

Abstract
Metabolic syndrome (MetSyn) is a group of metabolic conditions that occur together and promote the development of cardiovascular disease (CVD) and diabetes. Recent genome-wide association studies have identified several novel susceptibility genes for MetSyn traits, and studies in rodent models have provided important molecular insights. However, as yet, only a small fraction of the genetic component is known. Systems-based approaches that integrate genomic, molecular and physiological data are complementing traditional genetic and biochemical approaches to more fully address the complexity of MetSyn.
The common forms of type 2 diabetes and cardiovascular disease (CVD) are strongly associated with various common metabolic disturbances, including abdominal obesity, insulin resistance, dyslipidaemias and elevated blood pressure. Since Reaven noted in 1988 that insulin resistance could underlie much of this clustering1, a large body of work has supported the concept that these metabolic traits exhibit causal interactions and common etiologies (BOX 1). The clustering is now known as metabolic syndrome (MetSyn), although there is considerable controversy within the epidemiology and genetics communities as to whether MetSyn exists as a separate syndrome and how it should be defined (BOX 2). By applying a widely used clinical definition, individuals affected with MetSyn have at least a fivefold increased risk of type 2 diabetes and a twofold increased risk of CVD2.
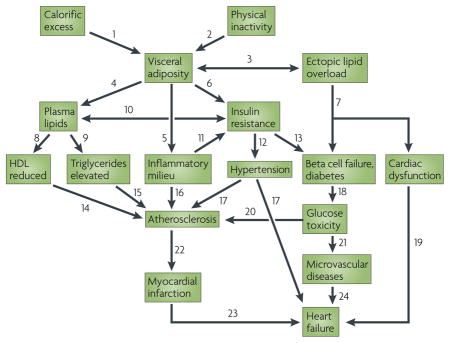
Box 1. Interactions of MetSyn traits in diabetes and cardiovascular diseases.
There are many interactions between components of metabolic syndrome (MetSyn) traits associated with diabetes and cardiovascular disease (CVD). These are shown in the figure and described below.
The response to calorific excess is influenced by genetic factors, as are other interactions2,7,75,80.
Effects of exercise include increased lipoprotein lipase, reduced plasma triglycerides, elevated high-density lipoprotein (HDL), improved glucose tolerance, heart function and oxygen uptake, and lower blood pressure2.
Excess lipids are stored largely in fat, but calorific excess can also promote storage by ectopic tissues, such as muscle and pancreatic beta cells, leading to a form of toxicity57,94.
Excess fat influences lipoprotein levels: for example, in obese individuals increased flux of free fatty acids from fat to the liver might stimulate production of triglyceride-rich lipoproteins7.
Excessive fat results in a proinflammatory state owing to altered production of inflammatory (for example, leptin) and anti-inflammatory (for example adiponectin) mediators and the recruitment of macrophages to adipose34,35,71,72.
Excessive adipose contributes to insulin resistance in part through increased release of free fatty acids and cytokines (for example, tumour necrosis factor a), and decreased production of adiponectin, an insulin sensitizer9,34,35,62,63.
Ectopic fat overload can result in dysfunction of cardiac muscle and pancreatic beta cells57,94,98.
Obesity can lead to decreased lipolysis of triglyceride-rich lipoproteins mediated by decreased lipoprotein lipase and to increased catabolism of HDL mediated by increased hepatic lipase7.
Obesity causes overproduction of very low-density lipoprotein (VLDL) by the liver. Also, insulin resistance results in an inability to suppress hepatic glucose production7.
Overproduction of triglyceride-rich lipoproteins, mediated in part by hepatic insulin resistance, is a common feature of MetSyn. Elevated fatty acids are also implicated in insulin resistance116.
Inter-organ interactions involving cytokines and inflammatory mediators produced by adipose tissue, liver or other tissues seem to contribute to insulin resistance6.
Obesity and insulin resistance can predict future development of hypertension. This might occur by activation of the sympathetic nervous system as well as the renin–angiotensin system. The succinate receptor GPR91 may explain the link between elevated glucose and renin117.
Insulin resistance almost always precedes type 2 diabetes. In diabetes the ability of beta cells to compensate for insulin resistance by both metabolic and mass changes is impaired3–7,9,58.
HDL levels are inversely related to CVD. HDL functions in transport of cholesterol from peripheral tissues to the liver for removal in bile and has anti-inflammatory properties7,91.
Triglyceride levels are a strong predictor of CVD, but the basis of this is still unclear2.
Obesity may exert direct and indirect effects on CVD, as proinflammatory and prothrombotic factors are produced by visceral fat, and a number of adipokines seem to contribute to CVD risk7
Elevated blood pressure has direct adverse effects on arteries, arterioles and the heart, and is strongly associated with stroke, myocardial infarction and myocardial hypertrophy4.
Decreased insulin production and increased insulin resistance result in elevated glucose levels, which can be toxic and proinflammatory92,93.
Poor cardiac function can combine with other MetSyn effects to cause heart failure.
Elevated glucose levels can contribute to atherosclerosis, for example via advanced glycation products92,93.
Probable causes of endothelial dysfunction include elevated glucose, altered lipid profiles, obesity-associated pro-inflammatory molecules such as interleukin-6, and, possibly, oxidative stress7.
Physical disruption or endothelial denudation account for about 75% and 25%, respectively, of myocardial infarction118.
Damage to the heart due to myocardial infarction promotes changes leading to heart failure.
Microvascular disease involves damage of the small arteries and can affect the functioning of the kidneys, brain and heart.
Box 2. A short history of metabolic syndrome.
In 1988 Gerald Reaven explained how insulin resistance and the compensatory hyperinsulinaemia could lead to diverse metabolic abnormalities as well as type 2 diabetes and cardiovascular disease (CVD)1. Reaven originally termed the clustering of abnormalities ‘syndrome X’ and it has also been called ‘insulin resistance syndrome’, but now it is commonly termed metabolic syndrome (MetSyn). The original definition of MetSyn component traits included impaired glucose tolerance, hyperinsulinaemia, reduced high-density lipoprotein (HDL) cholesterol, increased very low-density lipoprotein (VLDL) triglycerides and arterial hypertension, and it was suggested that insulin resistance was the underlying cause of this clustering1. Over the past two decades, it has become clear that these traits are not tied together exclusively by insulin resistance, although it is clearly a key component1,38,119. Additional definitions of MetSyn have been promoted, including alternative component traits such as central obesity (increased waist circumference), microalbuminuria and prothrombotic state (reviewed in REFS 120–122). Although the specific numbers obtained vary depending on the definition used123, based on the adult treatment panel III criteria2, MetSyn is incredibly prevalent worldwide; for example, in the United States about 25% of adults over age 20 and 40% over age 60 exhibit MetSyn124.
The controversy over a specific definition of MetSyn has complicated both clinical and genetic studies, and raises the question of whether there is value in studying the syndrome as a specific entity beyond that of studying the component traits125. However, there is no question that the concept of MetSyn has made both physicians and the public more aware of important interactions, such as the link between obesity and disease and the beneficial consequences of physical exercise for all MetSyn traits126. The concept of MetSyn has also greatly stimulated research into these relationships.
The MetSyn traits are highly heritable — they are shared among relatives according to the degree of relatedness. Recent genome-wide association (GWA) studies have clearly shown that these traits are the result of combinations of common and rare genetic variants, each of which contributes to a tiny fraction of risk. Traditional genetic approaches have identified some key genes and highlighted metabolic pathways that are dysregulated in MetSyn. However, it is becoming clear that new strategies, such as systems approaches that integrate the contributions of genetic variations in numerous genes simultaneously, will be required to elucidate the genetic factors underlying MetSyn in the majority of affected individuals (FIG. 1).
Figure 1. Approaches to identify gene variations that contribute to Metsyn.

Traditional methods for the genetic analysis of metabolic syndrome (MetSyn) (left) include identification of genetic variations and mutations underlying single-gene disorders with large effects on metabolic traits through genetic mapping and positional cloning, or, more recently, genome-wide association studies in which polymorphisms in specific genes are statistically linked with clinical traits of interest in large human cohorts. Systems approaches (right) are being adopted to allow the identification of contributions of small effects from genetic variations in many genes on traits, including levels of mRNA transcripts, proteins and metabolites, as well as clinical traits. The systems approach allows the generation of network models that suggest functional interactions between genes within the network. Genes identified with both approaches can be used to formulate hypotheses, and the corresponding mechanisms can be tested through the generation of genetically engineered mouse models, through the manipulation of gene expression levels in cell-based assays using RNAi or overexpression technologies, and through human clinical studies.
The MetSyn literature is vast and, in this Review, we have not attempted to be comprehensive. Many central issues, such as the relationships between insulin resistance, beta-cell function and the onset of diabetes, have been extensively reviewed elsewhere3–7 and are not addressed in detail here. Rather, we have focused on the current important research areas and challenges that are being addressed with genetic approaches. We first provide an overview of the genetic and environmental factors contributing to MetSyn. Second, we discuss some mostly unexplored but crucially important aspects, including sexspecific effects and maternal nutrition. Third, we outline current understanding of the underlying mechanisms, which is based mainly on studies in rodent models. Finally, we discuss the complexity of MetSyn, arguing that GWA studies and traditional biochemical approaches will not be capable of fully addressing the epistasis and other interactions that predominate in MetSyn, and that more integrative, systems-based approaches will be essential.
Genetic and environmental factors
All MetSyn traits are strongly influenced by genetic factors. Most have heritabilities above 40% and a few, such as obesity and high-density lipoprotein (HDL) levels, have heritabilities as high as 70% in some studies4. But heritability estimates are approximate and they generally make certain assumptions, such as the absence of gene– environment, gene–sex and gene–gene interactions as these are difficult to dissect in human populations. Also, estimates are specific for the particular population studied and will reflect both the diversity of the population and the diversity of the environment8.
Until recently, our understanding of the genetics of MetSyn came largely from studies of Mendelian traits in humans or of biochemically defined candidate genes. Although these studies were enormously informative in providing molecular insights into homeostatic mechanisms, they have not explained how genes interact with each other and with the environment.
Studies that began in the early 1990s to identify genes contributing to the common forms of MetSyn traits using linkage analysis were only modestly successful. This was primarily due to the low power and poor resolution of nonparametric linkage analyses, as well as the unexpected complexity of the traits. Whereas linkage analysis of the genes contributing to the traits in rats and mice is straightforward, at least for those genes contributing more than a few percent of the variance of the trait, resolution is very poor and loci generally contain 100 or more genes. As a consequence, successes in identifying genes underlying quantitative trait loci (QTLs) have been few.
GWA studies became feasible following the completion of the human genome sequence, the cataloguing of common variations in human populations and the development of improved SNP genotyping technologies. Association approaches are more powerful and have much better resolution than linkage approaches. Several large studies of traits relevant to MetSyn have been reported (BOX 3); these have confirmed a number of genes previously identified through candidate gene approaches and have identified many novel genes or loci (reviewed in REFS 9–11). These include: two common variants that affect fasting glucose levels (glucokinase (hexokinase 4) regulator (GCKR), and a genomic region containing glucose6phosphatase, catalytic, 2 (G6PC2) and ATP binding cassette, subfamily B, member 11 (ABCB11)); two obesity (that is, adiposity) variants (fat mass and obesity associated (FTO) and melanocortin 4 receptor (MC4R)); 19 type 2 diabetes loci; and many triglyceride, HDLcholesterol and lowdensity lipoprotein (LDL)-cholesterol loci12–24. None of the genes identified affect the entire spectrum of MetSyn traits, although some influence several of them (BOX 3). For example, studies of FTO show that, although its primary effect is on adiposity, it has secondary effects on insulin sensitivity, adipokine levels and resting metabolic rate25,26. Additional genetic studies, identification of rare functional variants by high-throughput sequencing and analysis of copy number variation should add to our knowledge24,27–29.
Box 3. Human genome-wide association studies: a view of the genetic architecture of MetSyn.
Several genome-wide association (GWA) studies relevant to metabolic syndrome (MetSyn), type 2 diabetes and coronary artery disease have confirmed candidate gene associations and have identified a number of novel genes and loci (discussed in the text and reviewed in REFS 9–11). Examples include:
Melanocortin 4 receptor, MC4R: this gene was identified in Asian-Indian and European populations for several MetSyn traits17, 127. Rare MC4R loss-of-function variants have previously been associated with hyperphagia and childhood obesity, and experimental studies have identified it as a key regulator of energy balance.
Fat mass and obesity associated, FTO: two GWA studies of Europeans have associated FTO with body mass index14, 23. Recent studies in rodents suggest that FTO might be co-regulated with an adjacent gene, FTM, and that it exhibits phenotypic overlap with Bardet-Biedl syndrome128.
MLX interacting protein-like, MLXIPL: in European and Indian–Asian populations this gene is linked to plasma triglyceride levels129. Its protein product coordinates transcriptional regulation of enzymes that channel glycolytic end-products into lipogenesis and energy storage.
Transcription factor 7-like 2, T-cell specific, HMG-box, TCF7L2: this is one of 19 susceptibility genes for type 2 diabetes. Although originally identified by genetic linkage followed by traditional genetic association130, this association was confirmed by GWA studies24. Many of the type 2 diabetes genes, including TCF7L2, seem to affect pancreatic beta-cell function.
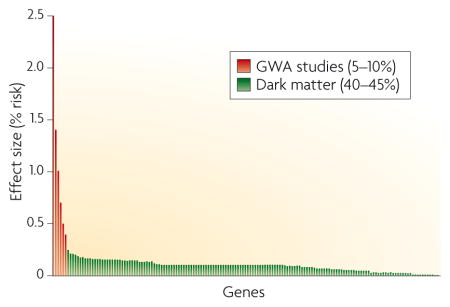
As yet, no genetic factors that encompass all MetSyn traits have been identified. This might simply reflect the lack of power of the analyses, as genes perturbing individual pathways might indirectly contribute to traits such as lipid levels and blood pressure. Some traits, such as blood pressure, have few or no loci that achieve genome-wide significance, and for the others the identified loci explain less than 10% of the variance of the trait. As most MetSyn traits have heritabilities of approximately 50%, genes detectable above the noise of GWA studies explain only a small fraction of the genetic component. This is illustrated in a hypothetical distribution of genes contributing to MetSyn (see figure). Genes identified thus far by GWA studies (shown at left) tend to be those exerting the largest effects, but account for only ~5–10% of the trait variance. The majority of remaining genes (so called ‘dark matter’) will be more difficult to identify owing to their modest effects on MetSyn traits, complex interactions and rare variations.
The loci identified in GWA studies frequently contain several genes in strong linkage disequilibrium, and biochemical or animal model studies might be required to definitively identify which gene is causal. One promising approach to validate susceptibility genes involves network modelling, which allows genes of unknown function to be related to known pathways or clinical traits. For example, the integration of human and mouse genotypic and expression data suggested that sortilin 1 (SORT1) and cadherin EGF LAG sevenpass G-type receptor 2 (CELSR2) are susceptibility genes for CVD and hyperlipidaemia30.
The loci identified thus far from GWA studies explain less than 10% of the population variance of MetSyn traits. Given the high heritabilities of MetSyn traits, it seems that the GWA study results reported so far have mapped a tiny fraction of their genetic components. This raises the possibility that MetSyn is underpinned by hundreds of genes, each with modest effects, and by many rare mutations not detected in GWA studies (BOX 2). Genes with such modest effects will be difficult to study using standard genetic approaches, and genetic heterogeneity, interactions and ethnic differences will complicate analyses. Recent sequencing studies designed to identify rare genetic variants involved in common disorders suggest that these are likely to contribute significantly to MetSyn traits (for example, REFS 27,28).
A central tenet of MetSyn has been the ‘thrifty gene’ hypothesis — the notion that the repeated famines in human history have selected for alleles that result in obesity during times of plentiful food. Thus, when a famine occurs those individuals with excess fat would be most likely to survive. However, recent data indicating that death from starvation results primarily from infection rather than depletion of fat stores has put the hypothesis into question31.
Environmental influences also play a major part in MetSyn: a high-calorie diet and a sedentary life-style are primary environmental contributors (BOX 1). Environmental factors are difficult to study in humans. Even if diet and exercise could be accurately assessed, interactions with genetic factors would be difficult to study because no two humans, with the exception of identical twins, share the same genetic background. Consequently, few human studies have as yet attempted to tackle gene–gene and gene–environment interactions, and have focused instead on single candidate genes. Whether classical genetic and molecular biology approaches can address these complex interactions is unclear. Alternative systems-based approaches are discussed later in this Review.
Important gaps in the understanding of MetSyn
Sex differences and MetSyn susceptibility
Men and women differ in susceptibility to MetSyn and its components, including obesity, insulin resistance, CVD and hypertension32. Differences between males and females in insulin resistance seem to be related to differences in the anatomical distribution of fat5,33,34. Males tend to have more visceral fat, which is linked to insulin resistance, whereas females typically carry more subcutaneous fat.
Several hypotheses have been put forward to explain the link between visceral fat and insulin resistance. One possibility is that the molecular characteristics of visceral versus subcutaneous fat differ, leading to increased visceral adiposetissue lipolysis, glucocorticoid receptor activity and inflammatory cytokine secretion, and to reduced secretion of the insulin sensitizing adipokine, adiponectin34,35. Alternatively, the physical location of the Mechanism visceral-fat compartment might allow release of free fatty acids, inflammatory cytokines and other adipose tissue metabolites directly into the portal circulation33. Recent gene expression profiling studies suggest that there are intrinsic molecular differences between visceral and subcutaneous adipose tissue depots in both humans and mice36. Furthermore, fat transplantation studies in the mouse revealed that transfer of subcutaneous adipose tissue to an intra-abdominal compartment led to an overall decrease in body fat and improved glucose homeostasis37,38. These results indicate that metabolic differences exist between fat deposited at subcutaneous versus visceral sites, at least in mice.
These studies raise the possibility that visceral and subcutaneous fat depots might be derived from distinct progenitor populations35. This is consistent with observations in human lipodystrophies caused by rare gene mutations. Individuals with Dunnigan-type lipodystrophy exhibit a dramatic loss of subcutaneous adipose tissue, but normal or increased fat accumulation in visceral, neck and facial areas39, and Hottentot (Khoisan) women show marked accumulation of gluteal–femoral adipose tissue40.
Given the important metabolic differences between visceral and subcutaneous fat depots, it is important to understand the genetic basis for their occurrence in males and females. Differences in levels of gonadal hormones are undoubtedly important. For example, the accumulation of excess abdominal adipose tissue in males is associated with low levels of gonadal androgen, and the reduced levels of oestrogen, progestins and androgens that occur in menopause are associated with increased central fat distribution in women41,42. Male and female mice exhibit marked gene expression differences in metabolic tissues, such as adipose tissue and liver43. Genetic studies in mice have revealed striking interactions between sex and adiposity, and some alleles affect adiposity in opposite directions between males and females. However, factors other than gonadal hormones also seem to have a role, as not all sex differences in adipose tissue gene expression are abolished by gonadectomy. For example, threefold higher adiponectin, leptin and resistin mRnA levels persist in female compared with male mice 3 weeks after castration at 9 weeks of age44. Differences occurring between males and females after gonadectomy can be attributed either to differences in previous steroid hormone history or to genetic differences that are due to the dosage of X and Y chromosome genes. Indeed, sex differences in embryonic growth of humans and mice are caused by direct effects of genes located on the sex chromosomes, as these differences precede the differentiation of gonads45. The contribution of sex-chromosome effects to obesity and MetSyn could be examined in, for example, mice that are engineered so that sex chromosome complement segregates independently of male and female gonadal sex46. By comparing normal male and female mice with animals that develop male gonads despite having an XX chromosome complement or female gonads despite an XY chromosome complement, it might be possible to assign MetSyn traits to hormonal or genetic factors.
Epigenetics and maternal nutrition
One potentially important environmental factor for MetSyn that has recently been experimentally validated is the quality of fetal nutrition. Extensive epidemiological studies originally revealed inverse correlations between birth weight and CVD47. Many additional studies have confirmed these findings and led to the hypothesis that early life stressors, such as poor maternal nutrition, maternal obesity and rapid postnatal weight gain, can programme metabolic adaptations for survival in a nutrient poor environment48. Thus, if the actual postnatal environment is not nutrient poor, such programming can lead to adult-onset MetSyn. Subsequent studies in rats and mice have confirmed these associations and have revealed evidence of both metabolic and structural programming48,49. For example, following intrauterine growth retardation (IUGR) in rats, the affected off-spring showed impaired insulin secretion and developed MetSyn traits with ageing50. Recently, studies in mice showed that the offspring of mothers maintained on a restricted diet exhibited a premature leptin surge and an increased density of hypothalamic nerve terminals49. CVD has also been associated with maternal hypercholesterolaemia in humans51, rabbits and mice52, raising the possibility of overlapping mechanisms. It should be noted that although the human studies could clearly involve genetic contributions, this cannot be the case for the animal studies owing to the use of inbred strains.
Recent studies suggest that epigenetic mechanisms might underlie intrauterine programming53. One of the molecular phenotypes associated with the aforementioned IUGR in rats is decreased expression of PDX1, a key transcription factor regulating pancreatic development. Recently, reduced PDX1 activity was associated with alterations in histone modification50. Similar findings were observed for the glucose transporter GLUT4 in the muscle of IUGR rats54. Interestingly, some metabolic traits resulting from low birth weights can be transmitted to subsequent generations, suggesting the possibility of epigenetic changes maintained during meiosis, as is observed for the agouti coat-colour variants in mice53. It will be of great interest to examine the effects of intrauterine programming using more global techniques for monitoring gene expression and chromatin structure.
Mechanistic insights
We now have at least a partial understanding of how molecular links between obesity, insulin resistance, dyslipidaemias and hypertension contribute to diabetes, atherosclerosis, heart failure and stroke (BOX 1). But the complexity and heterogeneity of MetSyn have made mechanistic studies in humans especially difficult, and most molecular details have come from studies with experimental models, particularly rodents3,7,55–57. A large number of MetSyn models, including naturally occurring, and dietarily and genetically induced, have been developed in rats and in mice55,56. Now the mouse is more widely used owing to its more advanced genetics and its ease of genetic manipulation.
Inbred strains of rodents provide a uniform genetic background, so the effects of genetic or environmental perturbations can be inferred. Also, the effects of genetic modifiers can be examined on the background of disease-causing alleles. Tools such as tissue-specific knockouts have enabled examination of the contributions of individual tissues or cell types to MetSyn. For example, although adipose tissue, pancreas and muscle have long been known to contribute to MetSyn, studies in rodents have indicated that several additional tissues, including brain, liver, intestine, kidney and haematopoietic cells, can have causal roles. Although genes and metabolic pathways are highly conserved between rodents and humans, and rodent models are particularly valuable for dissecting mechanistic interactions, human studies remain essential for determining their relevance to disease. For example, a significant factor in type 2 diabetes in humans but not rodents seems to be islet amyloid polypeptide, which contributes to beta cell endoplasmic reticulum (ER) stress and B-cell loss through apoptosis58.
Fuel partitioning
Insulin resistance is a central feature of MetSyn, and mouse models have had a major impact on our understanding of how dysregulated lipid and glucose metabolism contribute to its development. In 1963, Randle proposed a glucose–fatty acid cycle in an attempt to explain the inhibition of glucose oxidation by excess fatty acids59, and it is now known that in addition to their effect on glucose oxidation, fatty acids reduce insulin-stimulated glucose uptake into muscle60. Although Randle’s model did not address insulin signalling, it made several predictions that have since been tested in mouse models. For example, the fatty acid effect on insulin sensitivity is abrogated by deletion of fatty acid transport protein 1 (REF. 61), indicating that fatty-acid metabolites contribute to insulin resistance.
The question of why fatty acid uptake is linked to insulin resistance has been explored through genetic manipulation of enzymes that incorporate fatty acids into specific lipid species in mouse models. An initial hypothesis was that the incorporation of excess fatty acids into triacylglycerol (that is, fat) in tissues such as liver, skeletal muscle and pancreatic beta cells might cause insulin resistance. Manipulation of enzymes that convert fatty acids into various glycerolipids and eventually into triacylglycerol demonstrated that diacylglycerol, the immediate precursor of triacylglycerol and a lipid signalling molecule, is the culprit associated with reduced insulin sensitivity62. In addition, a structurally related lipid, ceramide, also modulates insulin sensitivity through the insulin signalling pathway63.
Mitochondria
Type 2 diabetes is associated with a decline in oxidative phosphorylation and commensurately diminished aerobic capacity64. A similar decline in oxidative capacity seen in elderly subjects65 is possibly linked to the sharp increase in diabetes that occurs with age. As mitochondria are essential for oxidative metabolism, attention has focused on impaired mitochondrial function in muscle and other tissues as a contributor to insulin resistance. Microarray analysis in muscle tissue of insulin resistant and diabetic subjects revealed a global, albeit modest, downregulation of genes encoding a subset of enzymes of oxidative phosphorylation and mitochondrial function66. In addition, insulin resistant non-diabetic offspring of patients with type 2 diabetes show a similar trait, implying that it is heritable and potentially causal for diabetes67.
The transcriptional co-activator peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PRGC1; also known as PGC1α) has a role in mitochondriogenesis and the regulation of oxidative phosphorylation genes. However, testing of PRGC1 in patient groups revealed that its expression levels are not necessarily altered in diabetes68, and a mouse model with muscle-specific deletion of PRGC1 had increased insulin sensitivity rather than the predicted insulin resistance69. These observations suggested that it is not reduced mitochondrial fatty acid oxidation rate, but inefficient oxidation in conjunction with lipid overload leading to an accumulation of oxidative intermediates, that is associated with insulin resistance70. In support of this hypothesis, mice deficient for malonyl-CoA decarboxylase, an enzyme that metabolizes an inhibitor of fatty acid uptake into the mitochondria, failed to accumulate fatty acid oxidation intermediates and were rescued from insulin resistance induced by a high-fat diet70.
Inflammation
MetSyn and its component traits, such as obesity and insulin resistance, have been associated with chronic inflammation, as evidenced by elevations in circulating levels of Creactive protein (CRP), tumour necrosis factor α (TNFα), fibrinogen, platelet activator inhibitor 1 and interleukin-6 (IL6)71,72. Although it was initially suspected that the liver contributed most of these cytokines, microarray studies of adipose tissue from lean versus obese mice revealed that adipose tissue from obese mice contains a substantial macrophage population, which correlates with adiposity and is responsible for the production of many of these cytokines73,74. Dietary cholesterol enhances the accumulation of adipose tissue macrophages in LDL-receptor-deficient mice, providing an additional link between obesity, insulin resistance and atherosclerosis — key components of MetSyn75.
Mouse models have shed light on factors that contribute to the recruitment of macrophages to adipose tissue in obesity. The monocyte chemoattractant proteins (MCPs), which are required for recruitment of monocytes to sites of injury, seem to play a part, as mice with genetic deficiency of MCP1 or its receptor CCR2 exhibit reduced macrophage accumulation in obesity76,77. Macrophages that are present in obese adipose tissue differ from resident macrophages. Resident tissue macrophages typically exist in a quiescent state (the alternative activation state) and do not actively secrete immune modulatory factors. However, macrophages in obese adipose tissue resemble those that are stimulated by foreign pathogens in conjunction with T lymphocytes, which become activated to secrete inflammatory cytokines (the classically activated state)78. The nuclear receptor peroxisome proliferator-activated receptor gamma (PPARG) might help regulate this process, as macrophage-specific disruption of the PPARG gene impairs alternative macrophage activation and promotes the development of diet-induced obesity and insulin resistance. Understanding the alterations that elicit inflammation in adipose tissue may provide promising new therapeutic targets.
Endoplasmic reticulum stress
Insulin resistance is also associated with oxidative stress that affects the ER in adipose and other tissues79. As the site of protein and lipid synthesis, the ER has a central metabolic role in integrating nutrient signals. Nutrient overload increases demand for adipose tissue expansion, requiring increased lipid and protein synthesis. This provokes an adaptive response in the ER known as the unfolded protein response (UPR), which activates several signalling pathways to re-establish homeostasis by limiting protein synthesis and inducing transcription of chaperone proteins that assist with the unfolded protein load80. As with the induction of macrophage infiltration and inflammation of adipose tissue described above, this adaptive response might have negative consequences when it is present chronically in conditions such as obesity, insulin resistance and atherosclerosis.
The UPR is activated in adipose tissue in mice by both dietarily and genetically induced obesity81. Consequences of the UPR include activation of the Jun N-terminal kinase (JNK) pathway with subsequent serine phosphorylation of insulin receptor substrate 1 (IRS1) and insulin resistance. Furthermore, haploinsufficiency for X-box binding protein 1 (XBP1), a transcription factor for chaperone proteins and ER biogenesis proteins, leads to increased JNK activation and insulin resistance. The UPR also activates nuclear factor-κB (NF-κB), a key inflammatory transcription factor, leading to increased expression of TNFα, IL6 and MCP1 and impaired insulin sensitivity82. The UPR is also induced in macrophages and endothelial cells83,84, indicating its key role in several key tissues in MetSyn.
Identification of ER stress and the UPR as inflammatory mechanisms in obesity, insulin resistance and atherosclerosis has also suggested potential therapeutic targets. For example, treatment of genetically obese mice with the chemical chaperones phenylbutyric acid and taurine-conjugated ursodeoxycholic acid decreases UPR signalling in adipose tissue and liver and improves systemic insulin sensitivity without affecting body weight85. Similarly, administration of salicylic acid, which inhibits the inhibitor of NF-κB kinase β (IKKβ)–NF-κB pathway, improves insulin signalling and reverses hyperglycaemia, hyperinsulinaemia and hyperlipidaemia in obese rodents86, and prevents free fatty acid-induced hepatic insulin resistance87.
Diabetes and cardiovascular disease
The causal relationships between MetSyn and diabetes have been extensively studied3–7. Although many obese individuals develop insulin resistance, only a fraction go on to develop diabetes. Obese rodent models, such as leptin-deficient mice or rats, all develop insulin resistance, but whether they develop diabetes is dependent upon the genetic background3,55. The beta cells of resistant strains proliferate to keep up with the increasing insulin demand, whereas the beta cells of susceptible strains undergo apoptosis. Several mechanisms have been proposed for increased beta-cell apoptosis, including oxygen free radicals, free fatty acid toxicity, interleukin-1β and formation of islet amyloid polypeptide toxic oligomers58. Positional cloning of a QTL in mice identified a novel gene, VPS10 domain receptor protein SORCS 1 (Sorcs1), that might be involved in the maintenance of islet vasculature and islet growth88. As suggested by recent GWA studies as well as studies with animal models, diabetes development is determined primarily by pancreatic beta-cell responses to insulin resistance.
MetSyn is also a strong susceptibility factor for CVD. A number of genetically engineered mouse models that develop hypercholesterolaemia and relatively advanced atherosclerotic lesions are now used for atherosclerosis research. The most widely used are apolipoprotein E null and LDL receptor null mice. The latter, when on the genetic background of strain C57BL/6J and fed a high-fat diet, develops all the MetSyn traits with the exception of hypertension89. Although these mice develop atherosclerotic lesions, they lack some features that are crucial in human disease, such as the rupture or erosion of lesions, which triggers thrombus formation and is the most common cause of myocardial infarction in humans90.
Atherosclerosis is a disease of the large arteries that is characterized by an accumulation of necrotic cell debris, cholesterol, fibrous tissue and inflammatory cells in the subendothelial space. The effects of obesity and insulin resistance on traditional risk factors such as HDL and triglyceride levels and blood pressure are important. Recent data have also revealed common variations in the anti-inflammatory properties of HDL91. There is emerging evidence that visceral fat secretes proinflammatory and prothrombotic factors, such as leptin, adiponectin, IL6, TNFα and plasminogen activator inhibitor, type I (PAI1), that exert direct effects on the vessel wall7. The interactions between diabetes and CVD have proven difficult to address owing to a paucity of animal models92. However, streptozotocin-induced elevations of glucose do accelerate atherosclerosis on a hyperlipidaemic background, mediated in part by the receptor for advanced glycation products93.
Obesity has been associated with structural and functional changes of the heart94. Interestingly, obese patients tend to have a better chance of survival once they are diagnosed with CVD, a phenomenon termed the ‘obesity paradox’95. Obesity in animal models is associated with coincident morbidities, including cardiac hypertrophy, cardiac adiposity and valvular dysfunction94. Obesity increases the use of fatty acids and decreases the use of glucose as myocardial substrates96, and recent studies have revealed that these changes are accompanied by increased myocardial oxygen consumption and decreased cardiac efficiency97. Mitochondrial numbers increase, in part due to PRGC1 and PPARA upregulation, but there is reduced oxygen consumption and ATP generation and increased superoxide production. Excess accumulation of lipids, such as ceramide, in cardiomyocytes can directly induce cardiomyopathy, a deterioration of the myocardium98.
Addressing the complexity of MetSyn
Only a small fraction of the genetic component of MetSyn is explained by known variations, and mechanistic insights have been based largely on qualitative analyses, such as studies of knockout mice, without regard to epistasis or other interactions. Indeed, it seems unlikely that the highly complex gene–gene and gene–environment interactions that are central to MetSyn can be easily modelled in transgenic mice. Recent studies imply that systems-based approaches might be able to better address such complex interactions.
Systems-based approaches
Systems biology uses technologies such as gene expression microarrays and mass spectrometry in combination with computational and statistical tools to address complex systems. MetSyn involves inputs from hundreds of genes, many environmental factors and a host of tissues. Therefore, analysing the individual components of a system is not sufficient, as it is important to know how these components interact with each other and how these interactions differ in disease states. Genetic and environmental factors influence clinical traits by perturbing molecular networks, and systems-based approaches have the potential to interrogate these molecular phenotypes and identify patterns associated with disease.
Currently, expression arrays provide the only quantitative, genome-wide window into molecular phenotypes, but high-throughput technologies for screening of proteins and metabolites, such as mass spectrometry, are also quite advanced. Microarray studies have revealed small alterations in expression levels of many genes, thus highlighting important pathways in MetSyn. These include altered macrophage-derived inflammatory gene expression in adipose tissue in obesity73 and, although not reproduced in other populations, altered oxidative phosphorylation gene expression in diabetic muscle66,99. Both of these findings spurred extensive investigation and have elucidated new aspects of the pathology of MetSyn.
Systems-based approaches seek to move beyond simple correlations of levels to determine how components interact. Such interactions can be based on known connections from the literature, experimental determination of physical interactions or experimental perturbations to test for co-regulation. Frequently, these interactions are described in terms of networks that consist of parts (nodes) and their connections (edges) (FIG. 2). The dynamics of the system can be mathematically modelled, allowing prediction of the response of the system to genetic or environmental perturbations100–102
Figure 2. Integration of DNA variation, gene expression and clinical phenotypes for analysis of complex traits.

a | The examination of molecular phenotypes, such as transcript levels, in genetically randomized mouse or human populations is a powerful strategy to identify molecular networks underlying complex traits. For example, genetic variations perturb both the molecular phenotypes (transcript levels) and clinical traits. Loci controlling transcript levels (termed expression QTLs, eQTLs) can be mapped and used to prioritize positional candidates111. Genes that vary together when subjected to multiple genetic perturbations are likely to share regulatory pathways, and the relationships thus obtained often closely parallel known biology, and have in some cases been validated by experimental perturbations (for example, REFS 83, 107, 111). b | A co-expression network of about 3,000 genes constructed from liver gene expression patterns of a segregating population of mice identifies modules of highly correlated genes133, which are indicated by the different colours. As some modules are highly correlated with clinical traits, they can explain much more of the total variation than any individual locus. Although it is possible that modules are due to the perturbing effects of the clinical traits themselves, this is unlikely because there is little overlap between the loci controlling the clinical traits and the loci perturbing the module genes. c | Correlations of the individual colour-coded liver modules from part b (correlations averaged over their genes) and body weight133. Strong correlation of particular network modules (for example, the module marked with an asterisk) with clinical traits provides a means of identifying new disease pathways. Although co-expression networks do not indicate the direction of interaction between nodes, causal modelling can be performed as discussed in BOX 5 to create ‘directed networks’. d | Sub-section of predicted causal interactions involving sortilin 1 (SORT1) and other genes in the liver30. Part d is reproduced from REF. 30.
The first biological networks were based on the large body of knowledge of metabolic pathways gained from over half of a century of biochemical studies103. Subsequent studies have extended such bibliomic data and combined it with genome annotation104. They have shown that biological circuitry is not random and that it tends to obey certain principles. The network concept has proven extremely useful in studies of metabolic traits, revealing ‘emergent properties’ not otherwise apparent as well as novel drug targets102–104.
Integrative genetics
One systems-based approach that has proven particularly powerful for analysis of MetSyn involves the integration of common DNA variation, global expression array analysis and clinical phenotypes (FIG. 2). In any natural population there will be thousands of polymorphisms that perturb gene expression. If transcript levels are quantified in genetically randomized individuals, such as patients with MetSyn or mice generated from a cross between strains differing in MetSyn traits, then these levels can be related to both DNA variation and to clinical traits by genetic mapping and correlation105,106.
Like other quantitative traits, transcript levels can be mapped using linkage or association. The resulting loci are termed expression QTLs (eQTLs) or expression SnPs (eSnPs), respectively. Using microarray technologies it is feasible to identify eQTLs for thousands of genes in genetic crosses in rodents, or in family or population studies in humans106–108. Databases of eQTLs are proving useful for prioritizing candidate genes for genetic traits. A good example of the use of eQTLs to help identify genes underlying MetSyn is the identification of osteoglycin (Ogn) in the control of left ventricular mass (LVM)109. Rat recombinant inbred strains segregating for complex variations of LVM were studied using eQTL analysis of the heart, and a number of cis-acting eQTLs localizing with a LVM QTL were identified. These were prioritized on the basis of the strength of the eQTL and correlation with LVM in both rats and human heart biopsies, and studies with Ogn knockout mice confirmed the identity of the gene109. Other findings include links between Cd36 and pressure elevation in rats110, and between Abcc6 and vascular calcification in mice111.
The fact that genetically randomized populations exhibit multiple perturbations influencing both molecular and clinical traits offers an opportunity to model causal interactions (BOX 4). In such modelling, the direction of the interactions between DNA and the traits can be inferred on the basis of conditional probabilities. This approach was recently applied to identify candidate genes involved in obesity and other MetSyn traits in a segregating mouse population, and several genes were experimentally validated using transgenic approaches107,112.
Box 4. Causal modelling of complex traits.
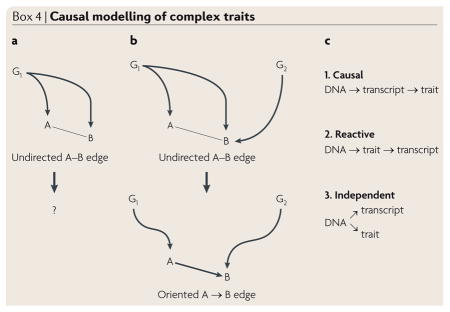
Integrative genetics can be used to model causal interactions between DNA variation and transcript levels, as well as clinical traits in genetically randomized populations. An important aspect of this approach is the complex nature of the genetic perturbations. Whereas single gene perturbations, such as transgenic mice, allow causality to be established, they have limited power to resolve the pleiotropic and homeostatic interactions resulting from the perturbation. Conversely, multiple perturbations, such as populations segregating for common genetic variations, allow detailed analyses of the interactions131.
The concept is illustrated in the figure. Panel a shows analysis of elements A and B that are perturbed by a single gene, G1. The relationship between A and B (whether G1 acts on A which then acts on B or vice versa, or whether G1 perturbs the two independently) cannot be determined. However, the introduction of a second perturbing gene, G2, shown in panel b, can clarify the relationship. For example, suppose that G2 acts solely on B, and G1 acts solely on A, which in turn perturbs B. In that case, the correlation between G1 and B would be expected to be the product of the correlation between G1 and A and the correlation between A and B. In the context of integrative genetics (FIG. 2), such relationships can be modelled using conditional probabilities, with the DNA variation serving as a causal anchor. For example, as illustrated in panel c, there are three likely relationships between DNA variation, the expression of a gene (transcript) and a clinical trait132. The likelihood of each model can be calculated and the one with the best fit chosen. Several algorithms for such causality modelling have been reported (for example, REF. 112), and recent studies with transgenic mice have validated the approach for the identification of genes for adiposity and insulin resistance107,112. More general methodologies, such as constrained Bayesian networks, can be used to model causal interactions among the elements of a biological network, as shown in FIG. 2d.
Data from such studies can be used to construct co-expression networks in which the nodes are transcript levels and the edges represent correlations between transcripts (FIG. 2b). Such modelling is based on the assumption that genes with correlated expression are likely to be functionally associated (although other explanations, such as linkage or linkage disequilibrium, or the impact of the clinical trait itself, could also result in correlations). It is also clear that many functionally associated genes would not be correlated, given that much regulation is post-transcriptional. Thus, such networks are clearly approximations of the underlying biology, and integration with other data sets and approaches is important. Nevertheless, groups of genes, or ‘modules’, identified by co-expression modelling are significantly enriched for functionally related genes. These modules have proven useful for annotating novel genes and revealing regulatory mechanisms (for example, REF. 113). Interestingly, certain modules correlate strongly with clinical traits (FIG. 2c). In some cases, the modules explain a much greater fraction of the variance of the clinical trait than any individual QTL, suggesting that they reflect a higher order network that integrates multiple genetic inputs. Co-expression networks can also be integrated with causal modelling to construct ‘directed’ networks (FIG. 2d). This approach was recently used to identify key genetic drivers in a macrophage-enriched subnetwork that is strongly associated with a number of MetSyn traits107.
Although it is difficult to obtain appropriate human tissues, recent systems-based analyses of liver and adipose tissue biopsies clearly show that these network modelling approaches are applicable to complex traits in humans30,114. Importantly, there is evidence that co-expression networks exhibit a degree of conservation between mice and humans107. Recently, such modelling has helped identify the likely susceptibility genes at several loci identified in human GWA studies30. Such integrative genetics approaches are now being expanded to include proteomic and metabolomic data. For example, analysis of levels of 67 metabolites in a cross between two strains of mice was used to construct a causal network linking gene expression and metabolic changes. The network was validated by examining responses to metabolic perturbations115.
Future directions
Recent advances in MetSyn have been impressive, but there remain large areas of ignorance, such as understanding details of the genetic and environmental interactions that are involved in MetSyn. Many successes in identifying genes for MetSyn traits have occurred within the last 2 years using GWA studies, and there are likely to be many additional findings in the next several years. These might help reveal the nature of the ‘dark matter’ (BOX 3) — that is, whether it results from complex interaction or simply from many variations, rare or common. These are of course important issues for the development of personalized medicine.
We believe that considerable effort now should be devoted to examining MetSyn from a broad perspective rather than focusing narrowly on individual pathways or metabolic components. This will require the application of interdisciplinary approaches, such as genetics, genomics, proteomics, metabolomics, physiology and mathematical modelling. This should eventually enable the development of a holistic picture of MetSyn, integrating information from multiple scales, including genes, transcripts, proteins, organelles, cells, tissues and organisms.
Despite great advances in mechanistic understanding, recent efforts to develop new therapies for MetSyn have a poor record of success. CVD has certainly not disappeared with the advent of powerful cholesterol-lowering drugs, and the widely used existing drugs — most notably the statins — are not without their side-effects. Thus, there are important unmet therapeutic needs. The identification of the metabolic pathways that are perturbed in MetSyn, and of the genetic networks that control them, might provide a clearer understanding of how the disease develops and how the components interact. This might suggest, in turn, the most rational targets for the development of effective and safe therapeutic strategies.
Acknowledgments
We thank our colleagues for valuable discussion, C. Farber and A. Ghazalpour for help with figures, and R. Chen for secretarial assistance.
- Insulin resistance
A condition in which normal amounts of insulin are inadequate to produce a normal response from muscle, fat, liver or other cells. Such insulin resistance can result in elevated glucose levels in the blood owing to decreased uptake by cells, as well as effects on glycogen storage and lipid metabolism
- Dyslipidaemia
An abnormal or atypical pattern of lipoproteins in the blood. Examples include low levels of high-density lipoprotein cholesterol (hypoalphalipoproteinaemia), or elevated levels of triglyceride (hypertriglyceridaemia) or cholesterol (hypercholesterolaemia)
- Genome-wide association study (GWA study)
An examination of common genetic variation across the genome designed to identify associations with traits such as common diseases. Typically, several hundred thousand SNPs are interrogated using microarray technologies
- High-density lipoprotein (HDL)
One of five classes of lipoproteins in the blood that transport cholesterol and triglycerides between tissues. HDL levels are inversely correlated with cardiovascular disease and thus are hypothesized to be protective, perhaps by removing cholesterol from atheroma
- Heritability
An estimate of the proportion of genetic variation in a population that is attributable to genetic variation among individuals
- Linkage analysis
Analysis of the segregation patterns of alleles or loci in families or experimental crosses. Such analysis is commonly used to map genetic traits by testing whether a trait co-segregates with genetic markers whose chromosomal locations are known
- Quantitative trait locus (QTL)
A genetic locus that influences complex and usually continuous traits, such as blood pressure or cholesterol levels. QTLs are identified using linkage analysis
- Linkage disequilibrium (LD)
In population genetics, LD is the nonrandom association of alleles. For example, alleles of SNPs that reside near one another on a chromosome often occur in nonrandom combinations owing to infrequent recombination. LD is useful in genome-wide association studies as it reduces the number of SNPs that must be interrogated to determine genotypes across the genome. Conversely, strong LD can complicate the identification of functional variants. LD should not be confused with genetic linkage, which occurs when genetic loci or alleles are inherited jointly, usually because they reside on the same chromosome
- Visceral fat
Fat that is located inside the peritoneal cavity, between internal organs, as opposed to subcutaneous fat, which is found under the skin, or intramuscular fat, which is interspersed in skeletal muscle
- Correlation
In statistics, a measure of the strength and direction of a linear relationship between two variables. Usually measured as a correlation coefficient
- Epigenetics
Changes in gene expression that are stable through cell division but do not involve changes in the underlying DNA sequence. The most common example is cellular differentiation, but it is clear that environmental factors, such as maternal nutrition, can influence epigenetic programming
- Ceramide
A family of lipid molecules composed of sphingosine and a fatty acid. In addition to being structural components of lipid bilayers, it is now clear that ceramides can act as signalling molecules
- Oxidative phosphorylation
A metabolic pathway that uses oxidation of nutrients to generate ATP. The electron transport chain in mitochondria is the site of oxidative phosphorylation in eukaryotes
- Haploinsufficiency
A condition in a diploid organism in which a single functional copy of a gene results in a phenotype, such as a disease. Genetically randomized population, A population in which genotypes are randomized owing to the random assortment of alleles during gametogenesis
- Conditional probability
The probability of an event, A, given the occurrence of some other event, B
Footnotes
DATABASES
Entrez Gene: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene
ABCB11 | CELSR2 | FTO | G6PC2 | GCKR | MC4R | SORT1
FURTHER INFORMATION
Attie laboratory diabetes database: http://diabetes.wisc.edu
Genomics of Lipid Associated Disorders Database: https://gold.tugraz.at/Main.jsp
Complex Trait Consortium: http://www.complextrait.org
The GeneNetwork: http://www.genenetwork.org
The Jackson Laboratory, Mouse Phenome Database: http://www.jax.org
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
References
- 1.Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–1607. doi: 10.2337/diab.37.12.1595. The first definition of MetSyn as an entity, defined by the clustering of abnormalities having insulin resistance as an underlying cause. Although the specific definition continues to evolve, the major insights presented here have remained important. [DOI] [PubMed] [Google Scholar]
- 2.Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365:1415–1428. doi: 10.1016/S0140-6736(05)66378-7. [DOI] [PubMed] [Google Scholar]
- 3.Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 4.King RA, Rotter JI, Motulsky AG. The Genetic Basis of Common Disease. Oxford University Press; New York: 2002. [Google Scholar]
- 5.Lazar MA. How obesity causes diabetes: not a tall tale. Science. 2005;307:373–375. doi: 10.1126/science.1104342. [DOI] [PubMed] [Google Scholar]
- 6.Muoio DM, Newgard CB. Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nature Rev Mol Cell Biol. 2008;9:193–205. doi: 10.1038/nrm2327. [DOI] [PubMed] [Google Scholar]
- 7.Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444:875–880. doi: 10.1038/nature05487. [DOI] [PubMed] [Google Scholar]
- 8.Visscher PM, Hill WG, Wray NR. Heritability in the genomics era — concepts and misconceptions. Nature Rev Genet. 2008;9:255–266. doi: 10.1038/nrg2322. [DOI] [PubMed] [Google Scholar]
- 9.Doria A, Patti ME, Kahn CR. The emerging genetic architecture of type 2 diabetes. Cell Metab. 2008;8:186–200. doi: 10.1016/j.cmet.2008.08.006. A review of our current understanding of factors contributing to monogenic and common forms of type 2 diabetes. The experimental approaches, the genes identified and implications for the development of diagnostics and therapeutics are discussed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kathiresan S, Musunuru K, Orho-Melander M. Defining the spectrum of alleles that contribute to blood lipid concentrations in humans. Curr Opin Lipidol. 2008;19:122–127. doi: 10.1097/MOL.0b013e3282f70296. [DOI] [PubMed] [Google Scholar]
- 11.Perry JR, Frayling TM. New gene variants alter type 2 diabetes risk predominantly through reduced beta-cell function. Curr Opin Clin Nutr Metab Care. 2008;11:371–377. doi: 10.1097/MCO.0b013e32830349a1. [DOI] [PubMed] [Google Scholar]
- 12.Bouatia-Naji N, et al. A polymorphism within the G6PC2 gene is associated with fasting plasma glucose levels. Science. 2008;320:1085–1088. doi: 10.1126/science.1156849. [DOI] [PubMed] [Google Scholar]
- 13.Chen WM, et al. Variations in the G6PC2/ABCB11 genomic region are associated with fasting glucose levels. J Clin Invest. 2008;118:2620–2628. doi: 10.1172/JCI34566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frayling TM, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–894. doi: 10.1126/science.1141634. A landmark GWA study that detected an association between a common variant in the FTO gene and type 2 diabetes, through effects on body mass index. Notably, the effects conferred by the susceptible allele are also apparent in children as well as in adults. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kathiresan S, et al. A genome-wide association study for blood lipid phenotypes in the Framingham Heart Study. BMC Med Genet. 2007;8(Suppl 1):S17. doi: 10.1186/1471-2350-8-S1-S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kathiresan S, et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nature Genet. 2008;40:189–197. doi: 10.1038/ng.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loos RJ, et al. Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nature Genet. 2008;40:768–75. doi: 10.1038/ng.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Orho-Melander M, et al. A common missense variant in the glucokinase regulatory protein gene (GCKR) is associated with increased plasma triglyceride and c-reactive protein but lower fasting glucose concentrations. Diabetes. 2008 Aug;4 doi: 10.2337/db08-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Samani NJ, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357:443–453. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sandhu MS, et al. LDL-cholesterol concentrations: a genome-wide association study. Lancet. 2008;371:483–491. doi: 10.1016/S0140-6736(08)60208-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saxena R, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 22.Scott LJ, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scuteri A, et al. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet. 2007;3:e115. doi: 10.1371/journal.pgen.0030115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeggini E, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nature Genet. 2008;40:638–645. doi: 10.1038/ng.120. A large GWA study that identified three novel susceptibility genes for type 2 diabetes, and that confirmed others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Do R, et al. Genetic variants of FTO influence adiposity, insulin sensitivity, leptin levels, and resting metabolic rate in the Quebec Family Study. Diabetes. 2008;57:1147–1150. doi: 10.2337/db07-1267. [DOI] [PubMed] [Google Scholar]
- 26.Freathy RM, et al. Common variation in the FTO gene alters diabetes-related metabolic traits to the extent expected given its effect on BMI. Diabetes. 2008;57:1419–1426. doi: 10.2337/db07-1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fahmi S, Yang C, Esmail S, Hobbs HH, Cohen JC. Functional characterization of genetic variants in NPC1L1 supports the sequencing extremes strategy to identify complex trait genes. Hum Mol Genet. 2008;17:2101–2107. doi: 10.1093/hmg/ddn108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frikke-Schmidt R, Nordestgaard BG, Jensen GB, Tybjaerg-Hansen A. Genetic variation in ABC transporter A1 contributes to HDL cholesterol in the general population. J Clin Invest. 2004;114:1343–1353. doi: 10.1172/JCI20361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jakobsson M, et al. Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451:998–1003. doi: 10.1038/nature06742. [DOI] [PubMed] [Google Scholar]
- 30.Schadt EE, et al. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008;6:e107. doi: 10.1371/journal.pbio.0060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Speakman JR. A nonadaptive scenario explaining the genetic predisposition to obesity: the ‘predation release’ hypothesis. Cell Metab. 2007;6:5–12. doi: 10.1016/j.cmet.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 32.Poirier P, et al. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss: an update of the 1997 American Heart Association Scientific Statement on Obesity and Heart Disease from the Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Circulation. 2006;113:898–918. doi: 10.1161/CIRCULATIONAHA.106.171016. [DOI] [PubMed] [Google Scholar]
- 33.Bergman RN, et al. Why visceral fat is bad: mechanisms of the metabolic syndrome. Obesity (Silver Spring) 2006;14(Suppl 1):16S–19S. doi: 10.1038/oby.2006.277. [DOI] [PubMed] [Google Scholar]
- 34.Wajchenberg BL, Giannella-Neto D, da Silva ME, Santos RF. Depot-specific hormonal characteristics of subcutaneous and visceral adipose tissue and their relation to the metabolic syndrome. Horm Metab Res. 2002;34:616–621. doi: 10.1055/s-2002-38256. [DOI] [PubMed] [Google Scholar]
- 35.Gesta S, Tseng YH, Kahn CR. Developmental origin of fat: tracking obesity to its source. Cell. 2007;131:242–256. doi: 10.1016/j.cell.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 36.Gesta S, et al. Evidence for a role of developmental genes in the origin of obesity and body fat distribution. Proc Natl Acad Sci USA. 2006;103:6676–6681. doi: 10.1073/pnas.0601752103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hocking SL, Chisholm DJ, James DE. Studies of regional adipose transplantation reveal a unique and beneficial interaction between subcutaneous adipose tissue and the intra-abdominal compartment. Diabetologia. 2008;51:900–902. doi: 10.1007/s00125-008-0969-0. [DOI] [PubMed] [Google Scholar]
- 38.Tran TT, Yamamoto Y, Gesta S, Kahn CR. Beneficial effects of subcutaneous fat transplantation on metabolism. Cell Metab. 2008;7:410–420. doi: 10.1016/j.cmet.2008.04.004. Fat transplantation studies in the mouse demonstrating that the observed detrimental effects of visceral adipose tissue on metabolism are related to intrinsic differences in the activity of visceral compared with subcutaneous fat tissue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Agarwal AK, Garg A. Genetic disorders of adipose tissue development, differentiation, and death. Annu Rev Genomics Hum Genet. 2006;7:175–199. doi: 10.1146/annurev.genom.7.080505.115715. [DOI] [PubMed] [Google Scholar]
- 40.Krut LH, Singer R. Steatopygia: The fatty acid composition of subcutaneous adipose tissue in the Hottentot. Am J Phys Anthropol. 1963;21:181–187. doi: 10.1002/ajpa.1330210210. [DOI] [PubMed] [Google Scholar]
- 41.Blouin K, Boivin A, Tchernof A. Androgens and body fat distribution. J Steroid Biochem Mol Biol. 2008;108:272–280. doi: 10.1016/j.jsbmb.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 42.Garaulet M, et al. Body fat distribution in pre- and post-menopausal women: metabolic and anthropometric variables. J Nutr Health Aging. 2002;6:123–126. [PubMed] [Google Scholar]
- 43.Wang S, et al. Genetic and genomic analysis of a fat mass trait with complex inheritance reveals marked sex specificity. PLoS Genet. 2006;2:e15. doi: 10.1371/journal.pgen.0020015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gui Y, Silha JV, Murphy LJ. Sexual dimorphism and regulation of resistin, adiponectin, and leptin expression in the mouse. Obes Res. 2004;12:1481–1491. doi: 10.1038/oby.2004.185. [DOI] [PubMed] [Google Scholar]
- 45.Bukowski R, et al. Human sexual size dimorphism in early pregnancy. Am J Epidemiol. 2007;165:1216–1218. doi: 10.1093/aje/kwm024. [DOI] [PubMed] [Google Scholar]
- 46.Arnold AP, Burgoyne PS. Are XX and XY brain cells intrinsically different? Trends Endocrinol Metab. 2004;15:6–11. doi: 10.1016/j.tem.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 47.Barker DJ. The developmental origins of well-being. Philos Trans R Soc Lond, B, Biol Sci. 2004;359:1359–1366. doi: 10.1098/rstb.2004.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gluckman PD, Hanson MA. Living with the past: evolution, development, and patterns of disease. Science. 2004;305:1733–1736. doi: 10.1126/science.1095292. [DOI] [PubMed] [Google Scholar]
- 49.Yura S, et al. Role of premature leptin surge in obesity resulting from intrauterine undernutrition. Cell Metab. 2005;1:371–378. doi: 10.1016/j.cmet.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 50.Park JH, Stoffers DA, Nicholls RD, Simmons RA. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of. Pdx1 J Clin Invest. 2008;118:2316–2324. doi: 10.1172/JCI33655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Napoli C, et al. Fatty streak formation occurs in human fetal aortas and is greatly enhanced by maternal hypercholesterolemia. Intimal accumulation of low density lipoprotein and its oxidation precede monocyte recruitment into early atherosclerotic lesions. J Clin Invest. 1997;100:2680–2690. doi: 10.1172/JCI119813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Napoli C, et al. Maternal hypercholesterolemia during pregnancy promotes early atherogenesis in LDL receptor-deficient mice and alters aortic gene expression determined by microarray. Circulation. 2002;105:1360–1367. doi: 10.1161/hc1102.106792. [DOI] [PubMed] [Google Scholar]
- 53.Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nature Rev Genet. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Raychaudhuri N, Raychaudhuri S, Thamotharan M, Devaskar SU. Histone code modifications repress glucose transporter 4 expression in the intrauterine growth-restricted offspring. J Biol Chem. 2008;283:13611–13626. doi: 10.1074/jbc.M800128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aitman TJ, et al. Progress and prospects in rat genetics: a community view. Nature Genet. 2008;40:516–522. doi: 10.1038/ng.147. [DOI] [PubMed] [Google Scholar]
- 56.Clee SM, Attie AD. The genetic landscape of type 2 diabetes in mice. Endocr Rev. 2007;28:48–83. doi: 10.1210/er.2006-0035. [DOI] [PubMed] [Google Scholar]
- 57.Wang MY, et al. Adipogenic capacity and the susceptibility to type 2 diabetes and metabolic syndrome. Proc Natl Acad Sci USA. 2008;105:6139–6144. doi: 10.1073/pnas.0801981105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang CJ, et al. Induction of endoplasmic reticulum stress-induced beta-cell apoptosis and accumulation of polyubiquitinated proteins by human islet amyloid polypeptide. Am J Physiol Endocrinol Metab. 2007;293:E1656–E1662. doi: 10.1152/ajpendo.00318.2007. [DOI] [PubMed] [Google Scholar]
- 59.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1:785–789. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 60.Magnan C, Gilbert M, Kahn BB. Chronic free fatty acid infusion in rats results in insulin resistance but no alteration in insulin-responsive glucose transporter levels in skeletal muscle. Lipids. 1996;31:1141–1149. doi: 10.1007/BF02524288. [DOI] [PubMed] [Google Scholar]
- 61.Kim JK, et al. Inactivation of fatty acid transport protein 1 prevents fat-induced insulin resistance in skeletal muscle. J Clin Invest. 2004;113:756–763. doi: 10.1172/JCI18917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chibalin AV, et al. Downregulation of diacylglycerol kinase delta contributes to hyperglycemia-induced insulin resistance. Cell. 2008;132:375–386. doi: 10.1016/j.cell.2007.12.035. [DOI] [PubMed] [Google Scholar]
- 63.Stratford S, Hoehn KL, Liu F, Summers SA. Regulation of insulin action by ceramide: dual mechanisms linking ceramide accumulation to the inhibition of Akt/protein kinase B. J Biol Chem. 2004;279:36608–36615. doi: 10.1074/jbc.M406499200. [DOI] [PubMed] [Google Scholar]
- 64.Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- 65.Petersen KF, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mootha VK, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 67.Petersen KF, Dufour S, Shulman GI. Decreased insulin-stimulated ATP synthesis and phosphate transport in muscle of insulin-resistant offspring of type 2 diabetic parents. PLoS Med. 2005;2:e233. doi: 10.1371/journal.pmed.0020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hancock CR, et al. High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc Natl Acad Sci USA. 2008:7815–7820. doi: 10.1073/pnas.0802057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Handschin C, et al. Abnormal glucose homeostasis in skeletal muscle-specific PGC-1 alpha knockout mice reveals skeletal muscle-pancreatic beta cell crosstalk. J Clin Invest. 2007;117:3463–3474. doi: 10.1172/JCI31785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Koves TR, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 71.Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest. 1995;95:2409–2415. doi: 10.1172/JCI117936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kressel G, et al. Systemic and vascular markers of inflammation in relation to metabolic syndrome and insulin resistance in adults with elevated atherosclerosis risk. Atherosclerosis. 2008 Apr;20 doi: 10.1016/j.atherosclerosis.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 73.Weisberg SP, et al. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. The use of expression profiling of adipose tissue in obese mouse models revealed a correlation between obesity and macrophage gene expression, focusing attention on inflammatory pathways that are activated in adipose tissue in obesity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu H, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Subramanian S, et al. Dietary cholesterol worsens adipose tissue macrophage accumulation and atherosclerosis in obese LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2008;28:685–691. doi: 10.1161/ATVBAHA.107.157685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kanda H, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116:1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Weisberg SP, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. 2006;116:115–124. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Odegaard JI, et al. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gregor MF, Hotamisligil GS. Thematic review series: adipocyte biology. Adipocyte stress: the endoplasmic reticulum and metabolic disease. J Lipid Res. 2007;48:1905–1914. doi: 10.1194/jlr.R700007-JLR200. [DOI] [PubMed] [Google Scholar]
- 80.Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol Rev. 2006;86:1133–1149. doi: 10.1152/physrev.00015.2006. [DOI] [PubMed] [Google Scholar]
- 81.Ozcan U, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 82.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gargalovic PS, et al. Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids. Proc Natl Acad Sci USA. 2006;103:12741–12746. doi: 10.1073/pnas.0605457103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tabas I. Apoptosis and efferocytosis in mouse models of atherosclerosis. Curr Drug Targets. 2007;8:1288–1296. doi: 10.2174/138945007783220623. [DOI] [PubMed] [Google Scholar]
- 85.Ozcan U, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yuan M, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkβ. Science. 2001;293:1673–1677. doi: 10.1126/science.1061620. [DOI] [PubMed] [Google Scholar]
- 87.Park E, Wong V, Guan X, Oprescu AI, Giacca A. Salicylate prevents hepatic insulin resistance caused by short-term elevation of free fatty acids in vivo. J Endocrinol. 2007;195:323–331. doi: 10.1677/JOE-07-0005. [DOI] [PubMed] [Google Scholar]
- 88.Clee SM, et al. Positional cloning of Sorcs1, a type 2 diabetes quantitative trait locus. Nature Genet. 2006;38:688–693. doi: 10.1038/ng1796. [DOI] [PubMed] [Google Scholar]
- 89.Schreyer SA, Vick C, Lystig TC, Mystkowski P, LeBoeuf RC. LDL receptor but not apolipoprotein E deficiency increases diet-induced obesity and diabetes in mice. Am J Physiol Endocrinol Metab. 2002;282:E207–E214. doi: 10.1152/ajpendo.2002.282.1.E207. [DOI] [PubMed] [Google Scholar]
- 90.Schwartz SM, Galis ZS, Rosenfeld ME, Falk E. Plaque rupture in humans and mice. Arterioscler Thromb Vasc Biol. 2007;27:705–713. doi: 10.1161/01.ATV.0000261709.34878.20. [DOI] [PubMed] [Google Scholar]
- 91.Navab M, Anantharamaiah GM, Fogelman AM. The role of high-density lipoprotein in inflammation. Trends Cardiovasc Med. 2005;15:158–161. doi: 10.1016/j.tcm.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 92.Hsueh W, et al. Recipes for creating animal models of diabetic cardiovascular disease. Circ Res. 2007;100:1415–1427. doi: 10.1161/01.RES.0000266449.37396.1f. [DOI] [PubMed] [Google Scholar]
- 93.Soro-Paavonen A, et al. Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes. 2008;57:2461–2469. doi: 10.2337/db07-1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Abel ED, Litwin SE, Sweeney G. Cardiac remodeling in obesity. Physiol Rev. 2008;88:389–419. doi: 10.1152/physrev.00017.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fonarow GC, Srikanthan P, Costanzo MR, Cintron GB, Lopatin M. An obesity paradox in acute heart failure: analysis of body mass index and inhospital mortality for 108,927 patients in the Acute Decompensated Heart Failure National Registry. Am Heart J. 2007;153:74–81. doi: 10.1016/j.ahj.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 96.Carley AN, Severson DL. What are the biochemical mechanisms responsible for enhanced fatty acid utilization by perfused hearts from type 2 diabetic db/db mice? Cardiovasc Drugs Ther. 2008;22:83–89. doi: 10.1007/s10557-008-6088-9. [DOI] [PubMed] [Google Scholar]
- 97.Boudina S, et al. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005;112:2686–2695. doi: 10.1161/CIRCULATIONAHA.105.554360. [DOI] [PubMed] [Google Scholar]
- 98.Park TS, et al. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J Lipid Res. 2008;49:2101–2112. doi: 10.1194/jlr.M800147-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Patti ME, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci USA. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Balling R. From mouse genetics to systems biology. Mamm Genome. 2007;18:383–388. doi: 10.1007/s00335-007-9044-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tegner J, Skogsberg J, Bjorkegren J. Thematic review series: systems biology approaches to metabolic and cardiovascular disorders. Multi-organ whole-genome measurements and reverse engineering to uncover gene networks underlying complex traits. J Lipid Res. 2007;48:267–277. doi: 10.1194/jlr.R600030-JLR200. [DOI] [PubMed] [Google Scholar]
- 102.Weiss JN, Yang L, Qu Z. Systems biology approaches to metabolic and cardiovascular disorders: network perspectives of cardiovascular metabolism. J Lipid Res. 2006;47:2355–2366. doi: 10.1194/jlr.R600023-JLR200. [DOI] [PubMed] [Google Scholar]
- 103.Barabasi AL, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nature Rev Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 104.Duarte NC, et al. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc Natl Acad Sci USA. 2007;104:1777–1782. doi: 10.1073/pnas.0610772104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Drake TA, Schadt EE, Lusis AJ. Integrating genetic and gene expression data: application to cardiovascular and metabolic traits in mice. Mamm Genome. 2006;17:466–479. doi: 10.1007/s00335-005-0175-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schadt EE, et al. Genetics of gene expression surveyed in maize, mouse and man. Nature. 2003;422:297–302. doi: 10.1038/nature01434. [DOI] [PubMed] [Google Scholar]
- 107.Chen Y, et al. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452:429–435. doi: 10.1038/nature06757. Molecular network analysis in a segregating mouse population identified gene networks that exhibit a causal relationship with MetSyn traits. The results revealed a macrophage-enriched network and novel obesity genes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Petretto E, Mangion J, Pravanec M, Hubner N, Aitman TJ. Integrated gene expression profiling and linkage analysis in the rat. Mamm Genome. 2006;17:480–489. doi: 10.1007/s00335-005-0181-1. [DOI] [PubMed] [Google Scholar]
- 109.Petretto E, et al. Integrated genomic approaches implicate osteoglycin (Ogn) in the regulation of left ventricular mass. Nature Genet. 2008;40:546–552. doi: 10.1038/ng.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pravenec M, et al. Identification of renal Cd36 as a determinant of blood pressure and risk for hypertension. Nature Genet. 2008;40:952–954. doi: 10.1038/ng.164. [DOI] [PubMed] [Google Scholar]
- 111.Meng H, et al. Identification of Abcc6 as the major causal gene for dystrophic cardiac calcification in mice through integrative genomics. Proc Natl Acad Sci USA. 2007;104:4530–4535. doi: 10.1073/pnas.0607620104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Schadt EE, et al. An integrative genomics approach to infer causal associations between gene expression and disease. Nature Genet. 2005;37:710–717. doi: 10.1038/ng1589. This study describes a method to distinguish causative versus reactive relationships between traits and gene expression levels. Here, this strategy was applied to identify novel obesity-associated genes, and will be applicable to many complex diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gargalovic PS, et al. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:2490–2496. doi: 10.1161/01.ATV.0000242903.41158.a1. [DOI] [PubMed] [Google Scholar]
- 114.Emilsson V, et al. Genetics of gene expression and its effect on disease. Nature. 2008;452:423–428. doi: 10.1038/nature06758. [DOI] [PubMed] [Google Scholar]
- 115.Ferrara CT, et al. Genetic networks of liver metabolism revealed by integration of metabolic and transcriptional profiling. PLoS Genet. 2008;4:e1000034. doi: 10.1371/journal.pgen.1000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Adiels M, Olofsson SO, Taskinen MR, Boren J. Overproduction of very low-density lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2008;28:1225–1236. doi: 10.1161/ATVBAHA.107.160192. [DOI] [PubMed] [Google Scholar]
- 117.Toma I, et al. Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. J Clin Invest. 2008;118:2526–2534. doi: 10.1172/JCI33293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111:3481–3488. doi: 10.1161/CIRCULATIONAHA.105.537878. [DOI] [PubMed] [Google Scholar]
- 119.Despres JP, et al. Abdominal obesity and the metabolic syndrome: contribution to global cardiometabolic risk. Arterioscler Thromb Vasc Biol. 2008;28:1039–1049. doi: 10.1161/ATVBAHA.107.159228. [DOI] [PubMed] [Google Scholar]
- 120.Brietzke SA. Controversy in diagnosis and management of the metabolic syndrome. Med Clin North Am. 2007;91:1041–1061. vii–viii. doi: 10.1016/j.mcna.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 121.Federspil G, Nisoli E, Vettor R. A critical reflection on the definition of metabolic syndrome. Pharmacol Res. 2006;53:449–456. doi: 10.1016/j.phrs.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 122.Gluckman PD. Evolving a definition of disease. Arch Dis Child. 2007;92:1053–1054. doi: 10.1136/adc.2007.126318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Reinehr T, de Sousa G, Toschke AM, Andler W. Comparison of metabolic syndrome prevalence using eight different definitions: a critical approach. Arch Dis Child. 2007;92:1067–1072. doi: 10.1136/adc.2006.104588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287:356–359. doi: 10.1001/jama.287.3.356. [DOI] [PubMed] [Google Scholar]
- 125.Kahn R. Metabolic syndrome: is it a syndrome? Does it matter? Circulation. 2007;115:1806–1810. doi: 10.1161/CIRCULATIONAHA.106.658336. discussion 1811. [DOI] [PubMed] [Google Scholar]
- 126.Grundy SM. Metabolic syndrome pandemic. Arterioscler Thromb Vasc Biol. 2008;28:629–636. doi: 10.1161/ATVBAHA.107.151092. [DOI] [PubMed] [Google Scholar]
- 127.Chambers JC, et al. Common genetic variation near MC4R is associated with waist circumference and insulin resistance. Nature Genet. 2008;40:716–8. doi: 10.1038/ng.156. [DOI] [PubMed] [Google Scholar]
- 128.Stratigopoulos G, et al. Regulation of Fto/Ftm gene expression in mice and humans. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1185–R1196. doi: 10.1152/ajpregu.00839.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kooner JS, et al. Genome-wide scan identifies variation in MLXIPL associated with plasma triglycerides. Nature Genet. 2008;40:149–151. doi: 10.1038/ng.2007.61. [DOI] [PubMed] [Google Scholar]
- 130.Grant SF, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nature Genet. 2006;38:320–3. doi: 10.1038/ng1732. [DOI] [PubMed] [Google Scholar]
- 131.Nadeau JH, et al. Pleiotropy, homeostasis, and functional networks based on assays of cardiovascular traits in genetically randomized populations. Genome Res. 2003;13:2082–2091. doi: 10.1101/gr.1186603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Willer CJ, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nature Genet. 2008;40:161–169. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ghazalpour A, et al. Integrating genetic and network analysis to characterize genes related to mouse weight. PLoS Genet. 2006;2:e130. doi: 10.1371/journal.pgen.0020130. [DOI] [PMC free article] [PubMed] [Google Scholar]