

Abstract
Bacterial ribonuclease P (RNase P) catalyzes cleavage of 5’ leader sequences from precursor tRNAs (pre-tRNAs). Previously, all known substrate nucleotide specificity in this system derived from RNA-RNA interactions with the RNase P RNA subunit (PRNA). Here, we demonstrate that pre-tRNA binding affinities for Bacillus subtilis and Escherichia coli RNase P are enhanced by sequence-specific contacts between the fourth nucleotide on the 5’ side of the cleavage site (N(−4)) and the RNase P protein subunit (P protein). B. subtilis RNase P has higher affinity for pre-tRNA with adenosine at N(−4) and this binding preference is amplified at physiological divalent ion concentrations. Measurements of pre-tRNA containing adenosine analogs at N(−4) indicate that specificity arises from a combination of hydrogen bonding to the N6 exocyclic amine of adenosine and steric exclusion of the N2 amine of guanosine. Mutagenesis of B. subtilis P protein indicates that F20 and Y34 contribute to selectivity at N(−4). The hydroxyl group of Y34 enhances selectivity, likely by forming a hydrogen bond with the N(−4) nucleotide. The sequence preference of E. coli RNase P is diminished, showing a weak preference for adenosine and cytosine at N(−4), consistent with the substitution of Leu for Y34 in the E. coli P protein. This is the first identification of a sequence-specific contact between the P protein and pre-tRNA that contributes to molecular recognition of RNase P. Additionally, sequence analyses reveal that a greater than expected fraction of pre-tRNAs from both E. coli and B. subtilis contain a nucleotide at N(−4) that enhances RNase P affinity. This suggests that specificity at N(−4) contributes to substrate recognition in vivo Furthermore, bioinformatic analyses suggest that sequence-specific contacts between the protein subunit and the leader sequences of pre-tRNAs may be common in bacterial RNase P and lead to species-specific substrate recognition.
Keywords: RNase P, sequence specificity, tRNA processing, substrate recognition
Introduction
Throughout phylogeny, the 5’ leaders of precursor tRNA (pre-tRNA) molecules are endonucleolytically removed by ribonuclease P (RNase P) (for review, see 1; 2; 3). In Bacteria, RNase P is comprised of a single ~400 nucleotide RNA subunit (PRNA) that is catalytic in vitro, and a single small (~120 amino acid) protein subunit (P protein) required for activity in vivo. The RNA and protein components function synergistically in both substrate recognition and catalysis, and exhibit broad substrate specificity 4; 5; 6; 7; 8; 9. In species such as Bacillus subtilis and Escherichia coli, RNase P recognizes and correctly processes the products of more than 80 pre-tRNA genes 10; 11. Intriguingly, the substrate pools in both of these species include pre-tRNAs that lack multiple known RNase P recognition elements, suggesting the possibility that additional recognition elements remain to be identified 5; 10; 11; 12.
Previous studies have established that both the PRNA and P protein subunits of RNase P contribute to molecular recognition of pre-tRNA substrates. PRNA contacts substrates through both base pairing and backbone interactions, including interactions with 2’ hydroxyls in the acceptor stem and D stem-loop 13. Proximal to the cleavage site, PRNA base pairs with the substrate nucleotide on the 5’ side of the cleavage site (N(−1)) 12; 14 and the 3’ RCCA substrate motif 15; 16, and recognizes functional groups in the closing base pair of the tRNA acceptor stem 2; 12; 17; 18; 19; 20. These interactions contribute to efficient catalysis, cleavage site specificity and high substrate binding affinity. Multiple lines of evidence indicate that the P protein subunit contacts the 5’ leader of pre-tRNA substrates 5; 17; 19; 21; 22; 23. Furthermore, the interaction between the B. subtilis P protein and the pre-tRNA leader increases binding affinity for substrates with increasing leader length 21; 24. Biochemical investigations indicate that the leader contacts side chains located in a large cleft formed by the central β-sheet and an α-helix of P protein (Figure 1A) 19; 22; 23; 24; 25. Time-resolved fluorescence resonance energy transfer (trFRET) experiments, affinity-cleavage assays, and molecular modeling indicate that this cleft is proximal to the second through seventh positions on the 5' side of the cleavage site (N(−2) through N(−7)) 19; 24. Importantly, unlike PRNA, no specific interactions between P protein and pre-tRNA have been identified or characterized to date.
Figure 1. Interaction of the 5’ leader of pre-tRNA with B. subtilis P protein in RNase P.

(A) Modeled structure of the interface between the pre-tRNA leader and P protein based on affinity cleavage 19, showing the backbone of PRNA (blue), P protein (red), the mature tRNA domain of the substrate (brown), and the pre-tRNA 5’ leader (black). The nucleotide at pre-tRNA position N(−4) is shown in green; P protein regions are labeled. (B) B. subtilis P protein crystal structure showing sites where single cysteine mutations alter the value of KD, obs for pre-tRNAAsp. The side chain color reflects the magnitude of the effect of the mutation on the value of KD, obs: yellow, ≤ 3-fold increase; blue, 3- to 25-fold increase; and green, > 25-fold increase. (C) The effect of alanine mutations on the binding selectivity for A(−4) relative G(−4) mapped onto the structure of the RNase P protein. Alanine mutations that do not alter the binding specificity ratio are highlighted in yellow and those that abolish the sequence preference at N(−4) are colored in green.
RNase P protein also modulates the cation requirements for RNase P function 26; 27. P protein enhances the apparent affinity of the RNase P holoenzyme for Mg2+ ions that contribute to substrate binding and catalysis 28 and alters the Mn2+ rescue of phosphorothioate substitutions in PRNA helix P4, a catalytically important divalent metal binding site 3; 29; 30; 31; 32. Biochemical studies suggest that the effects on metal affinity are related to the role of P protein as a structural linchpin in the RNase P holoenzyme - substrate complex, interacting with the pre-tRNA leader, helping to organize the active site, and enhancing reactivity under physiological conditions 19; 33; 34.
An additional function of P protein is to enhance the ability of RNase P to recognize a wide variety of naturally occurring, non-canonical pre-tRNA substrates 5. For E. coli RNase P, interactions between P protein and the 5’ leader of pre-tRNA increase both substrate affinity and single turnover cleavage rate constants in a sequence-dependent manner 5; 29; 33. The binding and catalytic enhancements specific to a 5’ leader sequence are transferable between substrates, independent of the tRNA sequence 5. Overall, these observations suggest that interactions between P protein and substrate leaders contribute to substrate specificity; however the molecular basis for this discrimination remains to be determined.
Here, we investigate the sequence specificity of interactions between bacterial RNase P and the 5’ leaders of pre-tRNA substrates using a combination of biochemical and genomic analyses. These data demonstrate that both the B. subtilis and E. coli RNase P holoenzymes display nucleotide preferences at position N(−4) of pre-tRNAAsp that contribute to substrate molecular recognition. Analysis of modified adenosine analogs at N(−4) demonstrate that the sequence preferences of B. subtilis RNase P at this position result from positive interactions with the N6 exocyclic amine of adenosine and unfavorable interactions with the N2 amine of guanosine. Furthermore, binding studies with P protein mutants reveal that the hydroxyl group of amino acid residue Y34 is required for sequence-specific recognition in B. subtilis RNase P. The nucleotide preferences observed in vitro for B. subtilis and E. coli RNase P are reflected in the sequences of pre-tRNA genes in these species, consistent with nucleotide selectivity at N(−4) contributing to substrate recognition in vivo. Extrapolation from tRNA gene sequence analysis suggests that such preferences are present in the majority of bacterial species, but are likely variable between species.
Results
Pre-tRNA affinities of B. subtilis and E. coli RNase P depend on the nucleotide at N(−4)
There is growing evidence suggesting that sequence-specific interactions between the 5’ leader of pre-tRNA and bacterial P protein enhance substrate binding affinity and/or cleavage rates, although no specific contacts have yet been identified. Previously, the thermodynamics and kinetics of pre-tRNA associating with B. subtilis RNase P suggested that the fourth (N(−4)) and fifth (N(−5)) nucleotides on the 5’ side of the cleavage site in the pre-tRNA leader interact with the P protein 21; 24. Consistent with this, structural models of the B. subtilis RNase P holoenzyme based on affinity cleavage and trFRET measurements place N(−4) and N(−5) near the central cleft of the protein (Figure 1A) 19; 24. For B. subtilis RNase P, interactions between the pre-tRNAAsp leader and P protein manifest primarily in substrate binding affinity 21. Therefore, to evaluate sequence-specific interactions between B. subtilis RNase P and the pre-tRNA leader, we measured binding affinities for pre-tRNAAsp with 4- and 5- nucleotide long 5' leaders with 2 mM free Ca2+ ([Ca2+]f) using centrifuge size-exclusion chromatography. Ca2+ was used in lieu of Mg2+ because it stabilizes RNase P folding and substrate binding with little activation of catalytic activity, allowing equilibrium measurements of substrate affinity 21; 35. In addition, this Ca2+ concentration approximates physiological concentrations of labile Mg2+ in Bacteria 33; 36; 37.
The binding affinity of B. subtilis RNase P for pre-tRNA bearing 4-nucleotide leader sequences varies with the nucleobase at N(−4), suggesting a sequence-specific interaction at this position (Table 1). In contrast, mutations at N(−5) in substrates with 5-nucleotide leaders alter the binding affinity by less than 2-fold (data not shown). B. subtilis RNase P binds A(−4) pre-tRNA 20-fold more tightly than the G(−4) substrate, and binds the C(−4) and U(−4) substrates with intermediate affinity (Table 1; Figure 2A). Similar measurements demonstrate that E. coli RNase P also exhibits a sequence preference at N(−4) of pre-tRNAAsp (Table 1); however, in this species the observed preference engenders higher binding affinity for A(−4) and C(−4) pre-tRNA relative to G(−4) and U(−4), and the overall preference is 5-fold. Thus recognition of the base at N(−4) contributes to substrate affinity in both species.
Table 1.
Effects of altering the nucleobase at N(−4) in B. subtilis pre-tRNAAsp on RNase P substrate affinity.
|
B. subtilis KD, obs (nM)b |
E. coli KD, obs (nM)b |
|||
|---|---|---|---|---|
| Substrate | Leader Sequencea |
2 mM [Ca2+]f |
3.5 mM [Ca2+]f |
2 mM [Ca2+]f |
| A(−4) | 5' - ACAU | 9 ± 2 | 5 ± 2 | 35 ± 8 |
| G(−4) | 5' - GCAU | 200 ± 20 | 11 ± 3 | 170 ± 60 |
| U(−4) | 5' - UCAU | 70 ± 10 | 8 ± 1 | 100 ± 30 |
| C(−4) | 5' - CCAU | 150 ± 50 | 6 ± 1 | 54 ± 9 |
| P(−4) | 5' - PCAU | 30 ± 7 | ndc | ndc |
| DAP(−4) | 5' - DAPCAU | 40 ± 7 | ndc | ndc |
| 2AP(−4) | 5' – 2APCAU | 170 ± 40 | ndc | ndc |
Position N(−4) is highlighted.
Conditions: 50 mM MES, 50 mM Tris, pH 6.0, 37°C, indicated [Ca2+]f. [KCl] was adjusted to maintain ionic strength as described in Materials and Methods.
Not determined
Figure 2. Dependence of pre-tRNA binding affinity on both the nucleotide at N(−4) and the calcium concentration.

Representative binding isotherms for RNase P binding to pre-tRNAAsp with varying sequence at N(−4) at 2 mM and 3.5 mM [Ca2+]f. Fraction substrate bound was measured using centrifuge gel-filtration chromatography in 50 mM MES, 50 mM Tris, pH 6.0, 37 °C, with KCl concentrations were adjusted to maintain ionic strength at 410 mM. Data in A and B for A(−4) (filled circles; solid line), U(−4) (open circles; solid line), C(−4) (open squares; dashed line) and G(−4) (filled squares, dashed line) are fit with a single binding isotherm. C KD,obs values for A(−4) (open circles) and G(−4) (filled circles) substrates as a function of [Ca2+]f. Error bars represent the standard deviation of at least three independent trials. The solid line is the fit of Equation 1 to the G(−4) data; a dashed line shows the best linear fit to the A(−4) data. Inset: Hill plot of data for G(−4) between 2 and 5 mM [Ca2+]f.
The affinity of RNase P for pre-tRNA is stabilized by multiple divalent cations and the P protein subunit has been shown to enhance the apparent affinities of these cations 28; 29. Therefore, we examined whether the sequence-specific interaction between P protein and N(−4) is influenced by the divalent metal ion concentration. The affinity of B. subtilis RNase P for A(−4) pre-tRNA increases (4-fold) as the Ca2+concentration increases from 2 to 5 mM (Figure 2C). In contrast, the affinity for the G(−4) substrate increases 65-fold over the same range (Figure 2C); the dependence of KD,obs on [Ca2+]f is well-described by a cooperative binding isotherm (Equation 1), yielding a Hill coefficient (nH) of 5.5 ± 0.5 and a midpoint for the transition (K1/2,Ca) of 4.5 ± 0.5 mM. The modest increase in the Hill coefficient for metal dependence of substrate affinity compared to previous measurements (nH = 4 ± 1 for pre-tRNAAsp with 2- and 3- nucleotide leaders 28) most likely results from differences in the substrate structure and solution conditions (e.g. Mg2+ vs. Ca2+). These results indicate that the affinity of the G(−4) substrate is coupled to the binding of multiple Ca2+ ions with dissociation constants in the mM range. If the same set of metal ions also stabilize the binding of the A(−4) substrate, the affinity of the metal ions must be significantly higher. Functionally, this dependence leads to a significant reduction in sequence selectivity at the higher divalent cation concentrations (10 – 25 mM) frequently used to measure RNase P activity (see 1; 3; 38 and references therein), although the overall pattern of nucleotide preference is retained (Figure 2B). In Bacteria the physiological concentration of available Mg2+ has been estimated at 2 mM or lower in vivo 37. Thus, the coupling of sequence specificity with divalent metal binding results in sequence selectivity at physiological cation concentrations.
RNase P recognizes two functional groups in the N(−4) nucleobase
To define the interactions between B. subtilis RNase P and the N(−4) nucleobase that contribute to sequence selectivity, we prepared pre-tRNA with adenosine analogs incorporated at N(−4). These analogs systematically alter the presentation of exocyclic amines along the adenosine Watson-Crick face (for structures see Figure S1, Supplementary data), including: purine (P(−4)), 2-aminopurine (2AP(−4)) and 2,6-diaminopurine (DAP(−4)) 39. The binding affinity of B. subtilis RNase P for pre-tRNA containing adenosine analogs at N(−4) indicates that recognition of adenosine involves two distinct functional groups (Table 1). Removal of the N6 exocyclic amine by incorporation of purine (P(−4)) decreases the binding affinity by 3-fold compared to A(−4) pre-tRNA, indicating a positive interaction, likely caused by the formation of a hydrogen bond with the N6 amine. Additionally, the DAP substitution at N(−4) decreases the binding affinity by 4-fold, demonstrating an unfavorable interaction with the N2 amine of this modified base, such as a steric clash, and, by extension, with the N2 exocyclic amine of a guanosine at this position. The 2AP analog combines these two structural changes, removal of the N6 exocyclic amine and addition of the N2 exocyclic amine, leading to a decrease in the affinity of RNase P for 2AP(4) pre-tRNA by 19-fold. The decrease in binding affinity for the 2AP substitution is consistent with an additive effect of each of the two individual interactions. Furthermore, the affinity of the 2AP(−4) substrate is comparable to that of G(−4) (Table 1), suggesting that the differential binding affinities for the A(−4) and G(−4) substrates can be accounted for by interactions with these two functional groups.
B. subtilis P protein side chains contribute to pre-tRNA affinity and sequence specificity
Structural models of the RNase P•pre-tRNA complex place nucleotides N(−2) through N(−7) of the pre-tRNA leader near the P protein central cleft and RNR motif 19; 22; 24 (Figure 1A). To identify P protein side chains that potentially contact pre-tRNA, we measured the pre-tRNA binding affinity of RNase P reconstituted with a library of mutant P proteins. In a preliminary screen, we measured dissociation constants for a single pre-tRNA construct (G(−4)) under one set of assay conditions (3.5 mM [Ca2+]f) using a previously prepared library of single-cysteine P protein mutants. This library includes mutations in the central cleft, RNR motif, and metal binding loop (Figure 1B) of P protein that do not disrupt RNase P catalytic activity 19; 21. Although it is possible that some of these mutations, such as F16C and F20C, could alter the structure of the P protein in the holoenzyme complex, the unperturbed single turnover cleavage rate constants19; 21 suggest that the structure of the RNase P•pre-tRNA complex is not significantly altered. Measurements using RNase P reconstituted with mutant P proteins (Table S1, Supplementary data) identified P protein residues that disrupt pre-tRNA affinity. Mapping the magnitude of the observed effects onto a structure of the P protein (Figure 1B) reveals that mutations in the P protein central cleft (K12, A27, V32, R45, S49, S51, and I86) and RNR motif (R62) decrease pre-tRNA binding affinity by 3- to 15-fold (Figure 1B). Additionally, three mutations increase the KD,obs for G(−4) pre-tRNA by at least 25-fold: F16C, F20C, and R68C. Both F16 and F20 are located on the α-helix that forms a portion of the central cleft, and are positioned suitably to interact with the 5’ leader, as inferred by structural models (Figure 1A) 19; 24. The third highly perturbing mutation, R68C, is in the RNR motif and is proximal to PRNA and N(−2) in structural models 19; 24. Overall, these data indicate that residues in the central cleft and the RNR motif are important for pre-tRNA affinity, consistent with cross-linking, affinity cleavage, and trFRET studies that place the 5’ leader near these regions of the protein 22; 24.
To test the importance of interactions between side chains in the central cleft and RNR motif with pre-tRNA, we prepared a series of alanine mutations in P protein and measured the binding affinities of the mutant RNase P enzymes for A(−4) and G(−4) pre-tRNA (Table 2). Alanine mutations were incorporated into P protein sites identified by the cysteine scanning mutagenesis (F16, F20, R68) as well as additional amino acids in the central cleft (S25, Y34) and the RNR motif (R60, N61, K64, R65). Pre-tRNA binding was evaluated in 3.5 mM [Ca2+]f to permit measurement of weak pre-tRNA affinities. However, under these conditions the A(−4) / G(−4) pre-tRNA selectivity ratio is only 2-fold for wild-type RNase P (Figure 2B), allowing only a qualitative analysis of mutations that affect selectivity.
Table 2.
Affinities of B. subtilis RNase P reconstituted with wild type and mutant P proteins for A(−4) and G(−4) pre-tRNAAsp.
| Protein |
KD, obs (nM) 3.5 mM [Ca2+]fa |
||
|---|---|---|---|
| G(−4) | A(−4) | G(−4) / A(−4) | |
| Wild Type | 11 ± 3 | 5 ± 1 | 2.2 |
| F16A | 380 ± 70 | 260 ± 90 | 1.5 |
| F16C | 285 ± 63 | 112 ± 54 | 2.5 |
| F20A | 39 ± 11 | 38 ± 13 | 1.0 |
| S25A | 43 ± 16 | 14 ± 5 | 3.1 |
| Y34A | 32 ± 11 | 57 ± 33 | 0.6 |
| R60A | 44 ± 13 | 68 ± 19 | 0.6 |
| N61A | 51 ± 21 | 35 ± 17 | 1.5 |
| K64A | 60 ± 21 | 33 ± 12 | 1.8 |
| R65A | 155 ± 40 | 73 ± 22 | 2.1 |
Conditions: 50 mM MES, 50 mM Tris, pH 6.0, 37°C, 3.5 [Ca2+]f. 400 mM [KCl] was used to maintain ionic strength as described in Materials and Methods.
All of the alanine point mutations in the P protein decrease the affinity of RNase P for both G(−4) and A(−4) pre-tRNA (Table 2); the effect of these mutations on pre-tRNA affinity is equal to (F16) or less than (F20, S25, R68) the decreases observed for the cysteine substitutions at the same position (Figure 1B). The affinity of RNase P for A(−4) pre-tRNA is decreased >10-fold by four mutations (F16A, Y34A, R60A and R65A) and 5- to 8-fold by four other mutations (F20A, N61A, K64A and R68A). These data are consistent with the cysteine scanning study, and confirm that the central cleft and the RNR motif of the P protein contribute to the pre-tRNA affinity of RNase P. Additionally, several P protein mutations alter the pre-tRNA substrate selectivity observed at N(−4). As expected for mutations that disrupt favorable contacts with adenosine at N(−4), three mutations (F20A, Y34A and R60A) cause a 2- to 3-fold larger decrease in the affinity of A(−4) compared to G(−4) pre-tRNA, causing the A(−4) / G(−4) selectivity ratio to decrease to ≤ 1. This loss in selectivity was confirmed by substrate affinity measurements at 2.0 mM [Ca2+]f. Under these conditions, the A(−4) / G(−4) selectivity ratio decreases from a value of 22 for wild-type RNase P to 2 for RNase P reconstituted with F20A and Y34A P protein (Table 3). The selectivity ratio at 3.5 mM calcium also decreases modestly (to 1.5) for two other mutations (F16A and N61A). However, changes in selectivity for these mutations are not confirmed by measurement of either the selectivity ratio of the F16C mutant (Table 2) or the affinity of the N61A mutant for DAP(−4)-pre-tRNA in 3.5 mM Ca2+ (134 ± 43 µM). Mapping the magnitude of the selectivity ratios onto the P protein structure (Figure 1C) reveals that the largest decreases in the selectivity ratio are caused by mutations in hydrophobic side chains located in the central cleft (F20 and Y34), presumably near the pre-tRNA leader 19; 34, and a conserved side chain in the RNR motif (R60), a region of the protein distal to the 5’ leader in structural models (Figure 1A). These side chains may play a role in the nucleobase selectivity of RNase P for N(−4) pre-tRNA.
Table 3.
Affinities of B. subtilis RNase P reconstituted with wild type and mutant P proteins for pre-tRNAAsp with nucleotide substitutions at N(−4).
| Protein |
KD, obs (nM) 2.0 mM [Ca2+]fa |
|||
|---|---|---|---|---|
| G(−4) | P (−4) | DAP(−4) | A(−4) | |
| WT | 200 ± 20 | 30 ± 7 | 40 ± 7 | 9 ± 2 |
| F20A | 400 ± 90 | 200 ± 30 | 210 ± 30 | 165 ± 25 |
| Y34A | 210 ± 70 | 100 ± 25 | 100 ± 25 | 140 ± 50 |
| Y34F | 110 ± 30 | 50 ± 5 | 50 ± 10 | 60 ± 15 |
Conditions: 50 mM MES, 50 mM Tris, pH 6.0, 37°C, 2 mM [Ca2+]f, 425 mM [KCl] was used to adjust maintain ionic strength as described in Materials and Methods.
To identify the mechanism of sequence selectivity we further characterized the properties of the mutations with the largest effects on the N(−4) selectivity (F20A, Y34A and R60A). Single turnover assays catalyzed by saturating RNase P (0.5 mM MgCl2, 2 mM Co[NH3]6•Cl3, pH 7) _indicate that mutations in the central cleft (F20A, Y34A and Y34F) of P protein alter the observed cleavage rate constant by ≤ 2-fold. Therefore these mutations affect only the pre-tRNA binding affinity. However, the R60A mutation in the RNR motif decreases the observed single turnover rate constant by 3-fold under these same conditions, suggesting that side chains in the RNR motif may contribute to pre-tRNA sequence specificity in a different manner than side chains in the central cleft.
To examine whether residues in the RNR motif and the central cleft enhance pre-tRNA affinity by interacting with the 5’ leader and/or tRNA, the binding affinity of B. subtilis RNase P mutants for mature tRNAAsp was measured. Wild-type RNase P has significantly weaker affinity for mature tRNAAsp, compared to pre-tRNAAsp, even at elevated calcium concentrations (KD,obs = 370 ± 80 nM at 3.5 mM Ca2+) 21. This binding affinity is largely unperturbed by four mutations in the central cleft of P protein (V32C, S51C, Y34A, and I86C; KD,obs = 250 – 400 nM) suggesting that the decrease in pre-tRNA affinity is due to disruption of an interaction with the pre-tRNA leader. Conversely, the R60A mutation in the RNR motif decreases the affinity of RNase P for mature tRNA by > 8-fold (R60A KD,obs > 1200 nM; WT KD,obs = 140 ± 100 nM at 5 mM [Ca2+]f). The fact that this decrease approaches the loss of A(−4) pre-tRNA affinity caused by the R60A mutation (14-fold; Table 2) suggests that the R60 side chain interacts mainly with the tRNA portion of the substrate, and not the 5’ leader. Overall, these data are consistent with amino acid residues in the central cleft of P protein contributing to affinity and N(−4) specificity through direct contacts to the 5' leader, while amino acids in the RNR motif likely influence selectivity through positioning of bound tRNA. These differential mechanisms are consistent with the locations of the two regions of the P protein in the structural model of the RNase P•pre-tRNA complex 24; 40.
Y34 and F20 contribute to recognition of adenosine functional groups at N(−4)
To further probe the possibility of direct interactions between the side chains of P protein residues F20 and Y34 and the N(−4) nucleobase, binding affinities were measured for RNase P reconstituted with F20A or Y34A P protein mutants for pre-tRNA containing adenosine analogs at N(−4) (Table 3). As described above, the affinity of wild type RNase P for A(−4)-pre-tRNA is 3-, 4- and 22-fold higher than P(−4)-, DAP(−4)- and G(−4)-pre-tRNA, respectively. Relative to the wild type enzyme, the affinity of F20A and Y34A RNase P enzymes for P(−4) and DAP(−4) pre-tRNA decreases by 3- to 6-fold. The resulting affinities are comparable to that of A(−4) pre-tRNA (Table 3), indicating a loss of both favorable interactions with the N6 position and unfavorable interactions with an N2 exocyclic amine of an adenosine at N(−4). The F20 and Y34 side chains may contribute to these interactions either through a direct contact with the N(−4) nucleobase or by contributing to the correct positioning of the 5' leaders in the enzyme-substrate complex. To distinguish between these possibilities, we examined the role of the hydroxyl of Y34, a possible hydrogen bonding partner for the N6 amine of an adenosine at N(−4). The Y34F mutation in P protein also decreases A(−4) pre-tRNA binding affinity, although to a smaller extent than the Y34A mutation (Table 3). Furthermore, removal of the Y34 hydroxyl group results in a loss of binding selectivity for A(−4) pre-tRNA compared to the P(−4), DAP(−4) and G(−4) pre-tRNA substrates (Table 3). Overall the A(−4) / G(−4) selectivity ratio is decreased from 22- to 2-fold for this mutant (Figure 3). This result suggests that Y34 may form a hydrogen bond with A(−4), perhaps as an acceptor for the N6 exocyclic amine, and that the energetic contribution of this contact may supercede the importance of any stacking interaction between the Y34 phenyl group and the A(−4) purine ring in conferring substrate specificity. Although RNase P is able to bind substrates with any nucleotide at position N(−4), the contact with Y34 confers an energetic preference for A over G at this position which, in turn, is transduced into the recognition of certain pre-tRNAs. This contact is one of many interactions that contribute to the specificity of RNase P for pre-tRNAs and a limited set of other substrates. However, if this sequence preference for A relative to G at N(−4) is also observed in pre-tRNA genes, then this interaction may be crucial for the specific recognition of pre-tRNA substrates by B. subtilis RNase P in vivo.
Figure 3. Binding preferences at N(−4) of pre-tRNA depend on Tyr34 in the P protein subunit.
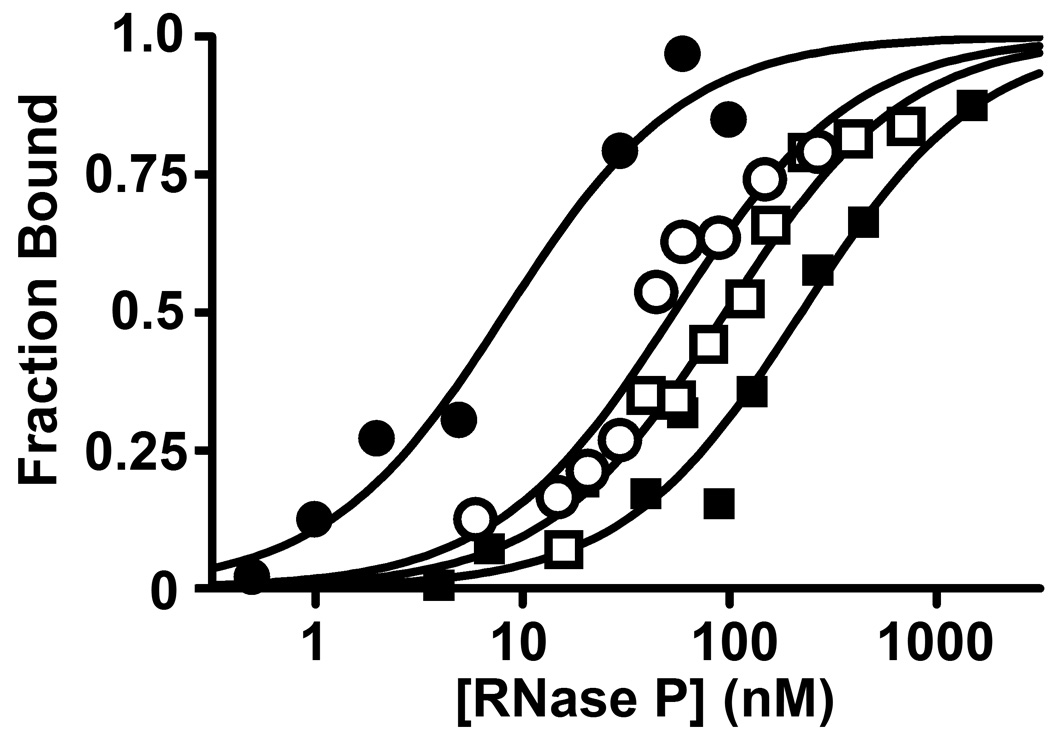
Isotherms for binding of B. subtilis RNase P to N(−4) pre-tRNAAsp were measured as described in the legend of Figure 2 at 2 mM [Ca2+]f for wild-type RNase P (closed symbols) and the Y34F mutant of RNase P (open symbols) with A(−4) pre-tRNA (circle) and G(−4) pre-tRNA (square).
Analysis of nucleotide enrichments in pre-tRNA gene sequences
In the absence of conflicting evolutionary constraints, sequence-specific interactions between RNase P and pre-tRNA leaders could influence the nucleotide composition of pre-tRNA genes at contacted positions. Similarly, in the presence of multiple factors that influence the selection of pre-tRNA leader sequences, RNase P and pre-tRNA substrate populations may evolve in parallel. In this case, RNase P and pre-tRNA substrates from a given species may form a "matched set" with the molecular recognition properties of the enzyme tuned to a specific substrate population and these nucleotide preferences may be observable in the sequences of pre-tRNA genes. Evidence that this type of mechanism links RNase P substrate recognition and pre-tRNA gene sequences comes from the interaction between a conserved nucleotide in the J5/15 region of PRNA and the N(−1) position of pre-tRNA. In vitro, this interaction favors a U at N(−1), and the pre-tRNA genes from diverse Bacteria and Archaea show significant enrichment of U at this position 14.
To explore the possibility that RNase P selectivity at N(−4) contributes to recognition of pre-tRNA substrates in vivo, we examined whether the nucleotide composition of pre-tRNA genes at N(−4) from B. subtilis and E. coli correlate with binding affinity preferences observed in vitro. Positions in pre-tRNA 5' leaders from B. subtilis and E. coli where one or more nucleotides are significantly enriched relative to the background nucleotide composition of the genome were identified using background-corrected information analysis 41; 42; 43. This method quantifies nucleotide enrichments in terms of information, expressed in units of bits. Results of this type of analysis are commonly represented as “sequence logo” plots, where the total bar height indicates the degree to which a position differs from background and the relative heights of letters represent the nucleotide composition at the position 41; 42; 43. Predicted tRNA genes were identified using tRNAscan SE 10, and leader nucleotides N(−15) through N(−1) were determined. The background nucleotide composition in regions of the genome proximal to pre-tRNA genes was estimated from the combined nucleotide frequencies at positions N(−1) through N(−15) for all tRNA genes. Uncertainties in information content were calculated based on this background distribution 42 and the statistical significance of observed nucleotide enrichments was determined using χ2 analysis.
As shown in Figure 4, pre-tRNA genes in B. subtilis and E. coli show statistically significant nucleotide enrichments at several positions, including N(−4). In B. subtilis, the base composition at position N(−4) is enriched in A and U with reduced C and low G content (N(−4): 37% A, 5% G, 13% C, 45% U; Background: 33% A, 11% G, 22% C, 34% U). These changes are significant (p = 0.02), even compared to the high A/U background nucleotide content characteristic of the B. subtilis genome. Qualitatively, increased A and U, and decreased G content are consistent with the relative binding affinities of B. subtilis RNase P for pre-tRNA substrates with varying sequence at N(−4) (A > U > C > G; Table 1). In E. coli, a statistically significant (p = 10−3) nucleotide enrichment is also observed in the pre-tRNA leader at N(−4) that results primarily from an increase in C content to 44% compared to the background level of 26%. This increase leads to a combined composition of 64% A and C at N(−4), the two nucleotides which confer the highest pre-tRNA binding affinity for E. coli RNase P (Table 1). The information content at N(−4) for E. coli pre-tRNA (R(−4) = 0.1 ± 0.04 bits) is less than that observed at N(−4) in B. subtilis (R(−4) = 0.22 ± 0.06 bits). This difference indicates a reduced level of sequence conservation at N(−4) in E. coli, relative to B. subtilis, and is consistent with the overall weaker preference observed at N(−4) for E. coli RNase P in vitro.
Figure 4. Sequence preferences in genomic pre-tRNA Leaders.

Sequence logos showing information content of 5’ leader in B. subtilis (A) and E. coli (B) pre-tRNA genes. Total bar height reflects increased information content relative to the background nucleotide content of the genome in the region of tRNA genes (See Materials and Methods). Error bars represent one standard deviation of the total bar height 42. C Percentage of 161 species examined with statistically significant (p ≤ 0.05) nucleotide enrichments at the indicated 5’ leader positions.
The pre-tRNA genes from both B. subtilis and E. coli also show additional positions in the leader with significant nucleotide enrichment, suggesting the possibility of additional sequence-specific contacts (Figure 4). As expected, both species show strong U enrichments at position N(−1) of pre-tRNA genes which manifests as an information content of R(−1) = 0.61 ± 0.06 bits (p = 10−10) for B. subtilis and R(−1) = 0.40 ± 0.03 bits (p = 10−13) for E. coli. In B. subtilis, an additional enrichment is observed at position N(−2) in the leader, primarily due to an increase in A content to 65%. This enrichment leads to information content and statistical significance similar to that observed at N(−1) (R(−2) = 0.58 ± 0.06 bits, p = 10−10), suggesting that a selective pressure, such as recognition by RNase P, influences pre-tRNA sequences at this position. In addition to N(−1) and N(−4), E. coli shows statistically significant nucleotide enrichments at N(−2) and N(−6). The increased G and A content at N(−2) of E. coli pre-tRNA genes suggests a purine preference at this position while the increased C content (47% compared to the 26% C content of the genome) suggests recognition at this position as well. Thus, while both E. coli and B. subtilis pre-tRNA genes display nucleotide enrichments at multiple positions, the observed patterns are markedly different, both with respect to the positions at which enrichments are observed and the specific nucleotide preferred. Like the preferences determined for substrate binding affinities, this result suggests the possibility of significant diversity in leader sequence recognition by RNase P from different species of Bacteria.
The observation of statistically significant nucleotide enrichments at several locations in the pre-tRNA leader sequences from E. coli and B. subtilis raises the intriguing possibility that sequence specificity in interactions with 5’ leaders may be a common element of substrate recognition for bacterial RNase P. To explore this possibility, we extended the information content analysis to 161 species of Bacteria with completely sequenced genomes available from GenBank 44. Overall, 96% of the species show one or more enriched nucleotides between N(−1) and N(−4) with p < 0.05 and 90% of the species contain at least one enrichment with p < 0.01. These results suggest that sequence-specific interactions between RNase P and pre-tRNA leaders are likely to be widespread in Bacteria as a whole. Furthermore, the frequency with which nucleotide enrichments are observed at a given leader position increases with proximity to the RNase P cleavage site (Figure 4C), increasing from 35% at N(−4) to 80% at N(−1). For positions further from the cleavage site, the number of species with a significant enrichment plateaus at approximately 15%, likely representing a statistical background in this analysis. The increasing prevalence of nucleotide enrichments for positions near the RNase P cleavage site suggests that recognition and cleavage site selection by RNase P may play a role as a driving force in the selection of pre-tRNA leader sequences.
Unexpectedly, the analysis of bacterial tRNA genes suggests that sequence preferences for positions N(−1) through N(−6) in the 5’ leaders of RNase P are diverse, varying in both the position and the identity of the enriched nucleotides. Specifically with regard to N(−4), 52 of the 161 species exhibit statistically significant nucleotide enrichments at this position (p < 0.05). The nucleotide composition at N(−4) for pre-tRNA genes from 25 representative species with statistically significant enrichments at this position are shown in Figure 5, and include species that exhibit strong U (e.g. Mycoplasma mycoides), A (e.g. Lactobacillus johnsonii), C (e.g. Salmonella typhimurium) and G (e.g. Thermus thermophilus) enrichments. This diversity suggests that the interaction between N(−4) and RNase P is variable between species and is therefore likely to involve one or more nonconserved enzyme residues, such as Y34 of B. subtilis P protein. In contrast, the composition of N(−1), which interacts with a conserved nucleotide in PRNA, retains an enrichment of U (> 25%) in 94% of the species examined. Overall the diversity of nucleotide enrichments observed for pre-tRNA genes from diverse species suggests that sequence-specific interactions between RNase P and 5' leaders likely vary markedly between species.
Figure 5. Distribution of Nucleotide Preferences at N(−4).

Nucleotide preferences at N(−4) for 20 representative bacterial species are shown. Bars show fractional composition of U (black), A (grey), G (white) and C (cross hatched). All species shown have statistically significant nucleotide enrichments (p < 0.05) preference at N(−4), and are arranged by overall A/U content at N(−4).
Discussion
Both B. subtilis and E. coli RNase P demonstrate selectivity for the nucleobase at the N(−4) position of the leader that contributes to molecular recognition of pre-tRNA substrates. For B. subtilis RNase P, this interaction results in a 1.9 kcal / mol binding preference for A(−4) over G(−4) pre-tRNAAsp at 2 mM Ca2+ in vitro, while the E. coli enzyme shows a moderate preference (0.6 – 0.9 kcal / mol) for A and C at this position (Table 1). In B. subtilis RNase P, sequence selectivity at N(−4) is consistent with previous analyses suggesting that nucleotides N(−2) to N(−7) interact with the P protein subunit 19; 21; 24. Furthermore, comparison of the KD,obs values for G(−4) and A(−4) pre-tRNA (Table 1) reveals that the previously observed enhanced affinity of pre-tRNA substrates with a 5-nucleotide leader (5’-GACAU; G(−5)) relative to a 4-nucleotide leader (5’-GCAU) 21 can be accounted for by the G to A change at N(−4), rather than an increase in leader length. In fact, comparison of affinities measured for A(−4) and G(−5) pre-tRNA 21 reveals that the addition of a G at N(−5) has a mild destabilizing, rather than strong stabilizing, effect. Recognition of the nucleobase at N(−4) pre-tRNA by B. subtilis RNase P is therefore consistent with the results of previous studies.
Interactions with P protein subunit lead to specificity at N(−4)
Structure-probing studies have previously suggested that nucleotides N(−3) through N(−7) of the pre-tRNA leader interact with the central cleft of the P protein subunit in the B. subtilis RNase P - substrate complex 5; 19; 21; 24. Consistent with this, mutagenesis studies indicate that protein residues in the central cleft interact with the nucleotide at N(−4) of pre-tRNA (Figure 1). In particular, three amino acids play an essential role in the preferential binding of pre-tRNA bearing an adenosine at N(−4): F20, Y34 and R60 (Table 2 and Table 3, Figure 1).
R60 is one of only two invariant residues in bacterial P proteins, and is located in the most highly conserved region of the protein, the RNR motif (Table 2, Figure 1) 45. The identification of R60 as important for the sequence specific recognition of the N(−4) position is unexpected; structural models of the B. subtilis RNase P•pre-tRNA complex place R60 near the tRNA moiety and distal to the leader sequence and near the P4 helix of PRNA (Figure 1) 19; 24. Consistent with this proposal, the R60A mutation decreases the affinity of RNase P for mature tRNA by more than 1.3 kcal / mol, accounting for the majority of the 1.6 kcal / mol decrease in pre-tRNA affinity. These biochemical data argue that R60 has an indirect influence on selectivity at N(−4). The side chain of R60 could either be important for aligning the 5’ leader in the RNase P - pre-tRNA complex such that the nucleobase at N(−4) is positioned to interact correctly with the P protein cleft, or be important for stabilizing local PRNA or P protein structure that enhances recognition of pre-tRNA.
The amino acids F20 and Y34 are located on the second and third strands of the central β-sheet with side chains oriented towards the central cleft that forms the binding pocket for the leader in the RNase P•pre-tRNA structural model (Figure 1) 19; 22; 24. Alanine substitutions at these residues decrease the binding affinity of A(−4) pre-tRNA by ~1.7 kcal / mol while having a small effect on the affinity of the G(−4) substrate (< 0.4 kcal / mol), thereby eliminating selectivity for the nucleobase at N(−4). These data suggest that the side chains of F20 and/or Y34 may form a direct contact with the N(−4) base (Figure 1) 19 either by base stacking (Phe and Tyr) or hydrogen bonding (Tyr). Substitution of Phe for Tyr34 has similar effects to the Y34A mutation, decreasing the binding affinity of A(−4)-pre-tRNA substantially (1.1 kcal / mol) while slightly increasing the affinity of the G(−4) substrate. These data highlight the importance of the hydrogen bonding potential of Y34 for sequence-specificity at N(−4). Additionally, while F20 is conserved in most species, variability is observed at position 34, including a Leu substitution in E. coli RNase P 45, perhaps contributing to the observed alterations in N(−4) specificity for this enzyme (Table 1).
Model of P protein – adenosine contacts
Substitution of adenosine analogs at N(−4) of pre-tRNA indicates that the A/G selectivity of B. subtilis RNase P involves interactions with two distinct functional groups (Table 1): a favorable interaction with the N6 exocyclic amine of adenosine (−0.7 kcal/mol), possibly due to the formation of a hydrogen bond with the hydoxyl group of Y34, and an unfavorable contact with the N2 exocyclic amine in guanosine (0.9 kcal/mol) (Table 1). Furthermore, RNase P containing the Y34F mutation binds A(−4) and P(−4) pre-tRNA with comparable affinity, consistent with the loss of a hydrogen bond between the N6 amine and the hydroxyl of Y34. However, this mutation also abrogates the unfavorable interaction with the N2 exocyclic amine (Table 3) leading to an almost complete lack of specificity for the nucleobase at this position. Therefore, removal of the hydroxyl group of Y34 presumably leads to repositioning of the N(−4) nucleobase and/or protein side chains in the RNase P•pre-tRNA complex, likely mediated by loss of a hydrogen bond interaction. The F20 mutation also significantly disrupts recognition of the N(−4) nucleobase since there is little difference between the binding affinity of RNase P containing this mutation for pre-tRNA with adenosine, purine or 2,6-diaminopurine at N(−4). These data indicate that the side chains of both F20 and Y34 are crucial for nucleobase recognition and suggest that both base stacking and hydrogen bonding contribute to selectivity.
One model consistent with these data is that the N6 amine of the base at N(−4) forms a hydrogen bond with the hydroxyl group of Y34 while the side chain of F20 orients the N(−4) base for maximal interaction with Y34, perhaps through stacking interactions. Alternatively, hydrogen bonding and hydrophobic interactions may stabilize the P protein fold to optimize interactions with the pre-tRNA leader. In other RNP systems, such as the RNA recognition motif (RRM) bound to the splicing protein Srp20 46, sequence-specific recognition has been linked to a direct contact between adenosine and a protein side chain, similar to the model proposed for RNase P. In this case, the exocyclic amine of adenosine forms a hydrogen bond with the side chain of serine and the base forms a stacking interaction with phenylalanine leading to a preference for recognizing adenosine relative to guanosine 46.
Metal dependence of the N(−4) interaction with RNase P
The magnitude of the selectivity of RNase P for the nucleotide at N(−4) is highly dependent on the concentration of divalent metal ions (Figure 2), indicating that metal binding and sequence selectivity are coupled. Previous studies have demonstrated that the affinity of the RNase P holoenzyme for pre-tRNA is cooperatively coupled to the binding of divalent cations with a Hill coefficient of ~4 28. Furthermore, the midpoint for this cooperativity has previously been shown to depend on the length of the leader sequence; pre-tRNA with a 2-nucleotide leader requires higher Mg2+ concentrations than a substrate with a 5-nucleotide leader 28. Here the data indicate that the midpoint for activation by divalent cations also depends on the formation of favorable contacts between the nucleobase at N(−4) and P protein. Pre-tRNA association with RNase P occurs in two steps: diffusion-controlled association followed by a conformational change that is stabilized by the leader-protein contact 47. The current data suggest that this conformation change is also stabilized by sequence-specific interactions with the leader sequence. In this context, the steep dependence of the G(−4)-pre-tRNA binding affinity on calcium could be explained by stabilization of a conformational change by divalent cations 48.
In practical terms, the calcium-dependence of substrate selectivity reduces the sequence preference under conditions commonly employed in the study of the RNase P holoenzyme in vitro (i.e. 10 to 20 mM divalent metal ions) 3. However, the physiological concentration of available Mg2+ in Bacteria has been estimated at 2 mM or lower in vivo 37; concentrations where sequence selectivity is significant (Table 1, Figure 2). Thus, the coupling of sequence specificity with divalent metal binding results in sequence selectivity at physiological cation concentrations. This coupling leads to the possibility that the sequence selectivity of RNase P in vivo could be modulated by changes in the magnesium concentration. Currently, the regulation of cellular Mg2+ is not well understood 49. However, intracellular concentrations of Mg2+ are affected by such factors as transport, sequestration in organelles, and binding, particularly to ATP 50. Therefore, it is possible that the specificity of RNase P is coupled to cellular metabolic activity via Mg2+ concentration.
tRNA gene analysis
The correlation between the genomic and biochemical data at the N(−4) and N(−1) positions in the leader of pre-tRNA for both B. subtilis and E. coli is consistent with binding affinity preferences observed in vitro playing a functional role in vivo 14. The interesting observation that, while adenosine is preferred at N(−4) in the B. subtilis genome, a number of pre-tRNAs possess nucleotides at N(−4) conferring reduced affinity (U, C, G) is consistent with previous observations and models of substrate recognition in this system. In bacterial RNase P, known interactions with the N(−1) nucleobase and 2' hydroxyl, the terminal base pair of the acceptor stem, and the 3' terminal substrate RCCA motif contribute to, but are not required for, recognition and processing 5; 10; 13; 14; 15. Recently, Sun and colleagues suggested a model of thermodynamic compensation between the 5' leader sequences and tRNA bodies of diverse substrates 5. These investigations revealed that the observed uniform affinity of RNase P for an array of pre-tRNA substrates is maintained by variations in the energetic contributions between RNase P and the 5’ leader that compensate for alterations in the affinity of RNase P for varied tRNAs. Thus, the importance of nucleotide specificity at N(−4) may be variable for pre-tRNA substrates within a given species.
Agreement between in vitro selectivity and pre-tRNA sequence preferences also suggests that examination of pre-tRNA leader sequences is a facile method for developing testable hypotheses regarding the recognition requirements of RNase P from diverse species. In this regard, the 5' leaders of tRNA genes from both B. subtilis and E. coli exhibit significant nucleotide enrichments at positions other than N(−1) and N(−4) (Figure 4), consistent with the possibility of sequence-specific contacts involving additional leader nucleotides. Intriguingly, aside from the increased U content at N(−1), B. subtilis and E. coli differ with respect to all other statistically significant leader nucleotide enrichments, suggesting that, like N(−4), molecular recognition of nucleotides at additional leader positions by RNase P differ between these species. Analysis of pre-tRNA leader sequences from 161 species further demonstrates that functionally important, sequence-specific interactions between RNase P and pre-tRNA leaders may exist at positions other than N(−4) and N(−1). The possibility of widespread sequence preferences in 5’ leader interactions represents a novel paradigm for understanding RNase P specificity across multiple species. Systematic examination of the impact of potential interactions between RNase P and additional 5’ leader positions therefore remains a key goal for better understanding substrate recognition by RNase P in vivo.
When combined with previous results for naturally occurring E. coli substrates 5, the correlation of biochemical and genetic data also suggests a model in which RNase P and its substrate population evolve in parallel. In this model, the enzyme and substrates from a given species form a matched set, optimized for uniform recognition and processing of a potentially diverse set of pre-tRNA and other substrates. The observation that species are more likely to have nucleotide enrichments in 5' leader sequences near the RNase P cleavage site suggests that recognition by RNase P may be a driving force in leader sequence selection. However, this need not be the only driving force in parallel evolution, and additional constraints on leader sequences, such as selection to prevent recognition by other ribonucleases, could influence the composition of the pre-tRNA substrate set and thereby RNase P recognition requirements. An important implication of this model is that, in the absence of a single overarching leader recognition sequence for all Bacteria, useful information regarding enzyme - substrate interactions is most likely to be gained from investigations of cognate RNase P / pre-tRNA pairings using naturally occurring 5’ leader sequences.
Conclusion
Sequence preferences observed at N(−4) for substrate affinity are reflected in the composition of pre-tRNA genes from E. coli and B. subtilis, indicating that specificity at N(−4) likely contributes to substrate recognition in vivo. We propose a model for the interaction between B. subtilis RNase and N(−4) in which the hydroxyl group of the B. subtilis protein residue Y34 hydrogen bonds with the N6 exocyclic amine of adenosine at N(−4). This is the first demonstration of a sequence-specific interaction between the P protein and pre-tRNA that contributes to molecular recognition. The correlation of genomic and biochemical data presented here suggests that analysis of pre-tRNA genes is a viable method for formulating testable hypotheses regarding RNase P substrate recognition in diverse species.
Materials and methods
RNA and protein preparation
B. subtilis RNase P RNA, E. coli RNase P RNA, and pre-tRNA substrates with 5’ terminal guanosine residues were prepared by in vitro transcription using linearized DNA templates and T7 RNA polymerase according to standard procedures 51. Substrates were 5’ end-labeled using γ-32P-ATP (MP Biomedical) and T4 polynucleotide kinase (New England Biolabs) 17. Pre-tRNA substrates with 5’ terminal nucleotides other than guanosine were prepared by RNA ligation. Synthetic 5’ oligoribonucleotides (Thermo / Dharmacon) comprising the 5’ leader and position +1 through +5 of pre-tRNAAsp were 5’ end-labeled using γ-32P-ATP and T4 polynucleotide kinase, and ligated to a 3’ tRNA fragment encoding positions +6 through +77 using splinted ligation with T4 DNA ligase (Fermentas) according to standard protocols 52; 53; 54. Experiments were carried out using B. subtilis pre-tRNAAsp, a widely used substrate that is canonical with respect to all known sequence-specific RNase P recognition elements.
P protein was expressed in E. coli (BL21(DE3) pLysS) and purified by CM-Sepharose ion-exchange chromatography in the presence of urea 55. E. coli P protein was prepared using the Impact expression system (New England Biolabs), as described previously 56. Variants of the B. subtilis P protein with site-specific cysteine, alanine, and phenylalanine mutations were prepared as described 19; 22. Prior to use, RNase P proteins were dialyzed overnight against the appropriate reaction buffer and final concentrations were determined by absorbance (B. subtilis P protein: ε280 = 5120 M−1 cm−1; E. coli C5 protein ε280 = 5500 M−1 cm−1; 55).
PRNA and pre-tRNAs were renatured by heating to 95 °C for 3 min in reaction buffer lacking divalent metal ions then incubated at 37 °C for 10 min. Divalent metals were added to the appropriate final concentration, and RNAs were incubated for 30 min. To obtain correct [Ca2+]f in the presence of high concentrations of PRNA, after renaturation, PRNA was diluted into reaction buffer with the desired [Ca2+]f and re-concentrated using Amicon ultra centrifugal filter units (10,000 molecular weight cut-off; Millipore) a minimum of three times. RNA concentrations were determined by absorbance. RNase P holoenzyme (0.5 to 10 µM) was formed by mixing equimolar concentrations of PRNA and P protein and incubating at 37 °C for a minimum of 1 hour.
Substrate Affinity Measurements
Substrate affinity measurements were carried out by centrifuge gel-filtration using Sephadex G-75 resin (Sigma-Aldrich) 5; 17 with total substrate concentrations ([Stot]) maintained at < 20% of total enzyme concentration. Binding affinity was measured in the presence of Ca2+, which supports RNase P substrate binding but decreases the rate at which substrate is converted to product 57. Enzyme and substrate were renatured separately, mixed and incubated at 37 °C for 5 min prior to loading onto preequilibrated columns. Columns were centrifuged at 6k rpm for 30 sec, and the counts in the filtrate and retentate measured. Dissociation constants were determined by fitting a single binding isotherm to the data. For [Ca2+]f titration experiments, the combined contribution of CaCl2 and KCl to ionic strength was held constant at 410 mM: at 2 mM [Ca2+] f, [KCl] = 405 mM; at 5 mM [Ca2+]f, [KCl] = 396 mM. Reactions also included 50 mM MES, 50 mM Tris, pH 6.0 (Tris: Tris-(hydroxymethyl)aminomethane; MES: β-morpholino-ethanesulfonate). For the G(−4) substrate, values for the midpoint of the transition (K1/2,Ca) and Hill coefficients (nH) were determined by fitting KD,obs values with Equation 1, where KD,∞ is the dissociation constant at saturating [Ca2+]f.
| Equation 1 |
Single-Turnover Kinetic Measurements
Single turnover experiments were performed in 2 mM Co[NH3]6•Cl3, 0.5 mM MgCl2, 200 mM NH4Cl, and 50 mM MES-Tris buffer, pH 7.0 at 37 °C using saturating RNase P holoenzyme (2 µM) as previously described 28. In 2 mM cobalt hexamine, PRNA folds, binds pre-tRNA and P protein but does not catalyze pre-tRNA cleavage 28, allowing for the measurement of the single turnover rate constant for pre-tRNA cleavage at low [Mg2+]. Substrate and product were separated by PAGE, and quantified by PhosphorImager (Amersham Bioscience Corp, Piscataway, NJ); observed rate constants (kobs) were determined by fitting a single exponential equation to the data.
5’ Leader Sequence Analysis of tRNA genes
Completely sequenced bacterial genomes were obtained from Genbank 44. Predicted tRNA genes were located using tRNAscan SE 10, and the first 15 nucleotides upstream of each tRNA, (N(−15) through N(−1)) were identified and compiled using custom AWK scripts. (Scripts available upon request.) Results for species shown in Figure 4 were adjusted to account for the 8-nucleotide acceptor stem of pre-tRNAHis. For each species, background nucleotide composition specific to the region of pre-tRNA genes was estimated from the total nucleotide composition of leader positions N(−1) to N(−15). Information content of individual leader positions relative to background, shown as sequence logos in Figure 4, was calculated according to Equation 2 41; 42,
| Equation 2 |
where RN(i) is the information content at position i, Hbkgd is the background entropy from positions N(−1) though N(−15), HN(i) is the entropy of position i, and en is a sample size correction factor. en was calculated as a function of the number of sequences (n) using Equation 3 42. Hbkgd and H(i) were calculated in units of bits using Equation 4, where fb is the observed frequency of a given base at position for Hi or for the combination of all 15 positions for Hbkgd.
| Equation 3 |
| Equation 4 |
Error bars in Figure 4, representing standard deviations for information content, were determined empirically using the CALHNB algorithm of Schneider et al. 42. Probabilities (p) that observed nucleotide frequencies appear due to random fluctuations in background nucleotide composition were determined for each position using standard χ2 analysis.
Supplementary Material
Acknowledgements
We would like to thank Dr. David Engelke and members of the Fierke group, including Dr. John Hsieh, Dr. James Hougland and Dr. Terry Watt for careful reading of this manuscript and helpful discussion. We also thank Dr. Hsieh for providing B. subtilis RNase P protein. This work was funded by National Institutes of Health (NIH) grants GM55387 to C.A.F and GM56742 to M.E.H. N.H.Z. was partially supported by NIH postdoctoral fellowship ES013881, and K.S.K. was partially supported by NIH training grant GM08353.
Abbreviations
- RNase P
ribonuclease P
- PRNA
RNase P RNA
- P protein
RNase P protein
- pre-tRNA
precursor tRNA
- N(−4)
fourth pre-tRNA nucleotide on the 5’ side of the RNase P cleavage site
- trFRET
time-resolved fluorescence resonance energy transfer
- P
purine
- 2AP
2-aminopurine
- DAP
2,6-diaminopurine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Smith JK, Hsieh J, Fierke CA. Importance of RNA-protein interactions in bacterial ribonuclease P structure and catalysis. Biopolymers. 2007;87:329–338. doi: 10.1002/bip.20846. [DOI] [PubMed] [Google Scholar]
- 2.Kirsebom LA. RNase P RNA mediated cleavage: substrate recognition and catalysis. Biochimie. 2007;89:1183–1194. doi: 10.1016/j.biochi.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 3.Christian EL, Zahler NH, Kaye NM, Harris ME. Analysis of substrate recognition by the ribonucleoprotein endonuclease RNase P. Methods. 2002;28:307–322. doi: 10.1016/s1046-2023(02)00238-4. [DOI] [PubMed] [Google Scholar]
- 4.Beebe JA, Fierke CA. A kinetic mechanism for cleavage of precursor tRNA(Asp) catalyzed by the RNA component of Bacillus subtilis ribonuclease P. Biochemistry. 1994;33:10294–10304. doi: 10.1021/bi00200a009. [DOI] [PubMed] [Google Scholar]
- 5.Sun L, Campbell FE, Zahler NH, Harris ME. Evidence that substrate-specific effects of C5 protein lead to uniformity in binding and catalysis by RNase P. EMBO J. 2006;25:3998–4007. doi: 10.1038/sj.emboj.7601290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peck-Miller KA, Altman S. Kinetics of the processing of the precursor to 4.5 S RNA, a naturally occurring substrate for RNase P from Escherichia coli. J Mol Biol. 1991;221:1–5. doi: 10.1016/0022-2836(91)80194-y. [DOI] [PubMed] [Google Scholar]
- 7.Kirsebom LA, Svard SG. The kinetics and specificity of cleavage by RNase P is mainly dependent on the structure of the amino acid acceptor stem. Nucleic Acids Res. 1992;20:425–432. doi: 10.1093/nar/20.3.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu F, Altman S. Differential evolution of substrates for an RNA enzyme in the presence and absence of its protein cofactor. Cell. 1994;77:1093–1100. doi: 10.1016/0092-8674(94)90448-0. [DOI] [PubMed] [Google Scholar]
- 9.Loria A, Pan T. Modular construction for function of a ribonucleoprotein enzyme: the catalytic domain of Bacillus subtilis RNase P complexed with B. subtilis RNase P protein. Nucleic Acids Res. 2001;29:1892–1897. doi: 10.1093/nar/29.9.1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sprinzl M, Vassilenko KS. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 2005;33:D139–D140. doi: 10.1093/nar/gki012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zahler NH, Christian EL, Harris ME. Recognition of the 5' leader of pre-tRNA substrates by the active site of ribonuclease P. RNA. 2003;9:734–745. doi: 10.1261/rna.5220703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan T, Loria A, Zhong K. Probing of tertiary interactions in RNA: 2'-hydroxyl-base contacts between the RNase P RNA and pre-tRNA. Proc Natl Acad Sci U S A. 1995;92:12510–12514. doi: 10.1073/pnas.92.26.12510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zahler NH, Sun L, Christian EL, Harris ME. The pre-tRNA nucleotide base and 2'-hydroxyl at N(−1) contribute to fidelity in tRNA processing by RNase P. J Mol Biol. 2005;345:969–985. doi: 10.1016/j.jmb.2004.10.080. [DOI] [PubMed] [Google Scholar]
- 15.Tallsjo A, Svard SG, Kufel J, Kirsebom LA. A novel tertiary interaction in M1 RNA, the catalytic subunit of Escherichia coli RNase P. Nucleic Acids Res. 1993;21:3927–3933. doi: 10.1093/nar/21.17.3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kirsebom LA, Svard SG. Base pairing between Escherichia coli RNase P RNA and its substrate. Embo J. 1994;13:4870–4876. doi: 10.1002/j.1460-2075.1994.tb06814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurz JC, Niranjanakumari S, Fierke CA. Protein component of Bacillus subtilis RNase P specifically enhances the affinity for precursor-tRNAAsp. Biochemistry. 1998;37:2393–2400. doi: 10.1021/bi972530m. [DOI] [PubMed] [Google Scholar]
- 18.Sun L, Campbell FE, Yandek LE, Harris ME. Binding of C5 protein to P RNA enhances the rate constant for catalysis for P RNA processing of pre-tRNAs lacking a consensus G(+1)/C(+72) pair. J Mol Biol. 2009 doi: 10.1016/j.jmb.2009.11.027. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Niranjanakumari S, Day-Storms JJ, Ahmed M, Hsieh J, Zahler NH, Venters RA, Fierke CA. Probing the architecture of the B. subtilis RNase P Holoenzyme active site by crosslinking and affinity cleavage. Rna. 2007;13:512–535. doi: 10.1261/rna.308707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loria A, Niranjanakumari S, Fierke CA, Pan T. Recognition of a pre-tRNA substrate by the Bacillus subtilis RNase P holoenzyme. Biochemistry. 1998;37:15466–15473. doi: 10.1021/bi9816507. [DOI] [PubMed] [Google Scholar]
- 21.Crary SM, Niranjanakumari S, Fierke CA. The protein component of Bacillus subtilis ribonuclease P increases catalytic efficiency by enhancing interactions with the 5' leader sequence of pre-tRNAAsp. Biochemistry. 1998;37:9409–9416. doi: 10.1021/bi980613c. [DOI] [PubMed] [Google Scholar]
- 22.Niranjanakumari S, Stams T, Crary SM, Christianson DW, Fierke CA. Protein component of the ribozyme ribonuclease P alters substrate recognition by directly contacting precursor tRNA. Proc Natl Acad Sci U S A. 1998;95:15212–15217. doi: 10.1073/pnas.95.26.15212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsai HY, Masquida B, Biswas R, Westhof E, Gopalan V. Molecular modeling of the three-dimensional structure of the bacterial RNase P holoenzyme. J Mol Biol. 2003;325:661–675. doi: 10.1016/s0022-2836(02)01267-6. [DOI] [PubMed] [Google Scholar]
- 24.Rueda D, Hsieh J, Day-Storms JJ, Fierke CA, Walter NG. The 5' Leader of Precursor tRNA(Asp) Bound to the Bacillus subtilis RNase P Holoenzyme Has an Extended Conformation. Biochemistry. 2005;44:16130–16139. doi: 10.1021/bi0519093. [DOI] [PubMed] [Google Scholar]
- 25.Stams T, Niranjanakumari S, Fierke CA, Christianson DW. Ribonuclease P protein structure: evolutionary origins in the translational apparatus. Science. 1998;280:752–755. doi: 10.1126/science.280.5364.752. [DOI] [PubMed] [Google Scholar]
- 26.Gardiner KJ, Marsh TL, Pace NR. Ion dependence of the Bacillus subtilis RNase P reaction. J Biol Chem. 1985;260:5415–5419. [PubMed] [Google Scholar]
- 27.Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell. 1983;35:849–857. doi: 10.1016/0092-8674(83)90117-4. [DOI] [PubMed] [Google Scholar]
- 28.Kurz JC, Fierke CA. The affinity of magnesium binding sites in the Bacillus subtilis RNase P x pre-tRNA complex is enhanced by the protein subunit. Biochemistry. 2002;41:9545–9558. doi: 10.1021/bi025553w. [DOI] [PubMed] [Google Scholar]
- 29.Sun L, Harris ME. Evidence that binding of C5 protein to P RNA enhances ribozyme catalysis by influencing active site metal ion affinity. RNA. 2007;13:1505–1515. doi: 10.1261/rna.571007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crary SM, Kurz JC, Fierke CA. Specific phosphorothioate substitutions probe the active site of Bacillus subtilis ribonuclease P. RNA. 2002;8:933–947. doi: 10.1017/s1355838202025025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Christian EL, Kaye NM, Harris ME. Helix P4 is a divalent metal ion binding site in the conserved core of the ribonuclease P ribozyme. RNA. 2000;6:511–519. doi: 10.1017/s1355838200000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaye NM, Christian EL, Harris ME. NAIM and site-specific functional group modification analysis of RNase P RNA: magnesium dependent structure within the conserved P1–P4 multihelix junction contributes to catalysis. Biochemistry. 2002;41:4533–4545. doi: 10.1021/bi012158h. [DOI] [PubMed] [Google Scholar]
- 33.Buck AH, Dalby AB, Poole AW, Kazantsev AV, Pace NR. Protein activation of a ribozyme: the role of bacterial RNase P protein. EMBO J. 2005;24:3360–3368. doi: 10.1038/sj.emboj.7600805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buck AH, Kazantsev AV, Dalby AB, Pace NR. Structural perspective on the activation of RNase P RNA by protein. Nat Struct Mol Biol. 2005;12:958–964. doi: 10.1038/nsmb1004. [DOI] [PubMed] [Google Scholar]
- 35.Smith D, Burgin AB, Haas ES, Pace NR. Influence of metal ions on the ribonuclease P reaction. Distinguishing substrate binding from catalysis. J Biol Chem. 1992;267:2429–2436. [PubMed] [Google Scholar]
- 36.Pan T, Sosnick TR. Intermediates and kinetic traps in the folding of a large ribozyme revealed by circular dichroism and UV absorbance spectroscopies and catalytic activity. Nat Struct Biol. 1997;4:931–938. doi: 10.1038/nsb1197-931. [DOI] [PubMed] [Google Scholar]
- 37.Froschauer EM, Kolisek M, Dieterich F, Schweigel M, Schweyen RJ. Fluorescence measurements of free [Mg2+] by use of mag-fura 2 in Salmonella enterica. FEMS Microbiol Lett. 2004;237:49–55. doi: 10.1016/j.femsle.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 38.Harris ME, Christian EL. Recent insights into the structure and function of the ribonucleoprotein enzyme ribonuclease P. Curr Opin Struct Biol. 2003;13:325–333. doi: 10.1016/s0959-440x(03)00069-1. [DOI] [PubMed] [Google Scholar]
- 39.Siew D, Zahler NH, Cassano AG, Strobel SA, Harris ME. Identification of adenosine functional groups involved in substrate binding by the ribonuclease P ribozyme. Biochemistry. 1999;38:1873–1883. doi: 10.1021/bi982329r. [DOI] [PubMed] [Google Scholar]
- 40.Radzicka A, Wolfenden R. A proficient enzyme. Science. 1995;267:90–93. doi: 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- 41.Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schneider TD, Stormo GD, Gold L, Ehrenfeucht A. Information content of binding sites on nucleotide sequences. J Mol Biol. 1986;188:415–431. doi: 10.1016/0022-2836(86)90165-8. [DOI] [PubMed] [Google Scholar]
- 43.Schneider TD. Consensus sequence Zen. Appl Bioinformatics. 2002;1:111–119. [PMC free article] [PubMed] [Google Scholar]
- 44.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank. Nucleic Acids Res. 2008;36:D25–D30. doi: 10.1093/nar/gkm929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gopalan V. Uniformity amid diversity in RNase P. Proc Natl Acad Sci U S A. 2007;104:2031–2032. doi: 10.1073/pnas.0611193104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hargous Y, Hautbergue GM, Tintaru AM, Skrisovska L, Golovanov AP, Stevenin J, Lian LY, Wilson SA, Allain FH. Molecular basis of RNA recognition and TAP binding by the SR proteins SRp20 and 9G8. EMBO J. 2006;25:5126–5137. doi: 10.1038/sj.emboj.7601385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hsieh J, Fierke CA. Conformational change in the Bacillus subtilis RNase P holoenzyme--pre-tRNA complex enhances substrate affinity and limits cleavage rate. RNA. 2009;15:1565–1577. doi: 10.1261/rna.1639409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hsieh J, Rueda D, Walter NG, Fierke CA. A high-affinity inner-sphere divalent cation stabilizes conformational change in the B. subtilis RNase P holoenzyme•pre-tRNA complex. in preparation. 2009 [Google Scholar]
- 49.Wolf FI, Torsello A, Fasanella S, Cittadini A. Cell physiology of magnesium. Mol Aspects Med. 2003;24:11–26. doi: 10.1016/s0098-2997(02)00088-2. [DOI] [PubMed] [Google Scholar]
- 50.Grubbs RD. Intracellular magnesium and magnesium buffering. Biometals. 2002;15:251–259. doi: 10.1023/a:1016026831789. [DOI] [PubMed] [Google Scholar]
- 51.Milligan JF, Uhlenbeck OC. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol. 1989;180:51–62. doi: 10.1016/0076-6879(89)80091-6. [DOI] [PubMed] [Google Scholar]
- 52.Moore MJ, Query CC. Joining of RNAs by splinted ligation. Methods Enzymol. 2000;317:109–123. doi: 10.1016/s0076-6879(00)17009-0. [DOI] [PubMed] [Google Scholar]
- 53.Moore MJ, Sharp PA. Site-specific modification of pre-mRNA: the 2'-hydroxyl groups at the splice sites. Science. 1992;256:992–997. doi: 10.1126/science.1589782. [DOI] [PubMed] [Google Scholar]
- 54.Loria A, Pan T. Recognition of the T stem-loop of a pre-tRNA substrate by the ribozyme from Bacillus subtilis ribonuclease P. Biochemistry. 1997;36:6317–6325. doi: 10.1021/bi970115o. [DOI] [PubMed] [Google Scholar]
- 55.Niranjanakumari S, Kurz JC, Fierke CA. Expression, purification and characterization of the recombinant ribonuclease P protein component from Bacillus subtilis. Nucleic Acids Res. 1998;26:3090–3096. doi: 10.1093/nar/26.13.3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guo X, Campbell FE, Sun L, Christian EL, Anderson VE, Harris ME. RNA-dependent folding and stabilization of C5 protein during assembly of the E. coli RNase P holoenzyme. J Mol Biol. 2006;360:190–203. doi: 10.1016/j.jmb.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 57.Smith D, Pace NR. Multiple magnesium ions in the ribonuclease P reaction mechanism. Biochemistry. 1993;32:5273–5281. doi: 10.1021/bi00071a001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.