

Abstract
Gene transfer to murine liver with vectors based on novel adeno-associated virus (AAV) serotypes is efficient, stable, and safe even in the setting of antigenic transgene products. We undertook a study in cynomolgus macaques to evaluate the relevance of these findings to primates. The vectors were based on AAV serotype 7 and expressed green fluorescence protein (GFP) from the cytomegalovirus enhanced β-actin promoter in both single-stranded and self-complementary genomes. Transduction efficiencies from the single-stranded vectors were similar to those observed in mice, although there was no advantage in primates with the self-complementary vectors. Primates elicited vibrant cytotoxic T cell responses to GFP that correlated with hepatitis and loss of transgene expression. There was no evidence of T cell activation in response to the AAV capsid. These studies indicate that under some conditions primates may activate more robust T cell responses to transgene products than is observed in mice.
Introduction
Liver has been considered an attractive target for gene transfer in the treatment of a variety of inherited and acquired diseases. Both ex vivo and in vivo approaches have been evaluated in preclinical and clinical models (Wilson et al., 1990; Grossman et al., 1992, 1994, 1995; Raper et al., 2002; Manno et al., 2006). Several aspects of hepatic biology impact on the design of liver-directed gene therapy. Liver serves as a site for control of many metabolic pathways and the production of several plasma proteins. Large fenestrae in the endothelium of the liver allow easy access of gene transfer vectors to hepatocytes after systemic administration. The liver is believed to be immunologically tolerant in nonpathologic conditions because it serves as a filter for potentially antigenic material absorbed from the gut into the mesenteric circulation.
The first attempts to deliver genes to the liver in vivo were based on recombinant adenoviruses, which in mice proved to be quite successful, transducing the majority of hepatocytes and correcting a variety of models of human diseases (Stratford-Perricaudet et al., 1990). The primary toxicity in mice was hepatitis due to activation of cytotoxic T lymphocytes (CTLs) in response to vector-encoded products (i.e., antigenic transgenes and viral open reading frames) (Yang et al., 1994). This toxicity was diminished by deleting some or all of the remaining viral open reading frames (ORFs) in third-generation and helper-dependent constructs (Gao et al., 1996; Segura et al., 2008). Clinical trials of adenovirus-mediated gene transfer to liver were first conducted in adults with liver cancer and the urea cycle defect ornithine transcarbamylase deficiency (OTCD) (Raper et al., 2002). A research subject in the OTCD trial experienced an unexpected and severe systemic inflammatory response to the vector and died of multiorgan failure. Further evaluation of this serious adverse event suggested the capsid proteins of the vector activated innate immunity (Schnell et al., 2001; Zhang et al., 2001; Raper et al., 2003). A subsequent clinical trial with a helper-dependent adenoviral vector in a hemophilia A subject elicited a similar but nonlethal inflammatory response, confirming that the capsid proteins alone are capable of activating innate immunity (Chuah et al., 2004). Several key questions remain unexplained, including why this adverse event was not predicted on the basis of animal studies, and why the response in the OTCD subject was so exaggerated in comparison with others in the trial.
Encouraging results have emerged from preclinical studies of AAV gene transfer to liver. Low-level but stable transgene expression has been demonstrated in mouse liver with vectors based on AAV serotype 2 encoding a variety of self and foreign transgenes (Xiao et al., 1998; Alexander et al., 2008). Several mechanisms have been proposed to explain the immunologic nonresponsiveness of mice to antigenic proteins expressed from AAV2 vectors including induction of regulatory T cells (T regs), anergy, and ignorance (Vandenberghe and Wilson, 2007; Zaiss and Muruve, 2008). Vectors based on AAV serotypes 7, 8, and 9 achieve much higher transduction efficiency in mouse liver than do AAV2 vectors while retaining favorable immunologic properties of tolerance to foreign transgenes in mice and low prevalence of neutralizing antibodies in humans (Gao et al., 2002; Gao et al., 2003, 2004, 2005; Calcedo et al., 2009). The improved efficiency of transduction with the novel AAVs observed in mice has been observed in larger animals such as dogs and nonhuman primates (NHPs), although the magnitude of the effects appears to be dependent on the transgene and the presence of preexisting antibodies (Davidoff et al., 2005; Gao et al., 2006a). Another approach for improving AAV transduction efficiency is to use genomes that are capable of self-annealing to overcome the need to replicate the input genome (called self-complementary [sc] vectors) (Hirata and Russell, 2000; McCarty et al., 2001; Wang et al., 2003; Gao et al., 2006b; Nathwani et al., 2006).
A clinical trial of liver-directed gene transfer was conducted with vectors based on AAV serotype 2 in subjects with hemophilia B. Several subjects who received the highest dose of vector demonstrated self-limited hepatitis that appeared at the time transgene expression was diminishing. The concurrent appearance of T cells to the AAV capsid led the investigators to propose that the input capsid is cross-presented into the MHC class I pathway to activate CTLs and to render transduced hepatocytes targets for CTL-mediated clearance (Manno et al., 2006). Attempts to model this hypothesis in animal models have been unsuccessful. Irrespective of the mechanism, it is concerning that the basic phenomenon of hepatitis, loss of transgene expression, and appearance of capsid T cells has not been observed after AAV gene transfer in any animal model including NHPs (Vandenberghe and Wilson, 2007; Zaiss and Muruve, 2008).
The goal of this study was to critically assess the biology of vectors based on AAV serotype 7 in NHP liver in terms of transduction efficiency with regular and sc genomes and to evaluate T cell responses to a foreign transgene product (green fluorescent protein [GFP]) and the input capsid proteins.
Materials and Methods
AAV vector production
The conventional single-stranded (ss) ssAAV2/7CBEGFP [with chicken β-actin [CB] promoter, woodchuck hepatitis virus posttranscriptional regulatory element [WPRE], and bovine growth hormone poly(A)], self-complementary (sc) scAAV2/7CBEGFP [with CB promoter and simian virus 40 (SV40) poly(A)], and sc1AAV2/7CBEGFP [with CB promoter and bovine growth hormone poly(A)] vectors were produced and purified by CsCl gradient sedimentation as previously described (Gao et al., 2006a). All vector preparations used for the NHP studies were subjected to extensive quality control tests including four repeated vector genome titrations by real-time polymerase chain reaction (PCR) (Applied Biosystems, Foster City, CA), sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) analysis for vector purity, Limulus amebocyte lysate (LAL) for endotoxin detection, and transgene expression analysis in mice. The assay for replication-competent AAV (rcAAV) in the AAV2/7 vector lots used for this study was carried out on 293 cells as previously described with modifications (Gao et al., 2000). Real-time PCR for quantification of DNase-resistant cap sequences in the vector lots was performed as previously described (Gao et al., 2003).
NHP experiments
Cynomolgus (Cyno) macaques (Philippine origin and captive bred, 2.10–3.85 kg; ssAAV2/7CBEGFP: 24499 and 24462; scAAV2/7CBEGFP: 24441, 24456, 24479 and 24481; sc1AAV2/7CBEGFP: 24466, 24527, and 24461) were treated and cared for at the NHP facility of the Gene Therapy Program of the University of Pennsylvania (Philadelphia, PA) during the study. The study was performed according to a study protocol approved by the Environmental Health and Radiation Safety Office, the Institutional Biosafety Committee, and the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania. Vectors were administered to the study animals intraportally at doses ranging from 0.7 × 1012 to 3.5 × 1012 GC/kg of animal weight as previously described (Table 1). Blood samples were taken weekly via venipuncture of the saphenous vein. Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood collected in EDTA-containing Vacutainer tubes after Percoll density-gradient centrifugation. Lymphocytes were isolated from bone marrow and liver by Percoll density-gradient centrifugation and from spleen, liver, and a variety of lymph nodes by crushing on 40-μm stainless steel meshes at the time of necropsies. At the time of necropsies (day 8: 24466, 24527, 24441, 24456, 24499, and 24462; day 37: 24461, 24479, and 24481), 11 tissues including the target tissue liver and 10 other distant organs (brain, bone marrow, colon, heart, gonadal tissue, kidney, lung, lymph nodes, small bowel, and spleen) were collected for histopathology and molecular analysis.
Table 1.
Summary of Study on AAV2/7CBEGFP Vector-Mediated Liver Gene Transfer to Cynomolgus Macaquesa
| |
|
Transcription |
|
|
|
|
|
||
|---|---|---|---|---|---|---|---|---|---|
| Animal no. | Vector | P | A+ | WPRE | Doseb(×1012) | Necropsy (day) | Percent GFP | GC/cell | 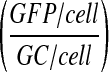 |
| 24466 | sc1 | CB | BGH | – | 3.5 | 8 | 9.1 ± 4.8 | 37 | 0.25 |
| 24527 | sc1 | CB | BGH | – | 0.7 | 8 | 0.07 ± 0.09 | 1 | 0.07 |
| 24441 | sc | CB | SV40 | – | 2.0 | 8 | 0.6 ± 0.3 | 11 | 0.006 |
| 24456 | sc | CB | SV40 | – | 2.0 | 8 | 4.1 ± 2.7 | 69 | 0.06 |
| 24499 | ss | CB | BGH | + | 3.5 | 8 | 10.1 ± 4.8 | 20 | 0.51 |
| 24462 | ss | CB | BGH | + | 3.5 | 8 | 24.5 ± 4.8 | 46 | 0.53 |
| 24461 | sc1 | CB | BGH | – | 3.5 | 37 | 0 | 2 | 0 |
| 24479 | sc | CB | SV40 | – | 2.0 | 37 | 0 | 4 | 0 |
| 24481 | sc | CB | SV40 | – | 2.0 | 37 | 0 | 27 | 0 |
Abbreviations: A+, polyadenylation signal; BGH, bovine growth hormone; CB, chicken β-actin; GC, genome copy; GFP, green fluorescent protein; P, promoter; sc, scAAV2/7.CBEGFP.SV40A+; sc1, scAAV2/7.CBEGFP.BGHA+; ss, ssAAV2/7.CBEGFP.WPRE.BGHA+; SV40, simian virus 40; WPRE, woodchuck hepatitis virus posttranscriptional regulatory element.
An estimate of activity of transduced genomes is attained by dividing the fraction of expressing hepatocytes by the copy number of genomes per cell. This is multiplied by 100 in the last column: [(GFP/cell)/(GC/cell)] × 100.
Dose expressed as GCs per kilogram of animal weight.
Sample analysis
All clinical pathology tests of blood samples were conducted by Laboratory Corporation of America (Burlington, NC), including complete blood counts and differentials, and complete clinical chemistries. The enzyme-linked immunospot (ELISPOT) assay was performed on either PBMCs or lymphocytes after stimulating with 15-mer peptide pools of either EGFP or AAV7 capsid protein, using anti-human monkey IFN-γ monoclonal antibody (mAb) GZ-4 as the coating antibody (Mabtech, Mariemont, OH), anti-human IFN-γ mAb 7-B6-1 as the detection antibody (Mabtech), horseradish peroxidase avidin-D as the third antibody (Vector Laboratories, Burlingame, CA), and an AEC substrate set for color development (BD Biosciences, San Jose, CA). Measurement of cytokine production by isolated lymphocytes was performed by combined surface and intracellular staining with monoclonal antibodies and subsequent five-color flow cytometric analysis. Lymphocytes (106) were incubated with GFP peptide library in the presence of purified antibodies to CD49d and CD28 (BD Biosciences) and brefeldin A (1 μg/ml) for 6 hr. Cells were washed and stained with energy-coupled dye (ECD)-conjugated anti-CD8 (Beckman Coulter, Fullerton, CA) or allophycocyanin (APC)-conjugated anti-CD4 (BD Biosciences). Cells were washed, permeabilized with 250 μl of Cytofix/Cytoperm solution at 4°C for 20 min, washed with Perm/Wash solution, and stained with anti-cytokine antibodies including fluorescein isothiocyanate (FITC)-conjugated anti-tumor necrosis factor (TNF)-α (BD Biosciences), phycoerythrin (PE)-conjugated anti-interleukin (IL)-2 (Beckman Coulter), and PE–cyanine 7 (Cy7)-conjugated anti-interferon (IFN)-γ (BD Biosciences) at 4°C for 30 min. Cells were washed and examined by flow cytometry (FC500; Beckman Coulter). Degranulation activity was measured with an FITC-labeled CD107a antibody (BD Biosciences), monensin (2 μM), and brefeldin A (1 μg/ml) at the time of stimulation. After 6 hr of stimulation, cells were stained with violet amine-reactive viability dye (ViViD)–Pacific Blue, Alexa 700-conjugated anti-CD4 (BD Biosciences), and Qdot 655-conjugated anti-CD8 (Department of Microbiology, University of Pennsylvania). Cells were washed, permeabilized with 250 μl of Cytofix/Cytoperm solution at 4°C for 20 min, washed with Perm/Wash solution, and stained with APC–Cy7-conjugated anti-CD3, APC-conjugated IFN-γ, and PE–Cy7-conjugated anti-TNF-α (BD Biosciences) at 4°C for 30 min. Cells were washed and examined by flow cytometry (LSRII; BD Biosciences). Data were analyzed with FlowJo software (Treestar, Ashland, OR).
To examine EGFP expression on liver sections, liver tissues harvested at necropsies were fixed in 10% formalin at room temperature overnight, frozen as O.C.T. compound (Sakura Finetek USA, Torrance, CA) blocks, sectioned at 8 μm and mounted on slides, and evaluated under a fluorescence microscope. To quantify expression levels of GFP in liver, for each animal 20 randomly chosen images from liver sections (5 images from each lobe) were taken at identical microscope and camera settings. The images were then analyzed with AnalySIS software (Soft Imaging System, Lakewood, CO) to determine the GFP-positive area of each image. Briefly, images were subjected to thresholding (i.e., determining the minimal brightness value that represents true GFP fluorescence) using GFP-negative control sections from untreated cynomolgus macaques as reference for background levels. The percentage of areas with brightness values equal to or exceeding the threshold value within each image was then calculated and averaged for all 20 images per animal.
For immunofluorescence against CD8, sections from unfixed frozen liver were fixed in acetone at −20°C for 10 min, air dried, blocked with 2% goat serum in phosphate-buffered saline (PBS) for 15 min, and stained with anti-CD8 antibodies from BD Biosciences (clone RPA-T8, diluted 1:25 in 2% goat serum) for 1 hr. After washing, tetramethylrhodamine isothiocyanate (TRITC)-labeled secondary antibodies against mouse IgG (diluted 1:150 in 2% goat serum; Sigma-Aldrich, St. Louis, MO) were applied for 30 min and sections were finally washed and mounted with VECTASHIELD (Vector Laboratories). Hematoxylin–eosin (H&E) staining was performed on formalin-fixed paraffin-embedded liver samples, followed by evaluation for the presence and size of mononuclear cell infiltrates as well as for the presence of mitotic hepatocytes, apoptotic cells, or any other abnormalities.
For molecular analysis, tissue DNAs were extracted as previously described. Copy numbers of the vector genomes in tissue samples were quantified by TaqMan PCR as previously described. Molecular status of vector genomes in tissue DNAs was analyzed by Southern blot as previously described, using a 750-bp NcoI–BsrGI EGFP fragment from the vector genome as the probe (Gao et al., 2006a). Signature PCR (SGPCR) assays for tissue DNA were performed as previously described (Gao et al., 2002). AAVrep2/cap7-allele-specific PCR was carried out with 5′ primer GTCGTCAAAAAGGCGTATCAGAAACTGTGCTAC to target AAV2 rep and with 3′ primer GCAGGTACGGATTGTCACCCGCTTTG to anneal to AAV7 cap under the same conditions as for SGPCR.
For real-time-based RT-PCR analysis of AAV cap transcripts in macaque tissues, total RNAs were extracted from macaque tissues with TRIzol reagent (Invitrogen, Carlsbad, CA), treated with RNase-free DNase I (Roche, Indianapolis, IN), and purified with an RNeasy Plus mini kit (Qiagen, Valencia, CA). Total RNA was reverse transcribed with a high-capacity cDNA reverse transcription kit (Applied Biosystems) according to the manufacturer's instructions. A combined absolute and relative quantification strategy was employed to assess the presence and level of AAV Cap sequences. The absolute quantification step included 10-fold serial dilutions of linearized AAV2/7 packaging plasmid as standard. The dynamic range of the assay covered 8 logs of standard input, with a limit of quantification of 10 copies per reaction and a limit of detection of 5 copies per reaction. The TaqMan assay against AAV Cap sequences was designed against a genomic DNA target derived from a multisequence alignment of wild-type AAV serotypes. The relative quantification step involved the detection of a reference mRNA target in sample cDNA against which all AAV Cap signals were normalized. The reference or housekeeping gene selected was the human ornithine carbamoyltransferase (OTC) gene. The human assay is 100% homologous to the simian target RNA used. Data were analyzed by the ΔCt method. Relative expression is estimated by simultaneous normalization against the calibrator sample and the reference gene.
Primers and probes used were as follows: CapR3 F2, 5′-CCAATGGCAGACAATAACGA-3′; CapR3 R2D, 5′-GTGCTGGTGGTRATGAC-3′; CapR3 probe, 6FAM-AATTGGCATTGCGATTCCACATG-TAMRA; hOTCg Fwd, 5′-AGGAACACTATAGCTCTCTGA-3′; hOTCg Rev, 5′-CTGCGCTCATCATGATGGA; and hOTCg probe, 6FAM-TCTTACCCTCAGCTGGAT-MGBNFQ. Fifty nanograms each of total RNA equivalent cDNA (5 μl of RT reaction) was used for TaqMan reactions. Reaction conditions were as follows: incubation at 95°C for 10 min, and 40 cycles of 95°C (15 sec) and 60°C (1 min).
Results
AAV2/7 efficiently transduces NHP hepatocytes
Table 1 summarizes details of the NHP studies. Nine cynomolgus macaques were dosed with various AAV2/7 vectors expressing GFP from a cytomegalovirus (CMV) enhanced β-actin promoter. Neutralizing antibody (NAb) to AAV7 was shown to be <1:20 in baseline sera of all animals. Three different genomes were used: (1) sc genome with bovine growth hormone (bGH) poly(A), called sc1; (2) sc genome with SV40 poly(A), called sc; and (3) regular single-stranded (ss) genome with bGH poly(A) and noncoding WPRE sequence from woodchuck hepatitis virus. Seven animals received constructs with an sc genome whereas two animals received vectors with a traditional ss genome. Doses ranged from 0.7 to 3.5 × 1012 GC/kg. Animals were killed at 8 days to evaluate transduction efficiency (n = 6) and at 37 days (n = 3) to evaluate stability of gene expression and immune responses.
An extensive comparison of the ss and sc1 vectors was performed in mouse liver at doses of 5 × 1011 and 5 × 1012 GC/kg (1 × 1010 and 1 × 1011 GC/mouse, respectively) and compared with results obtained with an adenoviral vector expressing GFP. On day 14 after vector administration, expression as measured by total fluorescence from the GFP transgene was 30-fold higher with the sc1 vector than with the ss vector as measured by percentage of GFP-positive area in liver sections (Fig. 1A). An early and transient peak of serum transaminases was observed after administration of the adenoviral vector that was not observed with the ss or sc vectors except for mild and variable elevations on day 14 with the high-dose sc vector (Fig. 1B). IFN-γ ELISPOT analyses of liver and spleen mononuclear cells demonstrated elevations of GFP-specific T cells after adenoviral vector but not AAV vector administration (Fig. 1C).
FIG. 1.

Performance of AAV7-GFP vectors in mouse liver. C57BL/6 male mice were injected intravenously with Ad5 (1011 particles/mouse), AAV2/7ss (1011 GC/mouse), or AAV2/7sc (1010 and 1011 GC/mouse) vector expressing GFP and were analyzed for (A) expression, (B) serum transaminases, and (C) GFP-specific T cells. (A) Comparison of percentages of GFP-positive liver area from tissues harvested 7, 14, 33, 42, and 90 days after gene transfer. Digital images of random areas from cryosections (three per animal, and four animals per time point) were acquired with a 10 × microscope objective under identical settings. Using AnalySIS software (Soft Imaging System, Lakewood, CO), the GFP-positive areas within each image were marked by “thresholding” and their percentage relative to the total area of each image was determined. Columns show the average values for each group. (B) Murine transaminase levels after vector administration. At 7, 14, 33, and 90 days postinjection mouse blood was collected to measure aspartate transaminase (AST) levels, expressed as units per liter (IU/L). AST levels for naive C57BL/6 male mice were also determined by blood collection at a time point 3 days preinjection, at the time of vector injection, and 14 days after vector administration to serve as a negative control. Data represent means ± SD for four mice per group. (C) Characterization of transgene-specific T cell responses in spleen and liver by IFN-γ ELISPOT assay. At 14 and 42 days after vector administration mice were killed and lymphocytes were harvested from spleen and liver. IFN-γ secretion in response to antigen stimulation was determined by IFN-γ ELISPOT. Lymphocytes were stimulated with the complete EGFP peptide library to determine the transgene-specific T cell response. Data represent means ± SD for four mice per group. Shaded columns, day 14; solid columns, day 42.
The pilot NHP study involved administration of AAV2/7 sc1 vector into single animals at doses of 0.7 × 1012 and 3.5 × 1012 GC/kg followed by tissue harvest on day 8. Expression data are summarized in Table 1 and actual micrographs are presented in Fig. 2. The higher dose was associated with transduction of 9% of hepatocytes; the number of transduced cells was reduced about 100-fold when vector dose was reduced approximately 5-fold, demonstrating a non-linear relationship of dose and transduction. Two additional animals that received AAV2/7 sc vector at an intermediate dose (2 × 1012 GC/kg) demonstrated transduction in 0.6 and 4.1% of hepatocytes on day 8 (Table 1 and Fig. 2). Two animals were dosed with the ss version of the AAV2/7 GFP vector at a dose of 3.5 × 1012 GC/kg; this vector also contained the WPRE cis sequence. Analysis of liver tissue harvested 8 days later showed impressive transduction, with GFP observed in 10 and 25% of all hepatocytes (Table 1 and Fig. 2). The improved efficiency of transduction observed in mice with the sc vectors compared with the ss vectors did not translate to the NHPs in terms of the numbers of GFP-expressing cells. Analysis of peripheral blood mononuclear cells by IFN-γ ELISPOT failed to reveal T cells specific to GFP or AAV7 capsid (data not shown).
FIG. 2.

Enhanced green fluorescent protein (EGFP) expression and histopathology in cynomolgus livers. Self-complementary (sc) and single-stranded (ss) AAV2/7CBEGFP vectors at various doses were administered to cynomolgus macaques via the mesenteric vein. (A) The animals were necropsied on either day 8 (animals 24466, 24527, 24441, 24456, 24499, and 24462) or day 37 (animals 24461, 24479, and 24481) and liver tissues were harvested for cryosectioning and examination under a fluorescence microscope. Representative images of liver sections from all day 8 animals are presented. However, a representative image of the liver sections from only one of the day 37 animals (animal 24461) is presented. The others harvested on day 37 had no detectable GFP expression. (B) Histopathology of liver sections of day 8 animal 24466 (normal) and day 37 animal 24461. The black arrows indicate infiltration sites and an apoptotic hepatocyte in H&E-stained paraffin sections and the white arrow points to immunofluorescently stained CD8+ T cells in a cryosection. These findings were present in all animals harvested on day 37 and not in the day 8 animals.
A formal biodistribution study was performed on all animals, using TaqMan PCR to quantify vector genomes. Each animal showed 100-fold more vector DNA in liver than in other tissues except for spleen as measured by genomes per nanogram of cellular DNA (Table 2). This analysis revealed high levels of vector genomes in liver, ranging from 1 to 37 genomes/cell (1 genome/cell =150 vector genomes/ng cellular DNA), that differed between animals but was similar among various liver lobes of the same animal (Tables 1 and 2). Vector genome abundance in liver as measured by PCR correlated with Southern blot analyses (Fig. 3A). The presence of vector DNA in liver did not correlate well with transduction efficiency, however. The ratio of GFP transduction efficiency (fraction of total hepatocytes transduced) to genome copies per cell ranged from 0.006 to 0.53 (Table 1). The highest extrahepatic dissemination of vector was found in spleen, followed by lung, gall bladder, duodenum, and kidney (Table 2). No significant laboratory, clinical, or pathological findings were observed in any animals necropsied on day 8 (data not shown).
Table 2.
Biodistribution of Vector Genomes and Adeno-associated Viral Capsid Sequences in Nonhuman Primate Tissues as Measured by Real-Time Polymerase Chain Reactiona
| |
Animal no. |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| |
24466 |
24527 |
24441 |
24456 |
24499 |
24462 |
24461 |
24479 |
24481 |
| Tissue | (Vector/Cap) | (Vector/Cap) | (Vector/Cap) | (Vector/Cap) | (Vector/Cap) | (Vector/Cap) | (Vector/Cap) | (Vector/Cap) | (Vector/Cap) |
| Gonadal tissue | 0.6/0 | 0.82/0 | 0.41/0 | 1.3/0 | 0.42/0 | 3.3/0 | 0.63/0 | 0.31/0 | 0.76/0 |
| Blood | 0.95/0.03 | 0.59/0 | 1.5/0 | 7.1/0 | 0.2/0 | 1.1/0 | 0.82/0 | 0.54/0 | 1.7/0 |
| Bone marrow | 47/0 | 1.3/0 | 23/0.06 | 35/0.05 | 10/0 | 57/0.08 | 8.1/0 | 4.3/0 | 41/0 |
| Brain | 0.68/0 | 0.02/0 | 0.32/0 | 4.4/0.02 | 0.3/0 | 1.8/0 | 0.97/0 | 1.2/0 | 4.7/0 |
| Lung, left | 180/0 | 1.8/0 | 6.3/0 | 44/0.06 | 8.3/0 | 71/0.06 | 10/0 | 5/0 | 16/0 |
| Lung, right | 34/0 | 0.43/0 | 9.4/0.03 | 13/0.03 | 6.5/0 | 71/0.09 | 7.2/0 | 6/0 | 8/0 |
| Heart | 5.4/0.03 | 0.22/0 | 2.7/0 | 41/0.18 | 2.4/0 | 5.4/0 | 5.6/0 | 3.9/0 | 6.9/0 |
| Skeletal muscle | 6/0 | 0.06/0 | 0.61/0 | 4.3/0 | 0.19/0 | 5.8/0 | 1.5/0 | 0.79/0 | 1.1/0 |
| Stomach | 3.7/0 | 2.9/0 | 8.8/0 | 12/0.03 | 33/0 | 5.4/0 | 5.2/0 | 0.87/0 | 13/0 |
| Duodenum | 24/0 | 4.9/0 | 1.5/0 | 16/0 | 26/0.05 | 20/0 | 14/0 | 14/0.1 | 10/0 |
| Colon | 20/0 | 2.6/0 | 4.6/0 | 6.1/0 | 2/0 | 8.8/0 | 2.2/0 | 6.4/1.1 | 7/0 |
| Pancreas | 11/0.06 | 0.08/0 | 2/0 | 13/0.07 | 100/0.02 | 12/0 | 0.71/0.04 | 0.71/0 | 3.7/0 |
| Gall bladder | 14/0 | 54/0 | 2.7/0 | 46/0 | 240/0 | 890/0 | 0.86/0 | 14/0 | 300/0 |
| Kidney, left | 14/0 | 0.95/0 | 5.7/0.04 | 20/0.12 | 79/0 | 40/0.09 | 6/0 | 6.7/0 | 22/0.02 |
| Kidney, right | 15/0 | 0.58/0 | 5.1/0.03 | 50/0.16 | 81/0.03 | 48/0.08 | 4.6/0 | 13/0 | 12/0 |
| Liver, left | 4,500/1.6 | 140/0.04 | 1,800/2.8 | 14,000/24 | 1,600/0.86 | 5,200/4.8 | 310/0.23 | 590/0.04 | 4,300/2.6 |
| Liver, right | 4,800/1.6 | 160/0.08 | 2,300/3.7 | 10,000/34 | 2,600/0.75 | 5,300/5.1 | 270/0.16 | 750/0.07 | 4,600/2.1 |
| Liver, middle | 5,000/2.5 | 280/0.09 | 1,900/3 | 14,000/15 | 2,400/0.73 | 7,400/5.3 | 320/0.21 | 710/0.07 | 4,300/2.2 |
| Liver, caudate | 4,100/3.7 | 280/0.055 | 1,500/2.7 | 8,500/25 | 1,900/0.9 | 6,500/7 | 320/0.17 | 640/0.05 | 4,400/2.3 |
| Spleen | 3,800/10 | 1,600/1.3 | 1,000/3.4 | 1,500/6.9 | 2,500/2.5 | 1,100/3.2 | 180/0.51 | 490/1.3 | 860/2.2 |
| Mesenteric lymph node | ND/ND | 840/49 | 83/0.33 | 1,100/3.8 | 350/0.64 | 370/0.78 | 130/0 | 250/0.33 | 550/4.2 |
Abbreviation: ND, not determined.
The primer–probe sets and PCR conditions are described in Materials and Methods. Data are presented as genome copies of either rAAV.CBEGFP (“Vector”) or wild-type AAV cap (“Cap”) gene per nanogram of total cellular DNA. The tissue source of DNA for analysis is described in the first column.
FIG. 3.

Molecular analysis of vector genomes, AAV cap, AAV2 rep/AAV7 cap junction sequences, and cap RNA transcripts. (A) Southern blot analysis of cellular DNAs from macaque livers. Cellular DNAs (10 μg) were digested with restriction endonucleases HpaI and SphI to release a 732-bp internal fragment, followed by Southern blot analysis. (B) Reverse transcription-PCR (RT-PCR) analysis of AAV cap transcripts in the macaque livers. Total RNAs were prepared from liver tissues of representative animals, and treated with RNase-free DNase and reverse transcriptase. cap-specific transcripts were quantified by real-time PCR using primers and probe that bind to the conserved regions of the cap gene. The quantitative data are presented in a bar graph. The RT-PCR products for the transcripts of cap and a single-copy cellular gene, OTC, were also examined on an ethidium bromide-stained 2% agarose gel. (–) ctr, negative control. (C) Signature PCR (SGPCR) analysis of AAV cap gene. Cellular DNAs from liver (L), spleen (S), and mesenteric lymph node (LN) tissues of the study animals were subjected to SGPCR amplification of hypervariable region II in the AAV cap gene as described previously. The SGPCR products were cloned (TOPO TA subcloning kit; Invitrogen) for sequencing characterization. NC, negative control. (D) Characterization of AAV sequences in liver tissues by AAV2 rep/AAV7 cap junction PCR (JNPCR). A set of primers designed specifically to amplify through the AAV2 rep/AAV7 cap junction of the AAV2/7 packaging plasmid was used for PCR of liver DNAs. Representative junction PCR fragments were cloned (TOPO TA subcloning kit) and sequenced to confirm detection of the AAV2 rep/AAV7 cap junction.
Expression of GFP is transient because of activation of destructive transgene-specific CTLs
Three animals dosed with the sc and sc1 vectors (3.5 × 1012 GC/kg [n = 1, sc] and 2 × 1012 GC/kg [n = 2, sc1]) were monitored for 37 days to evaluate stability of GFP expression. Surprisingly, none of the animals showed GFP expression at this late time point despite the fact that significant levels of vector genomes were retained in liver (2 to 27 genomes/cell; Table 1 and Fig. 3A). The animals appeared well during the in-life phase of the experiment (data not shown) although they all demonstrated an increase in serum transaminases that first appeared on day 14 and began to diminish on day 21 (Fig. 4). The only remarkable pathologic findings were in the livers. All animals showed mononuclear infiltrates (with some CD8+ cells) and apoptotic cells at the day 37 time point (Fig. 2B).
FIG. 4.

Correlation between detection of transgene (EGFP)-specific T cell responses and transient transaminitis in day 37 animals. (A) Time course of EGFP-specific T cell responses detected in PBMCs and transaminase elevations. Peripheral blood mononuclear cells (PBMCs) were collected from day 37 animals at various time points and subjected to EGFP-specific T cell analysis by ex vivo ELISPOT assay. The left y axis presents serum transaminase levels (IU/L). The right y axis shows ELISPOT IFN-γ results as SFUs per 106 cells. Diamonds, ALT results; squares, AST results; triangles, ELISPOT results. LFT, liver function test. (B) Characterization of transgene-specific T cell responses in lymphocytes isolated from various tissues. At the time of necropsy (day 37), tissue-specific lymphocytes were isolated from blood; axillary, inguinal, iliac, and mesenteric lymph nodes; spleen; bone marrow (B.M.); peritoneal lavage (P.L.); and liver for ex vivo ELISPOT analysis. Data are presented on the y axis as SFUs per 106 cells. Animal identifications are presented in each panel.
The time course of the transaminitis and liver histopathology was reminiscent of CTL-mediated targeting of hepatocytes that is observed with first-generation adenoviruses. We therefore evaluated PBMCs for the presence of T cells to GFP with an IFN-γ ELISPOT, using a peptide library spanning the entire transgene product. All three animals showed high levels of GFP-specific T cells (200 to 500 spots/106 cells) that peaked on day 14 or 21 and were coincident with the appearance of transaminases in the blood (Fig. 4A). Mononuclear cells were harvested from several tissues/organs at necropsy and evaluated by ELISPOT. The highest levels of GFP-specific T cells were observed in liver and spleen with frequencies ranging from 500 to 1200 spots/106 cells (Fig. 4B).
The quality and quantity of the GFP T cell response were further evaluated by intracellular cytokine staining (ICS) in some samples in which sufficient numbers of cells were available. PBMCs from one of the two animals that yielded the highest ELISPOT frequencies (animal 24461) were analyzed by ICS to determine the relative contribution of CD4+ and CD8+ T cells. As can be seen in Fig. 5A, both CD4+ and CD8+ GFP-specific T cells were observed from weeks 2 to 5. Mononuclear cells from liver and spleen of animal 24481 and from liver, spleen, and blood of animal 24479 were evaluated by ICS for expression of multiple cytokines (IL-2, IFN-γ, and TNF-α) in response to GFP peptides (Fig. 6A). The CD8 response was characterized by expression of TNF-α and IFN-γ and less expression of IL-2. The CD4 response demonstrated much higher levels of IL-2 expression, with the total number of GFP-specific CD4+ cells ranging from 6.5 to 36.3% of all CD4+ T cells (Fig. 6B). Further ICS studies of PBMCs of animal 24479 obtained at necropsy showed high degranulation activity (CD107a detection) in 80% of the IFN-γ-expressing GFP-specific T cells, indicating they are likely highly functional in terms of cytolytic function (Fig. 5B).
FIG. 5.

Time course and functional characterization of GFP-specific T cell response. (A) GFP-specific IFN-γ secretion of peripheral CD4+ and CD8+ T cells isolated from macaque 24461 after vector administration. (B) GFP-specific degranulation ability of CD8+ T cells isolated from blood (top) and spleen (bottom) from macaque 24479, 5 weeks after vector administration. Cells were stimulated with GFP peptide library (stim) or with medium (no stim) in the presence of FITC-labeled CD107a antibody (y axes) and stained with various surfaces marker (see Materials and Methods) including CD8 (x axes). Numbers represent the percentage of viable CD8+ T cells that express CD107a on the surface after antigen stimulation.
FIG. 6.

Polyfunctional analysis of GFP-specific T cells. Intracellular cytokine secretion profile of lymphocytes isolated from spleen (squares), liver (circles), and blood (diamonds) obtained from macaque 24481 (open symbols) and macaque 24479 (solid symbols) 3 weeks (blood) and 5 weeks after vector administration. Cytokine production of IFN-γ, TNF-α, and IL-2 by (A) CD8+ T cells and (B) CD4+ T cells after stimulation with the GFP peptide library was determined.
AAV2/7 fails to elicit capsid-specific T cells despite evidence of transduction of vector-derived cap genes
Our previous studies indicated that NHPs harbor endogenous AAV sequences. In fact, AAV7 was isolated from heart tissue harvested from one rhesus macaque. Tissues harvested for vector biodistribution were also analyzed for the presence of AAV cap sequences, using oligonucleotide primers to consensus sequences (Table 2 and Fig. 3C). Low levels of cap DNA were found in many tissues, although the highest frequencies were found in liver (5 ± 8 [mean ± 1 SD], ranging from 0.2 to 34 GC/ng of total cellular DNA) and spleen (4 ± 3 [mean ± 1 SD], ranging from 0.5 to 10 GC/ng of total cellular DNA) with slightly lower levels present in mesenteric lymph nodes (0.3 ± 1.7 [mean ± 1 SD], ranging from 0 to 4.2 GC/ng of total cellular DNA). Limited sequence analysis of the amplified cap sequence indicated it was a mix of AAV capsid variants, with a high proportion similar to AAV7, suggesting that at least some of this AAV DNA may have been derived from the vector preparations (data not shown). Direct analysis of the vector preparations by PCR did reveal the presence of cap DNA at a ratio of approximately 0.8% (cap to vector DNA ×100). We reevaluated DNA from liver and spleen of all animals for cap sequences derived from the vector preparations, using a PCR strategy with primers to AAV7 cap and the contiguous AAV2 rep sequence present in the trans-packaging plasmid used in AAV production. All tissues did demonstrate detectable AAV2–AAV7 junction fragments specific to the vector production plasmid, using a standard nonquantitative PCR assay (Fig. 3D); limited sequence analysis of these fragments confirmed their identity (data not shown). RT-PCR assays were developed to assess expression of cap genes in NHP tissues: RNA was isolated from liver, reverse transcribed, and amplified with primers specific to consensus cap sequences, and either run on a gel or subjected to TaqMan PCR analysis. Figure 3B shows the results of this experiment, in which the internal control was analyzed for expression of the liver-specific gene OTC. cap transcripts were detected in the three animals that harbored the higher levels of cap DNA; expression was proportional to cap DNA. Expression was not detected in the animal with only 0.001 cap DNA per cell.
Various sources of mononuclear cells from all NHPs were analyzed for the presence of capsid-specific T cells, using a peptide library spanning the AAV7 capsid (Table 3). Direct analysis of mononuclear cells from blood and liver in the ex vivo assay failed to detect any signal over background in terms of IFN-γ expression. Selected samples were also evaluated for capsid T cells after the samples were amplified ex vivo in the presence of capsid peptides in an attempt to expand a minor population of capsid T cells before ELISPOT analysis (called cultured ELISPOT). No animals demonstrated capsid T cells from PBMCs after culture from samples harvested before or after vector except animal 24481, which showed some signals in all samples harvested after vector. One animal showed signal after culture of liver-derived mononuclear cells although there was no baseline liver sample available for comparison.
Table 3.
T Cell Response to AAV7 Capsid in Peripheral Blood Mononuclear Cells and Liver-Derived Lymphocytes Detected by Either ex Vivo or Cultured Enzyme-Linked Immunospot Assaya
| |
|
T cell response to AAV capsid (SFU/106cells) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
|
Ex vivo |
|
|
|
|
|
|||||
| |
|
PBMCs |
|
Cultured PBMCs |
||||||||
| Animal no. | Necropsy (day) | Pre | Day 16 | Day 23 | Day 30 | End | Liver | Pre | Day 16 | Day 23 | Day 30 | End |
| 24466 | 8 | (–) | NA | NA | NA | (–) | (–) | (–) | NA | NA | NA | ND |
| 24527 | 8 | (–) | NA | NA | NA | (–) | (–) | (–) | NA | NA | NA | ND |
| 24441 | 8 | (–) | NA | NA | NA | (–) | (–) | (–) | NA | NA | NA | ND |
| 24456 | 8 | (–) | NA | NA | NA | (–) | (–) | (–) | NA | NA | NA | ND |
| 24499 | 8 | (–) | NA | NA | NA | (–) | (–) | (–) | NA | NA | NA | ND |
| 24462 | 8 | (–) | NA | NA | NA | (–) | 671b | (–) | NA | NA | NA | ND |
| 24461 | 37 | (–) | (–) | (–) | (–) | (–) | 345c | (–) | (–) | (–) | (–) | (–) |
| 24479 | 37 | (–) | (–) | (–) | (–) | (–) | (–) | (–) | 125b | (–) | (–) | ND |
| 24481 | 37 | (–) | (–) | (–) | (–) | (–) | (–) | (–) | 135b | 454d | 570d | ND |
Abbreviations: AAV, adeno-associated virus: NA, not applicable; ND, not determined; PBMCs, peripheral blood mononuclear cells; SFU, spot-forming unit.
Isolation of PBMCs and liver-derived lymphocytes and ELISPOT assays are described in Materials and Methods. A peptide library specific for the full length of AAV7-VP1 was synthesized as 15-mers with 11-amino acid overlaps (Mimotopes, Clayton, Victoria, Australia). The entire lyophilized library, consisted of 146 peptides, was reconstituted with dimethyl sulfoxide and grouped into pools A (50 peptides). B (50 peptides), and C (46 peptides) for use in cell stimulation. Samples were harvested before injection (Pre), at necropsy (End), and, for the day 37 animals, on days 16, 23, and 30. Please see additional footnotes in Table 2.
AAV7 peptide pool C.
AAV7 peptide pool A.
AAV7 peptide pool A + B + C.
Discussion
Initial preclinical and clinical models of AAV gene transfer to liver used vectors based on AAV serotype 2. Important limitations of these vectors emerged, such as poor transduction efficiency, activation of T cells against the capsid, and substantial quantities of preexisting neutralizing antibodies in human populations (Grimm and Kay, 2003; Vandenberghe et al., 2006; Wang et al., 2007; Calcedo et al., 2009). Vectors based on natural and engineered AAV capsid variants have been identified that appear to overcome many of the barriers associated with AAV2. Our work has focused on vectors created from capsid of AAV endogenous to primates, including AAV serotypes 7 and 8, which have low seroprevalence in humans, diminished activation of capsid T cells, and improved transduction efficiency of hepatocytes in most preclinical models (Gao et al., 2002; Vandenberghe et al., 2006; Wang et al., 2007; Calcedo et al., 2009). The goal of this study was to critically evaluate several key measures of performance of AAV2/7 vectors in nonhuman primates. GFP was selected as a reporter gene to facilitate the direct assessment of transduction efficiency and to critically assess the potential of T cell responses against antigenic transgene products.
It became apparent that the transduction efficiency of AAV2/7 vectors containing ss genomes was the same as that achieved in mice when dosed on a per-kilogram basis. What was surprising is that AAV2/7 vectors with sc AAV genomes were not substantially more efficient than ss vectors in NHPs, which was different from what was observed in mice. It is possible that total expression is higher with sc vectors in NHPs because of more expression per cell rather than because of more transduced cells, although quantitative morphometry of tissue sections failed to support this hypothesis (data not shown). In fact, we showed previously that the expression of a secreted reporter gene from AAV2/7 vectors delivered to NHP liver as measured by serum levels was higher with sc vectors as compared with ss vectors (Gao et al., 2006b). Other differences between the sc and ss constructs used in the current study could have influenced expression, such as the WPRE in the ss vector (size limitations of the sc vectors precluded inclusion of these sequences). There was much more animal-to-animal variation of transduction efficiency in primates than in mice, which is not surprising because monkeys are outbred whereas the mice are not. We suspect a significant contribution to variation in NHPs is the existence of neutralizing antibodies that escape detection by our assay but are meaningful in vivo. We eliminated macaques as candidates for this study that had preexisting NAbs to AAV7, based on the in vitro transduction inhibition assay.
Molecular studies indicated that the abundance of vector genomes in liver was far in excess of the number of transduced hepatocytes, ranging from 1 transduced cell per 200 to 20,000 genome copies. Several possible mechanisms could explain this, including the following: (1) all the genomes are sequestered in the subpopulation of hepatocytes that express; (2) genomes are distributed throughout the majority of hepatocytes, but only a subpopulation is transcriptionally active; and (3) significant numbers of genomes are sequestered in nonparenchymal liver cells and are not expressed. A sensitive in situ hybridization assay to detect vector genomes would help sort out this conundrum.
The most dramatic result was the qualitative difference in T cell responses to GFP observed in NHPs, in which a robust CTL response was formed as compared with mice, which are nonresponsive to GFP. The absence of functional GFP-specific T cells in mice is not due to MHC restriction or a paucity of T cell epitopes because adenovirus elicits robust GFP-specific T cells. In our NHP study, analysis of mononuclear cells from liver, spleen, and blood clearly showed T cell responses to GFP including CD8+ T cells expressing multiple cytokines and the degranulation marker CD107a; and CD4+ T cells expressing all three cytokines (IL-2, IFN-γ, and TNF-α), with more than 30% expressing IL-2. The concurrent appearance of GFP-specific T cells with elevated serum transaminases coupled with the loss of GFP expression during this time interval suggests in vivo CTL activity. It is also interesting to note that ELISPOT analysis of PBMCs was predictive of liver-associated GFP-specific CD8+ T and CD4+ T cells, which will be useful in the evaluation of host immune responses in clinical trials where PBMCs are the only practical source of cells for T cell analyses.
It was again surprising that the presence and abundance of vector genomes in liver did not reflect the status of GFP expression in the animals necropsied on day 37; in these animals expression was not detected but vector genomes persisted at relatively high levels. This may be due to mechanisms of transgene extinction other than killing of GFP-expressing cells by CTLs, such as inactivation of expression as has been proposed by Guidotti and Chisari in the T cell response of the host to hepatitis B virus (Guidotti and Chisari, 2006). This phenomenon may also be related to the observation of excess vector genomes detected at shorter time points before CTLs are mobilized. The presence of transcriptionally inactive genomes in nonexpressing parenchymal and nonparenchymal cells would not be recognized by CTLs and therefore would persist despite complete elimination of all GFP-expressing cells by CTLs.
The development of transaminitis with loss of transgene expression is reminiscent of what was observed in a clinical trial of AAV2-mediated gene transfer to liver of subjects with hemophilia B, where the investigators proposed CTLs against the input capsid rather than the transgene product (Manno et al., 2006). However, our animals failed to develop T cells to the AAV7 capsid as measured by a sensitive ex vivo ELISPOT assay. During the course of these studies we evaluated the animals for endogenous production of Cap that could efficiently load MHC class I via the direct pathway and activate CD8+ T cells. We showed low-level expression of cap gene in liver and the presence of both endogenous genomes and those derived from the preparations of the vectors due to contamination with the trans-packaging plasmid. It is not possible to determine which cap genome contributes to expression although it appears irrelevant in terms of activating T cells or rendering hepatocytes targets for CTLs because no CTLs were activated.
We conclude that AAV2/7 ss-based vectors do indeed confer highly efficient transduction in NHP liver. The immunologic studies suggest that the immune privilege afforded AAV in mouse liver may not be relevant to NHP liver, although the limited number of primates available for this study precluded a systematic evaluation of all parameters. Induction of destructive transgene-specific CTLs was demonstrated with sc vectors only; additional studies are needed to determine whether this is relevant to ss vectors. The transgene product GFP is presumably highly antigenic in primates, which may not be the case in clinical situations where the transgene product is only minimally different from self-antigens, such as in gene replacement therapy for recessive diseases caused by missense mutations. The mechanism(s) responsible for qualitatively different T cell responses in mice versus nonhuman primates in the current setting have yet to be defined, although transgene expression in primate antigen-presenting cells may play a role. If so, attempts to restrict transgene expression to hepatocytes with tissue-specific promoters and microRNA targets could help (Franco et al., 2005; Brown et al., 2007). Finally, these studies fail to demonstrate the activation of T cells to capsid of AAV2/7 despite the presence of capsid expression in vivo. This may be because AAV7 capsid does not bind heparin and therefore is not efficiently cross-presented.
Acknowledgments
This work was funded by the following grants to J.M.W.: NIDDK P30 DK47757, NHLBI P01 HL59407, NICHD P01 HD57247, and GlaxoSmithKline.
Author Disclosure Statement
G.G. and L.H.V. are inventors on patents licensed to various companies. J.M.W. is an inventor on patents licensed to various biopharmaceutical companies including ReGenX, in which he has equity, for which he consults, and from which he receives a grant.
References
- Alexander I.E. Cunningham S.C. Logan G.J. Christodoulou J. Potential of AAV vectors in the treatment of metabolic disease. Gene Ther. 2008;15:831–839. doi: 10.1038/gt.2008.64. [DOI] [PubMed] [Google Scholar]
- Brown B.D. Gentner B. Cantore A. Colleoni S. Amendola M. Zingale A. Baccarini A. Lazzari G. Galli C. Naldini L. Endogenous microRNA can be broadly exploited to regulate transgene expression according to tissue, lineage and differentiation state. Nat. Biotechnol. 2007;25:1457–1467. doi: 10.1038/nbt1372. [DOI] [PubMed] [Google Scholar]
- Calcedo R. Vandenberghe L.H. Gao G. Lin J. Wilson J.M. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J. Infect. Dis. 2009;199:381–390. doi: 10.1086/595830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuah M.K. Collen D. Vandendriessche T. Clinical gene transfer studies for hemophilia A. Semin. Thromb. Hemost. 2004;30:249–256. doi: 10.1055/s-2004-825638. [DOI] [PubMed] [Google Scholar]
- Davidoff A.M. Gray J.T. Ng C.Y. Zhang Y. Zhou J. Spence Y. Bakar Y. Nathwani A.C. Comparison of the ability of adeno-associated viral vectors pseudotyped with serotype 2, 5, and 8 capsid proteins to mediate efficient transduction of the liver in murine and nonhuman primate models. Mol. Ther. 2005;11:875–888. doi: 10.1016/j.ymthe.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Franco L.M. Sun B. Yang X. Bird A. Zhang H. Schneider A. Brown T. Young S.P. Clay T.M. Amalfitano A. Chen Y.T. Koeberl D.D. Evasion of immune responses to introduced human acid α-glucosidase by liver-restricted expression in glycogen storage disease type II. Mol. Ther. 2005;12:876–884. doi: 10.1016/j.ymthe.2005.04.024. [DOI] [PubMed] [Google Scholar]
- Gao G. Yang Y. Wilson J.M. Biology of adenovirus vectors with E1 and E4 deletions for liver-directed gene therapy. J. Virol. 1996;70:8934–8943. doi: 10.1128/jvi.70.12.8934-8943.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G. Qu Q. Bumham M.S. Huang J. Chirmule N. Joshi B. Yu Q.C. Marsh J.A. Conceicao C.M. Wilson J.M. Purification of recombinant adeno-associated virus vectors by column chromatography and its performance in vivo. Hum. Gene Ther. 2000;11:2079–2091. doi: 10.1089/104303400750001390. [DOI] [PubMed] [Google Scholar]
- Gao G. Alvira M.R. Wang L. Calcedo R. Johnston J. Wilson J.M. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. U.S.A. 2002;99:11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G. Alvira M.R. Somanathan S. Lu Y. Vandenberghe L.H. Rux J.J. Calcedo R. Sanmiguel J. Abbas Z. Wilson J.M. Adeno-associated viruses undergo substantial evolution in primates during natural infections. Proc. Natl. Acad. Sci. U.S.A. 2003;100:6081–6086. doi: 10.1073/pnas.0937739100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G. Vandenberghe L.H. Alvira M.R. Lu Y. Calcedo R. Zhou X. Wilson J.M. Clades of adeno-associated viruses are widely disseminated in human tissues. J. Virol. 2004;78:6381–6388. doi: 10.1128/JVI.78.12.6381-6388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G. Vandenberghe L.H. Wilson J.M. New recombinant serotypes of AAV vectors. Curr. Gene Ther. 2005;5:285–297. doi: 10.2174/1566523054065057. [DOI] [PubMed] [Google Scholar]
- Gao G. Lu Y. Calcedo R. Grant R.L. Bell P. Wang L. Figueredo J. Lock M. Wilson J.M. Biology of AAV serotype vectors in liver-directed gene transfer to nonhuman primates. Mol. Ther. 2006a;13:77–87. doi: 10.1016/j.ymthe.2005.08.017. [DOI] [PubMed] [Google Scholar]
- Gao G. Lu Y. Sun X. Johnston J. Calcedo R. Grant R. Wilson J.M. High-level transgene expression in nonhuman primate liver with novel adeno-associated virus serotypes containing self-complementary genomes. J. Virol. 2006b;80:6192–6194. doi: 10.1128/JVI.00526-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm D. Kay M.A. From virus evolution to vector revolution: Use of naturally occurring serotypes of adeno-associated virus (AAV) as novel vectors for human gene therapy. Curr. Gene Ther. 2003;3:281–304. doi: 10.2174/1566523034578285. [DOI] [PubMed] [Google Scholar]
- Grossman M. Raper S.E. Wilson JM. Transplantation of genetically modified autologous hepatocytes into nonhuman primates: Feasibility and short-term toxicity. Hum. Gene Ther. 1992;3:501–510. doi: 10.1089/hum.1992.3.5-501. [DOI] [PubMed] [Google Scholar]
- Grossman M. Raper S.E. Kozarsky K. Stein E.A. Engelhardt J.F. Muller D. Lupien P.J. Wilson J.M. Successful ex vivo gene therapy directed to liver in a patient with familial hypercholesterolaemia. Nat. Genet. 1994;6:335–341. doi: 10.1038/ng0494-335. [DOI] [PubMed] [Google Scholar]
- Grossman M. Rader D.J. Muller D.W. Kolansky D.M. Kozarsky K. Clark B.J., III Stein E.A. Lupien P.J. Brewer H.B., Jr. Raper S.E. Wilson J.M. A pilot study of ex vivo gene therapy for homozygous familial hypercholesterolaemia. Nat. Med. 1995;1:1148–1154. doi: 10.1038/nm1195-1148. [DOI] [PubMed] [Google Scholar]
- Guidotti L.G. Chisari F.V. Immunobiology and pathogenesis of viral hepatitis. Annu. Rev. Pathol. 2006;1:23–61. doi: 10.1146/annurev.pathol.1.110304.100230. [DOI] [PubMed] [Google Scholar]
- Hirata R.K. Russell D.W. Design and packaging of adeno-associated virus gene targeting vectors. J. Virol. 2000;74:4612–4620. doi: 10.1128/jvi.74.10.4612-4620.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manno C.S. Pierce G.F. Arruda V.R. Glader B. Ragni M. Rasko J.J. Ozelo M.C. Hoots K. Blatt P. Konkle B. Dake M. Kaye R. Razavi M. Zajko A. Zehnder J. Rustagi P.K. Nakai H. Chew A. Leonard D. Wright J.F. Lessard R.R. Sommer J.M. Tigges M. Sabatino D. Luk A. Jiang H. Mingozzi F. Couto L. Ertl H.C. High K.A. Kay M.A. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- McCarty D.M. Monahan P.E. Samulski R.J. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001;8:1248–1254. doi: 10.1038/sj.gt.3301514. [DOI] [PubMed] [Google Scholar]
- Nathwani A.C. Gray J.T. Ng C.Y. Zhou J. Spence Y. Waddington S.N. Tuddenham E.G. Kemball-Cook G. McIntosh J. Boon-Spijker M. Mertens K. Davidoff A.M. Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver. Blood. 2006;107:2653–2661. doi: 10.1182/blood-2005-10-4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raper S.E. Yudkoff M. Chirmule N. Gao G.P. Nunes F. Haskal Z.J. Furth E.E. Propert K.J. Robinson M.B. Magosin S. Simoes H. Speicher L. Hughes J. Tazelaar J. Wivel N.A. Wilson J.M. Batshaw M.L. A pilot study of in vivo liver-directed gene transfer with an adenoviral vector in partial ornithine transcarbamylase deficiency. Hum. Gene Ther. 2002;13:163–175. doi: 10.1089/10430340152712719. [DOI] [PubMed] [Google Scholar]
- Raper S.E. Chirmule N. Lee F.S. Wivel N.A. Bagg A. Gao G.P. Wilson J.M. Batshaw M.L. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol. Genet. Metab. 2003;80:148–158. doi: 10.1016/j.ymgme.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Schnell M.A. Zhang Y. Tazelaar J. Gao G.P. Yu Q.C. Qian R. Chen S.J. Varnavski A.N. Leclair C. Raper S.E. Wilson J.M. Activation of innate immunity in nonhuman primates following intraportal administration of adenoviral vectors. Mol. Ther. 2001;3:708–722. doi: 10.1006/mthe.2001.0330. [DOI] [PubMed] [Google Scholar]
- Segura M.M. Alba R. Bosch A. Chillon M. Advances in helper-dependent adenoviral vector research. Curr. Gene Ther. 2008;8:222–235. doi: 10.2174/156652308785160647. [DOI] [PubMed] [Google Scholar]
- Stratford-Perricaudet L.D. Levrero M. Chasse J.F. Perricaudet M. Briand P. Evaluation of the transfer and expression in mice of an enzyme-encoding gene using a human adenovirus vector. Hum. Gene Ther. 1990;1:241–256. doi: 10.1089/hum.1990.1.3-241. [DOI] [PubMed] [Google Scholar]
- Vandenberghe L.H. Wilson J.M. AAV as an immunogen. Curr. Gene Ther. 2007;7:325–333. doi: 10.2174/156652307782151416. [DOI] [PubMed] [Google Scholar]
- Vandenberghe L.H. Wang L. Somanathan S. Zhi Y. Figueredo J. Calcedo R. Sanmiguel J. Desai R.A. Chen C.S. Johnston J. Grant R.L. Gao G. Wilson J.M. Heparin binding directs activation of T cells against adeno-associated virus serotype 2 capsid. Nat. Med. 2006;12:967–971. doi: 10.1038/nm1445. [DOI] [PubMed] [Google Scholar]
- Wang L. Figueredo J. Calcedo R. Lin J. Wilson J.M. Cross-presentation of adeno-associated virus serotype 2 capsids activates cytotoxic T cells but does not render hepatocytes effective cytolytic targets. Hum. Gene Ther. 2007;18:185–194. doi: 10.1089/hum.2007.001. [DOI] [PubMed] [Google Scholar]
- Wang Z. Ma H.I. Li J. Sun L. Zhang J. Xiao X. Rapid and highly efficient transduction by double-stranded adeno-associated virus vectors in vitro and in vivo. Gene Ther. 2003;10:2105–2111. doi: 10.1038/sj.gt.3302133. [DOI] [PubMed] [Google Scholar]
- Wilson J.M. Chowdhury N.R. Grossman M. Wajsman R. Epstein A. Mulligan R.C. Chowdhury , Jr. Temporary amelioration of hyperlipidemia in low density lipoprotein receptor-deficient rabbits transplanted with genetically modified hepatocytes. Proc. Natl. Acad. Sci. U.S.A. 1990;87:8437–8441. doi: 10.1073/pnas.87.21.8437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W. Berta S.C. Lu M.M. Moscioni A.D. Tazelaar J. Wilson J.M. Adeno-associated virus as a vector for liver-directed gene therapy. J. Virol. 1998;72:10222–10226. doi: 10.1128/jvi.72.12.10222-10226.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y. Nunes F.A. Berencsi K. Furth E.E. Gönczöl E. Wilson J.M. Cellular immunity to viral antigens limits E1-deleted adenoviruses for gene therapy. Proc. Natl. Acad. Sci. U.S.A. 1994;91:4407–4411. doi: 10.1073/pnas.91.10.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaiss A.K. Muruve D.A. Immunity to adeno-associated virus vectors in animals and humans: A continued challenge. Gene Ther. 2008;15:808–816. doi: 10.1038/gt.2008.54. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Chirmule N. Gao G.P. Qian R. Croyle M. Joshi B. Tazelaar J. Wilson J.M. Acute cytokine response to systemic adenoviral vectors in mice is mediated by dendritic cells and macrophages. Mol. Ther. 2001;3:697–707. doi: 10.1006/mthe.2001.0329. [DOI] [PubMed] [Google Scholar]