

Abstract
Abnormal hyperphosphorylation and aggregation of microtubule-associated protein tau play a crucial role in neurodegeneration of Alzheimer’s disease (AD). Anesthesia has been associated with cognitive impairment and the risk for AD. Here we investigated the effects of anesthesia on site-specific tau phosphorylation and the possible mechanisms. We found that anesthesia for short periods (30 sec to 5 min) induced tau phosphorylation at Thr181, Ser199, Thr205, Thr212, Ser262, and Ser404 to small, but significant, extents, which appeared to result from anesthesia-induced activation of stress-activated protein kinases. Anesthesia for a longer time (1 h) induced much more dramatic phosphorylation of tau at the above sites, and the further phosphorylation may be associated with hypothermia induced by anesthesia. Anesthesia-induced tau phosphorylation appears to be specific because the increased phosphorylation was only seen at half of the tau phosphorylation sites studied and was not observed in global brain proteins. These studies clarified the dynamic changes of tau phosphorylation at various sites and, thus, served as a fundamental guide for future studies on tau phosphorylation by using brains of anesthetized experimental animals. Our findings also provide a possible mechanism by which anesthesia may cause postoperative cognitive impairment and increase the risk for AD.
Keywords: Alzheimer’s disease, anesthesia, phosphorylation, tau, tau kinases, tau phosphatases
INTRODUCTION
Alzheimer disease (AD) is a neurodegenerative disease and is the most common cause of dementia in adults. Less than 5% of AD cases are caused by mutations of amyloid-β precursor protein, presenilin-1, or presenilin-2. The majority of AD cases are sporadic in origin, and the exact causes of sporadic AD are currently unknown. Several etiological factors, including genetic susceptibility, metabolic alterations, and environmental factors, have been proposed to contribute to AD. One key neuronal protein involved in neurodegeneration in AD is the microtubule-associated protein tau. Tau is abnormally hyperphosphorylated and aggregated into neurofibrillary tangles in AD brain [1,2]. Many studies have demonstrated that abnormal hyperphosphorylation and aggregation of tau are crucial to neurodegeneration in AD [3–5].
It has been reported that anesthesia may be associated with cognitive impairment [6–8] and increased risk for AD [9–11]. However, the molecular mechanisms underlying these associations are not understood. In this study, we investigated the effect of anesthesia on phosphorylation of tau and its possible mechanisms in mouse brains. We found that anesthesia of mice for short periods (30 sec to 5 min) induced tau phosphorylation at some selective phosphorylation sites to small extents, which may be caused by activation of stress-activated protein kinases, whereas anesthesia for a longer time (1 h) induced further phosphorylation at the same sites, which also appeared to be associated with hypothermia induced by anesthesia.
MATERIALS AND METHODS
Animals and methods of anesthesia
Female C57BL/6J mice (14–15 weeks old) obtained from the Jackson Laboratory (Bar Harbor, ME) were used for this study. Animal use was in full compliance with the NIH guidelines and was approved by our institutional Animal Care and Use Committee. Anesthesia was induced by inhalation of ether vapors or by intraperitoneal injection of sodium pentobarbital (50 mg/kg body weight). Control mice were sacrificed by cervical dislocation without anesthesia. For all animals, brains were removed immediately after sacrifice, frozen in dry ice, and stored at −70°C till used.
Antibodies
Primary antibodies used in this study include polyclonal antibody 92e [12] to total tau; polyclonal tau antibodies pT181, pS199, pS202, pT205, pT212, pS214, pT217, pS262, pS396, and pS404 (Biosource, Carlsbad, CA) to tau phosphorylated at the specific sites indicated by the amino acid residues and numbers; polyclonal pS422 to tau phosphorylated at Ser422 (Quality Controlled Biochemicals, Hopkinton, IL); monoclonal antibody AT180 (Pierce Biotechnology, Inc., Rockford, IL) to tau phosphorylated at Thr231; monoclonal antibody 12E8 (Athena Neuroscience, San Francisco, CA) to tau phosphorylated at Ser262 or Ser356; polyclonal antibody to glycogen synthase kinase-3β (GSK-3β) [13]; polyclonal anti-GSK-3β(pS9) (Cell Signaling Technology, Danvers, MA) that recognizes the inactive form of GSK-3β (i.e., GSK-3β phosphorylated at Ser9); polyclonal anti-GSK-3β(pY216) (Biosource) that recognizes GSK-3β if Tyr216 is phosphorylated and GSK-3α if Tyr279 is phosphorylated; monoclonal anti-cdk5 and polyclonal anti-p25/p35 from Santa Cruz Biotechnology (Santa Cruz, CA); polyclonal anti-AKT, polyclonal anti–active P-AKT (phosphorylated at Ser473), polyclonal antibodies against p85 and the activated p85 (phosphorylated at Tyr450) of phosphatidylinositol 3-kinase (PI3K), polyclonal antibodies against c-Jun N-terminal kinase (JNK) and the activated JNK (i.e. P-JNK that is phosphorylated at Thr183/Tyr185), and polyclonal antibodies against mitogen-activated protein kinase (MAPK or Erk) and the active P-MAPK (phosphorylated at Thr202/Tyr204 of Erk1 or Thr183/Tyr185 of Erk2) from Cell Signaling Technology; and monoclonal anti-β-actin from Sigma (St. Louis, MO). Secondary antibodies were purchased from Jackson Immuno Research Laboratories, Inc. (West Grove, PA).
Western blots and immunodot-blots
Mouse forebrains were homogenized in buffer consisting of 50 mM Tris-HCl (pH 7.4), 2.0 mM ED-TA, 10 mM β-mercaptoethanol, 2.0 μg/ml aprotinin, 25 μg/ml leupeptin, 1.0 μg/ml pepstatin, and 8.5% sucrose. Aliquots of the homogenates were immediately mixed with the same volume of 2-fold concentrated Laemmeli buffer (125 mM Tris-HCl, pH 6.8, 4% SDS, 20% glycerol, 2% β-mercaptoethanol, and 0.005% bromophenal blue), followed by heating in boiling water for 5 min. Protein concentrations of the samples were determined by using modified Lowry method [14].
The levels of specific brain proteins and their phosphorylation extents were determined by Western blots using 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Phosphorylation-dependent and site-specific tau antibodies were used to determine the extent of tau phosphorylation at the corresponding individual phosphorylation sites. Phosphorylation-dependent antibodies against pSer9 and pTyr216 of GSK-3β, pSer473 of AKT, pTyr458 of PI3K, pThr183/Tyr185 of JNK, and pThr202/Tyr204 of MAPK were used to determine the inactivation (in case of pSer9 of GSK-3β) or activation (in all other cases) of these kinases, because the phosphorylation at these sites switches the kinase activities. The blots were developed by using enhanced chemiluminescence kit (Pierce Biotechnology, Rockford, IL). The immunore-activities of the blots were quantified densitometrically. For quantitation, the levels of specific proteins were normalized by the level of β-actin, and phosphorylation levels of tau were normalized by the level of total tau. Quantification of global changes in phosphorylation of proteins was carried out by immunodot-blots of brain crude extracts, which were developed with anti-phosphothreonine antibody, as described previously [15]. Comparisons between groups were analyzed statistically by using two-tail Student t-test.
Measurement of body temperature
Body temperature of mice was monitored by using a rectal probe connected to a homeothermic blanket control unit (Harvard Apparatus, Holliston, MA) according to the manufacturer’s instructions.
RESULTS
Anesthesia induces phosphorylation of tau
To study the effects of anesthesia on tau phosphorylation in rodent brains, we determined the levels of tau phosphorylation at several phosphorylation sites in brain homogenates of mice sacrificed after inhalation of ether vapor for 30 sec or sacrificed 5 min after intraperitoneal injection of pentobarbital (50 mg/kg body weight), which are the most commonly used anesthetic methods for euthanasia of laboratory rodents. The control mouse brains were collected immediately after the mice were sacrificed by cervical dislocation without anesthesia. We found that anesthesia induced tau phosphorylation at 6 o f the 12 phosphorylationsites examined. These phosphorylation sites included Thr181, Ser199, Thr205, Thr212, Ser262, and Ser404 (Fig. 1).
Fig. 1.

Site-specific phosphorylation of tau induced by anesthesia. Mouse brains were removed immediately after being sacrificed by cervical dislocation (control), after inhalation of ether vapor for 30 sec, or 5 min after intraperitoneal injection of pentobarbital (50 mg/kg body weight). Homogenates of the forebrains were analyzed by Western blots developed with phosphorylation-dependent tau antibodies for measuring tau phosphorylation at specific sites or with a phosphorylation-independent tau antibody 92e that measures total tau level (A). Each lane represents an individual brain sample. The blots were also quantitated densitometrically (B). The relative immunoreactivities with each phospho-tau antibody, after being normalized with 92e immunoreactivity (total tau), are shown as mean ± SD. *p < 0.05 vs. controls.
Anesthesia induces alterations in brain protein kinases
Several protein kinases have been implicated in the regulation of tau phosphorylation and in the abnormal hyperphosphorylation of tau in AD. These kinases include GSK-3β, cyclin-dependent protein kinase 5 (cdk5), and stress-activated protein kinases (e.g., JNK and MAPK) (reviewed by [4,16]). Therefore, we studied the effect of anesthesia on the levels and the activation of GSK-3β and its upstream regulating kinases (AKT and PI3K), cdk5 and its activators (p35 and p25), JNK, and MAPK. We found that anesthesia had no significant effect on the levels of these kinases or of cdk5 activators (Fig. 2). However, the level o f Ser9 phosphorylation of GSK-3β, which represents inactivation of the kinase, was significantly increased in the anesthetized mouse brains, whereas the Tyr216 phosphorylation, which is required for the kinase activity, remained unchanged. The increase in Ser9 phosphorylation of GSK-3β was consistent with the activation of AKT that phosphorylates Ser9 of GSK-3β, a s indicated by the increased AKT phosphorylation. However, the phosphorylation/activation of PI3K that phosphorylates and then activates AKT was not changed during anesthesia. On the other hand, the phosphorylation/activation of MAPK and JNK was increased in the ether-anesthetized and pentobarbital-anesthetized mouse brains, respectively. Since these two kinases are stimulated by stress, these results suggest that the anesthesia-induced elevation of tau phosphorylation might be mediated by stress-activated protein kinases.
Fig. 2.

Effects of anesthesia on the levels and phosphorylation/activation of various protein kinases. (A) Brain homogenates of mice anesthetized by ether inhalation for 30 sec or 5 min after intraperitoneal injection of pentobarbital were analyzed by Western blots developed with antibodies against several protein kinases and their phosphorylated/activated counterparts. Mice sacrificed without anesthesia (by cervical dislocation) were included as controls. Anti-GSK-3β (pY216) also immunoreacted with phosphorylated GSK-3α (upper band). The anti-JNK is known to detect both 46-kDa JNK1 and 54-kDa JNK2/3. Actin blot was included as a loading control. (B) Densitometric quantitation of blots as shown in (A). Actin blot was used for normalization for quantitation of the kinase levels. Each kinase blot was used for normalization of quantitation of the phosphorylated/activated form of the corresponding kinase. *p < 0.05 vs. controls.
Two phases of tau phosphorylation during anesthesia
Planel and colleagues [17] recently reported that 1 h after intraperitoneal injection of pentobarbital, anesthesia led to tau phosphorylation at several sites due to hypothermia. To investigate whether the above observed increase in tau phosphorylation could also be attributed to hypothermia, we measured the rectal temperatures of mice during anesthesia under the conditions used. We found no significant change in body temperature when the mice were anesthetized with ether for 30 sec (Fig. 3A) or with pentobarbital for 5 min (Fig. 3B). These results indicate that the induction of tau phosphorylation does not result from hypothermia under these conditions.
Fig. 3.

Body temperature of mice during anesthesia. Rectal temperature of mice during anesthesia with ether inhalation (A) or intraperitoneal injection of pentobarbital (50 mg/kg body weight) (B, C). Each line represents an individual mouse.
When we anesthetized mice for longer time, up to 2 h, we found that the body temperature of the mice dropped significantly (Fig. 3C), which is consistent with previous observations [17]. We then compared the extent of site-specific tau phosphorylation in mouse brains between 5 min (when body temperature was not significantly decreased; see Fig. 3B) and 1 h (when body temperature was decreased to 26–30°C; see Fig. 3C) after pentobarbital injection. We found that anesthesia for 1 h (when body temperature was decreased) induced tau phosphorylation at Thr181, Thr205, Thr212, and Ser262/Ser356(12E8 sites) to a much larger extent than did anesthesia for 5 min (when body temperature was not significantly decreased) (Fig. 4). Anesthesia for a longer time only slightly induced further tau phosphorylation at Ser404 and did not induce further tau phosphorylation at Ser199 or Ser202. When the protein kinases were compared between 5 min and 1 h anesthesia, we did not find any significant differences in the levels or the activation of GSK-3β, AKT, PI3K, CDK5, JNK, or MAPK (data not shown). These results indicate that anesthesia for short periods induces a small extent of tau phosphorylation in a hypothermia-independent mechanism, whereas for longer periods anesthesia induces further tau phosphorylation at certain sites that may be attributed partially to hypothermia.
Fig. 4.

Alterations of site-specific tau phosphorylation during anesthesia with pentobarbital. Brain homogenates of mice either without anesthesia (sacrificed by cervical dislocation) or after anesthesia with pentobarbital for 5 min or 1 h were analyzed by quantitative Western blots for tau phosphorylation at several individual phosphorylation sites. Data were calculated after normalization with the total tau level and are presented as mean ± SD. *p < 0.05 vs. controls.
Phosphorylation of brain proteins during anesthesia
To investigate whether anesthesia also induces phosphorylation of other brain proteins, we quantified the global phosphorylation of brain proteins by using immunodot-blots developed with anti-phosphothreonine. We found that anesthesia by pentobarbital did not alter the overall phosphorylation of brain proteins significantly either at 5 min or 1 h after pentobarbital injection (Fig. 5).
Fig. 5.
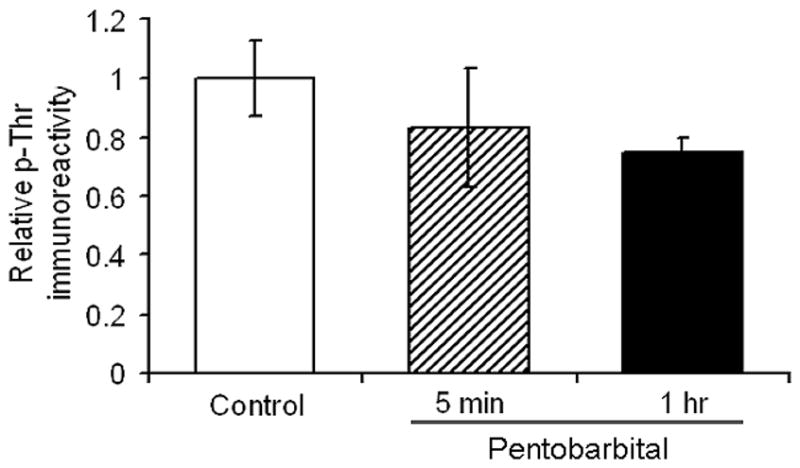
Levels of phosphothreonine of brain proteins in mice after anesthesia with pentobarbital for 5 min or 1 h.
DISCUSSION
In this study, we investigated the effects of anesthesia on site-specific phosphorylation of tau and a possible underlying mechanism. We found that anesthesia for short periods (30 sec to 5 min) induced tau phosphorylation at some of the phosphorylation sites, which is probably due to activation of stress-activated protein kinases and is unrelated to hypothermia. Anesthesia for a longer time (1 h) induced much more dramatic phosphorylation of tau, which may be attributed to hypothermia in addition to activation of the stress-activated protein kinases. Because longer anesthesia induced up to ten-fold more phosphorylation at the affected phosphorylation sites than did anesthesia for shorter times (up to 5 min; Fig. 4), the latter only induced tau phosphorylation to very limited extents. When anesthesia is used for euthanasia of rodents, animals are normally sacrificed after 30 sec of anesthesia with ether or within 5 min of anesthesia with pentobarbital. Thus, our findings suggest that short-term anesthesia has little impact on studies of tau phosphorylation if the time periods of anesthesia are identical for experimental and control animals. Anesthesia for a longer time may jeopardize studies on tau phosphorylation because dramatic tau phosphorylation is induced by anesthesia itself. If subtle changes of tau phosphorylation are investigated, the present study suggests that cervical dislocation is required for sacrifice of rodents.
To understand the possible mechanisms leading to tau phosphorylation during anesthesia, we studied the major tau kinases. Among the kinases studied, only JNK and MAPK were found to be activated during anesthesia, suggesting that anesthesia may induce tau phosphorylation via activation of these stress-activated protein kinases. Anesthesia induced tau phosphorylation at Thr181, Ser199, Thr205, Thr212, Ser262, and Ser404. These sites have been reported to be phosphorylated by JNK and MAPK in various systems [18–20], which is consistent with our conclusions. Both JNK and MAPK have been implicated in hyperphosphorylation of tau and the pathogenesis of AD [21–24]. Interestingly, GSK-3β, which is a major tau kinase [25,26], was found to be inactivated rather than activated during anesthesia, suggesting that the anesthesia-induced tau phosphorylation is unrelated to GSK-3β. Inactivation of GSK-3β was also found previously during anesthesia and cold water stress [17,27,28]. Our further studies on the upstream kinases of GSK-3β pathway indicate that the inactivation of GSK-3β may be the result of AKT activation, the latter of which has been shown in the neurons affected by tau pathology in AD brain [29]. However, activation of PI3K, which is the upstream kinase of AKT, was not observed in the brains of anesthetized mice, suggesting that AKT is activated by other mechanisms instead of PI3K activation during anesthesia. Because activation of JNK and MAPK can cause neuron-specific inhibition of PP2A [30], and PP2A is a negative regulator of AKT [31,32], it is possible that AKT is activated by anesthesia-induced activation of stress-activated protein kinases via PP2A inhibition. Inhibition of PP2A has been reported in the brains of anesthetized mice [17]. Activation of AKT might also contribute to hyperphosphorylation of tau during anesthesia and in AD brain [29].
We have demonstrated in this study that anesthesia led to two phases of tau phosphorylation. In the early phase (up to 5 min), tau was phosphorylated at Thr181, Ser199, Thr205, Thr212, Ser262, and Ser404. Because the body temperature was not decreased in the early phase, tau phosphorylation must result from a hypothermia-independent mechanism. Since stress-activated protein kinases such as JNK and MAPK were activated during this phase, the early-phase phosphorylation of tau likely results from activation of these kinases. When mice were anesthetized for a longer time (1 h), we found that tau was phosphorylated to a much larger extent at Thr181, Thr205, Thr212, and Ser262/Ser356 (12E8 sites) and that the body temperature dropped significantly. Under similar conditions, Planel and colleagues [17] also observed tau hyperphosphorylation at some phosphorylation sites and hypothermia. These authors further suggested that tau hyperphosphorylation was not caused by anesthesia per se, but by anesthesia-induced hypothermia that, in turn, leads to PP2A inhibition. Therefore, as compared to tau phosphorylation during early-phase anesthesia, tau phosphorylation induced by 1 h anesthesia appears to result from an additional mechanism that involves in hypothermia and PP2A inhibition. Two distinct mechanisms leading to tau hyperphosphorylation were also reported in the brain of insulin-deficient mice [33]. Consistent with our conclusions, hyperphosphorylation of tau was also observed in brains of arctic ground squirrels during hibernation [34].
Anesthesia has been associated with cognitive impairment [6–8]. Several studies have demonstrated that anesthesia may increase the risk for AD [9–11]. Now, we have shown that anesthesia induced tau phosphorylation at Thr181, Ser199, Thr205, Thr212, Ser262, and Ser404. These phosphorylation sites are among the hyperphosphorylation sites of tau seen in the early stage of tau pathology in AD brain. Because abnormal hyperphosphorylation of tau and its consequent aggregation play a critical role in neurodegeneration of AD [3–5], the present study provides a possible explanation of the molecular mechanism by which anesthesia associates with cognitive impairment in, and increases the risk for, AD. Our further observation that anesthesia did not induce a global hyperphosphorylation of brain proteins, but instead a specific hyperphosphorylation of tau protein at the AD-related abnormal hyperphosphorylation sites suggests that tau hyperphosphorylation might be a mechanistic bridge linking anesthesia and the risk of cognitive impairment as well as AD.
Acknowledgments
This work was supported in part by funds from the New York State Office of Mental Retardation and Developmental Disabilities, NIH grants (AG027429 and AG019158), a U.S. Alzheimer’s Association grant (IIRG-05-13095), and NSFC grants (30500475 and 30701092). We thank Ms. Janet Murphy for secretarial assistance, and Ms. Maureen Marlow for editorial suggestions.
References
- 1.Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. 1986;261:6084–6089. [PubMed] [Google Scholar]
- 2.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hernandez F, Avila J. Tauopathies. Cell Mol Life Sci. 2007;64:2219–2233. doi: 10.1007/s00018-007-7220-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iqbal K, Alonso A del C, Chohan MO, El-Akkad E, Gong C, Khatoon S, Liu F, Grundke-Iqbal I. Molecular basis of tau protein pathology: role of abnormal hyperphosphorylation. In: Dawbarn D, Allen SJ, editors. Neurobiology of Alzheimer’s Disease. Oxford University Press; New York: 2007. pp. 111–131. [Google Scholar]
- 5.Wen Y, Planel E, Herman M, Figueroa HY, Wang L, Liu L, Lau LF, Yu WH, Duff KE. Interplay between cyclin-dependent kinase 5 and glycogen synthase kinase 3 beta mediated by neuregulin signaling leads to differential effects on tau phosphorylation and amyloid precursor protein processing. J Neurosci. 2008;28:2624–2632. doi: 10.1523/JNEUROSCI.5245-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson T, Monk T, Rasmussen LS, Abildstrom H, Houx P, Korttila K, Kuipers HM, Hanning CD, Siersma VD, Kristensen D, Canet J, Ibanaz MT, Moller JT. Postoperative cognitive dysfunction in middle-aged patients. Anesthesiology. 2002;96:1351–1357. doi: 10.1097/00000542-200206000-00014. [DOI] [PubMed] [Google Scholar]
- 7.Ritchie K, Polge C, de Roquefeuil G, Djakovic M, Ledesert B. Impact of anesthesia on the cognitive functioning of the elderly. Int Psychogeriatr. 1997;9:309–326. doi: 10.1017/s1041610297004468. [DOI] [PubMed] [Google Scholar]
- 8.Xie Z, Tanzi RE. Alzheimer’s disease and postoperative cognitive dysfunction. Exp Gerontol. 2006;41:346–359. doi: 10.1016/j.exger.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 9.Bohnen N, Warner MA, Kokmen E, Kurland LT. Early and midlife exposure to anesthesia and age of onset of Alzheimer’s disease. Int J Neurosci. 1994;77:181–185. doi: 10.3109/00207459408986029. [DOI] [PubMed] [Google Scholar]
- 10.Bone I, Rosen M. Alzheimer’s disease and anaesthesia. Anaesthesia. 2000;55:592–593. doi: 10.1046/j.1365-2044.2000.01479-5.x. [DOI] [PubMed] [Google Scholar]
- 11.Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, Stenzel TT, Speer M, Pericak-Vance MA, Diamond A, Guttmacher AE, Jackson CE, Attisano L, Kucherlapati R, Porteous ME, Marchuk DA. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13:189–195. doi: 10.1038/ng0696-189. [DOI] [PubMed] [Google Scholar]
- 12.Grundke-Iqbal I, Vorbrodt AW, Iqbal K, Tung YC, Wang GP, Wisniewski HM. Microtubule-associated polypeptides tau are altered in Alzheimer paired helical filaments. Brain Res. 1988;464:43–52. doi: 10.1016/0169-328x(88)90017-4. [DOI] [PubMed] [Google Scholar]
- 13.Pei JJ, Tanaka T, Tung YC, Braak E, Iqbal K, Grundke-Iqbal I. Distribution, levels, and activity of glycogen synthase kinase-3 in the Alzheimer disease brain. J Neuropathol Exp Neurol. 1997;56:70–78. doi: 10.1097/00005072-199701000-00007. [DOI] [PubMed] [Google Scholar]
- 14.Bensadoun A, Weinstein D. Assay of proteins in the presence of interfering materials. Anal Biochem. 1976;70:241–250. doi: 10.1016/s0003-2697(76)80064-4. [DOI] [PubMed] [Google Scholar]
- 15.Liu F, Zaidi T, Iqbal K, Grundke-Iqbal I, Gong CX. Aberrant glycosylation modulates phosphorylation of tau by protein kinase A and dephosphorylation of tau by protein phosphatase 2A and 5. Neuroscience. 2002;115:829–837. doi: 10.1016/s0306-4522(02)00510-9. [DOI] [PubMed] [Google Scholar]
- 16.Mazanetz MP, Fischer PM. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat Rev Drug Discov. 2007;6:464–479. doi: 10.1038/nrd2111. [DOI] [PubMed] [Google Scholar]
- 17.Planel E, Richter KE, Nolan CE, Finley JE, Liu L, Wen Y, Krishnamurthy P, Herman M, Wang L, Schachter JB, Nelson RB, Lau LF, Duff KE. Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia. J Neurosci. 2007;27:3090–3097. doi: 10.1523/JNEUROSCI.4854-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu R, Pei JJ, Wang XC, Zhou XW, Tian Q, Winblad B, Wang JZ. Acute anoxia induces tau dephosphorylation in rat brain slices and its possible underlying mechanisms. J Neurochem. 2005;94:1225–1234. doi: 10.1111/j.1471-4159.2005.03270.x. [DOI] [PubMed] [Google Scholar]
- 19.Yoshida H, Hastie CJ, McLauchlan H, Cohen P, Goedert M. Phosphorylation of microtubule-associated protein tau by isoforms of c-Jun N-terminal kinase (JNK) J Neurochem. 2004;90:352–358. doi: 10.1111/j.1471-4159.2004.02479.x. [DOI] [PubMed] [Google Scholar]
- 20.Reynolds CH, Betts JC, Blackstock WP, Nebreda AR, Anderton BH. Phosphorylation sites on tau identified by nanoelectrospray mass spectrometry: differences in vitro between the mitogen-activated protein kinases ERK2, c-Jun N-terminal kinase and P38, and glycogen synthase kinase-3beta. J Neurochem. 2000;74:1587–1595. doi: 10.1046/j.1471-4159.2000.0741587.x. [DOI] [PubMed] [Google Scholar]
- 21.Pei JJ, Braak E, Braak H, Grundke-Iqbal I, Iqbal K, Winblad B, Cowburn RF. Localization of active forms of C-jun kinase (JNK) and p38 kinase in Alzheimer’s disease brains at different stages of neurofibrillary degeneration. J Alzheimers Dis. 2001;3:41–48. doi: 10.3233/jad-2001-3107. [DOI] [PubMed] [Google Scholar]
- 22.Pei JJ, Braak H, An WL, Winblad B, Cowburn RF, Iqbal K, Grundke-Iqbal I. Up-regulation of mitogen-activated protein kinases ERK1/2 and MEK1/2 is associated with the progression of neurofibrillary degeneration in Alzheimer’s disease. Brain Res Mol Brain Res. 2002;109:45–55. doi: 10.1016/s0169-328x(02)00488-6. [DOI] [PubMed] [Google Scholar]
- 23.Ferrer I, Gomez-Isla T, Puig B, Freixes M, Ribe E, Dalfo E, Avila J. Current advances on different kinases involved in tau phosphorylation, and implications in Alzheimer’s disease and tauopathies. Curr Alzheimer Res. 2005;2:3–18. doi: 10.2174/1567205052772713. [DOI] [PubMed] [Google Scholar]
- 24.Zhu X, Lee HG, Raina AK, Perry G, Smith MA. The role of mitogen-activated protein kinase pathways in Alzheimer’s disease. Neurosignals. 2002;11:270–281. doi: 10.1159/000067426. [DOI] [PubMed] [Google Scholar]
- 25.Takashima A. GSK-3 is essential in the pathogenesis of Alzheimer’s disease. J Alzheimers Dis. 2006;9:309–317. doi: 10.3233/jad-2006-9s335. [DOI] [PubMed] [Google Scholar]
- 26.Avila J, Hernandez F. GSK-3 inhibitors for Alzheimer’s disease. Expert Rev Neurother. 2007;7:1527–1533. doi: 10.1586/14737175.7.11.1527. [DOI] [PubMed] [Google Scholar]
- 27.Ikeda Y, Ishiguro K, Fujita SC. Ether stress-induced Alzheimer-like tau phosphorylation in the normal mouse brain. FEBS Lett. 2007;581:891–897. doi: 10.1016/j.febslet.2007.01.064. [DOI] [PubMed] [Google Scholar]
- 28.Li X, Friedman AB, Roh MS, Jope RS. Anesthesia and post-mortem interval profoundly influence the regulatory serine phosphorylation of glycogen synthase kinase-3 in mouse brain. J Neurochem. 2005;92:701–704. doi: 10.1111/j.1471-4159.2004.02898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pei JJ, Khatoon S, An WL, Nordlinder M, Tanaka T, Braak H, Tsujio I, Takeda M, Alafuzoff I, Winblad B, Cowburn RF, Grundke-Iqbal I, Iqbal K. Role of protein kinase B in Alzheimer’s neurofibrillary pathology. Acta Neuropathol (Berl) 2003;105:381–392. doi: 10.1007/s00401-002-0657-y. [DOI] [PubMed] [Google Scholar]
- 30.Kins S, Kurosinski P, Nitsch RM, Gotz J. Activation of the ERK and JNK signaling pathways caused by neuron-specific inhibition of PP2A in transgenic mice. Am J Pathol. 2003;163:833–843. doi: 10.1016/S0002-9440(10)63444-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ugi S, Imamura T, Maegawa H, Egawa K, Yoshizaki T, Shi K, Obata T, Ebina Y, Kashiwagi A, Olefsky JM. Protein phosphatase 2A negatively regulates insulin’s metabolic signaling pathway by inhibiting Akt (protein kinase B) activity in 3T3-L1 adipocytes. Mol Cell Biol. 2004;24:8778–8789. doi: 10.1128/MCB.24.19.8778-8789.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuo YC, Huang KY, Yang CH, Yang YS, Lee WY, Chiang CW. Regulation of phosphorylation of Thr-308 of Akt, cell proliferation, and survival by the B55alpha regulatory subunit targeting of the protein phosphatase 2A holoenzyme to Akt. J Biol Chem. 2008;283:1882–1892. doi: 10.1074/jbc.M709585200. [DOI] [PubMed] [Google Scholar]
- 33.Planel E, Tatebayashi Y, Miyasaka T, Liu L, Wang L, Herman M, Yu WH, Luchsinger JA, Wadzinski B, Duff KE, Takashima A. Insulin dysfunction induces in vivo tau hyperphosphorylation through distinct mechanisms. J Neurosci. 2007;27:13635–13648. doi: 10.1523/JNEUROSCI.3949-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Su B, Wang X, Drew KL, Perry G, Smith MA, Zhu X. Physiological regulation of tau phosphorylation during hibernation. J Neurochem. 2008;105:2098–2108. doi: 10.1111/j.1471-4159.2008.05294.x. [DOI] [PMC free article] [PubMed] [Google Scholar]