

Abstract
The opioidergic system, an endogenous stress pathway, modulates cardiac function. Furthermore, opioid peptide and receptor expression is altered in a number of cardiac pathologies. However, whether the response of myocardial opioid receptor signaling is altered in heart failure progression is currently unknown. Elucidating possible alterations in and effects of opioidergic signaling in the failing myocardium is of critical importance as opioids are commonly used for pain management, including in patients at risk for cardiovascular disease. A hamster model of cardiomyopathy and heart failure (Bio14.6) was used to investigate cardiac opioidergic signaling in heart failure development. This study found an augmented negative inotropic and lusitropic response to administration of agonists selective for the kappa opioid receptor and delta opioid receptor in the failing heart that was mediated by a pertussis toxin-sensitive G-protein. The augmented decrease in cardiac function was manifested by increased inhibition of cAMP accumulation and the amplitude of the systolic Ca2+ transient. Furthermore, increased depression of cardiac function and of two important second messengers, cAMP and intracellular Ca2+, were independent of changes in cardiac opioid peptide or receptor expression. Thus, the cardiomyopathy-induced failing heart experiences increased cardiac depressant effects following opioid receptor stimulation which could exacerbate diminished cardiac function in end-stage heart failure. As cardiac function is already depressed in heart failure patients, administration of opioids could exacerbate the degree of cardiac dysfunction and worsen disease progression.
Keywords: cardiomyopathy, opioids, cardiac physiology, work-performing heart
INTRODUCTION
Hypertrophic cardiomyopathy (HCM) is defined as myocardial hypertrophy in the absence of a sufficient accompanying hemodynamic stress [1]. HCM in humans has a prevalence of 1 in 500 and is the leading cause of sudden cardiac death in young individuals [1]. Disease pathology shows considerable variation ranging from little to no phenotype to severe cardiac remodeling and heart failure [1]. Currently 12 genes have been found to possess mutations that lead to HCM, ten of which involve sarcomeric proteins [1]. Mutations in the sarcomeric protein delta-sarcoglycan have been linked to both human familial and sporadic forms of HCM [2]. Furthermore, a delta-sarcoglycan gene mutation is also found in a hamster model of HCM, the Bio14.6 cardiomyopathic hamster [3]. This hamster model demonstrates the characteristic cardiac hypertrophy and fibrotic infiltration of HCM [3]. In addition, the Bio14.6 presents a defined progression to heart failure. Necrosis is first observed around 8 weeks, hypertrophy initiates around 18 weeks, with cardiac decompensation beginning around 24 weeks, and heart failure as early as 40 weeks [4]. Previous studies with this model demonstrated decreased contractile function [5], impaired Ca2+ kinetics, and elevated cytosolic Ca2+ concentrations [6] in individual myocytes, which were hypertrophied compared to control cardiomyocytes[6].
Centrally mediated opioidergic influence on cardiac function has long been known. Yet, discovery of opioid receptors and peptides in the heart [7] suggests opioids may act locally to influence cardiac function. Furthermore, studies show altered protein [8] and message [9] for opioid receptors and peptides in cardiac pathology including hemorrhagic shock, hypertension, and cardiomyopathy. Specifically, elevated prodynorphin [10] and preproenkephalin [11] mRNA as well as dynorphin B protein content [8] were detected in ventricular myocytes from cardiomyopathic hamsters as early as 60 days. However, whether myocardial opioidergic function is altered in heart failure caused by genetically-derived HCM awaits elucidation. To this end, Bio14.6 cardiomyopathic hamsters and age-matched controls were investigated prior to development of pathology and in heart failure to elucidate alterations in opioidergic expression or signaling that may be inherent to, or the result of, the cardiomyopathic, cardiac failure phenotype. Findings reported here show increased negative inotropic and lusitropic responses to kappa (KOR) and delta opioid receptor (DOR) selective agonists, mediated via a pertussis toxin (PTX)-sensitive G-protein to inhibit cAMP accumulation and the amplitude of the systolic Ca2+ transient.
METHODS
Hamster Model
Hamsters were housed in a pathogen free environment and handled in accordance with standard use protocols, animal welfare regulations, and the NIH Guide for the Care and Use of Laboratory Animals. All protocols were approved by the University of Cincinnati Institutional Animal Care and Use Committee. The Bio14.6 cardiomyopathic hamster, used as a heart failure model in this investigation, is well-studied and has defined pathological endpoints through disease progression [3]. The delta-sarcoglycan gene mutation, determined to be responsible for the cardiomyopathic phenotype in these hamsters [3], is also observed in some human familial cardiomyopathies [3], making this a highly relevant and translational model of heart disease. One and nine months old male cardiomyopathic hamsters (Bio14.6) and controls (F1B) were purchased from BioBreeders Inc. Exclusion criteria for working heart studies were +dP/dt less than 2000 mmHg/s and/or −dP/dt less than 1000 mmHg/s, and/or mitral valve dysfunction as indicated by significantly elevated atrial pressure and irregular pressure tracings, and/or the presence of a leak from the aorta or pulmonary vein. Based on these criteria, a total of 3 one month F1B, 5 one month Bio14.6, 3 nine month F1B, and 34 nine month Bio14.6 hamster hearts were excluded from these studies. A significantly greater number of nine month Bio14.6 hamsters had to be excluded from working heart studies due to the surgical manipulation and the severity of pathology in this end-stage heart failure model. Hearts were excluded from cardiomyocyte studies if isolation procedure resulted in less than 60% cell viability, as determined by cells being rod-shaped and exhibiting clearly distinguishable striations, resulting in 2 F1B and 3 Bio14.6 hearts being excluded from these studies. Animals were not excluded from peptide evaluation.
Ex vivo Perfused, Working Heart
The isolated, perfused work performing heart is a valuable diagnostic tool that allows direct analysis of cardiac function, including the effects of pharmacological or physiological manipulation, independent of neuronal or humoral influences. Thus, this system enables direct evaluation of changes in heart function elicited by cardiac opioidergic activity, independent of centrally-mediated effects. Utilizing this methodology, the ability of opioid agonists to decrease cardiac inotropy and lusitropy was measured prior to the development of cardiac pathology and in the failing heart, as well as in age-matched controls.
Isolated, perfused work-performing heart preparation was accomplished as previously described [12]. Briefly, the aorta and pulmonary vein were cannulated, allowing control over cardiac work, and the left ventricle catheterized for measurement of ventricular function. Hearts were paced and subject to anterograde perfusion with a 95% O2 and 5% CO2 modified Krebs-Henseleit solution.
Opioid Receptor Agonist Concentration Response Curves
Concentrations of opioid receptor selective agonists, U50488H for the kappa opioid receptor (KOR), Tan-67 for the delta opioid receptor (DOR), and fentanyl for the mu opioid receptor (MOR) were infused for ten minutes to produce a cumulative semi-logarithmic concentration response curve. U50488H, was given at a range between 10 nM and 5 μM. Tan-67, administered at a range from 100 nM to 10 μM. Fentanyl, concentrations were between 100 pM and 10 μM. Agonist concentrations were based on receptor selectivity [13, 14] to assure that drugs would act preferentially at the desired receptor. In addition, preliminary concentration response curves were performed to insure an effective range of concentrations. Functional measurements and oxygen consumption were evaluated at 5 and 10 minutes into perfusion of each drug concentration as well as prior to the initiation of drug treatment (baseline).
Pertussis Toxin Studies
Additional studies were performed to determine whether decreased cardiac contractility observed upon opioid agonist administration was mediated through coupling of opioid receptors to the inhibitory G-protein, Gi. PTX (10 μg/kg) was administered intraperitoneally three days before concentration response studies were performed. Work-performing hearts from one and nine months old saline- or PTX-treated hamsters were challenged by opioid agonists at concentrations that induced a 50% decrease from baseline function for 10 minutes each in a cumulative concentration response curve. Agonist concentrations used were 500 nM and 1 μM for U50488H in one month hamsters and nine month Bio14.6 and 2 and 5 μM in nine month F1B; 300 nM, 1 μM, and 3 μM for Tan-67; and as fentanyl failed to elicit a response except at 10 μM, full concentration response curves were performed.
Measurement of Opioid Peptides
Hearts and plasma were collected from Bio14.6 and F1B hamsters at one and nine months and processed and assayed for β-endorphin, dynorphin A, and leu-enkephalin as previously described [12].
Cardiomyocyte Isolation
Cardiomyocytes were isolated from nine month Bio14.6 and F1B hamsters as previously described [12], with the exception that following digestion and removal of the atria and right ventricle, the left ventricle was placed in 10 mL of 25 μM Ca2+-Tyrode. Cardiomyocytes were made Ca2+ tolerant by allowing them to settle for 5 minutes, removing the supernatant and resuspending the cells in 100 μM Ca2+ Tyrode. This was repeated as the cells were reintroduced to Ca2+ concentrations of 200 μM and 1 mM Ca2+ at which they were maintained until measurement.
Opioid Receptor Western Blots
Left ventricular cardiomyocytes isolated from one and nine month F1B and Bio14.6 hamsters were homogenized using a glass-teflon homogenizer. Protein content was determined by Lowry assay and opioid receptor expression determined by Western blot analysis as previously described [12]. Antibodies specific for the KOR (1:1000), DOR (1:5000), and MOR (1:1000) were utilized. Intensity of bands was normalized to a standard curve generated on a per gel basis consisting of a pooled sample of control hearts, while equal protein loading was verified by Ponceau S staining.
Gα Subunit Western Blots
Left ventricular cardiomyocytes were homogenized and separated by polyacrylamide gel electrophoresis as previously described [12]. Following blocking, membranes were probed with antibodies selective for the α-subunits of Gi2, Go, and Gq (1:500, Santa Cruz Biotech). Membranes were then probed with secondary antibodies and detection and densitometry performed as previously described [12]. Intensity of bands was normalized to a standard curve generated on a per gel basis consisting of a pooled sample of control hearts, and equal protein loading was verified by Ponceau S staining.
Cyclic Adenosine-3′, 5′-Monophosphate (cAMP) Assay
The ability of opioid agonists to inhibit isoproterenol-induced cAMP production was tested in cardiomyocytes isolated from the left ventricle of Bio14.6 and F1B hamsters at nine months of age as previously described [12]. Cardiomyocytes were pretreated with opioid agonist (U50488H or Tan-67) with or without antagonist (nor-binaltorphimine [nor-BNI, KOR selective antagonist] or naloxone [non-selective OR antagonist]) or left untreated prior to administration of isoproterenol. Accumulation of cAMP was determined by competition assay for binding to PKA. Inhibition of cAMP accumulation was evaluated as cAMP concentration normalized to protein content of cell lysate for an individual treatment as a percentage of isoproterenol treatment alone from same heart also normalized to protein content.
Calcium Transients
Ca2+ transients were measured in the presence or absence of 5 μM U50488H with and without 5 μM nor-BNI, or 5 μM DPDPE with and without 5 μM naloxone, dissolved in 1.8 mM Ca2+-Tyrode. For detection of Ca2+ transients, 1 mL of cells in Tyrode were incubated with 2 μL of 1 mM Fura-2 and 2 μL of 20% pluronic acid for 20 minutes on a rocker. The cells were then allowed to settle for 5 minutes and washed twice with 1 mM Ca2+-Tyrode. Fura-loaded cells were placed in a stimulation chamber with 1.8 mM Ca2+-Tyrode in the absence (no treatment) or presence of opioid agonists or agonists with antagonists. Cells were electrically stimulated at 0.5 Hz and the Ca2+ tracing was recorded. From this the amplitude of the Ca2+ transient, basal Ca2+ concentration, and tau (rate of decline for Ca2+ amplitude) were measured.
Drugs and chemicals
Fentanyl, U50488H, DPDPE, PKA, IBMX, and isoproterenol were purchased from Sigma-Aldrich. Tan-67 and pluronic acid were purchased from Tocris. Pertussis toxin was purchased from Calbiochem. Fura-2 was purchased from Molecular Probes. [3H]-cAMP was purchased from Perkin-Elmer. All drugs were dissolved in milli-Q water and diluted to required initial concentration in Krebs, Tyrode, or Tris/EDTA depending on the experiment.
Statistical Analysis
All values shown are mean ± SEM. Cardiac functional data was analyzed using two-way analysis of variance with Bonferroni’s post-test. Opioid peptide and receptor levels, Ca2+ transients, and cAMP levels were analyzed by Student’s t test. Statistical differences were considered significant at p<0.05.
Results
Cardiac hypertrophy in Bio14.6 hamsters
The Bio14.6 cardiomyopathic hamster exhibited signs of cardiac hypertrophy and heart failure. This included pulmonary edema (wet lung-to-dry lung ratio: 4.8±0.1 for control versus 5.1±0.1 in Bio14.6 at nine months, p<0.05, Table 1) in the failing heart that was not evident prior to pathology (1 month of age). Cardiac hypertrophy was indicated by increased heart weight-to-tibia length ratio (20.8±0.5 mg/mm for control versus 31.6±1 mg/mm in Bio14.6, p<0.05, Table 1) and increased left ventricular weight-to-tibia length ratio (14.5±0.4 mg/mm for control versus 20.6±0.7 mg/mm in Bio14.6, p<0.05, Table 1). There was no evidence of cardiac hypertrophy in one month Bio14.6 hamsters.
Table 1.
Cardiac functional and morphological parameters of Bio14.6 and F1B hamsters
| LVSP (mmHg) | LVEDP (mmHg) | +dP/dt (mmHg/s) | −dP/dt (mmHg/s) | wL/dL | HW/tibia (mg/mm) | LV/tibia (mg/mm) | |
|---|---|---|---|---|---|---|---|
|
1 mo F1B |
94±3 | 7.8±1.2 | 3451±142 | −2393±141 | 4.7±0.1 | 15.6±0.3 | 10.4±0.3 |
|
1 mo Bio14.6 |
86±1* | 7.4±0.7 | 3033±101* | −2253±90 | 4.9±0.1 | 15.4±1.1 | 10.5±0.8 |
|
9 mo F1B |
77±1 | 7.0±0.8 | 2858±122 | −1835±76 | 4.8±0.1 | 20.8±0.5 | 14.5±0.3 |
|
9 mo Bio14.6 |
67±2* | 12.7±1.5* | 2156±128* | −1409±78* | 5.1±0.1* | 31.6±1* | 20.6±0.7* |
LVSP- left ventricular systolic pressure, LVEDP- left ventricular end-diastolic pressure, +dP/dt- positive change in left ventricular pressure over time, −dP/dt- negative change in left ventricular pressure over time, wL/dL- wet lung-to-dry lung ratio, HW/tibia- heart weight-to-tibia length ratio, LV/tibia- left ventricular weight-to-tibia length ratio.
Data are mean±SEM.
p<0.05 vs. age-matched F1B.
n=10–15.
Decreased cardiac inotropy and lusitropy in cardiomyopathic hearts
Baseline cardiac inotropy and lusitropy (as determined by the positive and negative changes in left ventricular pressure over time, respectively) were evaluated in hearts from cardiomyopathic hamsters as well as age-matched control hearts (Table 1). A slight but significant decrease in basal cardiac inotropy was observed in hearts from hamsters predisposed to cardiomyopathy compared to hearts from control hamsters; however, there was no difference in basal cardiac lusitropy at one month (Table 1). In comparison, there was a significant and robust decrease in parameters of basal cardiac inotropy and lusitropy in failing hearts compared to age-matched control hearts (Table 1).
KOR- and DOR-induced negative inotropy and lusitropy is augmented in the failing heart
To elucidate whether the ability of the myocardial opioidergic system to modulate cardiac function is altered in heart failure, parameters of cardiac inotropy and lusitropy were measured using the isolated, work performing heart following administration of receptor selective agonists. Cumulative concentration response curves were generated using the kappa agonist U50488H, the delta agonist Tan-67, and the mu agonist fentanyl at one month, prior to cardiac pathology, and at nine months, in the heart failure stage. Kappa and delta agonists were administered as these receptor types are known to be expressed in the juvenile and adult heart [15], while the mu agonist was utilized to test the hypothesis that the MOR which is expressed in neonatal myocardium [15] would be recapitulated in heart failure similar to that of other cardiac fetal genes such as β-myosin heavy chain and α-smooth muscle actin [16]. Administration of agonists selective for the KOR or DOR decreased cardiac inotropy and lusitropy in a concentration-dependent manner in both Bio14.6 and F1B hamster hearts at one and nine months. U50488H, the KOR selective agonist, induced a similar decrease in cardiac function in 1 month Bio14.6 and F1B hamster hearts (Figures 1 and 2, panel A). However, the cardiac depressant effect of U50488H was significantly more pronounced in 9 month Bio14.6 hamster hearts compared to F1B (p<0.05, Figures 1 and 2, panel B), indicating enhanced KOR signaling in the cardiomyopathic heart.
Figure 1.

Kappa agonist, U50488H, caused a greater decrease in cardiac inotropy in the failing heart. Acute U50488H administration decreased cardiac inotropy in Bio14.6 (■, dashed lines) and F1B (◆, solid lines) hamster hearts at (A) one and (B) nine months. This response was mediated by a PTX-sensitive G-protein as PTX pretreatment attenuated the effect of agonist concentrations that produced a 50% decrease from baseline function (0.5 and 1 μM for (C) one month F1B and (D) one and (F) nine months Bio14.6 and 2 and 5 μM for (E) nine months F1B). Data are mean±SEM. *p<0.05 vs. F1B or No PTX treatment. n=4–10 hamster hearts.
Figure 2.

Kappa agonist, U50488H, caused a greater decrease in cardiac lusitropy in the failing heart. Acute U50488H administration decreased cardiac lusitropy in Bio14.6 (■, dashed lines) and F1B (◆, solid lines) hamster hearts at (A) one and (B) nine months. This response was mediated by a PTX-sensitive G-protein as PTX pretreatment attenuated the effect of agonist concentrations that produced a 50% decrease from baseline function (0.5 and 1 μM for (C) one month F1B and (D) one and (F) nine months Bio14.6 and 2 and 5 μM for (E) nine months F1B). Data are mean±SEM. *p<0.05 vs. F1B or No PTX treatment. n=4–10 hamster hearts.
Tan-67, a DOR selective agonist, produced an equivalent decrease in cardiac inotropy and lusitropy in 1 month Bio14.6 and F1B hearts (Figures 3 and 4, panel A). Similar to the KOR response, Bio14.6 hamsters in heart failure were significantly more sensitive to Tan-67 than were age-matched controls. Hearts from nine month Bio14.6 hamsters demonstrated a greater decrease in cardiac contractility and relaxation (p<0.05, Figures 3 and 4, panel B) at intermediate agonist concentrations despite equal efficacy at the highest concentration administered.
Figure 3.

Delta agonist, Tan-67, caused a greater decrease in cardiac inotropy and was PTX-sensitive in the failing heart. Acute Tan-67 administration decreased cardiac inotropy in Bio14.6 (■, dashed lines) and F1B (◆, solid lines) hamster hearts at (A) one and (B) nine months. Agonist concentrations producing a 50% decrease from baseline function (300 nM, 1, and 3 μM) were not attenuated by PTX pretreatment in either line at (C and D) one month or in (E) nine month control hearts. However, PTX did antagonize the negative inotropic effect of Tan-67 in (F) the failing heart. Data are mean±SEM. *p<0.05 vs. F1B or No PTX treatment. n=3–6 hamster hearts.
Figure 4.

Tan-67 caused a greater decrease in cardiac lusitropy and was PTX-sensitive in the failing heart. Acute Tan-67 administration decreased cardiac lusitropy in Bio14.6 (■, dashed lines) and F1B (◆, solid lines) hamster hearts at (A) one and (B) nine months. Agonist concentrations producing a 50% decrease from baseline function (300 nM, 1, and 3 μM) were not attenuated by PTX pretreatment in either line at (C and D) one month or in (E) nine month F1B hearts. However, PTX did antagonize the negative lusitropic effect of Tan-67 in (F) the failing heart. Data are mean±SEM. *p<0.05 vs. F1B or No PTX treatment. n=3–6 hamster hearts.
The MOR selective agonist, fentanyl, induced a negative inotropic and lusitropic response only at the 10 μM concentration (Figures 5 and 6, panels A and B), a concentration beyond the range of receptor specificity [14]. Failure to elicit a response at lower concentrations, suggests a lack of MOR in cardiac tissue.
Figure 5.

Administration of the MOR-selective agonist fentanyl decreased cardiac contractility only at non-selective concentrations. Acute fentanyl administration only decreased cardiac inotropy in (A) one and (B) nine month Bio14.6 (■, dashed lines) and F1B (◆, solid lines) hamster hearts at 10 μM. The depressant effect of this high fentanyl concentration was not antagonized by PTX pretreatment at (C and D) one or (E and F) nine months. Data are mean±SEM. *p<0.05 vs. F1B. n=3–4 hamster hearts.
Figure 6.

Administration of the MOR-selective agonist fentanyl decreased cardiac relaxation only at non-selective concentrations. Acute fentanyl administration only decreased cardiac lusitropy in (A) one and (B) nine month Bio14.6 (■, dashed lines) and F1B (◆, solid lines) hamster hearts at 10 μM. The depressant effect of this high fentanyl concentration was not antagonized by PTX pretreatment at (C and D) one or (E and F) nine months. Data are mean±SEM. *p<0.05 vs. F1B. n=3–4 hamster hearts.
Enhanced KOR and DOR effects in the failing heart are mediated by PTX-sensitive G-protein
Pertussis toxin was administered intraperitoneally three days prior to opioid agonist concentration response studies to determine whether the cardiac depressant effects observed were receptor-mediated, signaling through the PTX-sensitive G-proteins, Gi/o. Inhibition of Gi/o signaling by PTX significantly attenuated the ability of U50488H to elicit a depression in cardiac contractility or relaxation in hearts from Bio14.6 and F1B hamsters at one and nine months (p<0.05, Figures 1 and 2, panel C–F). Thus, the KOR is coupled to a PTX-sensitive G-protein in the heart, mediating the cardiac depressant response to U50488H.
PTX administration had no effect on the cardiac depressant effects of Tan-67 in either model at 1 month of age or in 9 month F1B hearts (Figures 3 and 4, panels C, D, and E). Prior PTX administration significantly attenuated the negative inotropic and lusitropic effects of Tan-67 in 9 month Bio14.6 hamsters in the heart failure stage (p<0.05, Figures 3 and 4, panel F). This finding suggests enhanced delta signaling in genetically-derived cardiomyopathy and heart failure is mediated by a PTX-sensitive pathway while delta signaling is PTX-insensitive in the healthy hamster heart.
PTX administration did not block the decreased cardiac function observed upon administration of high, receptor non-selective, concentrations of fentanyl (Figures 5 and 6, panels C–F), suggesting that fentanyl’s effect at 10 μM is opioid receptor-independent.
Opioid peptides unaltered in cardiomyopathic hamster
To determine whether enhanced opioidergic function in the cardiomyopathic-induced failing heart was in response to alterations in the endogenous opioid peptides, cardiac and plasma levels of a member of each family of endogenous opioid peptides were investigated. There were no differences detected in either blood or cardiac tissue between Bio14.6 and F1B hamsters for the opioid peptides measured (leu-enkephalin, β-endorphin, or dynorphin A) at either age investigated (Table 2).
Table 2.
Cardiac and plasma opioid peptide levels.
| Plasma β-endorphin (ng βE/mg protein) | Cardiac β-endorphin (ng βE/mg protein) | Plasma Dynorphin A (ng DA/mg protein) | Cardiac Dynorphin A (ng DA/mg protein) | Plasma Leu-enkephalin (ng LE/mg protein) | Cardiac Leu-enkephalin (ng LE/mg protein) | |
|---|---|---|---|---|---|---|
|
1mo F1B |
0.080±0.01 | 0.296±0.16 | 0.048±0.1 | 0.198±0.11 | 4.031±1.34 | 1.879±0.55 |
|
1mo Bio14.6 |
0.109±0.4 | 0.264±0.16 | 0.086±0.03 | 0.139±0.06 | 3.396±0.98 | 2.069±1.13 |
|
9mo F1B |
0.176±0.06 | 0.398±0.15 | 0.106±0.03 | 0.106±0.03 | 9.243±2.57 | 2.934±0.49 |
|
9mo Bio14.6 |
0.135±0.04 | 0.266±0.11 | 0.144±0.04 | 0.144±0.04 | 9.643±3.85 | 3.637±0.65 |
Data are mean±SEM.
n=9–15.
Bio14.6 hamsters at risk for (1 month) and in heart failure (9 months) as well as age-matched controls (F1B).
KOR unaltered, DOR decreased in failing heart
Cardiac opioid receptor expression was investigated by Western blot analysis to determine whether enhanced cardiac opioidergic function could be explained by alterations in receptor expression. No differences were detected in protein expression of KOR in left ventricular cardiomyocytes from Bio14.6 or F1B hamsters at 1 or 9 months of age (Figure 7). Similarly, there was no difference in DOR protein expression in 1 month left ventricular cardiomyocytes from Bio14.6 and F1B hamster hearts (Figure 7). However, DOR protein expression was significantly decreased in left ventricular cardiomyocytes from the failing heart compared to age-matched control, despite increased DOR-induced cardiac depression in Bio14.6 hearts at this age (p<0.05, Figure 7). The MOR was not detected in cardiomyocytes from Bio14.6 or F1B hamsters at either age investigated (data not shown).
Figure 7.

Opioid receptor and Gαi2 expression in left ventricular cardiomyocytes. Expression of (B) KOR, (C) DOR, and (D) the α-subunit of Gi2 was measured by Western immunoblot analysis in left ventricular cardiomyocytes from 1 and 9 month F1B (solid bars) and Bio14.6 (open bars) hamsters. (A) Representative immunoblots are shown for KOR, DOR, and Gαi2. Data are mean±SEM. *p<0.05 vs. 9 month F1B. n=8–9 hamster hearts.
Gαi2 increased, Go and Gq α-subunits unaltered in failing heart
Enhanced opioidergic function could not be explained by changes in receptor expression. Since increased opioid-induced cardiac depression was PTX-sensitive, expression of the α-subunits of the G-proteins through which opioid receptors commonly signal was investigated. Protein content of the α-subunit of Gi2, a PTX-sensitive G-protein, was increased in the failing heart (p<0.05), but was not different from control at one month (Figure 7). Go and Gq content did not differ significantly between Bio14.6 and F1B left ventricular cardiomyocytes at 1 or 9 months (data not shown).
Opioid receptor inhibition of cAMP accumulation enhanced in failing heart
It was determined that enhanced opioidergic cardiac depression was PTX-sensitive, and that there was increased expression of the α-subunit of the inhibitory, PTX-sensitive G-protein, Gi, in the genetically-derived cardiomyopathic, failing heart. Therefore, the effect of opioid receptor activity on cAMP accumulation was evaluated in cardiomyocytes from nine month Bio14.6 and F1B as a possible mechanism. The ability of isoproterenol to increase the cellular cAMP content in Bio14.6 and F1B left ventricular cardiomyocytes was significantly inhibited by U50488H (p<0.05, Figure 8). The ability of U50488H to antagonize isoproterenol-induced cAMP accumulation was greater in cardiomyocytes from Bio14.6 hamsters than those from F1B. This effect was significantly attenuated when nor-BNI was administered (p<0.05, Figure 8), indicating it was mediated by the KOR. Tan-67 only significantly inhibited the isoproterenol-induced increase in cAMP content in Bio14.6 cardiomyocytes (p<0.05, Figure 8). This finding supports the observation that the Tan-67-induced decrease in cardiac function in control hearts was not mediated by a PTX-sensitive mechanism (Figures 3 and 4). The increased inhibition of cAMP by Tan-67 in cardiomyopathic hearts was significantly blocked by the addition of naloxone (p<0.05, Figure 8), showing it was receptor-mediated.
Figure 8.
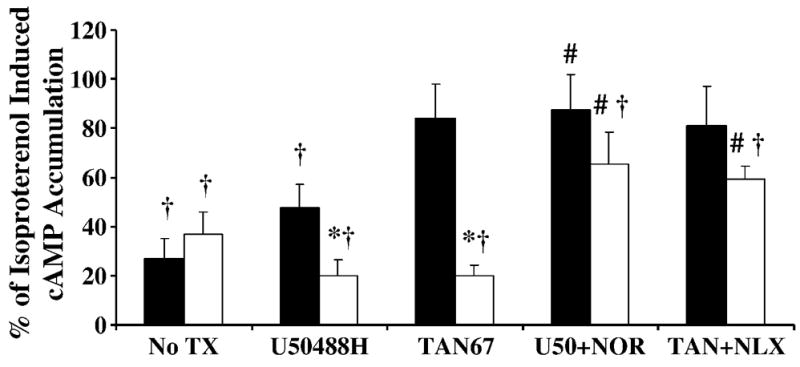
Inhibition of cAMP accumulation by selective opioid receptor agonists. The KOR agonist U50488H (100 μM) inhibited the isoproterenol-induced accumulation of cAMP to a greater extent in left ventricular cardiomyocytes from Bio14.6 (open bars) compared to F1B (solid bars) hamsters, while the DOR agonist Tan-67 (100 μM) only inhibited cAMP accumulation in Bio14.6 cardiomyocytes. Agonist effects were blocked by the antagonists nor-BNI (100 μM) (KOR) and naloxone (100 μM) (non-selective OR). Data was normalized to protein content and represented as percentage of isoproterenol alone in same heart. No TX is basal [cAMP]. Data are mean±SEM. *p<0.05 vs. F1B with same treatment, #p<0.05 vs. agonist-treated cohort, †p<0.05 vs. Iso treatment alone. n=6–9 hamster hearts.
Opioid receptor-mediated decrease in calcium transient augmented in failing heart
Opioidergic signaling is known to influence calcium cycling in a variety of manners [17–19]. Therefore, calcium cycling was investigated as an end effector of enhanced cardiac opioidergic activity. The KOR agonist U50488H decreased the amplitude of the electrically stimulated Ca2+ transient in both Bio14.6 and F1B left ventricular cardiomyocytes, and this effect was abolished by co-administration of the kappa antagonist nor-BNI, indicating a KOR-mediated effect (p<0.05, Figure 9). Furthermore, the inhibitory effect of U50488H on the Ca2+ transient was greater in Bio14.6 left ventricular cardiomyocytes compared to F1B cardiomyocytes (p<0.05, Figure 9). The DOR agonist DPDPE significantly inhibited the amplitude of the electrically-stimulated Ca2+ transient only in Bio14.6 cardiomyocytes (p<0.05, Figure 9). This effect was blocked by the non-selective OR antagonist naloxone (p<0.05, Figure 9), indicating a receptor-mediated effect. There was no difference in basal cytosolic Ca 2+ concentration or the rate of Ca 2+ decline following opioid agonist treatment (data not shown).
Figure 9.
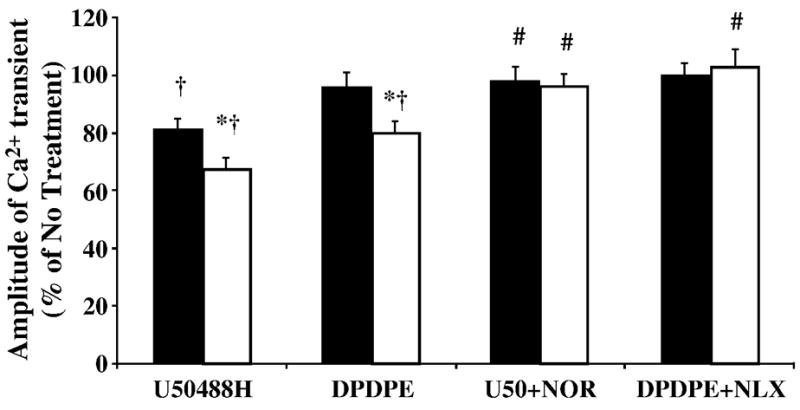
Selective KOR and DOR agonists decreased the amplitude of the systolic calcium transient. Cardiomyocytes were either incubated in Tyrode alone (no treatment, No TX), or agonists selective for the KOR (U50488H, 5 μM) or the DOR (DPDPE, 5 μM) with and without the antagonists nor-BNI (5 μM) or naloxone (NLX, 5 μM). The KOR agonist U50488H inhibited the electrically stimulated rise in intracellular calcium to a greater extent in left ventricular cardiomyocytes from Bio14.6 (open bars) compared to F1B (solid bars) hamsters, while the selective DOR agonist DPDPE only inhibited the amplitude of the calcium transient in Bio14.6 cardiomyocytes. Agonist effects were blocked by the antagonists nor-BNI (5 μM) (KOR) and naloxone (5 μM) (non-selective OR). Data represented as percentage of electrical stimulation alone (no treatment). Data are mean±SEM. *p<0.05 vs. F1B with same treatment, #p<0.05 vs. agonist-treated cohort, †p<0.05 vs. no treatment. n=9–11 hamster hearts.
Discussion
Results of this study showed, for the first time, an augmented ability of cardiac opioid receptor stimulation to depress cardiac function in the genetically-derived cardiomyopathic heart, a common etiology leading to heart failure. This intensified opioidergic response was PTX-sensitive and involved enhanced inhibition of cAMP accumulation, resulting in a diminished Ca2+ transient. Furthermore, this functional alteration is not the product of increased opioid receptor protein expression, but may result from increased receptor coupling as expression of the PTX-sensitive G-protein, Gi, was increased.
Opioid agonists have been previously shown to decrease cardiac function in the non-diseased Langendorff heart [20] and contractility in healthy cardiomyocytes [8]. However, whether this capability is altered in heart failure remains unknown and was the focus of this investigation. Similar to previous observations [7, 12], administration of the KOR selective agonist, U50488H, induced a concentration-dependent decrease in cardiac function in Bio14.6 and F1B hearts at one and nine months. The effects of U50488H administration were similar in Bio14.6 and F1B hearts at one month of age. However, in the cardiomyopathic-induced failing heart the negative inotropic and lusitropic responses were significantly greater than in control. The cardiac depressant response to U50488H was significantly antagonized by prior administration of PTX, indicating these effects were mediated by the KOR coupled to a PTX-sensitive G-protein, Gi/o. The augmented decrease in cardiac function upon U50488H administration was also observed in individual left ventricular cardiomyocytes as indicated by greater inhibition of isoproterenol-induced cAMP accumulation as well as Ca2+ transient amplitude. Effects of U50488H in LV cardiomyocytes were blocked by the KOR antagonist nor-BNI, indicating a receptor-mediated response. These results show that the cardiac depressant effects of U50488H were mediated by the KOR, signaling through an inhibitory G-protein, acting directly on the cardiomyocytes.
The DOR agonist Tan-67 also decreased cardiac inotropy and lusitropy in a concentration-dependent manner in hearts from Bio14.6 and F1B hamsters at one and nine months of age. The response to Tan-67 administration was similar in Bio14.6 and F1B hearts prior to pathology; however the cardiac depressant effects were augmented in the genetically-derived cardiomyopathic, failing heart. Interestingly, PTX pretreatment failed to antagonize the negative inotropic and lusitropic response to Tan-67 in one month Bio14.6 and F1B hamster hearts or in nine month F1B hearts, while PTX did antagonize the agonist-induced cardiac depression in the failing heart. This suggests a switch in DOR coupling in the failing heart from a PTX-insensitive signaling mechanism to a PTX-sensitive G-protein. The inability of PTX pretreatment to influence delta agonist-induced cardiac depression in the healthy hamster heart was unexpected as Gi/o signaling is the predominant opioidergic pathway [17]. Yet, opioid receptor-mediated cardiac depression may occur following PTX pretreatment for a variety of reasons. It has been previously shown in the rat that this concentration of PTX (10 μg/kg) was sufficient to block cardiac preconditioning despite only a relatively small number of Gi α-subunits being ADP-ribosylated [21]. Therefore, the concentration of PTX used, though sufficient to inhibit opioid receptor mediated signal transduction, may not completely abolish opioid receptor signaling as non-ribosylated Gi subunits would remain which could couple to opioid receptors and decrease cardiac function. It is also possible that opioidergic cardiac depression may involve multiple signaling pathways. In addition to the prototypical Gi-mediated opioidergic pathway, opioid receptors also couple to Gq [18] and the PKC pathway is a critical mediator of some opioidergic effects [8, 10], including cardioprotection which is a DOR-mediated phenomenon [22]. For example, DOR-induced cardioprotection mediated by the delta agonist, Tan-67, involved PKC activation [23]. In fact, a number of biological responses to KOR stimulation are PKC-dependent [24], resulting from PLC-mediated production of DAG following activation of Gq-coupled receptors. Thus multiple signaling pathways are likely involved in cardiac opioid receptor functional responses. Furthermore, opioid receptors are thought to be fairly promiscuous in regards to the G-protein to which they couple [17], and it has been shown that opioid receptors can switch the G-protein to which they typically couple and signal through within an organ system; however normally this is observed in response to chronic stimulation [25]. Therefore, PTX-sensitive DOR signaling in the genetically-derived cardiomyopathic, failing heart, in contrast to the PTX-insensitive response in hearts from healthy or at risk hamsters, could result from increased availability of the α-subunit of Gi and the inherent promiscuity of the DOR-G-protein interaction. Another explanation for the switch in DOR coupling could involve opioid receptor dimerization in heart failure. DOR has been shown to form both homo-oligomers and heterodimers with the KOR, MOR, and β-adrenergic receptors (βAR), among other receptors, in vitro [26, 27]. Furthermore, dimerization results in altered cellular localization, agonist affinity, and signaling properties [26, 27]. Thus dimerization of the DOR with other DOR, KOR, βAR, or another receptor may induce a conformational change in DOR structure resulting in altered coupling and signaling efficiency. Yet another mechanism by which DOR signaling could switch from PTX-insensitive in the normal hamster heart to PTX-sensitive in the failing heart pertains to receptor localization to plasma microdomains. It has previously been shown that opioid receptors localize to microdomains on the plasma membrane [28], particularly to caveolin-rich membrane domains [28], and that caveolin microdomains contain Gq but not Gi [28]. It has further been shown that caveolin and cholesterol levels are highly correlative, with decreases in membrane cholesterol causing decreased caveolin mRNA and protein content [29]. Decreased cholesterol content of the plasma membrane has also been reported in the Bio14.6 hamster [30] and caveolin null mice develop cardiomyopathy [31]. Thus, the DOR may be localized to caveolin-rich microdomains in the healthy hamster heart and couple to the α-subunits of Gq, as it is more highly expressed in these regions, resulting in PTX-insensitive signaling. However, with the decline in caveolin microdomains in cardiomyopathy, perhaps due to decreased membrane cholesterol, the DOR may be localized to the more fluid plasma membrane and can bind to its preferential G-protein, Gi, which is also more highly expressed in the heart failure stage. Furthermore, decreased membrane cholesterol was correlated with an increase in DOR-mediated GTPγS binding [28], providing another mechanism for increased DOR signaling in the genetically-derived cardiomyopathic, failing heart. However, whether the switch from PTX-insensitive DOR signaling in the healthy hamster heart to PTX-sensitive signaling in the failing heart is merely a response to increased Gi α-subunit expression, receptor dimerization, or loss of localization to microdomains remains to be elucidated. Further evidence for Gi-mediated DOR signaling in the cardiomyopathic heart was provided by studies investigating the effects of DOR stimulation on cAMP accumulation and Ca2+ transients in LV cardiomyocytes. Tan-67 attenuated isoproterenol-induced cAMP accumulation in LV cardiomyocytes from nine month Bio14.6 hamster hearts, but had no effect on cardiomyocytes from F1B hamsters. Similarly, DPDPE, a DOR agonist, decreased the amplitude of the Ca2+ transient in LV cardiomyocytes from failing hearts, but not from control. Both these effects were antagonized by co-administration of naloxone, an opioid receptor blocker. These results show that there is altered coupling of the DOR in the cardiomyopathic-induced failing heart and that this change takes place in the cardiomyocyte itself, resulting in enhanced cardiac DOR activity.
The MOR-selective agonist, fentanyl, did not alter cardiac function except at concentrations above the range of receptor specificity. In addition, PTX pretreatment did not attenuate fentanyl-induced cardiac depression at 10 μM. Fentanyl is highly potent at the MOR with a binding affinity in the picomolar range [14]. That fentanyl did not alter cardiac function until micromolar concentrations were administered and that this response was not PTX-sensitive indicates the effects were likely not receptor-mediated but the product of a non-specific, receptor independent mechanism. This also suggests that the MOR is not expressed in cardiac tissue, and Western blot analysis confirmed that cardiomyocytes are devoid of MOR, including in the failing heart. It had been proposed that as the MOR is present in the fetal and neonatal, but not juvenile or adult heart [15], it may be recapitulated in heart failure similar to other fetal genes such as the β-myosin heavy chain and α-smooth muscle actin [16]. However, the results of this study indicate the MOR is not part of the cardiac fetal gene program in cardiomyopathy; whether this is true for other etiologies of heart failure remains to be elucidated. However, recent work from our laboratory [12] demonstrated that the MOR is not expressed in the heart following chronic untreated hypertension, another common underlying etiology of heart failure.
Increased opioidergic tone in heart failure was previously indicated in studies using canine heart failure models as well as human heart failure patients. In these instances, administration of opioid antagonists resulted in increased parameters of cardiac function and peripheral blood flow [32, 33]. These results suggest increased opioidergic signaling could be responsible for the cardiac insufficiency that is the very definition of heart failure; however these studies did not look specifically at the cardiac opioidergic system. In addition, Redfern and colleagues showed, using a modified KOR, that sustained cardiac Gi activation could induce a lethal cardiomyopathy in the mouse [34]. These results indicated that cardiac opioidergic signaling could be a sufficient stimulus to induce heart failure; however whether cardiac opioidergic function or signaling is altered in the failing heart was not elucidated. Therefore, these studies showed, for the first time, that cardiac opioidergic function is enhanced in cardiomyopathy, a common etiology leading to heart failure, and further elucidated the mechanism responsible for enhanced function, showing it involved Gi-mediated inhibition of cAMP accumulation and the amplitude of the rise in cytosolic calcium upon systole.
This study showed an enhanced ability of both the cardiac kappa and delta opioid receptors to decrease cAMP accumulation and the amplitude of the calcium transient in cardiomyocytes from failing hearts indicating that the opioidergic system is hyperactive in cardiomyopathy and could be a mediating factor in heart failure progression. Furthermore, decreased myocardial cAMP content has been previously reported in patients with cardiac disease [34] while abnormal calcium handling is a hallmark of cardiac dysfunction [35]. Opioids have been shown to influence calcium handling in a variety of manners [19] including inhibition of cAMP [17]. That decreased calcium cycling lies at the root of cardiac insufficiency gives further credence to opioidergic signaling playing a pivotal role in the transition from cardiac decompensation to heart failure.
The finding that both myocardial KOR and DOR activities were enhanced in the cardiomyopathic-derived failing heart has significant implications for the role that the opioidergic system plays in cardiac pathology as different opioid receptor types have been shown to have opposing [36] or synergistic effects [27]. Furthermore, these two opioid receptor types have been shown to form heterodimers in vitro which produced a synergistic decrease in cAMP accumulation [27]. Yet, whether these two opioid receptors dimerize in vivo and specifically in the heart remains unknown. Still, that multiple opioid receptors exhibit enhanced function in the cardiomyopathic-induced failing heart provides the potential for a synergistic decrease in cardiac function that could occur in either the monomeric or dimerized state. It also suggests that enhanced opioidergic function could be the product of receptor heterodimerization, which has been shown to alter ligand binding properties and receptor signaling [27]. This is of further importance as the common opioidergic analgesic morphine (which is used clinically to alleviate breathlessness in heart failure patients [37]) has significant activities at both these receptors [14], and thus could produce a synergistic decrease in cardiac function in heart failure patients. A profound opioid-induced decrease in heart function in a patient population that already exhibits compromised cardiac output could have drastic effects, even acutely, including hypotension and cardiogenic shock. This is especially true in patients who also use β-blockers as opioid agonists have been shown to inhibit adrenergic function at agonist concentrations below that which induced a response alone [38].
In conclusion, this study was the first to show increased ability of myocardial opioid receptors to decrease cardiac function in the failing heart caused by genetically-derived cardiomyopathy. Furthermore, the enhanced cardiac opioidergic response in the cardiomyopathic, failing heart is PTX-sensitive, mediated by inhibition of cAMP accumulation and the intracellular calcium transient, and involves both the KOR and DOR. These findings have direct clinical significance as opioids are a common therapy for pain including in populations at risk for cardiovascular disease. This is particularly true as opioid agonists have recently been suggested for use to treat arthritis [39] and an AHA recommendation proposed opioids be considered firstline treatment for management of chronic pain in patient populations at risk for or having known cardiovascular disease [40]. As the cardiac depressant effects of opioid agonists are enhanced in cardiomyopathic-derived heart failure, it may prove detrimental to administer such an agent to a patient population that already has compromised cardiac function. This treatment option may induce severe hypotension that could lead to cardiogenic shock in the acute setting and may exacerbate the rate or degree of cardiac pathology when administered chronically. Therefore, the direct cardiac effects of opioid/opiate treatment must be considered when these agents are administered clinically, particularly in patients at risk for or having known cardiovascular disease.
Acknowledgments
This work was supported by grants from the American Heart Association (SDG 23004N), the Pharmaceutical Research and Manufacturers of America (Research Starter Grant), NIH/NHLBI R01 (HL075633) and NIH/NIDA R21 (DA0804) to Jo El J. Schultz.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jurynec J. Hypertrophic cardiomyopathy: a review of etiology and treatment. J Cardiovasc Nurs. 2007;22(1):65–73. doi: 10.1097/00005082-200701000-00010. quiz 74-5. [DOI] [PubMed] [Google Scholar]
- 2.Tsubata S, Bowles KR, Vatta M, Zintz C, Titus J, Muhonen L, Bowles NE, Towbin JA. Mutations in the human delta-sarcoglycan gene in familial and sporadic dilated cardiomyopathy. J Clin Invest. 2000;106(5):655–62. doi: 10.1172/JCI9224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawada T, Nakatsuru Y, Sakamoto A, Koizumi T, Shin WS, Okai-Matsuo Y, Suzuki J, Uehara Y, Nakazawa M, Sato H, Ishikawa T, Toyo-oka T. Strain- and age-dependent loss of sarcoglycan complex in cardiomyopathic hamster hearts and its re-expression by delta-sarcoglycan gene transfer in vivo. FEBS Lett. 1999;458(3):405–8. doi: 10.1016/s0014-5793(99)01164-3. [DOI] [PubMed] [Google Scholar]
- 4.Bajusz E. Hereditary cardiomyopathy: a new disease model. Am Heart J. 1969;77(5):686–96. doi: 10.1016/0002-8703(69)90556-0. [DOI] [PubMed] [Google Scholar]
- 5.Kasper E, Ventura C, Ziman BD, Lakatta EG, Weisman H, Capogrossi MC. Effect of U-50, 488H on the contractile response of cardiomyopathic hamster ventricular myocytes. Life Sci. 1992;50(26):2029–35. doi: 10.1016/0024-3205(92)90568-a. [DOI] [PubMed] [Google Scholar]
- 6.Sen LY, O’Neill M, Marsh JD, Smith TW. Inotropic and calcium kinetic effects of calcium channel agonist and antagonist in isolated cardiac myocytes from cardiomyopathic hamsters. Circ Res. 1990;67(3):599–608. doi: 10.1161/01.res.67.3.599. [DOI] [PubMed] [Google Scholar]
- 7.Barron BA. Cardiac opioids. Proc Soc Exp Biol Med. 2000;224(1):1–7. doi: 10.1046/j.1525-1373.2000.22358.x. [DOI] [PubMed] [Google Scholar]
- 8.Ventura C, Pintus Gianfranco, Tadolini Bruna. Opioid Peptide Gene Expression in the Myocardial Cell. TCM. 1998;8(3):102–10. doi: 10.1016/S1050-1738(97)00140-0. [DOI] [PubMed] [Google Scholar]
- 9.Feuerstein G, Siren AL. The opioid peptides. A role in hypertension? Hypertension. 1987;9(6):561–5. doi: 10.1161/01.hyp.9.6.561. [DOI] [PubMed] [Google Scholar]
- 10.Ventura C, Pintus G, Fiori MG, Bennardini F, Pinna G, Gaspa L. Opioid peptide gene expression in the primary hereditary cardiomyopathy of the Syrian hamster. I. Regulation of prodynorphin gene expression by nuclear protein kinase C. J Biol Chem. 1997;272(10):6685–92. doi: 10.1074/jbc.272.10.6685. [DOI] [PubMed] [Google Scholar]
- 11.Ouellette M, Brakier-Gingras L. Increase in the relative abundance of preproenkephalin A messenger RNA in the ventricles of cardiomyopathic hamsters. Biochem Biophys Res Commun. 1988;155(1):449–54. doi: 10.1016/s0006-291x(88)81107-0. [DOI] [PubMed] [Google Scholar]
- 12.Bolte C, Newman G, Schultz Jel J. Hypertensive state, independent of hypertrophy, exhibits an attenuated decrease in systolic function on cardiac kappa-opioid receptor stimulation. Am J Physiol Heart Circ Physiol. 2009;296(4):H967–75. doi: 10.1152/ajpheart.00909.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knapp RJ, Landsman R, Waite S, Malatynska E, Varga E, Haq W, Hruby VJ, Roeske WR, Nagase H, Yamamura HI. Properties of TAN-67, a nonpeptidic delta-opioid receptor agonist, at cloned human delta- and mu-opioid receptors. Eur J Pharmacol. 1995;291(2):129–34. doi: 10.1016/0922-4106(95)90134-5. [DOI] [PubMed] [Google Scholar]
- 14.Raynor K, Kong H, Chen Y, Yasuda K, Yu L, Bell GI, Reisine T. Pharmacological characterization of the cloned kappa-, delta-, and mu-opioid receptors. Mol Pharmacol. 1994;45(2):330–4. [PubMed] [Google Scholar]
- 15.Zimlichman R, Gefel D, Eliahou H, Matas Z, Rosen B, Gass S, Ela C, Eilam Y, Vogel Z, Barg J. Expression of opioid receptors during heart ontogeny in normotensive and hypertensive rats. Circulation. 1996;93(5):1020–5. doi: 10.1161/01.cir.93.5.1020. [DOI] [PubMed] [Google Scholar]
- 16.Hefti MA, Harder BA, Eppenberger HM, Schaub MC. Signaling pathways in cardiac myocyte hypertrophy. J Mol Cell Cardiol. 1997;29(11):2873–92. doi: 10.1006/jmcc.1997.0523. [DOI] [PubMed] [Google Scholar]
- 17.Clark MJ, Harrison C, Zhong H, Neubig RR, Traynor JR. Endogenous RGS protein action modulates mu-opioid signaling through Galphao. Effects on adenylyl cyclase, extracellular signal-regulated kinases, and intracellular calcium pathways. J Biol Chem. 2003;278(11):9418–25. doi: 10.1074/jbc.M208885200. [DOI] [PubMed] [Google Scholar]
- 18.Standifer KM, Pasternak GW. G proteins and opioid receptor-mediated signalling. Cell Signal. 1997;9(3–4):237–48. doi: 10.1016/s0898-6568(96)00174-x. [DOI] [PubMed] [Google Scholar]
- 19.Ventura C, Spurgeon H, Lakatta EG, Guarnieri C, Capogrossi MC. Kappa and delta opioid receptor stimulation affects cardiac myocyte function and Ca2+ release from an intracellular pool in myocytes and neurons. Circ Res. 1992;70(1):66–81. doi: 10.1161/01.res.70.1.66. [DOI] [PubMed] [Google Scholar]
- 20.Ventura C, Muscari C, Spampinato S, Bernardi P, Caldarera CM. Effects of naloxone on the mechanical activity of isolated rat hearts perfused with morphine or opioid peptides. Peptides. 1987;8(4):695–9. doi: 10.1016/0196-9781(87)90045-3. [DOI] [PubMed] [Google Scholar]
- 21.Schultz JE, Hsu AK, Barbieri JT, Li PL, Gross GJ. Pertussis toxin abolishes the cardioprotective effect of ischemic preconditioning in intact rat heart. Am J Physiol. 1998;275(2 Pt 2):H495–500. doi: 10.1152/ajpheart.1998.275.2.H495. [DOI] [PubMed] [Google Scholar]
- 22.Schultz JE, Gross GJ. Opioids and cardioprotection. Pharmacol Ther. 2001;89(2):123–37. doi: 10.1016/s0163-7258(00)00106-6. [DOI] [PubMed] [Google Scholar]
- 23.Fryer RM, Wang Y, Hsu AK, Gross GJ. Essential activation of PKC-delta in opioid-initiated cardioprotection. Am J Physiol Heart Circ Physiol. 2001;280(3):H1346–53. doi: 10.1152/ajpheart.2001.280.3.H1346. [DOI] [PubMed] [Google Scholar]
- 24.Bian JS, Zhang WM, Pei JM, Wong TM. The role of phosphodiesterase in mediating the effect of protein kinase C on cyclic AMP accumulation upon kappa-opioid receptor stimulation in the rat heart. J Pharmacol Exp Ther. 2000;292(3):1065–70. [PubMed] [Google Scholar]
- 25.Peart JN, Gross GJ. Cardioprotective effects of acute and chronic opioid treatment are mediated via different signaling pathways. Am J Physiol Heart Circ Physiol. 2006;291(4):H1746–53. doi: 10.1152/ajpheart.00233.2006. [DOI] [PubMed] [Google Scholar]
- 26.Angers S, Salahpour A, Bouvier M. Dimerization: an emerging concept for G protein-coupled receptor ontogeny and function. Annu Rev Pharmacol Toxicol. 2002;42:409–35. doi: 10.1146/annurev.pharmtox.42.091701.082314. [DOI] [PubMed] [Google Scholar]
- 27.Jordan BA, Devi LA. G-protein-coupled receptor heterodimerization modulates receptor function. Nature. 1999;399(6737):697–700. doi: 10.1038/21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang P, Xu W, Yoon SI, Chen C, Chong PL, Liu-Chen LY. Cholesterol reduction by methyl-beta-cyclodextrin attenuates the delta opioid receptor-mediated signaling in neuronal cells but enhances it in non-neuronal cells. Biochem Pharmacol. 2007;73(4):534–49. doi: 10.1016/j.bcp.2006.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hailstones D, Sleer LS, Parton RG, Stanley KK. Regulation of caveolin and caveolae by cholesterol in MDCK cells. J Lipid Res. 1998;39(2):369–79. [PubMed] [Google Scholar]
- 30.Pogue DH, Moravec CS, Roppelt C, Disch CH, Cressman MD, Bond M. Effect of lovastatin on cholesterol content of cardiac and red blood cell membranes in normal and cardiomyopathic hamsters. J Pharmacol Exp Ther. 1995;273(2):863–9. [PubMed] [Google Scholar]
- 31.Hnasko R, Lisanti MP. The biology of caveolae: lessons from caveolin knockout mice and implications for human disease. Mol Interv. 2003;3(8):445–64. doi: 10.1124/mi.3.8.445. [DOI] [PubMed] [Google Scholar]
- 32.Fontana F, Bernardi P, Pich EM, Capelli M, Bortoluzzi L, Spampinato S, Canossa M. Relationship between plasma atrial natriuretic factor and opioid peptide levels in healthy subjects and in patients with acute congestive heart failure. Eur Heart J. 1993;14(2):219–25. doi: 10.1093/eurheartj/14.2.219. [DOI] [PubMed] [Google Scholar]
- 33.Yatani A, Imai N, Himura Y, Suematsu M, Liang CS. Chronic opiate-receptor inhibition in experimental congestive heart failure in dogs. Am J Physiol. 1997;272(1 Pt 2):H478–84. doi: 10.1152/ajpheart.1997.272.1.H478. [DOI] [PubMed] [Google Scholar]
- 34.Redfern CH, Degtyarev MY, Kwa AT, Salomonis N, Cotte N, Nanevicz T, Fidelman N, Desai K, Vranizan K, Lee EK, Coward P, Shah N, Warrington JA, Fishman GI, Bernstein D, Baker AJ, Conklin BR. Conditional expression of a Gi-coupled receptor causes ventricular conduction delay and a lethal cardiomyopathy. Proc Natl Acad Sci U S A. 2000;97(9):4826–31. doi: 10.1073/pnas.97.9.4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bers DM, Despa S. Cardiac myocytes Ca2+ and Na+ regulation in normal and failing hearts. J Pharmacol Sci. 2006;100(5):315–22. doi: 10.1254/jphs.cpj06001x. [DOI] [PubMed] [Google Scholar]
- 36.Aitchison KA, Baxter GF, Awan MM, Smith RM, Yellon DM, Opie LH. Opposing effects on infarction of delta and kappa opioid receptor activation in the isolated rat heart: implications for ischemic preconditioning. Basic Res Cardiol. 2000;95(1):1–10. doi: 10.1007/s003950050001. discussion 11. [DOI] [PubMed] [Google Scholar]
- 37.Johnson MJ, McDonagh TA, Harkness A, McKay SE, Dargie HJ. Morphine for the relief of breathlessness in patients with chronic heart failure--a pilot study. Eur J Heart Fail. 2002;4(6):753–6. doi: 10.1016/s1388-9842(02)00158-7. [DOI] [PubMed] [Google Scholar]
- 38.Pepe S, Xiao RP, Hohl C, Altschuld R, Lakatta EG. ‘Cross talk’ between opioid peptide and adrenergic receptor signaling in isolated rat heart. Circulation. 1997;95(8):2122–9. doi: 10.1161/01.cir.95.8.2122. [DOI] [PubMed] [Google Scholar]
- 39.Gibofsky A, Barkin RL. Chronic pain of osteoarthritis: considerations for selecting an extended-release opioid analgesic. Am J Ther. 2008;15(3):241–55. doi: 10.1097/MJT.0b013e3181727f68. [DOI] [PubMed] [Google Scholar]
- 40.Antman EM, Bennett JS, Daugherty A, Furberg C, Roberts H, Taubert KA. Use of nonsteroidal antiinflammatory drugs: an update for clinicians: a scientific statement from the American Heart Association. Circulation. 2007;115(12):1634–42. doi: 10.1161/CIRCULATIONAHA.106.181424. [DOI] [PubMed] [Google Scholar]