2008; Akki et al., 2009). Because tetrandrine is an antioxidant, we examined the effects of tetrandrine on the production of ROS in response to hypertrophic stress. In agreement with previous reports, in the present study, we demonstrated that both Ang II and pressure overload increased the production of ROS and this effect was abolished by tetrandrine treatment. Our results further showed that tetrandrine attenuated the oxidation of the myofibrillar proteins tropomyosin and α-sarcomeric actin. Collectively, these data suggest, for the first time, that tetrandrine suppresses cardiac hypertrophy by blocking the excessive generation of ROS and oxidation of myofibrillar proteins, and then disrupting the activation of ERK1/2-dependent NF-κB and NFAT.
In summary, our present work demonstrates that tetrandrine inhibits cardiac hyertrophy in mice. We have shown that tetrandrine can prevent the development of cardiac hypertrophy by blocking ROS generation and ERK1/2-dependent NF-κB and calcineurin/NFAT signallings. This study is highly relevant to the understanding of the inhibitory effect of tetrandrine on cardiac hypertrophy and related molecular mechanisms. It also serves to elucidate the dominant signalling pathways leading to cardiac hypertrophy, inflammation and fibrosis in response to hypertrophic stimuli.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (30900524, 30972954 and 30770733) and by the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry (2006-331).
Glossary
Abbreviations:
- Ang II
angiogensin II
- BrdU
bromodeoxyuridine
- BW
body weight
- CSA
cardiomyocyte cross-sectional area
- DCFH-DA
2′,7′-dichlorofluorescein dilacerate
- ESR
electron spin resonance
- FS
fractional shortening
- GAPDH
glyceraldehydes-3-phosphate dehydrogenase
- HR
heart rate
- HW
heart weight
- HW/BW
heart weight/body weight
- hydroxy-TEMPO
4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl
- IKK
IκB kinase
- LVEDD
left ventricular end-diastolic diameter
- LVESD
left ventricular end-systolic diameter
- LVPWD
left ventricular posterior wall, diastolic
- LVSD
left ventricular septum, diastolic
- LW
lung weight
- LW/BW
lung weight/body weight
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NAC
N-acetylcysteine
- NFAT
nuclear factor of activated T cells
- NF-κB
nuclear factor-κB
- PWT
posterior wall thickness
- ROS
reactive oxygen species
- SBP
systolic blood pressure
Conflict of interest
The authors state no conflict of interest.
References
- Ago T, Liu T, Zhai P, Chen W, Li H, Molkentin JD, et al. A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell. 2008;133:978–993. doi: 10.1016/j.cell.2008.04.041. [DOI] [PubMed] [Google Scholar]
- Akki A, Zhang M, Murdoch C, Brewer A, Shah AM. NADPH oxidase signaling and cardiac myocyte function. J Mol Cell Cardiol. 2009;47:15–22. doi: 10.1016/j.yjmcc.2009.04.004. [DOI] [PubMed] [Google Scholar]
- Barbato R, Menabò R, Dainese P, Carafoli E, Schiaffino S, Di Lisa F. Binding of cytosolic proteins to myofibrils in ischemic rat hearts. Circ Res. 1996;78:821–828. doi: 10.1161/01.res.78.5.821. [DOI] [PubMed] [Google Scholar]
- Barry SP, Davidson SM, Townsend PA. Molecular regulation of cardiac hypertrophy. Int J Biochem Cell Biol. 2008;40:2023–2039. doi: 10.1016/j.biocel.2008.02.020. [DOI] [PubMed] [Google Scholar]
- Bian Z, Cai J, Shen DF, Chen L, Yan L, Tang Q, et al. Cellular repressor of E1A-stimulated genes attenuates cardiac hypertrophy and fibrosis. J Cell Mol Med. 2008;13:1302–1313. doi: 10.1111/j.1582-4934.2008.00633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Yi FF, Bian ZY, Shen DF, Yang L, Yan L, et al. Crocetin protects against cardiac hypertrophy by blocking MEK-ERK1/2 signalling pathway. J Cell Mol Med. 2009a;13:909–925. doi: 10.1111/j.1582-4934.2008.00620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Yi FF, Yang L, Shen DF, Yang Q, Li A, et al. Targeted expression of receptor-associated late transducer inhibits maladaptive hypertrophy via blocking epidermal growth factor receptor signaling. Hypertension. 2009b;53:539–548. doi: 10.1161/HYPERTENSIONAHA.108.120816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canton M, Neverova I, Menabò R, Van Eyk J, Di Lisa F. Evidence of myofibrillar protein oxidation induced by postischemic reperfusion in isolated rat hearts. Am J Physiol Heart Circ Physiol. 2004;286:H870–H877. doi: 10.1152/ajpheart.00714.2003. [DOI] [PubMed] [Google Scholar]
- Canton M, Skyschally A, Menabò R, Boengler K, Gres P, Schulz R, et al. Oxidative modification of tropomyosin and myocardial dysfunction following coronary microembolization. Eur Heart J. 2006;27:875–881. doi: 10.1093/eurheartj/ehi751. [DOI] [PubMed] [Google Scholar]
- Chen H, Chen SC, Zhang TH, Tian HC, Guan Y, Su DF. Protective effects of silybin and tetrandrine on the outcome of spontaneously hypertensive rats subjected to acute coronary artery occlusion. Int J Cardiol. 1993;41:103–108. doi: 10.1016/0167-5273(93)90148-a. [DOI] [PubMed] [Google Scholar]
- Chen LY, Chen X, Tian XL, Yu XH. Effects of tetrandrine on calcium transport, protein fluorescences and membrane fluidity of sarcoplasmic reticulum. Br J Pharmacol. 2000;131:530–536. doi: 10.1038/sj.bjp.0703582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Chen JC, Tseng SH. Tetrandrine suppresses tumor growth and angiogenesis of gliomas in rats. Int J Cancer. 2007;124:2260–2269. doi: 10.1002/ijc.24208. [DOI] [PubMed] [Google Scholar]
- Clerk A, Cullingford TE, Fuller SJ, Giraldo A, Markou T, Pikkarainen S, et al. Signaling pathways mediating cardiac myocyte gene expression in physiological and stress responses. J Cell Physiol. 2007;212:311–322. doi: 10.1002/jcp.21094. [DOI] [PubMed] [Google Scholar]
- Dalla Libera L, Ravara B, Gobbo V, Danieli Betto D, Germinario E, Angelini A, et al. Skeletal muscle myofibrillar protein oxidation in heart failure and the protective effect of Carvedilol. J Mol Cell Cardiol. 2005;38:803–807. doi: 10.1016/j.yjmcc.2005.02.023. [DOI] [PubMed] [Google Scholar]
- Dhalla NS, Saini-Chohan HK, Rodriguez-Leyva D, Elimban V, Dent MR, Tappia PS. Subcellular remodelling may induce cardiac dysfunction in congestive heart failure. Cardiovasc Res. 2009;81:429–438. doi: 10.1093/cvr/cvn281. [DOI] [PubMed] [Google Scholar]
- Dorn GW., 2nd Novel pharmacotherapies to abrogate postinfarction ventricular remodeling. Nat Rev Cardiol. 2009;6:283–291. doi: 10.1038/nrcardio.2009.12. [DOI] [PubMed] [Google Scholar]
- Duncan JG, Ravi R, Stull LB, Murphy AM. Chronic xanthine oxidase inhibition prevents myofibrillar protein oxidation and preserves cardiac function in a transgenic mouse model of cardiomyopathy. Am J Physiol Heart Circ Physiol. 2005;289:H1512–H1518. doi: 10.1152/ajpheart.00168.2005. [DOI] [PubMed] [Google Scholar]
- Fang LH, Zhang YH, Ma JJ, Du GH, Ku BS, Yao HY, et al. Inhibitory effects of tetrandrine on the serum- and platelet-derived growth factor-BB-induced proliferation of rat aortic smooth muscle cells through inhibition of cell cycle progression, DNA synthesis, ERK1/2 activation and c-fos expression. Atherosclerosis. 2004;174:215–223. doi: 10.1016/j.atherosclerosis.2004.01.036. [DOI] [PubMed] [Google Scholar]
- Feng D, Mei Y, Wang Y, Zhang B, Wang C, Xu L. Tetrandrine protects mice from concanavalin A-induced hepatitis through inhibiting NF-kappaB activation. Immunol Lett. 2008;121:127–133. doi: 10.1016/j.imlet.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Hall G, Hasday JD, Rogers TB. Regulating the regulator: NF-kappaB signaling in heart. J Mol Cell Cardiol. 2006;41:580–591. doi: 10.1016/j.yjmcc.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Ho LJ, Juan TY, Chao P, Wu WL, Chang DM, Chang SY, et al. Plant alkaloid tetrandrine downregulates IkappaBalpha kinases-IkappaBalpha-NF-kappaB signaling pathway in human peripheral blood T cell. Br J Pharmacol. 2004;143:919–927. doi: 10.1038/sj.bjp.0706000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YC, Chiu YT, Lee CY, Wu CF, Huang YT. Anti-fibrotic effects of tetrandrine on bile-duct ligated rats. Can J Physiol Pharmacol. 2006;84:967–976. doi: 10.1139/y06-050. [DOI] [PubMed] [Google Scholar]
- Jenkins CM, Cedars A, Gross RW. Eicosanoid signalling pathways in the heart. Cardiovasc Res. 2009;82:240–249. doi: 10.1093/cvr/cvn346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan CY, Achike FI. Tetrandrine and related bis-benzylisoquinoline alkaloids from medicinal herbs: cardiovascular effects and mechanisms of action. Acta Pharmacol Sin. 2002;23:1057–1068. [PubMed] [Google Scholar]
- Li HL, Wang AB, Huang Y, Liu DP, Wei C, Williams GM, et al. Isorhapontigenin, a new resveratrol analog, attenuates cardiac hypertrophy via blocking signaling transduction pathways. Free Radic Biol Med. 2005;38:243–257. doi: 10.1016/j.freeradbiomed.2004.10.020. [DOI] [PubMed] [Google Scholar]
- Li HL, Huang Y, Zhang CN, Liu G, Wei YS, Wang AB, et al. Epigallocathechin-3 gallate inhibits cardiac hypertrophy through blocking reactive oxidative species-dependent and -independent signal pathways. Free Radic Biol Med. 2006;40:1756–1775. doi: 10.1016/j.freeradbiomed.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Li HL, She ZG, Li TB, Wang AB, Yang Q, Wei YS, et al. Overexpression of myofibrillogenesis regulator-1 aggravates cardiac hypertrophy induced by angiotensin II in mice. Hypertension. 2007a;49:1399–1408. doi: 10.1161/HYPERTENSIONAHA.106.085399. [DOI] [PubMed] [Google Scholar]
- Li HL, Zhuo ML, Wang D, Wang AB, Cai H, Sun LH, et al. Targeted cardiac overexpression of A20 improves left ventricular performance and reduces compensatory hypertrophy after myocardial infarction. Circulation. 2007b;115:1885–1894. doi: 10.1161/CIRCULATIONAHA.106.656835. [DOI] [PubMed] [Google Scholar]
- Liou JT, Chen ZY, Ho LJ, Yang SP, Chang DM, Liang CC, et al. Differential effects of triptolide and tetrandrine on activation of COX-2, NF-kappaB, and AP-1 and virus production in dengue virus-infected human lung cells. Eur J Pharmacol. 2008;589:288–298. doi: 10.1016/j.ejphar.2008.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo JD, Xie F, Zhang WW, Ma XD, Guan JX, Chen X. Simvastatin inhibits noradrenaline-induced hypertrophy of cultured neonatal rat cardiomyocytes. Br J Pharmacol. 2001;132:159–164. doi: 10.1038/sj.bjp.0703792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCubrey JA, Bäsecke J, Cervello M, Martelli AM, Franklin RA. GSK-3beta is a critical mediator of tetrandrine induced cell cycle arrest and cytotoxicity. Cancer Biol Ther. 2008;7:1079. doi: 10.4161/cbt.7.7.6519. [DOI] [PubMed] [Google Scholar]
- Machackova J, Barta J, Dhalla NS. Myofibrillar remodeling in cardiac hypertrophy, heart failure and cardiomyopathies. Can J Cardiol. 2006;22:953–968. doi: 10.1016/s0828-282x(06)70315-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkentin JD. Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc Res. 2004;63:467–475. doi: 10.1016/j.cardiores.2004.01.021. [DOI] [PubMed] [Google Scholar]
- Olson EN. A decade of discoveries in cardiac biology. Nat Med. 2004;10:467–474. doi: 10.1038/nm0504-467. [DOI] [PubMed] [Google Scholar]
- Rao MR. Effects of tetrandrine on cardiac and vascular remodeling. Acta Pharmacol Sin. 2002;23:1075–1085. [PubMed] [Google Scholar]
- Schulz RA, Yutzey KE. Calcineurin signaling and NFAT activation in cardiovascular and skeletal muscle development. Dev Biol. 2004;266:1–16. doi: 10.1016/j.ydbio.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Tang Q, Cai J, Shen D, Bian Z, Yan L, Wang YX, et al. Lysosomal cysteine peptidase cathepsin L protects against cardiac hypertrophy through blocking AKT/GSK3beta signaling. J Mol Med. 2009;87:249–260. doi: 10.1007/s00109-008-0423-2. [DOI] [PubMed] [Google Scholar]
- Vega RB, Bassel-Duby R, Olson EN. Control of cardiac growth and function by calcineurin signaling. J Biol Chem. 2003;278:36981–36984. doi: 10.1074/jbc.R300023200. [DOI] [PubMed] [Google Scholar]
- Wang G, Lemos JR, Iadecola C. Herbal alkaloid tetrandrine: fron an ion channel blocker to inhibitor of tumor proliferation. Trends Pharmacol Sci. 2004;25:120–123. doi: 10.1016/j.tips.2004.01.009. [DOI] [PubMed] [Google Scholar]
- Xu Y, Rao MR. Effects of tetrandrine on left ventricle hypertrophy in deoxycorticosterone acetate-salt hypertensive rats. Eur J Pharmacol. 1995;278:1–7. doi: 10.1016/0014-2999(95)00055-p. [DOI] [PubMed] [Google Scholar]
- Yao WX, Jiang MX. Effects of tetrandrine on cardiovascular electrophysiologic properties. Acta Pharmacol Sin. 2002;23:1069–1074. [PubMed] [Google Scholar]
- Yin MF, Lian LH, Piao DM, Nan JX. Tetrandrine stimulates the apoptosis of hepatic stellate cells and ameliorates development of fibrosis in a thioacetamide rat model. World J Gastroenterol. 2007;13:1214–1220. doi: 10.3748/wjg.v13.i8.1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka J, Imahashi K, Gabel SA, Chutkow WA, Burds AA, Gannon J, et al. Targeted deletion of thioredoxin-interacting protein regulates cardiac dysfunction in response to pressure overload. Circ Res. 2007;101:1328–1338. doi: 10.1161/CIRCRESAHA.106.160515. [DOI] [PubMed] [Google Scholar]