

Abstract
Mouse models are useful for studying genes involved in behavior, but whether they are relevant for human behavior is unclear. Here, we identified parallel phenotypes in mice and humans resulting from a common single-nucleotide polymorphism in the brain-derived neurotrophic factor (BDNF) gene, which is involved in anxiety-related behavior. An inbred genetic knock-in mouse strain expressing the variant BDNF recapitulated the phenotypic effects of the human polymorphism. Both were impaired in extinguishing a conditioned fear response, which was paralleled by atypical frontoamygdala activity in humans. Thus, this variant BDNF allele may play a role in anxiety disorders showing impaired learning of cues that signal safety versus threat, and in the efficacy of treatments that rely on extinction mechanisms such as exposure therapy.
Genetically modified mice provide useful model systems for testing the role of candidate genes in behavior. The extent to which such genetic manipulations in the mouse and the resulting phenotype can be translated across species, from mouse to human, is less clear. In this report we focused on identifying biologically valid phenotypes across species. We utilized a common single nucleotide polymorphism (SNP) in the brain-derived neurotrophic factor (BDNF) gene that leads to a valine (Val) to methionine (Met) substitution at codon 66 (Val66Met). In an inbred genetic knock-in mouse strain that expresses the variant BDNF allele to recapitulate the specific phenotypic properties of the human polymorphism in vivo, we found the BDNF Val66Met genotype was associated with treatment resistant forms of anxiety-like behavior (1). The objective of this study was to test if the Val66Met genotype could impact extinction learning in our mouse model, and if such findings could be generalized to human populations.
BDNF mediates synaptic plasticity associated with learning and memory (2, 3) specifically in fear learning and extinction (4, 5). BDNF-dependent forms of fear learning have known biological substrates, and lie at the core of a number of clinical disorders (6, 7) associated with the variant BDNF (8–10). Fear learning paradigms require the ability to recognize and remember cues that signal safety or threat and to extinguish these associations when they no longer exist. These abilities are impaired in anxiety disorders such as posttraumatic stress disorder and phobias (11, 12). Behavioral treatments for these disorders such as exposure therapy rely on basic principles of extinction learning (13) in which an individual is repeatedly exposed to an event that was previously associated with aversive consequences. Understanding the effect of the BDNF Met allele on these forms of learning can provide insight into the mechanism of risk for anxiety disorders, refine existing treatments, and may lead to genotype-based personalized medicine.
We examined the impact of the variant BDNF on classic fear conditioning and extinction paradigms adapted to be suitable for each species and that are associated with well known underlying biological substrates (14, 15, 16). Fear conditioning consisted of pairing a neutral cue with an aversive stimulus. With repeated pairings, the cue itself takes on properties of the aversive stimulus as it predicts threat of an impending aversive event. Extinction consisted of presenting the cue alone following conditioning, whereby the association is diminished with repeated exposure to empty threat.
We tested 68 mice (17 BDNFVal/Val, 33 BDNFVal/Met and 18 BDNFMet/Met) and 72 humans group-matched on age, gender and ethnic background (36 Met allele carriers: 31 BDNFVal/Met and 5 BDNFMet/Met, and 36 nonMet allele carriers: BDNFVal/Val –Table S1). We found no effect of the BDNF Met allele on fear conditioning in mice as measured by percentage of time spent freezing in response to the conditioned stimulus (F(2,65) = 1.58, p < 0.22) (Fig. S1A) or on general fear arousal as measured by freezing during the intertrial interval (ITI) (Fig. S2). We grouped human Met allele carriers together (Val/Met and Met/Met) for analyses because the rarity of human Met allele homozygotes prevents enough observations for meaningful analysis. Similar to the mouse findings, we found no effect of the BDNF Met allele on fear conditioning in humans as measured by skin conductance response to the cue predicting the aversive stimulus relative to a neutral cue (F(1,70) = 0.67, p < 0.42) (Fig. S1B).
Analysis of extinction trials showed a main effect of genotype for both mice ((F(2,65) = 6.55, p < 0.003); Val/Val: 48.8 ± 2.3; Val/Met: 53.2 ± 1.8; Met/Met: 61.3 ± 2.8) and humans ((F(1,70) = 4.86, p < 0.03); Val/Val: 0.32 ± 0.03; Val/Met: 0.42 ± 0.04), such that extinction learning was impaired in Met allele carriers relative to nonMet allele carriers. The Met allele carriers showed slower extinguishing as indicated by an interaction of time X genotype for the mouse (F(2,65) = 6.51, p < 0.003) (Fig. 1) with no differences in freezing initially, but a dose response of the Met allele on percent freezing behavior during late trials (Val/Val vs Val/Met: t(48) = − 2.62, p < 0.01; Val/Val vs Met/Met: t(33) = −4.78, p < 0.0001; Val/Met vs Met/Met: t(49) = −2.90, p < 0.006) (Fig. 1A). Humans showed a similar pattern to the mice with no genotypic difference in the initial human skin conductance response during early trials of extinction (t(70) = −1.57, p < 0.12), but significant differences by late trials (t(70) = −2.43, p < 0.02, corrected for time) (Fig. 1B). These data demonstrate slower or impaired extinction related to the Met allele in both mouse and human.
Figure 1. Altered extinction in mice and humans with BDNF Val66Met.

Impaired extinction in Met allele carriers (Val/Met and Met/Met) as a function of time in 68 mice (A) and 72 humans (B) as indexed by percent time freezing in mice and skin conductance response (SCR) in humans to the conditioned stimulus when it was no longer paired with the aversive stimulus. All results are presented as a mean ± SEM. *p < 0.01, Student’s t test. **p < 0.02, Student’s t test. VV = Val/Val; VM = Val/Met; MM= Met/Met.
The learning paradigm for humans included a conditioned stimulus paired with the aversive stimulus and a neutral stimulus that was not paired with the aversive stimulus. This design allowed for distinguishing between effects due to impaired learning versus a general effect of heightened anxiety, as generalized heightened anxiety would lead to a similar response to both the conditioned and neutral cues. Met allele carriers had an overall heightened response to both conditioned and neutral cues [main effect of genotype (F (1,70) = 7.21, p < 0.009)], but overall differentiated between the conditioned and neutral cues similar to the nonMet allele carriers (Fig. S1B). Yet, when examining these effects over time, Met allele carriers took longer to recognize that the neutral cue was not associated with the aversive stimulus, as evidenced by significant genotypic differences during late trials (t(70) = −3.46, p < 0.001, corrected for time) but not early trials (t(70) = −1.44, p < 0.16) (Fig. 2). Thus, the skin conductance response to the neutral cue during fear conditioning, showed a similar pattern as that observed during extinction trials (16).
Figure 2. Impaired learning of neutral cue in human Met allele carriers.
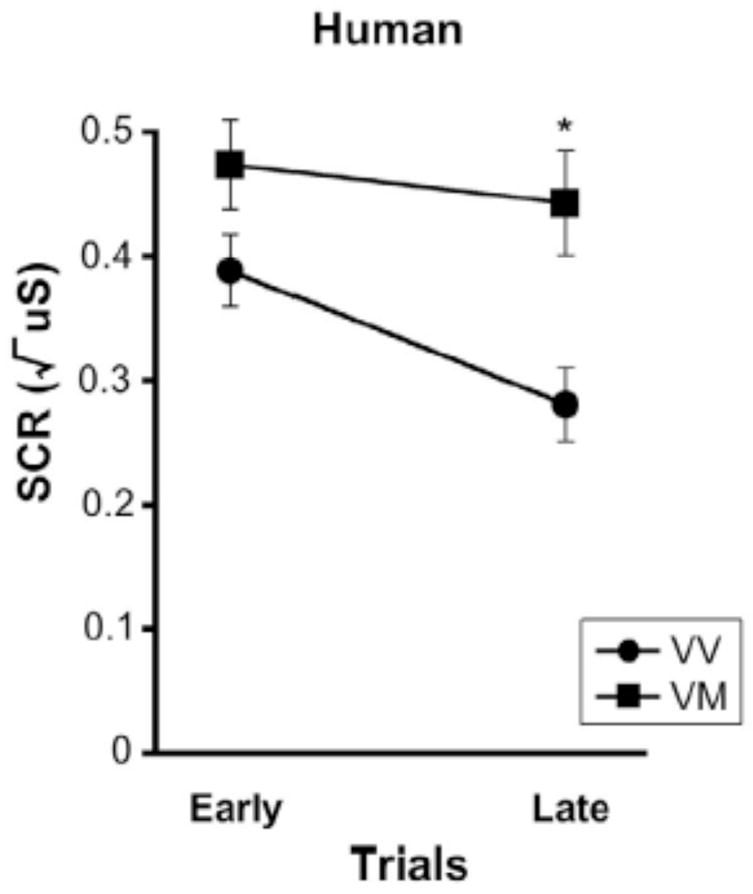
Elevated skin conductance response (SCR) to the cue never paired with the aversive stimulus during fear conditioning as a function of time in Met allele carriers (VM) relative to nonMet allele carriers (VV). All results are presented as a mean ± SEM. *p < 0.001, Student’s t test. VV = Val/Val; VM = Val/Met.
The genetic findings for both fear conditioning and extinction suggest that learning about cues that signal threat of an impending aversive event is intact in Met allele carriers. However, learning that cues no longer signal threat (e.g., extinction) or do not predict threat (cues not paired with an aversive stimulus) is impaired in Met allele carriers, leading to exaggerated and longer retention of aversive responses where they are not warranted.
To provide neuroanatomical evidence to validate of our cross-species translation, we used human functional magnetic resonance imaging (MRI) to define the underlying neural circuitry of the behavioral effects of BDNF Val66Met and map them to known circuits involved in fear learning in the rodent (Table S2). We targeted frontoamygdala circuitry that has been demonstrated to support fear conditioning and extinction in both rodent (17–20) and human (21–26) studies. Whereas portions of the amygdala have been shown to be essential for fear conditioning (27, 28), ventral prefrontal cortical regions have been shown to be important for modifying previously learned associations and extinction (19, 29). Thus, based on our behavioral findings in the mouse and human, we hypothesized that ventromedial prefrontal regions, important in extinction, would be less active in Met allele carriers relative to nonMet allele carriers and that amygdala activity may be enhanced.
To test this hypothesis we examined the main effect of genotype on brain activity during extinction of the previously conditioned stimulus. The analysis directly parallels the observed behavioral main effects of genotype on extinction as measured by mean percent time freezing in the mice (Fig. 3A) and mean skin conductance response in humans (Fig. 3B) (16) with Met allele carriers showing weaker extinction. The imaging results showed significantly less ventromedial prefrontal cortical (vmPFC) activity during extinction in Met allele carriers relative to nonMet allele carriers (t(68) = −3.78, p < 0.05, corrected), (Fig. 3C) (16). In contrast, Met allele carriers show greater amygdala activity relative to nonMet allele carriers during extinction (t(68) = 2.23, p < 0.05, corrected) (Fig. 3D). These findings indicate that cortical regions previously shown to be essential for extinction (vmPFC) in both rodent and human (19, 26, 30) are hyporesponsive in Met allele carriers relative to nonMet allele carriers. Moreover, Met allele carriers show continued recruitment of the amygdala, a region that should show diminished activity during the extinction trials of the experiment (26). These findings are most likely due to the SNP biasing activity-dependent learning rather than affecting CNS development per se, as there was no evidence of genotypic developmental effects on brain structure in this ethnicity-, age- and gender- matched sample using MRI-based brain morphometry (supporting online text). Furthermore, an association between vmPFC activity and strength of fibers connecting frontolimbic regions is consistent with more effective extinction learning as a result of better vmPFC modulation of the amygdala (supporting online text, Fig. S3 and S4).
Figure 3. Neural circuitry of the behavioral effect of BDNF Val66Met during extinction.

(A) Average percent freezing during extinction by genotype in 68 mice. (B) Average skin conductance response (SCR) during extinction by genotype in 72 humans. (C) Brain activity as indexed by percent change in MR signal during extinction in the ventromedial prefrontal cortex (vmPFC) by genotype (xyz = −4, 24, 3), with Met allele carriers having significantly less activity than Val/Val homozygotes [VM < VV = blue], image threshold p < 0.05, corrected. (D) Genotypic differences in left amygdala activity during extinction (xyz = −25, 2, −20) in 70 humans, with Met allele carriers having significantly greater activity than Val/Val homozygotes [VM > VV = orange], image threshold p < 0.05, corrected. *p < 0.05. **MM were included in the analysis with VM, but plotted separately to see dose response. All results are presented as a mean ± SEM. VV = Val/Val; VM = Val/Met; MM = Met/Met; MR = magnetic resonance.
These experiments identify a behavioral phenotype related to BDNF Val66Met across species providing evidence for translation from mouse to human. The mouse model provides the opportunity to test dose-dependent effects of the BDNF Met allele in both a controlled genetic and environmental background not feasible in humans. These features allow for reliable assignment of behavioral differences to the effects of the Val66Met polymorphism. The human behavioral and imaging findings provide confidence that cross-species translation is biologically valid, by defining the underlying neural circuitry of the behavioral effects of BDNF Val66Met that can be mapped onto known circuits involved in fear learning and extinction. The robustness of our findings across species and paradigms is evidenced by work showing slower extinction coupled with decreased neuronal dendritic complexity in vmPFC in the BDNFMet/Met mice in a conditioned taste aversion task compared with wild-type counterparts (31). Furthermore BDNFMet/Met mice exhibit a trend towards blunted expression of c-Fos in the vmPFC as compared to wildtype mice after the fear extinction paradigm (supporting online text, Fig. S5).
Impaired extinction learning has been implicated in anxiety disorders, including phobias and posttraumatic stress disorder, whereby the individual has difficulty recognizing an event as safe (32). Our neuroimaging findings of diminished ventromedial prefrontal activity and elevated amygdala activity during extinction are reminiscent of those reported in patients with anxiety disorders and depression when presented with empty threat or aversive stimuli (e.g., fearful faces) (33, 34). Understanding the effect of the BDNF Met allele on specific components of a simple form of learning provides insight into risk for anxiety disorders and has important implications for the efficacy of treatments for these disorders that rely on extinction mechanisms. One such treatment is exposure therapy whereby an individual is repeatedly exposed to a traumatic event in order to diminish the significance of that event. Our findings suggest that the BDNF Val66Met SNP may play a key role in the efficacy of such treatments and may ultimately guide personalized medicine for related clinical disorders.
Supplementary Material
Acknowledgments
We acknowledge two anonymous reviewers for their thoughtful comments, resources and staff at the Biomedical Imaging Core Facility of the Citigroup Biomedical Imaging Center at Weill Cornell Medical College, Rafael Oania, a generous gift by the Dr. Mortimer D. Sackler family and support from NIH grants MH079513 (BJC, FSL), MH060478 (BJC), NS052819 (FSL), HD055177 (BJC, SSP), GM07739 (FS), and UNCF/Merck Graduate Science Research Dissertation Fellowship (FS), Burroughs Wellcome Foundation (FSL), International Mental Health Research Organization (FSL).
References and Notes
- 1.Chen ZY, et al. Science. 2006 Oct 6;314:140. doi: 10.1126/science.1129663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Egan MF, et al. Cell. 2003 Jan 24;112:257. doi: 10.1016/s0092-8674(03)00035-7. [DOI] [PubMed] [Google Scholar]
- 3.Hariri AR, et al. J Neurosci. 2003 Jul 30;23:6690. doi: 10.1523/JNEUROSCI.23-17-06690.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chhatwal JP, Stanek-Rattiner L, Davis M, Ressler KJ. Nat Neurosci. 2006 Jul;9:870. doi: 10.1038/nn1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rattiner LM, Davis M, Ressler KJ. Neuroscientist. 2005 Aug;11:323. doi: 10.1177/1073858404272255. [DOI] [PubMed] [Google Scholar]
- 6.Charney DS, Manji HK. Sci STKE 2004. 2004 Mar 23;:re5. doi: 10.1126/stke.2252004re5. [DOI] [PubMed] [Google Scholar]
- 7.Nestler EJ, et al. Neuron. 2002 Mar 28;34:13. [Google Scholar]
- 8.Gratacos M, et al. Biol Psychiatry. 2007 Apr 1;61:911. doi: 10.1016/j.biopsych.2006.08.025. [DOI] [PubMed] [Google Scholar]
- 9.Gatt JM, et al. Biol Psychol. 2008 Oct;79:275. doi: 10.1016/j.biopsycho.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 10.Jiang X, et al. Neuropsychopharmacology. 2005 Jul;30:1353. doi: 10.1038/sj.npp.1300703. [DOI] [PubMed] [Google Scholar]
- 11.Rauch SL, Shin LM, Phelps EA. Biol Psychiatry. 2006 Aug 15;60:376. doi: 10.1016/j.biopsych.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 12.Lissek S, et al. Behav Res Ther. 2005 Nov;43:1391. doi: 10.1016/j.brat.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 13.Rothbaum BO, Davis M. Ann N Y Acad Sci. 2003 Dec;1008:112. doi: 10.1196/annals.1301.012. [DOI] [PubMed] [Google Scholar]
- 14.Delgado MR, Olsson A, Phelps EA. Biol Psychol. 2006 Jul;73:39. doi: 10.1016/j.biopsycho.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 15.Sotres-Bayon F, Cain CK, LeDoux JE. Biol Psychiatry. 2006 Aug 15;60:329. doi: 10.1016/j.biopsych.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 16.Materials and methods are available as supporting material on Science Online.
- 17.Myers KM, Davis M. Neuron. 2002 Nov 14;36:567. doi: 10.1016/s0896-6273(02)01064-4. [DOI] [PubMed] [Google Scholar]
- 18.Quirk GJ, Likhtik E, Pelletier JG, Pare D. J Neurosci. 2003 Sep 24;23:8800. doi: 10.1523/JNEUROSCI.23-25-08800.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Milad MR, Quirk GJ. Nature. 2002 Nov 7;420:70. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- 20.LeDoux JE. Annu Rev Neurosci. 2000;23:155. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- 21.LaBar KS, Gatenby JC, Gore JC, LeDoux JE, Phelps EA. Neuron. 1998 May;20:937. doi: 10.1016/s0896-6273(00)80475-4. [DOI] [PubMed] [Google Scholar]
- 22.Schiller D, Levy I, Niv Y, LeDoux JE, Phelps EA. J Neurosci. 2008 Nov 5;28:11517. doi: 10.1523/JNEUROSCI.2265-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Delgado MR, Nearing KI, LeDoux JE, Phelps EA. Neuron. 2008 Sep 1;59:829. doi: 10.1016/j.neuron.2008.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalisch R, et al. J Neurosci. 2006 Sep 13;26:9503. doi: 10.1523/JNEUROSCI.2021-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gottfried JA, Dolan RJ. Nat Neurosci. 2004 Oct;7:1144. doi: 10.1038/nn1314. [DOI] [PubMed] [Google Scholar]
- 26.Phelps EA, Delgado MR, Nearing KI, LeDoux JE. Neuron. 2004 Sep 16;43:897. doi: 10.1016/j.neuron.2004.08.042. [DOI] [PubMed] [Google Scholar]
- 27.Nader K, LeDoux J. Behav Neurosci. 1999 Feb;113:152. doi: 10.1037//0735-7044.113.1.152. [DOI] [PubMed] [Google Scholar]
- 28.Kim M, Davis M. Behav Neurosci. 1993 Aug;107:580. doi: 10.1037//0735-7044.107.4.580. [DOI] [PubMed] [Google Scholar]
- 29.Lebron K, Milad MR, Quirk GJ. Learn Mem. 2004 Sep-Oct;11:544. doi: 10.1101/lm.78604. [DOI] [PubMed] [Google Scholar]
- 30.Milad MR, et al. Biol Psychiatry. 2007 Sep 1;62:446. doi: 10.1016/j.biopsych.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 31.Yu H, et al. J Neurosci. 2009 Apr 1;29:4056. doi: 10.1523/JNEUROSCI.5539-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Charney DS. Am J Psychiatry. 2004 Feb;161:195. doi: 10.1176/appi.ajp.161.2.195. [DOI] [PubMed] [Google Scholar]
- 33.Liberzon I, et al. Biol Psychiatry. 1999 Apr 1;45:817. doi: 10.1016/s0006-3223(98)00246-7. [DOI] [PubMed] [Google Scholar]
- 34.Rauch SL, et al. Biol Psychiatry. 2000 May 1;47:769. doi: 10.1016/s0006-3223(00)00828-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.