

Abstract
The neurodegenerative disease Huntington’s Disease (HD) is caused by an expanded polyglutamine (polyQ) tract in the protein huntingtin (htt). Although the gene encoding htt was identified and cloned more than 15 years ago, and in spite of impressive efforts to unravel the mechanism(s) by which mutant htt induces nerve cell death, these studies have so far not led to a good understanding of pathophysiology or an effective therapy. Set against a historical background, we review data supporting the idea that metabolites of the kynurenine pathway (KP) of tryptophan degradation provide a critical link between mutant htt and the pathophysiology of HD. New studies in HD brain and genetic model organisms suggest that the disease may in fact be causally related to early abnormalities in KP metabolism, favoring the formation of two neurotoxic metabolites, 3-hydroxykynurenine and quinolinic acid, over the related neuroprotective agent kynurenic acid. These findings not only link the excitotoxic hypothesis of HD pathology to an impairment of the KP but also define new drug targets and therefore have direct therapeutic implications. Thus, pharmacological normalization of the imbalance in brain KP metabolism may provide clinical benefits, which could be especially effective in the early stages of the disease.
Keywords: Excitotoxicity, 3-Hydroxykynurenine, Kynurenines, Microglia, Neuroprotection
1. Excitotoxins and Huntington’s disease: the beginning
Huntington’s disease (HD), originally termed Huntington’s chorea because of the characteristic involuntary movements shown by affected individuals, is a chronic neurodegenerative disorder, which is inherited in an autosomal dominant fashion. Overt motor symptoms usually develop in mid-life, and patients succumb to the disease after another 15–20 years on average. Long believed to be an exclusive basal ganglia disease because of the substantial and progressive neostriatal shrinkage and the massive dilation of the adjacent lateral ventricles, the neuropathology of HD is now known to involve a large number of brain regions and neuronal populations (Jackson et al., 1995; Rosas et al., 2008; Vonsattel et al., 1985).
For a century following the first description of the disease by George Huntington in 1872 (Huntington, 1872), speculations about the cause of neuronal loss in HD were sporadic and mostly based on clinical observations, occasionally supported by insights from microscopic studies of HD brains (Barbeau et al., 1973). Several of these hypotheses, which focused, for example, on the excessive glucose consumption of patients and on abnormal iron deposits in the basal ganglia, remain attractive to this day and may eventually turn out to be useful for a comprehensive understanding of HD pathophysiology. It was not until the 1970s, however, that a new concept provided a plausible mechanism for the characteristic nerve cell death seen in the disease.
As is often the case in the biomedical sciences, this breakthrough in HD research was made possible by disparate and seemingly unrelated findings. The first clue came from neurochemical post-mortem studies of the HD striatum, which revealed a peculiar sparing of dopaminergic afferent fibers even in the final stages of the disease, when intrinsic striatal neurons, identified by measuring GABA and its biosynthetic enzyme glutamate decarboxylase, were dramatically depleted (Bernheimer et al., 1973; Bird and Iversen, 1974; McGeer et al., 1973). This at the time unprecedented neuropathological feature was reminiscent of the “axon-sparing” lesions caused by glutamate and other excitatory amino acids, which John Olney had described in a series of publications in the late 1960s and early 1970s (Olney et al., 1971; Olney and Sharpe, 1969). In the course of these studies, which were mainly concerned with neuronal damage in the hypothalamus and the retina, Olney made two seminal observations: a) axons of extrinsic origin do not degenerate at the lesion site; and b) the neurotoxic potency of acidic amino acids parallels their neuroexcitatory efficacy. Subsequently, he introduced the operational term “excitotoxicity” to describe neuronal lesions that were characterized by the survival of myelinated axons “en passage” and afferent axon terminals (Olney, 1974). The corresponding excitotoxins ranged from endogenous metabolites such as glutamic, aspartic and cysteine sulfinic acid to more potent exogenous compounds, including the seaweed-derived kainic acid, which was found to be approximately 500 times more effective than glutamate both as a toxin and as an excitant (Olney et al., 1971; Olney et al., 1974). Notably, however, the excitotoxic concept remained merely correlative in nature. No attempts to study the mechanism underlying the characteristic features of excitotoxic lesions were made in those early years when excitatory amino acid receptors were still beyond the reach of neurochemists and molecular biologists.
Interest in excitotoxicity increased substantially in the mid-1970s following the demonstration by Coyle and Schwarcz (Coyle and Schwarcz, 1976), and shortly thereafter by McGeer and McGeer (McGeer and McGeer, 1976), that stereotaxic microinjections of kainic acid cause selective, axon-sparing lesions in the rat striatum. These studies not only introduced kainate as a distinct lesioning tool that was easy to use and applicable to a wide variety of experimental situations (McGeer et al., 1978) but also, based on the duplication of the earlier post-mortem findings, provided a novel animal model of HD (Figure 1). In relatively quick succession, a number of laboratories described further morphological and neurochemical features of the kainate-lesioned striatum and also described motor and other behavioral abnormalities in these animals (Divac et al., 1978; Mason et al., 1978; Sanberg et al., 1979). Jointly, these studies resulted in a remarkably consistent catalog of data, which not only confirmed the similarities between HD and the new animal model (Coyle et al., 1983) but also prompted investigators to study additional human neurodegenerative diseases based on excitotoxic lesions of other brain structures (Bergman et al., 1990; Flicker et al., 1983; Wenk et al., 1984).
Figure 1.

Schematic representation of the “axon-sparing”, excitotoxic nerve cell loss in the HD neostriatum. Degenerated neurons in this simplistic model are indicated by dotted lines.
2. The Swedish connection: rotational behavior and ibotenic acid
2.1. A dopaminergic component of excitotoxicity
The postdoctoral tenure of one of us (R.S.) at the Karolinska Institute in the late 1970s coincided with two significant advances in the excitotoxic model of HD. The first innovation was the development of a rapid, behavioral in vivo test for assessing excitotoxic striatal damage in unilaterally lesioned animals. The underlying idea was an offshoot of the work of Ungerstedt, who had demonstrated a few years earlier that systemic administration of dopaminergic agonists in rats with a unilateral striatal dopamine depletion causes the animals to rotate towards the hemisphere that shows lesser dopamine receptor activity (Ungerstedt, 1971). Since a focal kainate injection kills intrinsic neurons carrying dopamine receptors (Schwarcz et al., 1978), unilaterally lesioned rats have a hemispheric imbalance of dopamine function. Consequently, systemic administration of a dopamine receptor agonist such as apomorphine causes these animals to circle towards the side containing the lesioned striatum (Figure 2). The speed of these rotations is roughly proportional to the lesion size and therefore provides a useful quantitative measure of excitotoxic striatal neurodegeneration (Schwarcz et al., 1979a; cf. also Herrera-Marschitz et al., this volume).
Figure 2.

Rotational behavior in rodents with unilateral striatal excitotoxic lesions. Degenerated neurons are indicated by dotted lines. The arrow indicates the rotational direction following the systemic administration of a dopamine receptor agonist.
2.2. Ibotenate: a new and improved excitotoxin
The second advance from this period was triggered by the realization that the kainic acid model of HD had substantial shortcomings, which were related to the pronounced convulsive properties of the toxin. Thus, intrastriatal kainate injections not only cause epileptiform activity, which is rarely observed in adult-onset HD (Bruyn, 1968), but also result in seizure-induced neurodegeneration in the hippocampus, the olfactory cortex and other limbic structures, which suffer only little or no cell loss in HD (Olney and de Gubareff, 1978a; Schwarcz and Coyle, 1977). Studies at the Karolinska Institute revealed that another exogenous excitotoxin, ibotenic acid, was devoid of these unbecoming properties (Figure 3). Originally isolated from the mushroom Amanita muscaria (Eugster, 1968), ibotenate caused well-circumscribed neuronal loss upon focal injection into various brain regions and was therefore soon recognized as a superior tool for producing excitotoxic lesions in rats (Schwarcz et al., 1979b) and other experimental animals (Ciaramitaro et al., 1997; Wenk, 1993). Ibotenate-induced striatal lesions also created an improved animal model of HD, which became widely used – maybe most famously by Björklund and his collaborators in their first attempts to develop cell transplantation therapies for HD (Isacson et al., 1984).
Figure 3.

Chemical structures of kainic, ibotenic, glutamic and quinolinic acids.
The differences between kainate and ibotenate were not limited to the advantages that the latter provided as a research tool. For example and in contrast to kainate (Biziere and Coyle, 1979; McGeer et al., 1978), ibotenate-induced lesions were not dependent on the integrity of glutamatergic fibers, suggesting that endogenous glutamate was a critical component of kainate – but not ibotenate – neurotoxicity (Köhler et al., 1979). Moreover, while the developing striatum was found to be remarkably resistant to kainate (Campochiaro and Coyle, 1978), no such age-dependent vulnerability was seen with ibotenate (Steiner et al., 1984). These and other distinctions between the two toxins, which included different susceptibilities of neuronal populations throughout the brain (Schwarcz and Köhler, 1983), prompted researchers to explore the nature of excitotoxic neuron death in greater detail. In addition to scientific curiosity, it was also hoped, of course, that a better understanding of the mechanisms underlying diverse excitotoxic phenomena might provide critical clues regarding the pathophysiology of HD and, possibly, other neurodegenerative diseases (Coyle et al., 1977).
3. Glutamate receptors and neuroprotection
3.1. Glutamate receptor subtypes
The development of increasingly selective antagonists in the 1970s allowed the study and categorization of ionotropic (i.e. cation-permeable) excitatory amino acid receptors, which had been previously defined exclusively by electrophysiological means (Curtis et al., 1972; Curtis and Watkins, 1960). By the end of the decade, work by the teams of McLennan and Watkins had identified three distinct receptor subtypes, which were named after their model agonists kainate, quisqualate and N-methyl-D-aspartate (NMDA) (Davies et al., 1979; Hall et al., 1979). This classification was subsequently supplanted by the use of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) in place of the less specific quisqualate (Honoré et al., 1982) and further refined by the molecular revolution of the 1990s (Seeburg, 1993). Awareness of these three classes of pharmacologically defined glutamate receptors stimulated research into their physiological differences and led to first attempts to describe the anatomical localization of the individual receptors using experimental lesions and the recently developed methods of receptor binding and autoradiography. These studies revealed the existence of both pre- and postsynaptic receptors and, in particular, a heterogeneous distribution of the different receptor subtypes (Biziere and Coyle, 1979; Greenamyre et al., 1984; Monaghan et al., 1983; Schwarcz and Fuxe, 1979; Vincent and McGeer, 1979). This, in turn, led to the idea that the subtyping of glutamate receptors might be used to provide a rational explanation for the qualitative differences in the excitotoxic properties of kainate, which selectively activates its own receptor, and ibotenate, which at the time was believed to be a specific NMDA receptor agonist (Curtis et al., 1972). For example, it was proposed that the prevention of striatal kainate neurotoxicity by prior surgical removal of cortical input was causally related to the presence of functional kainate receptors on these glutamatergic afferents (Biziere and Coyle, 1979; Ferkany et al., 1982). The demonstration that the addition of glutamate can partly restore kainate-induced excitotoxicity in the deafferentated striatum further supported the relevance of these presynaptic receptors and at the same time suggested an adjuvant role of endogenous glutamate in the toxic effects of kainic acid (Biziere and Coyle, 1979). In contrast, NMDA receptors are mainly localized on intrinsic striatal neurons (Samuel et al., 1990), which could account for the fact that ibotenate-induced lesions are not affected by the prior removal of glutamatergic nerve terminals of distant origin (Köhler et al., 1979). These findings not only confirmed the need to elaborate the link between excitatory amino acid-mediated excitation and neurotoxicity in greater depth but also raised the question which, if any, of these experimental toxins provided the best animal model of HD.
3.2. Mechanism(s) of excitotoxic neurodegeneration
It soon became obvious that a better understanding of the cellular and molecular mechanisms underlying selective, axon-sparing neuron death was needed to test the construct validity of the excitotoxic HD model. In the early days, these studies were dominated not only by receptor pharmacologists (see above) but also by microscopists, who set out to define the sequence of degenerative events that are initiated after neural tissue is exposed to an excitotoxic insult. These analyses revealed that acute swelling of dendritic spines and branches constitute the first detectable signs of structural damage (Olney et al., 1979a), suggesting the critical importance of glutamate receptors situated on dendritic elements. Since asymmetric, i.e. excitatory, axo-dendritic synapses are placed in close apposition to presynaptic glutamatergic axon terminals (Sharpe and Tepper, 1998), however, these observations did not resolve the question whether the neurotoxic cascade is triggered by presynaptic receptors stimulating glutamate release or by direct activation of postsynaptic receptors. At the same time, it also became quite clear that a deeper awareness of the molecular events following both normal and excessive glutamate receptor activation was needed to properly evaluate the clinical ramifications of the excitotoxic concept.
In the many years since, these mechanisms have been elaborated in detail using various in vitro preparations and, with increasing frequency, informative in vivo paradigms. The consensus reached by these studies, which have become exceedingly complex in nature and now increasingly focus on a plethora of intracellular processes, is that pathology is initiated by a glutamate receptor-mediated, excessive influx of Ca2+ ions (Choi, 1985), which over a period of hours leads to hyperphysiological Ca2+ concentrations within the cell. Elevated Ca2+ levels then set off a cascade of events – both destructive and protective –, which eventually decide the fate of the affected neuron. These events have been dissected studiously and found to depend to a significant extent on the circumstances and conditions of the excitotoxic insult, and on the energy status and developmental stage of the susceptible neurons. Researchers also identified remarkable and poorly understood species and sex differences, and demonstrated that the mechanism of excitotoxic cell death can change, sometimes significantly, in abnormal (i.e. sick) organisms. Description of these complex events, their relation to apoptotic cell death, contributions of additional membrane proteins, and discussion of differentiating features, are beyond the scope of this article. For further information, the interested reader is referred to authoritative summaries and reviews of the subject (Aarts and Tymianski, 2004; Beal, 1992; Besancon et al., 2008; Bossy-Wetzel et al., 2004; Fagni et al., 1994; Greene and Greenamyre, 1996; Johnston, 2005; Leist et al., 1997; Mattson, 2007; Nicotera and Lipton, 1999).
3.3. An offshoot of the excitotoxic concept: developing the idea of neuroprotection
If excitotoxic mechanisms are indeed causally involved in pathological nerve cell loss, any intervention – pharmacological or otherwise – targeting the chain of events leading to neurodegeneration should prevent or arrest the neuronal demise. Although this conclusion seems trivial in hindsight, its therapeutic implications are substantial and have guided many drug discovery programs and clinical strategies during the past decades. First proposed at a Wenner-Gren symposium in Stockholm in 1978 (Schwarcz et al., 1979c), the idea was experimentally verified by demonstrating that the weak NMDA receptor-preferring antagonist D-α-aminoadipic acid and the more potent D-aminophosphonopentanoic acid (AP5) and D-aminophosphonoheptanoic acid (AP7) prevent the neurotoxic effects of systemically administered N-methyl-DL-aspartate (Olney et al., 1979b; Olney et al., 1981) and of intracerebrally applied ibotenate but not kainate (Schwarcz et al., 1982). Subsequently, Meldrum and co-workers tested the anti-excitotoxic concept in animal models of epilepsy (Croucher et al., 1982) and cerebral ischemia (Simon et al., 1984) and showed remarkable anticonvulsant and neuroprotective efficacy, respectively, of NMDA receptor blockade. These two “Science” publications, together with the demonstration that AP7 prevents hypoglycemia-induced neurodegeneration (also published in “Science”; Wieloch, 1985), convinced both laboratory-based and clinical neuroscientists that the targeting of NMDA receptors provided unprecedented, exciting opportunities for the treatment of neurological disorders (Schwarcz and Meldrum, 1985). During the following years, all major pharmaceutical firms with a presence in central nervous system (CNS) research became engaged in the concept of neuroprotection and, together with medicinal chemists in academia, produced an invaluable armamentarium of glutamatergic compounds for pre-clinical and clinical use. These efforts in chemistry were not restricted to targeting the NMDA receptor. By the end of the 1980s, Danish scientists had succeeded in synthesizing the first selective AMPA (“non-NMDA”) receptor antagonists (Honoré et al., 1988), which, interestingly, also afforded impressive neuroprotection in an animal model of cerebral ischemia (Sheardown et al., 1990). It was later shown that NMDA and non-NMDA receptor interact both functionally and physically (Jonas and Spruston, 1994; O’Brien et al., 1998). Moreover, this interplay is influenced by metabotropic glutamate receptors and additional endogenous factors (Bruno et al., 2001; Caldeira et al., 2007; Salt and Eaton, 1996; Stoll et al., 2007) and is especially complex under pathological conditions (Besancon et al., 2008; Choi, 1993; Pellegrini-Giampietro et al., 1994). Thus, in the case of cerebral ischemia, as well as in HD and a host of other acute and chronic pathologies where anti-excitotoxic interventions are indicated on theoretical grounds, translation into clinically useful strategies is complicated, and predictions of success are difficult (Kalia et al., 2008; Sacco et al., 2001; Villmann and Becker, 2007). The problem is further augmented by the fact that glutamate is the major excitatory neurotransmitter in the mammalian CNS, so that any direct manipulation of its receptors might compromise important physiological functions. These and related considerations prompted investigators to examine excitotoxicity from a different perspective, i.e. to ask questions about upstream events leading to excessive glutamate receptor activation. Initially, these studies were designed to identify endogenous agonists of excitatory amino acid receptors and to evaluate their neurotoxic potential.
4. Endogenous excitotoxins and HD
4.1. Glutamate and other acidic amino acids
Based on Olney’s pioneering findings, glutamate was originally regarded the prime suspect in the search for (an) endogenous excitotoxic agent(s). Present in the mammalian brain at millimolar levels, this simple amino acid could be readily envisioned to accumulate in sufficiently high concentrations to overstimulate its receptors and initiate excitotoxic cell death under adverse conditions. However, since the ambient extracellular concentration of glutamate is held in the low micromolar range by powerful neuronal and glial re-uptake processes (Danbolt, 2001), and in view of the old observation that focal injections of even massive amounts of glutamate cause surprisingly little neuron loss in the normal rat brain (McGeer and McGeer, 1976; Olney and de Gubareff, 1978b), it appears unlikely that relatively brief surges in extracellular glutamate would cause irreparable structural damage to the brain. But it is entirely possible that glutamate, maybe in concert with additional endogenous factors, indeed functions as an excitotoxin in situations where its effectiveness is unduly enhanced. Examples, selected here from the earlier literature, include glutamate receptor supersensitivity (Tsai et al., 1995), impairment of glutamate transport mechanisms (Coyle et al., 1977; Cross et al., 1982; McBean and Roberts, 1985) and abnormal glutamate metabolism (Plaitakis et al., 1982).
The question if endogenous acidic amino acids other than glutamate should also be considered possible endogenous excitotoxins with links to human CNS pathology remains unresolved to this day. Although such a connection, namely a causative role of the obscure excitotoxin cysteine-S-sulfate in sulfite oxidase deficiency, was already postulated in the mid-1970s (Olney et al., 1975), only sporadic efforts have been made so far to examine the possibility that pathophysiology is caused by more abundant excitatory amino acids such as aspartate (Bradford and Nadler, 2004), cysteine sulfinate (Iwata et al., 1982) and homocysteate (Klancnik et al., 1992; Olney et al., 1987). Individually or jointly, and especially under conditions favoring their excitotoxic efficacy, these acidic α-amino acids may yet turn out to play important pathogenic roles in HD and other neurological disorders (May and Gray, 1985).
In order to test the possible involvement of glutamate in HD, a flurry of studies in the early 1980s examined its status and effects in patients using cerebrospinal fluid, blood and skin cells, and post-mortem brain tissue. Analyses included the determination of glutamate levels (Bonilla et al., 1988; Ellison et al., 1987; Kim et al., 1980), glutamate uptake (Comings et al., 1981; Mangano and Schwarcz, 1981), glutamate receptors (Greenamyre et al., 1985), intermediary glutamate metabolism (Mangano and Schwarcz, 1982) and glutamate toxicity (Archer and Mancall, 1983; Stahl et al., 1984). In spite of this rather extensive scrutiny, which is still in progress (Hassel et al., 2008), no convincing evidence of glutamate dysfunction was found, leaving the field in a state of confusion (Perry and Hansen, 1990) and, at the same time, stimulating the quest for alternate links between excitotoxicity and HD pathology.
4.2. Quinolinate
A brief communication reporting NMDA receptor-mediated excitation following the ionophoretic application of the tryptophan metabolite quinolinic acid (pyridine-2,3-dicarboxylic acid; QUIN) to rat cortical neurons (Stone and Perkins, 1981) attracted our attention during our own search for novel endogenous excitotoxins. Together with the earlier demonstration of the powerful convulsant effects of QUIN following intracerebroventricular injection in mice (Lapin, 1978), this finding immediately suggested that the compound might have neurotoxic properties, prompting us to examine the consequences of an intrastriatal injection in rats. Indeed, QUIN proved to be a potent striatal excitotoxin, producing the classic, axon-sparing neurodegeneration seen after a focal injection of kainate or ibotenate (cf. above; Schwarcz et al., 1983). In line with a selective activation of NMDA receptors, QUIN-induced lesions were readily prevented by the co-administration of AP7 (Foster et al., 1983). Moreover, like lesions generated by the NMDA receptor agonist ibotenate, neuron loss caused by QUIN was limited to a well-circumscribed area around the injection site.
It soon became apparent, however, that QUIN’s neurotoxic properties were not identical to those of ibotenate and in fact differed qualitatively from those of any other known excitotoxin. Thus, in spite of its lack of affinity for kainate receptors, QUIN shares a number of features with kainate, including diminished excitotoxic potency in the developing striatum (Foster et al., 1983), prevention of striatal neurotoxicity by cortical deafferentation (Schwarcz et al., 1984), and, in the hippocampus, preferential degeneration of pyramidal cells (Schwarcz et al., 1983). Interestingly, QUIN, like kainate but unlike ibotenate, glutamate and cysteine sulfinate – which serve as precursors of muscimol, GABA and taurine, respectively –, does not yield an inhibitory compound upon decarboxylation. Several of its characteristics may indeed be related to the fact that the pyridine derivative QUIN, again like kainate but in contrast to many other excitotoxins, does not contain a free amino group in α position to a carboxylic acid residue and therefore does not strictly qualify as an amino acid.
The QUIN-lesioned rat striatum bears an unusually close resemblance to the HD neostriatum. This was first recognized by Beal and co-workers (Beal et al., 1986), who described the relative sparing of a small population of medium-sized, aspiny neurons containing the peptides somatostatin and neuropeptide Y in the markedly neuron-depleted striatum. These cells, which also contain NADPH-diaphorase, survive in the HD brain (Ferrante et al., 1987a; Ferrante et al., 1987b) but degenerate in experimental animals following a local injection of kainate, ibotenate or NMDA (Beal et al., 1986). These studies were later complemented by other parallels between the HD neostriatum and the QUIN-lesioned rat striatum, such as the resistance of large cholinergic neurons (Davies and Roberts, 1988; Ferrante and Kowall, 1987) and – less unexpectedly – the presence of hypertrophic astrocytes (Björklund et al., 1986; Vonsattel et al., 1985). Since QUIN also produces a HD-like lesion in the primate striatum (Ferrante et al., 1993), and since the chronically QUIN-lesioned rat striatum maintains the characteristic features of the HD neostriatum (Beal et al., 1991), intrastriatal QUIN injections became the experimental model of choice to test novel HD treatments (Beal et al., 1988) and to elaborate the pathological and functional changes seen in the disease (DiFiglia, 1990). Notably, the QUIN model has remained attractive following the identification of the HD gene (Huntington’s Disease Collaborative Research Group, 1993) and is now increasingly considered in conjunction with genetic factors that determine the etiology and pathophysiology of the disease (Pérez-De La Cruz and Santamaria, 2007; see below).
The most interesting conjecture of these studies from a pathogenic viewpoint was the possibility that QUIN might be the long sought-after endogenous excitotoxin. In human and rat brain, QUIN is present at concentrations in the high nanomolar range (Wolfensberger et al., 1983), and prolonged exposure of rat organotypic cortico-striatal cultures to as little as 100 nM QUIN results in characteristic excitotoxic damage (Whetsell and Schwarcz, 1989). We and others therefore argued that a pathological elevation of QUIN levels may produce excitotoxic neurodegeneration in HD. Disappointingly, this hypothesis was initially not corroborated by QUIN measurements in HD. Thus, QUIN concentrations in patients were found to be unaffected (Reynolds et al., 1988) or even reduced (Heyes et al., 1991) in brain tissue, and remained unchanged in the cerebrospinal fluid (Heyes et al., 1991; Schwarcz et al., 1988a). HD patients were also found to excrete normal amounts of QUIN (Heyes et al., 1985). These results significantly dampened enthusiasm for the QUIN hypothesis of HD, especially since they contrasted with parallel reports of dramatic QUIN increases in the cerebrospinal fluid of AIDS patients (Heyes et al., 1989). Unknown to researchers at the time, however, the hypothesis had not been tested with sufficient rigor and was slated to re-emerge a few years later based on new biological and chemical insights.
5. The kynurenine pathway of tryptophan degradation
5.1. Kynurenines
The kynurenine pathway (KP), named after L-kynurenine, the pivotal metabolite placed at a branching point of the enzymatic cascade (Figure 5), is the major catabolic route of dietary tryptophan in mammals. As one of the downstream metabolites in the KP, QUIN is synthesized by a succession of enzymatic steps, which have long been identified and thoroughly examined in peripheral tissues (Schlossberger et al., 1984). In turn, QUIN is broken down further, eventually forming the common cofactor NAD+. This “QUIN branch” of the KP is implicated in a variety of immune functions and may play a particularly important role in innate and adaptive immunosuppression (Belladonna et al., 2007; Gonzalez et al., 2008). In spite of the fact that all individual KP enzymes responsible for the conversion of tryptophan to QUIN and beyond are well characterized, however, their involvement in biological processes is just beginning to be understood (Belladonna et al., 2007; Lopez et al., 2006). This active area of research is currently primarily concerned with indoleamine-2,3-dioxygenase (IDO), one of the two enzymes responsible for the initial oxidative opening of tryptophan’s indole ring (the other one being the more substrate-specific tryptophan-2,3-dioxygenase) (Schröcksnadel et al., 2006; Xu et al., 2008).
Figure 5.

The kynurenine pathway of tryptophan degradation in mammalian cells.
Although the link between KP metabolism, the immune system and non-CNS diseases is increasingly appreciated, most speculations in the literature revolve around the question if the KP plays a role in brain physiology and pathology. This focus is justified considering the fact that the KP contains at least two neuroactive metabolites in addition to QUIN, i.e. the QUIN precursor 3-hydroxykynurenine (3-HK) and, in a sidearm of the pathway, kynurenic acid (KYNA) (Figure 5). The (neuro)biological properties of these compounds, which include the generation of free radicals by 3-HK (Vazquez et al., 2000; cf 7.2) and the inhibition of NMDA and α7 nicotinic acetylcholine receptors by KYNA (Hilmas et al., 2001; Kessler et al., 1989), suggest that they might participate actively in a wide range of brain functions that are unrelated to pathological events (Carpenedo et al., 2001; Leipnitz et al., 2007; Rassoulpour et al., 2005; Valko et al., 2007). Like QUIN, however, both 3-HK and KYNA have until now received more attention for their possible involvement in neurological and psychiatric diseases (Moroni, 1999; Müller and Schwarz, 2007; Németh et al., 2006; Oxenkrug, 2007; Ruddick et al., 2006; Schwarcz and Pellicciari, 2002; Stone and Darlington, 2002). Unless they are relevant to the pathophysiology of HD, these pathological consequences of abnormal brain KP metabolism will not be discussed here.
One of the major challenges facing neuroscientists involved in in vivo studies of brain 3-HK, QUIN and KYNA relates to the presumed interplay between peripheral and central kynurenines (the term used to describe all KP metabolites collectively). Whereas tryptophan, kynurenine and 3-HK readily penetrate the blood-brain barrier, no such transport mechanisms exist for the acidic metabolites QUIN and KYNA (Fukui et al., 1991; Gál and Sherman, 1980). This raises the important question to what extent and by which mechanism(s) fluctuations in the levels of circulating kynurenines affect brain function and, in the case of pathological changes, dysfunction (Kita et al., 2002; Wu et al., 2000). This issue has obvious pharmacological and therapeutic relevance since systemic interventions targeting specific KP enzymes in the periphery may not have the desired effects on neuroactive kynurenines in the brain (Reinhard, 2004). Unfortunately, comparatively little is known about the short- and, especially, long-term effects of such peripheral KP manipulations. Since these operations modulate circulating levels of kynurenine in a predictable fashion, however, some useful inferences can be drawn from studies that examine the fate of blood-derived kynurenine in the brain.
5.2. Kynurenine pathway metabolism in the brain
In spite of the fact that the KP accounts for >95% of dietary tryptophan degradation in the periphery (Schlossberger et al., 1984), intracerebral administration of tryptophan normally has very little effect on the flux through the cerebral KP (Heyes et al., 1993). This implies either that the brain activities of IDO or tryptophan-2,3-dioxygenase are exceedingly low or that exogenously applied tryptophan does not have sufficient access to these oxidative enzymes under physiological conditions. In contrast, cerebral KP metabolism is quite responsive to systemically applied tryptophan (During et al., 1989), suggesting, together with the data from Gál and Sherman (Gál and Sherman, 1980), that kynurenine – rather than tryptophan – initiates the metabolic chain leading to the synthesis of neuroactive KP metabolites within the brain. This was verified experimentally in rats by focal intrastriatal injections of radiolabelled kynurenine, which was first taken up (Speciale and Schwarcz, 1990) and then promptly incorporated into several kynurenines, including 3-HK, QUIN and KYNA (Guidetti et al., 1995). In line with expectations, systemic administration of kynurenine, too, increases the brain levels of KP metabolites (Reinhard and Erickson, 1994; Swartz et al., 1990).
Pioneering studies by Heyes and his collaborators in the 1990s revealed that cerebral KP metabolism is qualitatively impaired in several situation of immune compromise. In a series of elegant experiments, the investigators traced some of the impairments to the induction of IDO (Heyes et al., 1992; Heyes et al., 1993; Saito et al., 1991), though they also reported inflammation-related increases in the activity of the downstream KP enzymes kynurenine-3-monooxygenase (KMO), kynureninase and 3-hydroxyanthranilic acid dioxygenase (3-HAD) (Figure 4; Heyes et al., 1993; Saito et al., 1993). Notably, these enzymatic changes were associated with often dramatic increases in the tissue levels of metabolites of the QUIN branch of the KP including, prominently, QUIN itself. On the other hand, the affected brains showed only comparatively minor or no changes in the levels of KYNA and its biosynthetic enzyme, kynurenine aminotransferase. These findings therefore revealed an interesting dissociation between the two KP branches under conditions of immunological compromise.
Figure 4.
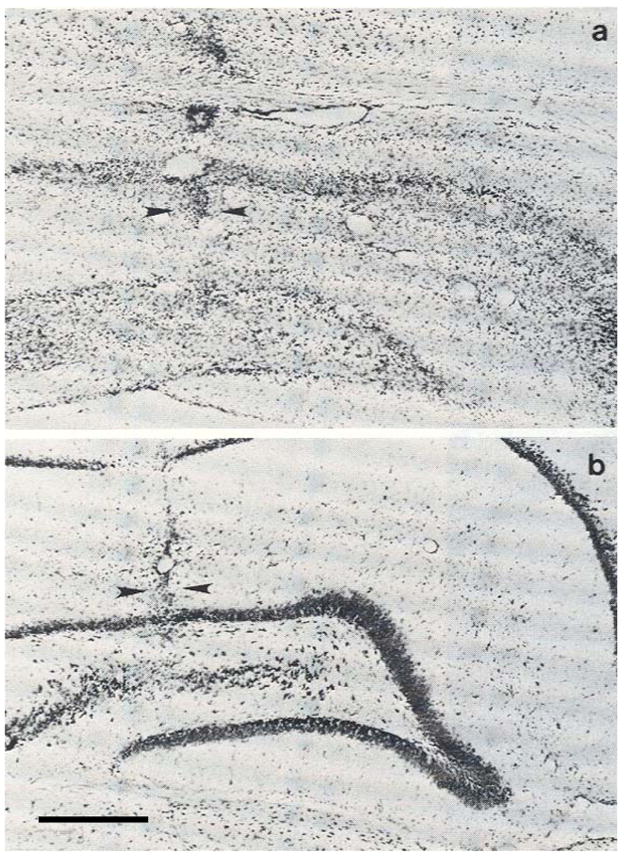
Anti-excitotoxic neuroprotection: effects of an intrahippocampal ibotenate injection (5 μg) in the absence (a) or presence (b) of D-amino-phosphonoheptanoic acid (5 μg) in rats. Arrowheads demarcate the tracks of the injection needles. Bar: 500 μM. (From Schwarcz et al., 1982).
Our laboratory had previously identified the synthetic and degradative enzymes of QUIN, 3-HAD and quinolinic acid phosphoribosyltransferase (QPRT), in the brain (Foster et al., 1985a; Foster et al., 1986). Based on studies in the neuron-depleted striatum, where we found substantial increases in both 3-HAD and QPRT activities, we proposed subsequently that the enzymes had to be localized primarily in glial or other non-neuronal cells (Schwarcz et al., 1989). In light of the results of Heyes’ group, it was likely that the QUIN branch of the KP was mainly localized in microglial cells, which are robustly activated by neuronal damage and degeneration, and in response to immunological challenges (Gehrmann et al., 1995). This was later verified directly, using human microglia in vitro (Heyes et al., 1996). Complementary studies revealed that the KYNA branch of the KP, in contrast, was not present in microglia but largely contained in astrocytes (Guillemin et al., 2001; Kiss et al., 2003; Roberts et al., 1992). This physical segregation of the two pathway branches, which can be traced to a lack of KMO in astrocytes (Guillemin et al., 2001), might have important functional ramifications in CNS physiology and pathology.
5.3. Experimental manipulation of kynurenine pathway metabolism
Selective pharmacological or genetic tools are necessary to explore the intricacies of cerebral KP function and to assess the possible role of kynurenines in brain diseases. Examples of specific inhibitors of mammalian KP enzymes had been reported decades ago (Decker et al., 1963; Kido and Noguchi, 1975), and it was shown, in Drosophila, that genetic deletions of individual KP enzymes resulted in phenotypic changes implying neuronal abnormalities (Savvateeva et al., 2000). Beginning in the 1990s and with increasing frequency, medicinal chemists in academia and industry produced new inhibitors, which specifically targeted several mammalian KP enzymes. Specificity was most often achieved by modifying the chemical structure of the natural substrate of an enzyme, and enzyme inhibition was therefore mostly competitive in nature (for review; Schwarcz and Pellicciari, 2002; Stone and Darlington, 2002). In some cases, however, interesting novel structures were discovered by approaches that were not guided by substrate analogy (Lima et al., 2009; Röver et al., 1997). Moreover, endogenous agents such as steroid hormones and cytokines were found to stimulate the activity of individual KP enzymes and may therefore have distinct effects on the function of neuroactive KP metabolites (Andre et al., 2008; Heyes et al., 1997; O’Connor et al., 2008; Oxenkrug, 2007).
Almost without exception, the usefulness of the new synthetic compounds for the study of brain KP metabolism in vivo is restricted by their difficulty to cross the blood-brain barrier after systemic administration (cf. Németh et al., 2005). Experimental studies are also complicated by the fact that pharmacologically induced changes in the function of peripheral KP enzymes can have secondary effects on brain KP metabolism. Probably the best example so far, and one that is of relevance to HD research (see Section 8.), is the substantial increase in brain KYNA levels that is reliably seen after the peripheral application of a KMO inhibitor (Moroni et al., 1991; Pellicciari et al., 1994; Röver et al., 1997; Speciale et al., 1996). As expected, this effect is associated with an accumulation of the enzyme substrate kynurenine in the circulation and, because of the metabolite’s ready penetration from the blood, in the brain. Without further experimentation, it is therefore impossible to unequivocally trace the rise in brain KYNA levels to either peripherally or centrally derived kynurenine, i.e. to determine whether the initial KMO inhibition had occurred intra- or extracerebrally (or both). To circumvent this complexity and its formidable interpretative challenges, which are even more daunting when peripheral or central kynurenines are implicated in neuropathological events, agents targeting individual KP enzymes have been applied directly into the brain. These studies, which have so far been limited to intracerebral injections of enzyme inhibitors, have met with some success (Hamann et al., 2008; Miranda et al., 1997; Walsh et al., 1994) and are beginning to reveal information about the functional interactions of the two KP branches within the brain (Amori et al., in press). However, newly synthesized pharmacological probes have so far not transformed our understanding of cerebral KP metabolism and function, or of the relationship between central and circulating kynurenines. Knowledge in this regard is therefore still rather rudimentary, and improved chemical tools are clearly needed.
Genetic approaches in mammals have met with some success but their impact has so far also been reduced by the fact that brain enzymes were not targeted specifically. For example, mice with a genomic elimination of kynurenine aminotransferase II (a major enzyme of cerebral KYNA synthesis) have remarkable characteristics, which might be causally related to abnormal functions of kynurenines in the brain (Alkondon et al., 2004). Of particular interest with regard to HD pathology, these animals, which have a diminished ability to form the neuroprotective metabolite KYNA, show enhanced neuronal vulnerability to an intrastriatal QUIN injection. This lesion enlargement is not observed when the striatal KYNA deficit in mutant mice is neutralized by pharmacological means. These results indicate that endogenous KYNA controls the vulnerability of striatal neurons to QUIN (Coyle, 2006; Sapko et al., 2006).
6. The kynurenine pathway in Huntington’s disease
6.1. Studies in humans
Although several studies had failed to reveal increased QUIN accumulation in HD (see 4.2), there were early indications of impaired cerebral KP metabolism in the disease. This included our demonstration of elevated 3-HAD and QPRT activity (Foster et al., 1985b; Schwarcz et al., 1988b) as well as contradictory reports of abnormal KYNA levels (Beal et al., 1992; Beal et al., 1990; Connick et al., 1989) in the HD brain. Most interesting in view of later developments, Reynolds and colleagues reported increased brain tissue levels of 3-HK in HD, which were not, however, accompanied by changes in the activity of 3-HK’s degradative enzyme, kynureninase (Pearson et al., 1995; Pearson and Reynolds, 1992). Notably, no metabolic abnormalities indicative of an overactive QUIN branch of the KP have so far been found in the periphery (Foster and Schwarcz, 1985; Heyes et al., 1985; Stoy et al., 2005).
Triggered by evocative studies in genetic mouse models of HD (see below), we showed more recently that the brain levels of both 3-HK and QUIN are substantially elevated – by three to four-fold – in low-grade (grade 0/1) HD brain (Guidetti et al., 2004; Fig. 1). These changes were seen in the neocortex and in the neostriatum but not in the cerebellum, which suffers only little damage in HD. Compared to age-matched controls, brain 3-HK and QUIN levels were either unchanged or even tended to decrease in grade 2 and advanced grade (grades 3–4) HD brain, and cerebral KYNA levels fluctuated only modestly as the illness progressed. These results suggested a brain region-specific involvement of 3-HK and QUIN especially in the early phases of HD pathophysiology, raising the possibility that timely, targeted interventions aimed at reducing flux through the QUIN branch of the KP might slow or prevent disease progression.
6.2 Studies in genetic mouse models
The discovery that HD is triggered by a polyglutamine expansion in Exon 1 of a protein termed huntingtin (htt) (Huntington’s Disease Collaborative Research Group, 1993) allowed the generation of genetic model organisms that can be used to test potential links between mutant htt and KP dysregulation. We therefore examined the tissue levels of the KP metabolites 3-HK and QUIN longitudinally in four informative brain areas (striatum, cortex, cerebellum and hippocampus) in well-established transgenic (R6/2 and YAC128; Mangiarini et al., 1996; Slow et al., 2003) and knock-in (HdhQ92/HdhQ111; Wheeler et al., 2000) mouse models. These analyses, as well as a more limited, earlier study showing a large increase in brain 3-HK levels in another full-length transgenic mouse model (Guidetti et al., 2000), revealed remarkable biochemical abnormalities in all cases (Guidetti et al., 2006).
In R6/2 mice up to 3 weeks of age, 3-HK and QUIN levels were unchanged in all four brain regions. From 4 through 12 weeks of age, i.e. from the time of earliest phenotypic changes until the animals became moribund, mutant mice had significantly higher 3-HK levels in the cortex, striatum and cerebellum than age-matched wild-type controls, though no changes were observed in the hippocampus. These region-specific elevations in 3-HK were recently shown to be associated with, and may in fact be caused by, increases in KMO and reductions in kynureninase activity (Stachowski et al., 2008). In YAC128 mice, which show slower disease onset and a less dramatic phenotype, 3-HK levels in all four brain regions were indistinguishable from controls at 2 and 5 months but were elevated in cortex, striatum and cerebellum (but again not in the hippocampus) at 8 and 12 months of age. This increase was particularly pronounced in the cortex, where 3-HK levels were increased 3–4 times at 12 months of age. In 8- and 12-month-old YAC128 mice, QUIN levels were also significantly elevated, but this increase was restricted to striatum and cortex, i.e. the regions most affected in HD. Finally, both 3-HK and QUIN levels were also significantly elevated in cortex and striatum of 15–17-month-old HdhQ92/HdhQ111 knock-in mice, which show the most modest phenotypic changes of the three mutants tested.
Collectively, these studies supported the idea that mutant htt induces cellular alterations, which consistently result in the up-regulation of the neurotoxic branch of the KP in striatum and cortex, i.e. the two brain regions that are most prominently affected in HD. The parallels to the human disease are not likely to be accidental and suggest that genetic HD mouse models can be used for the further elaboration of the role of KP metabolites in pathogenesis.
6.3. Lessons from yeast
The yeast Saccharomyces cerevisiae, which has been used to brew beer and wine for millennia, is commonly used in the laboratory, because numerous intracellular processes are well conserved between yeast and higher organisms, in many cases including humans. In addition, this simple eukaryotic microorganism lends itself to easy genetic manipulation including selective insertion or deletion of genes of interest. Using a simple bioassay (cell toxicity), we recently performed a large-scale screen in yeast that identified mutations in 28 genes that suppress toxicity of a mutant htt fragment (Giorgini et al., 2005). One of the most potent of these mutations was in the gene encoding Bna4, the yeast counterpart of mammalian KMO. We therefore proceeded to measure the formation of 3-HK and QUIN in yeast cells. Compared to cells expressing wild-type htt, and in remarkable agreement with the studies in humans and mice described above, 3-HK levels were ~2.2-fold higher in cells expressing mutant htt. QUIN levels, too, were significantly increased. Moreover, as expected, 3-HK and QUIN were not detected in cells lacking Bna4.
We next assessed the viability of mutant htt-expressing strains in which several additional yeast orthologs of KP enzymes were individually deleted. Elimination of BNA1, the yeast equivalent of 3-HAD (the immediate biosynthetic enzyme of QUIN), partially suppressed mutant htt-mediated toxicity. Biochemical analyses of these mutant cells showed that the deletion of BNA1 had prevented QUIN production as predicted and had caused an accumulation of the upstream metabolite 3-HK. This indicated that the remaining toxicity of mutant mtt in this strain might be related to the presence of excessive concentrations of 3-HK (see 7.2. for further discussion).
Elimination of two other KP genes, ARO9 and NPT1, enhanced mutant htt toxicity in yeast. Deletion of ARO9, the yeast equivalent of kynurenine aminotransferase, should block the irreversible formation of KYNA from kynurenine and therefore increase the levels of kynurenine and its toxic downstream metabolites 3-HK and QUIN. In addition, toxicity might be aggravated because the absence of KYNA deprives the cells of a protective free-radical scavenger (Goda et al., 1999). Deletion of NPT1 blocks the formation of NAD+ from nicotinic acid, forcing all synthesis of NAD+ to occur via the QUIN branch of the KP. Consistent with these explanations, 3-HK levels were increased 90- and 15-fold, respectively, and QUIN was elevated 10-fold in Npt1-deficient cells compared to the parental strain expressing mutant htt. Although 3-HK levels were also higher in Aro9-deficient yeast carrying an empty vector, expression of mutant htt in these cells increased 3-HK levels more than 40-fold.
Of the 28 gene deletions identified in the original suppressor screen in yeast (Giorgini et al., 2005), BNA4 was the only one that affected the KP directly. Subsequent studies revealed, remarkably, that 22 of the remaining 27 suppressors also affected KP metabolism, albeit indirectly. Compared with wild-type cells expressing mutant htt, 61% of the suppressors showed significant reductions in 3-HK alone, 75% in QUIN alone, 82% in 3-HK or QUIN, and 54% in both 3-HK and QUIN (Table 1). These results strongly suggested a central role for the KP in mediating the toxicity of mutant htt in yeast. Additional experiments indicated that the reductions in 3-HK and QUIN were controlled by the activity of histone deacetylases on KP promoters. Finally, histone deacetylase-dependent regulation of the KP was also observed in a mouse model of HD, in which treatment with a neuroprotective histone deacetylase inhibitor blocked KP activation in microglia expressing a mutant htt fragment both in vitro and in vivo (Giorgini et al., 2008). These studies provided further support for the hypothesis that KP impairment might play a pivotal role in HD pathology and, in particular, suggest that regulatory mechanisms might be targeted to normalize brain KP metabolism and provide clinical benefits.
Table 1.
Levels of 3-hydroxykynurenine (3-HK) and quinolinic acid (QUIN) in toxicity suppressor yeast strains expressing mutant huntingtion (103Q). Numbers listed refer to the relative percentage of 3-HK and QUIN levels in deletion strains as compared to the parental strain (BY4741). bna4Δ: deletion of the yeast ortholog of kynurenine 3-monooxygenase.
| Strain | 3-HK | QUIN |
|---|---|---|
| wild-type (BY4741) | 100.0 | 100.0 |
| Vesicular transport, vacuolar protein sorting and import | ||
| bfr1Δ | 49.0* | 51.0* |
| cyk3Δ | 46.8* | 53.6* |
| def1Δ | 49.6* | 87.6* |
| mso1Δ | 60.2* | 69.8* |
| sna2Δ | 21.7* | 48.2* |
| vps53Δ | 298.3* | 57.9* |
| Transcription or maintenance of chromatin architecture | ||
| mbf1Δ | 32.5* | 77.5* |
| nhp6bΔ | 87.3 | 51.6* |
| paf1Δ | 102.9 | 23.7* |
| rxt3Δ | 98.4 | 91.3 |
| ume1Δ | 13.2* | 31.0* |
| ylr278cΔ | 18.0* | 42.6* |
| Known and putative yeast prion genes | ||
| def1Δ | 49.6* | 87.6* |
| rnq1 Δ | 116.0* | 63.3* |
| ybr016wΔ | 132.0* | 62.8* |
| yir003wΔ | 60.2* | 62.2* |
| ylr278cΔ | 18.0* | 42.6* |
| Other cellular processes | ||
| arg7Δ | 133.0 | 96.4 |
| bna4 Δ | 0* | 0* |
| ecm37Δ | 51.6* | 67.8* |
| fmp27Δ | 128.2 | 96.6 |
| hsp104Δ | 96.9 | 58.4* |
| mgt1Δ | 61.4* | 73.3* |
| pho87Δ | 16.8* | 85.4 |
| rdh54Δ | 64.4* | 63.6* |
| ydr287wΔ | 53.3* | 62.1* |
| yer185wΔ | 26.0* | 81.8* |
| Genes with unknown function | ||
| smy2Δ | 54.4* | 182.2* |
| ymr082cΔ | 171.3* | 96.5 |
| ymr244c-aΔ | 74.8 | 79.8 |
P < 0.05 (Student’s t-test). (From Giorgini et al., 2008).
7. Mechanism of quinolinate toxicity
7.1. Quinolinate is not a conventional excitotoxin
The new evidence supporting a pathophysiologically significant role of QUIN in HD rekindled interest in the neurodegenerative properties of the metabolite. As mentioned earlier, the sequelae of an intracerebral QUIN injection are distinct from those of the classic excitotoxins kainate and ibotenate. Surprisingly, in spite of the fact that QUIN is a selective NMDA receptor agonist, its neurotoxic effects also differ substantively from those of NMDA. Thus, unlike QUIN, NMDA is an potent neurotoxin in the cerebellum (Garthwaite et al., 1986), does not distinguish between neuronal populations in the hippocampus (Nadler et al., 1981), is more effective in the immature than in the adult brain (McDonald et al., 1988), and produces nerve cell loss that is significantly less susceptible to blockade by KYNA than QUIN-induced lesions (Foster et al., 1984). The reasons for these remarkable qualitative differences between two supposedly very similar excitotoxins have been tentatively related to the fact that QUIN preferentially activates only a subgroup of NMDA receptors in the forebrain (De Carvalho et al., 1996; Monaghan and Beaton, 1991), whereas NMDA itself, by definition, targets all receptor subtypes. This interpretation would be consistent with the observation that QUIN lesions are always preventable by broad spectrum NMDA receptor antagonists such as AP5 and AP7. It would also be compatible with a role of a subset of NMDA receptors in the pathophysiology of HD (see Fan and Raymond, 2007, for review).
A study by Rios and Santamaria first suggested that the neurotoxic characteristics of QUIN might also be related to its ability to stimulate lipid peroxidation under physiological conditions (Rios and Santamaria, 1991). Although this effect was observed in cell-free brain homogenate, it was blocked by KYNA or specific NMDA receptor antagonists such as MK-801 (Santamaría and Ríos, 1993) and therefore appears to be dependent on receptor activation. Induction of lipid peroxidation, monitored by measuring peroxinitrite production (Pérez-De La Cruz et al., 2005), is iron (II)-dependent (Stípek et al., 1997) and probably related to the formation of specific QUIN-iron complexes. Upon auto-oxidation, these complexes generate reactive hydroxyl radicals through the Fenton reaction (Pláteník et al., 2001; Santamaría et al., 2001). This mechanism may explain the remarkable neurotoxic potency of the weak NMDA receptor agonist QUIN in vivo. In support of this idea, QUIN-induced striatal lesions are significantly attenuated by treatment with agents that enhance peroxynitrite decomposition, bind ferrous iron and/or scavenge reactive oxygen species (ROS) (Müller et al., 2007; Nakao et al., 1996; Santiago-López et al., 2004). It is also tempting to speculate that these protective interventions may be especially effective vis-à-vis QUIN, because they target Fe (II), which is an essential co-factor of 3-HAD and other oxygenases responsible for the biosynthesis of QUIN.
7.2. A cytotoxic role for 3-hydroxykynurenine?
The cytotoxic properties of 3-HK, which is present in the mammalian brain in nanomolar concentration (Pearson and Reynolds, 1991), were first described 20 years ago (Eastman and Guilarte, 1989). The compound readily auto-oxidizes and thereby generates hydrogen peroxide and other ROS, which appear to be singularly responsible for neurotoxicity. Thus, 3-HK has no known direct effect on NMDA receptors, and cell damage in vitro and in vivo can be prevented by co-administration of metal chelators and radical scavengers (Chiarugi et al., 2001; Eastman and Guilarte, 1990; Goldstein et al., 2000; Nakagami et al., 1996; Okuda et al., 1996, 1998). Of particular interest for HD research, exposure to as little as 1 μM 3-HK causes the death of spiny striatal neurons in organotypic cultures, yet is relatively innocuous towards the same small population of medium-sized aspiny neurons that is spared in the HD neostriatum (Okuda et al., 1996; see 4.2). Again in analogy to the HD brain, cerebellar neurons in culture are remarkably resistant to 3-HK-induced oxidative stress (Okuda et al., 1998).
These studies, as well as the increasing appreciation that ROS might play a critical role in HD pathology (Fagni et al., 1994; Wyttenbach et al., 2002), prompted us a few years ago to examine the possibility that 3-HK might exacerbate striatal lesions caused by its downstream metabolite QUIN in rats. Intrastriatal co-application of 3-HK, at a concentration that did not produce neuronal damage on its own, indeed potentiated QUIN-induced neurodegeneration several-fold while maintaining the excitotoxic, i.e. axon-sparing, nature of the lesion (Guidetti and Schwarcz, 1999). Confirming the unique characteristics of QUIN as an excitotoxin, 3-HK was unable to similarly increase the efficacy of intrastriatal NMDA injections. Moreover, we showed that the effect of 3-HK was not due to its in situ conversion to QUIN and demonstrated pharmacologically that the potentiation by 3-HK, which can also be observed in vitro (Chiarugi et al., 2001), was caused by ROS generation. These experiments coincided chronologically with new evidence of enhanced 3-HK levels in vulnerable brain regions of HD patients and HD mouse models (see above) and gave credence to the concept that the two closely related KP metabolites, rather than QUIN alone, should be considered joint pathogens in the disease.
Further support for a role of 3-HK-derived ROS in HD came from additional experiments in yeast. Thus, ROS levels in yeast cells expressing mutant htt were approximately eight-fold higher than in control cells expressing wild-type htt, but this difference was neutralized with the elimination of the KMO ortholog Bna4 in both cells (see 6.3). In a complementary pharmacological study, both the elevated 3-HK and the high ROS levels in mutant htt-expressing cells were reduced by more than 50% by treatment with the selective KMO inhibitor Ro 61–8048 (Röver et al., 1997). This drug, like the deletion of BNA4, also enhanced the viability of yeast cells expressing mutant htt (Giorgini et al., 2005). Thus, either genetic or pharmacological interference with 3-HK synthesis abolishes or significantly attenuates the toxic consequences of mutant htt expression and the excessive formation of both 3-HK and ROS. Together with the finding that deletion of BNA1, the yeast ortholog of 3-HAD, partially suppresses mutant htt-mediated toxicity in yeast (Giorgini et al., 2005; see 6.3), these results indicate a pathophysiologically significant role of ROS, generated by both 3-HK and QUIN, in HD.
7.3. Non-cell-autonomous neurodegeneration
Increased immunostaining for microglia and astrocytes is observed specifically in affected brain regions of HD patients (Myers et al., 1991; Sapp et al., 2001; Simmons et al., 2007; Singhrao et al., 1999; Singhrao et al., 1998; Vonsattel et al., 1985), and prominent glial abnormalities are also seen in HD mouse models (Ma et al., 2003; Minghetti et al., 2007; Reiner et al., 2007; Simmons et al., 2007). Moreover, recent positron emission tomography (PET) studies using a radiotracer specific to activated microglia documented distinct abnormalities in presymptomatic HD gene carriers. Most interestingly, microglial activation in these preclinical HD cases correlated inversely with striatal neuronal dysfunction, as determined by loss of dopamine receptor binding (Tai et al., 2007). These and related studies indicate that glial cells, and especially microglia, might be more than innocent bystanders and mere markers of ongoing or completed neuronal destruction. They may, in fact, actively participate in the pathological process by mobilizing toxic KP metabolites.
Most KP enzymes are constitutively expressed in both microglia and astrocytes (Guillemin et al., 2001; Heyes et al., 1996). However, microglial cells contain far more KMO and 3-HAD than astrocytes (Giorgini et al., 2008; Lehrmann et al., 2001) and are therefore the major source of 3-HK and QUIN, while transamination of kynurenine to KYNA occurs mainly in astrocytes (Guillemin et al., 2001; Kiss et al., 2003). As KP metabolites are deranged in HD, we propose that KMO and other KP enzymes may contribute to neurodegeneration in a non-cell autonomous manner. As illustrated schematically in Figure 6, we speculate that the toxic cascade is likely to be initiated by mutant htt expression in neurons. Perhaps in response to mitochondrial impairment and/or a plethora of other abnormalties, dysfunctional neurons and neuronal activity then create a pro-inflammatory environment leading to the activation of surrounding microglial cells. This, in turn, would lead to an up-regulation of microglial KP metabolism, enhanced microglial release of 3-HK and QUIN and, finally, the demise of already weakened neurons by a combination of excessive ROS production and NMDA receptor activation. As indicated, mutant htt in microglia may also play an active role in these processes (Figure 6).
Figure 6.

Schematic representation of non-cell-autonomous neurodegeneration in HD. (From Guidetti et al., 2006).
8. Kynurenine 3-monooxygenase as a therapeutic target
Inhibition of KMO (E.C. 1.14.13.9) appears to be the best strategy to reduce both 3-HK and QUIN formation in HD. Not only is the enzyme an optimal target for attenuating flux through the entire QUIN branch of the pathway, but reduced KMO activity might also shift KP metabolism towards enhanced KYNA synthesis and thereby boost neuroprotection (cf. Figure 5). Located in the outer mitochondrial membrane (Okamoto and Hayaishi, 1969), KMO is a NADPH-dependent simple flavoprotein, which, as a classic oxygenase, uses molecular oxygen as a co-substrate. Mammalian – including human – KMO has been purified, cloned and functionally expressed in vitro (Alberati-Giani et al., 1997; Breton et al., 2000; Uemura and Hirai, 1998), and its catalytic properties are similar in brain, which contains comparatively low enzyme activity, and peripheral organs (Erickson et al., 1992). Of relevance to the present discourse, the Km value of 10–20 μM for the substrate kynurenine, determined in vitro, also appears to apply to brain KMO activity in vivo (Guidetti et al., 1995). This implies that the enzyme is not saturated in vivo and will produce increasing amounts of 3-HK when kynurenine levels rise above their ambient tissue concentration (~2 μM; Beal et al., 1990) or when enzyme activity is stimulated by (an) endogenous activator(s) (Connor et al., 2008). One of these possibilities likely accounts for the elevation in 3-HK levels seen in vulnerable regions of the HD brain.
Since KMO knockouts, i.e. genetically engineered animals in which the KMO gene has been inactivated, are just beginning to be investigated (Giorgini et al., in preparation), pharmacological agents that inhibit brain KMO activity are presently the best experimental tools for testing the 3-HK/QUIN hypothesis of HD. After systemic administration, selective KMO inhibitors indeed show impressive neuroprotective efficacy, prevent or ameliorate neurological symptoms, and modulate neurotransmitter release in the brain (Carpenedo et al., 1994; Clark et al., 2005; Cozzi et al., 1999; Erhardt et al., 2001; Pellicciari et al., 1994; Rassoulpour et al., 2005; Wu et al., 2000). As mentioned earlier, however, currently available compounds do not effectively penetrate the blood-brain barrier, so that these effects cannot be unambiguously attributed to the inhibition of the brain enzyme (cf. 5.3). Still, brain KMO is a valid drug target, as evidenced by the fact that enzyme inhibition reduces excitotoxic damage in organotypic hippocampal cultures, and direct intracranial applications of a selective KMO inhibitor attenuate QUIN-induced neurodegeneration in vivo (Carpenedo et al., 2002; Harris et al., 1998; Miranda et al., 1997). However, the synthesis of novel KMO inhibitors with good brain access will clearly be instrumental for studying these neuroprotective phenomena in greater depth.
Proof-of-concept studies will undoubtedly also be aided by KMO knockout animals. Both heterozygous and homozygous KMO mutants will be useful for studying the physiological role(s) of the KP, and for identifying compensatory mechanisms and possible functional isoforms of the enzyme. Experiments with conditional and brain region-specific knockouts should, additionally, clarify the relationship between peripheral and central KP metabolism, the role of the KP in development, and the function of KMO the normal brain. Finally, crosses between HD mice and KMO knockout mice will be especially informative with regard to pathophysiological mechanisms specific to HD.
Jointly, the new genetic and pharmacological tools will make it possible to determine the impact of long-lasting decreases in 3-HK and QUIN levels in the normal and the dysfunctional brain. These studies will not only assess the potential therapeutic value of cerebral KMO inhibition in HD but will also provide information regarding possible chronic treatment hazards. For example, although the two KP branches are acutely segregated in the brain (Amori et al., in press), persistent down-regulation of the QUIN branch may with time increase the bioavailability of KMO’s substrate, kynurenine, for the synthesis of KYNA. The resulting benefit of enhanced neuroprotection would then have to be weighed against the cognitive side effects, which can be expected as a consequence of increased inhibition of NMDA and α7 nicotinic receptors (Hilmas et al., 2001; Kessler et al., 1989). These and similar considerations will help in the design of future clinical trials in which the KP is chronically manipulated in humans.
9. Conclusions and future challenges
The work summarized here suggests that the KP metabolites 3-HK and QUIN, by a combination of excessive NMDA receptor stimulation and ROS production, play a causal role in HD pathology. This hypothesis, which builds on and expands the idea that excitotoxic mechanisms are central to the disease, is supported by studies in model organisms and analyses of brain tissue specimens from HD patients. Since microglial cells produce hyperphysiological amounts of 3-HK and QUIN under pathological conditions, this concept implies an active participation of these neuroimmune cells in the neurodegenerative process. Consequently, the 3-HK/QUIN hypothesis posits that interventions leading to a normalization of microglial KP metabolism will provide therapeutic benefits in HD patients (cf. also Giorgini, 2008).
Several formidable challenges remain. In addition to the development of KMO inhibitors with good brain penetration and possible clinical utility, our laboratories are most interested in the cellular and molecular mechanisms linking mutant htt to impaired cerebral KP metabolism. Our results from studies in animals and humans suggest that KP abnormalities occur early in the disease process (Guidetti et al., 2006; Guidetti et al., 2004), and experiments in yeast indicate that KP dysfunction constitutes a central common mechanism connecting the mutant gene to cell death (Giorgini et al., 2008) (cf. Section 6.). These findings suggest that microglial KP dysfunction could involve currently unknown, yet druggable, regulatory proteins converging on the KP, and also imply that interventions aimed at reducing flux through the QUIN branch of the KP are likely to be most effective in the early stages of HD. With the appropriate experimental tools and paradigms, these hypotheses are testable and should soon reveal interesting novel insights regarding the cell biology and treatment of HD.
Acknowledgments
We dedicate this paper to the memory of our dear friend Paolo Guidetti, who sadly passed away on December 28, 2007. Parts of the work described here were supported by USPHS grants NS47237, NS57715 and AG22074 and a grant from the HighQ Foundation.
Abbreviations
- 3-HK
3-Hydroxykynurenine
- 3-HAD
3-Hydroxyanthranilic acid dioxygenase
- AMPA
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- AP5
D-Amino-phosphonopentanoic acid
- AP7
D-Amino-phosphonoheptanoic acid
- CNS
Central nervous system
- HD
Huntington’s disease
- htt
Huntingtin
- IDO
Indoleamine-2,3-dioxygenase
- KMO
Kynurenine-3-monooxygenase
- KP
Kynurenine pathway
- KYNA
Kynurenic acid
- NMDA
N-methyl-D-aspartic acid
- QUIN
Quinolinic acid
- QPRT
Quinolinic acid phosphoribosyltransferase
- ROS
Reactive oxygen species
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aarts MM, Tymianski M. Molecular mechanisms underlying specificity of excitotoxic signaling in neurons. Curr Mol Med. 2004;4:137–147. doi: 10.2174/1566524043479202. [DOI] [PubMed] [Google Scholar]
- Alberati-Giani D, Cesura AM, Broger C, Warren WD, Rover S, Malherbe P. Cloning and functional expression of human kynurenine 3-monooxygenase. FEBS Lett. 1997;410:407–412. doi: 10.1016/s0014-5793(97)00627-3. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Yu P, Arruda EZ, Almeida LE, Guidetti P, Fawcett WP, Sapko MT, Randall WR, Schwarcz R, Tagle DA, Albuquerque EX. Targeted deletion of the kynurenine aminotransferase II gene reveals a critical role of endogenous kynurenic acid in the regulation of synaptic transmission via alpha7 nicotinic receptors in the hippocampus. J Neurosci. 2004;24:4635–4648. doi: 10.1523/JNEUROSCI.5631-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amori L, Guidetti P, Pellicciari R, Kajii Y, Schwarcz R. On the relationship between the two branches of the kynurenine pathway in the rat brain in vivo. J Neurochem. doi: 10.1111/j.1471-4159.2009.05893.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre C, O’Connor JC, Kelley KW, Lestage J, Dantzer R, Castanon N. Spatio-temporal differences in the profile of murine brain expression of proinflammatory cytokines and indoleamine 2,3-dioxygenase in response to peripheral lipopolysaccharide administration. J Neuroimmunol. 2008;200:90–99. doi: 10.1016/j.jneuroim.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer FJ, Mancall EL. Cultured fibroblasts in Huntington’s disease I. Effects of L-glutamic acid. Arch Neurol. 1983;40:19–23. doi: 10.1001/archneur.1983.04050010039009. [DOI] [PubMed] [Google Scholar]
- Barbeau A, Chase TN, Paulson GW. Huntington’s Chorea. Raven Press; New York: 1973. [Google Scholar]
- Beal M, Kowall N, Ellison D, Mazurek M, Swartz K, Martin J. Replication of the neurochemical characteristics of Huntington’s disease by quinolinic acid. Nature. 1986;321:168–171. doi: 10.1038/321168a0. [DOI] [PubMed] [Google Scholar]
- Beal MF. Mechanisms of excitotoxicity in neurologic diseases. FASEB J. 1992;6:3338–3344. [PubMed] [Google Scholar]
- Beal MF, Ferrante RJ, Swartz KJ, Kowall NW. Chronic quinolinic acid lesions in rats closely resemble Huntington’s disease. J Neurosci. 1991;11:1649–1659. doi: 10.1523/JNEUROSCI.11-06-01649.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF, Kowall NW, Swartz KJ, Ferrante RJ, Martin JB. Systemic approaches to modifying quinolinic acid striatal lesions in rats. J Neurosci. 1988;8:3901–3908. doi: 10.1523/JNEUROSCI.08-10-03901.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF, Matson WR, Storey E, Milbury P, Ryan EA, Ogawa T, Bird ED. Kynurenic acid concentrations are reduced in Huntington’s disease cerebral cortex. J Neurol Sci. 1992;108:80–87. doi: 10.1016/0022-510x(92)90191-m. [DOI] [PubMed] [Google Scholar]
- Beal MF, Matson WR, Swartz KJ, Gamache PH, Bird ED. Kynurenine pathway measurements in Huntington’s disease striatum: evidence for reduced formation of kynurenic acid. J Neurochem. 1990;55:1327–1339. doi: 10.1111/j.1471-4159.1990.tb03143.x. [DOI] [PubMed] [Google Scholar]
- Belladonna ML, Puccetti P, Orabona C, Fallarino F, Vacca C, Volpi C, Gizzi S, Pallotta MT, Fioretti MC, Grohmann U. Immunosuppression via tryptophan catabolism: the role of kynurenine pathway enzymes. Transplantation. 2007;84:S17–20. doi: 10.1097/01.tp.0000269199.16209.22. [DOI] [PubMed] [Google Scholar]
- Bergman H, Wichmann T, DeLong MR. Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science. 1990;249:1436–1438. doi: 10.1126/science.2402638. [DOI] [PubMed] [Google Scholar]
- Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F. Brain dopamine and the syndromes of Parkinson and Huntington: clinical, morphological and neurochemical correlations. J Neurol Sci. 1973;20:415–455. doi: 10.1016/0022-510x(73)90175-5. [DOI] [PubMed] [Google Scholar]
- Besancon E, Guo S, Lok J, Tymianski M, Lo EH. Beyond NMDA and AMPA glutamate receptors: emerging mechanisms for ionic imbalance and cell death in stroke. Trends Pharmacol Sci. 2008;29:268–275. doi: 10.1016/j.tips.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Bird ED, Iversen LL. Huntington’s chorea. Post-mortem measurement of glutamic acid decarboxylase, choline acetyltransferase and dopamine in basal ganglia. Brain. 1974;97:457–472. doi: 10.1093/brain/97.1.457. [DOI] [PubMed] [Google Scholar]
- Biziere K, Coyle JT. Effects of cortical ablation on the neurotoxicity and receptor binding of kainic acid in striatum. J Neurosci Res. 1979;4:383–398. doi: 10.1002/jnr.490040507. [DOI] [PubMed] [Google Scholar]
- Björklund H, Olson L, Dahl D, Schwarcz R. Short- and long-term consequences of intracranial injections of the excitotoxin, quinolinic acid, as evidenced by GFA immunohistochemistry of astrocytes. Brain Res. 1986;371:267–277. doi: 10.1016/0006-8993(86)90362-8. [DOI] [PubMed] [Google Scholar]
- Bonilla E, Prasad ALN, Arrieta A. Huntington’s disease: studies on brain free amino acids. Life Sci. 1988;42:1153–1158. doi: 10.1016/0024-3205(88)90610-8. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Schwarzenbacher R, Lipton SA. Molecular pathways to neurodegeneration. Nat Med. 2004;10(Suppl):S2–9. doi: 10.1038/nm1067. [DOI] [PubMed] [Google Scholar]
- Bradford SE, Nadler JV. Aspartate release from rat hippocampal synaptosomes. Neuroscience. 2004;128:751–765. doi: 10.1016/j.neuroscience.2004.06.065. [DOI] [PubMed] [Google Scholar]
- Breton J, Avanzi N, Magagnin S, Covini N, Magistrelli G, Cozzi L, Isacchi A. Functional characterization and mechanism of action of recombinant human kynurenine 3-hydroxylase. Eur J Biochem. 2000;267:1092–1099. doi: 10.1046/j.1432-1327.2000.01104.x. [DOI] [PubMed] [Google Scholar]
- Bruno V, Battaglia G, Copani A, D’Onofrio M, Di Iorio P, De Blasi A, Melchiorri D, Flor PJ, Nicoletti F. Metabotropic glutamate receptor subtypes as targets for neuroprotective drugs. J Cereb Blood Flow Metab. 2001;21:1013–1033. doi: 10.1097/00004647-200109000-00001. [DOI] [PubMed] [Google Scholar]
- Bruyn GW. Huntington’s chorea. In: Vinken PJ, Bruyn GW, editors. Handbook of Clinical Neurology. North-Holland: Amsterdam: 1968. pp. 298–377. [Google Scholar]
- Caldeira MV, Melo CV, Pereira DB, Carvalho R, Correia SS, Backos DS, Carvalho AL, Esteban JA, Duarte CB. Brain-derived neurotrophic factor regulates the expression and synaptic delivery of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor subunits in hippocampal neurons. J Biol Chem. 2007;282:12619–12628. doi: 10.1074/jbc.M700607200. [DOI] [PubMed] [Google Scholar]
- Campochiaro P, Coyle JT. Ontogenetic development of kainate neurotoxicity: correlates with glutamatergic innervation. Proc Natl Acad Sci (USA) 1978;75:2025–2029. doi: 10.1073/pnas.75.4.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenedo R, Chiarugi A, Russi P, Lombardi G, Carla V, Pellicciari R, Mattoli L, Moroni F. Inhibitors of kynurenine hydroxylase and kynureninase increase cerebral formation of kynurenate and have sedative and anticonvulsant activities. Neuroscience. 1994;61:237–244. doi: 10.1016/0306-4522(94)90227-5. [DOI] [PubMed] [Google Scholar]
- Carpenedo R, Meli E, Peruginelli F, Pellegrini-Giampietro DE, Moroni F. Kynurenine 3-mono-oxygenase inhibitors attenuate post-ischemic neuronal death in organotypic hippocampal slice cultures. J Neurochem. 2002;82:1465–1471. doi: 10.1046/j.1471-4159.2002.01090.x. [DOI] [PubMed] [Google Scholar]
- Carpenedo R, Pittaluga A, Cozzi A, Attucci S, Galli A, Raiteri M, Moroni F. Presynaptic kynurenate-sensitive receptors inhibit glutamate release. Eur J Neurosci. 2001;13:2141–2147. doi: 10.1046/j.0953-816x.2001.01592.x. [DOI] [PubMed] [Google Scholar]
- Chiarugi A, Cozzi A, Ballerini C, Massacesi L, Moroni F. Kynurenine 3-mono-oxygenase activity and neurotoxic kynurenine metabolites increase in the spinal cord of rats with experimental allergic encephalomyelitis. Neuroscience. 2001;102:687–695. doi: 10.1016/s0306-4522(00)00504-2. [DOI] [PubMed] [Google Scholar]
- Chiarugi A, Meli E, Moroni F. Similarities and differences in the neuronal death processes activated by 3OH-kynurenine and quinolinic acid. J Neurochem. 2001;77:1310–1318. doi: 10.1046/j.1471-4159.2001.00335.x. [DOI] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci Lett. 1985;58:293–297. doi: 10.1016/0304-3940(85)90069-2. [DOI] [PubMed] [Google Scholar]
- Choi DW. NMDA receptors and AMPA/kainate receptors mediate parallel injury in cerebral cortical cultures subjected to oxygen-glucose deprivation. Prog Brain Res. 1993;96:137–143. doi: 10.1016/s0079-6123(08)63263-x. [DOI] [PubMed] [Google Scholar]
- Ciaramitaro VM, Wallace SF, Rosenquist AC. Ibotenic acid lesions of the substantia nigra pars reticulata ipsilateral to a visual cortical lesion fail to restore visual orienting responses in the cat. J Comp Neurol. 1997;377:596–610. [PubMed] [Google Scholar]
- Clark CJ, Mackay GM, Smythe GA, Bustamante S, Stone TW, Phillips RS. Prolonged survival of a murine model of cerebral malaria by kynurenine pathway inhibition. Infect Immun. 2005;73:5249–5251. doi: 10.1128/IAI.73.8.5249-5251.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comings DE, Goetz IE, Holden J, Holtz J. Huntington disease and Tourette syndrome. II Uptake of glutamic acid and other amino acids by fibroblasts. Am J Hum Genet. 1981;33:175–186. [PMC free article] [PubMed] [Google Scholar]
- Connick JH, Carlá V, Moroni F, Stone TW. Increase in kynurenic acid in Huntington’s disease motor cortex. J Neurochem. 1989;52:985–987. doi: 10.1111/j.1471-4159.1989.tb02552.x. [DOI] [PubMed] [Google Scholar]
- Connor TJ, Starr N, O’Sullivan JB, Harkin A. Induction of indolamine 2,3-dioxygenase and kynurenine 3-monooxygenase in rat brain following a systemic inflammatory challenge: a role for IFN-gamma? Neurosci Lett. 2008;441:29–34. doi: 10.1016/j.neulet.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Coyle JT. Glial metabolites of tryptophan and excitotoxicity: coming unglued. Exp Neurol. 2006;197:4–7. doi: 10.1016/j.expneurol.2005.10.021. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Ferkany JW, Zaczek R. Kainic acid: insights from a neurotoxin into the pathophysiology of Huntington’s disease. Neurobehav Toxicol Teratol. 1983;5:617–624. [PubMed] [Google Scholar]
- Coyle JT, Schwarcz R. Lesion of striatal neurones with kainic acid provides a model for Huntington’s chorea. Nature. 1976;263:244–246. doi: 10.1038/263244a0. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Schwarcz R, Bennett JP, Campochiaro P. Clinical, neuropathologic and pharmacologic aspects of Huntington’s disease: correlates with a new animal model. Prog Neuropsychopharmacol. 1977;1:13–30. doi: 10.1016/0364-7722(77)90025-x. [DOI] [PubMed] [Google Scholar]
- Cozzi A, Carpenedo R, Moroni F. Kynurenine hydroxylase inhibitors reduce ischemic brain damage: studies with (m-nitrobenzoyl)-alanine (mNBA) and 3,4-dimethoxy-[-N-4-(nitrophenyl)thiazol-2yl]-benzenesulfonamide (Ro 61–8048) in models of focal or global brain ischemia. J Cereb Blood Flow Metab. 1999;19:771–777. doi: 10.1097/00004647-199907000-00007. [DOI] [PubMed] [Google Scholar]
- Cross AJ, Slater P, Reynolds GP. Reducted high-affinity glutamate uptake sites in the brains of patients with Huntington’s disease. Neurosci Lett. 1982;67:198–202. doi: 10.1016/0304-3940(86)90397-6. [DOI] [PubMed] [Google Scholar]
- Croucher MJ, Collins JF, Meldrum BS. Anticonvulsant action of excitatory amino acid antagonists. Science. 1982;216:899–901. doi: 10.1126/science.7079744. [DOI] [PubMed] [Google Scholar]
- Curtis DR, Duggan AW, Felix D, Johnston GAR, Tebecis AK, Watkins JC. Excitation of mammalian central neurones by acidic amino acids. Brain Res. 1972;41:283–301. doi: 10.1016/0006-8993(72)90503-3. [DOI] [PubMed] [Google Scholar]
- Curtis DR, Watkins JC. The excitation and depression of spinal neurones by structurally related amino acids. J Neurochem. 1960;6:117–141. doi: 10.1111/j.1471-4159.1960.tb13458.x. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Davies J, Evans RH, Francis AA, Watkins JC. Excitatory amino acid receptors and synaptic excitation in the mammalian central nervous system. J Physiol. 1979;75:641–654. [PubMed] [Google Scholar]
- Davies SW, Roberts PJ. Sparing of cholinergic neurons following quinolinic acid lesions of the rat striatum. Neuroscience. 1988;26:387–393. doi: 10.1016/0306-4522(88)90156-x. [DOI] [PubMed] [Google Scholar]
- De Carvalho LP, Bochet P, Rossier J. The endogenous agonist quinolinic acid and the non endogenous homoquinolinic acid discriminate between NMDAR2 receptor subunit. Neurochem Int. 1996;28:445–452. doi: 10.1016/0197-0186(95)00091-7. [DOI] [PubMed] [Google Scholar]
- Decker RH, Brown RR, Price JM. Studies on the biological activity of nicotinylalanine, an analogue of kynurenine. J Biol Chem. 1963;238:1049–1053. [PubMed] [Google Scholar]
- DiFiglia M. Excitotoxic injury of the neostriatum: a model for Huntington’s disease. Trends Neurosci. 1990;13:286–289. doi: 10.1016/0166-2236(90)90111-m. [DOI] [PubMed] [Google Scholar]
- Divac I, Markowitsch HJ, Pritzel M. Behavioral and anatomical consequences of small intrastriatal injections of kainic acid in the rat. Brain Res. 1978;151:523–532. doi: 10.1016/0006-8993(78)91084-3. [DOI] [PubMed] [Google Scholar]
- During MJ, Freese A, Heyes MP, Swartz KJ, Markey SP, Roth RH, Martin JB. Neuroactive metabolites of L-tryptophan, serotonin and quinolinic acid, in striatal extracellular fluid. FEBS Lett. 1989;247:438–444. doi: 10.1016/0014-5793(89)81387-0. [DOI] [PubMed] [Google Scholar]
- Eastman CL, Guilarte TR. Cytotoxicity of 3-hydroxykynurenine in a neuronal hybrid cell line. Brain Res. 1989;495:225–231. doi: 10.1016/0006-8993(89)90216-3. [DOI] [PubMed] [Google Scholar]
- Eastman CL, Guilarte TR. The role of hydrogen peroxide in the in vitro cytotoxicity of 3-hydroxykynurenine. Neurochem Res. 1990;15:1101–1107. doi: 10.1007/BF01101711. [DOI] [PubMed] [Google Scholar]
- Ellison DW, Beal MF, Mazurek MF, Malloy JR, Bird ED, Martin JB. Amino acid neurotransmitter abnormalities in Huntington’s disease and the quinolinic acid animal model of Huntington’s disease. Brain. 1987;110:1657–1673. doi: 10.1093/brain/110.6.1657. [DOI] [PubMed] [Google Scholar]
- Erhardt S, Oberg H, Engberg G. Pharmacologically elevated levels of endogenous kynurenic acid prevent nicotine-induced activation of nigral dopamine neurons. Naunyn Schmiedeberg’s Arch Pharmacol. 2001;363:21–27. doi: 10.1007/s002100000325. [DOI] [PubMed] [Google Scholar]
- Erickson JB, Flanagan EM, Russo S, Reinhard JF., Jr A radiometric assay for kynurenine 3-hydroxylase based on the release of 3H2O hydroxylation of L-[3,5–3H]kynurenine. Anal Biochem. 1992;205:257–262. doi: 10.1016/0003-2697(92)90432-7. [DOI] [PubMed] [Google Scholar]
- Eugster CH. Active substances from the toadstool. Naturwissenschaften. 1968;55:305–313. doi: 10.1007/BF00600445. [DOI] [PubMed] [Google Scholar]
- Fagni L, Lafon-Cazal M, Rondouin G, Manzoni O, Lerner-Natoli M, Bockaert J. The role of free radicals in NMDA-dependent neurotoxicity. Prog Brain Res. 1994;103:381–390. doi: 10.1016/s0079-6123(08)61152-8. [DOI] [PubMed] [Google Scholar]
- Fan MM, Raymond LA. N-methyl-D-aspartate (NMDA) receptor function and excitotoxicity in Huntington’s disease. Prog Neurobiol. 2007;81:272–293. doi: 10.1016/j.pneurobio.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Ferkany JW, Zaczek R, Coyle JT. Kainic acid stimulates excitatory amino acid neurotransmitter release at presynaptic receptors. Nature. 1982;298:757–759. doi: 10.1038/298757a0. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Beal MF, Kowall NW, Richardson EP, Martin JB. Sparing of acetylcholinesterase-containing striatal neurons in Huntington’s disease. Brain Res. 1987a;411:162–166. doi: 10.1016/0006-8993(87)90694-9. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW. Tyrosine hydroxylase-like immunoreactivity is distributed in the matrix compartment of normal human and Huntington’s disease striatum. Brain Res. 1987;416:141–146. doi: 10.1016/0006-8993(87)91506-x. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW, Beal MF, Martin JB, Bird ED, Richardson EPJ. Morphologic and histochemical characteristics of a spared subset of striatal neurons in Huntington’s disease. J Neuropathol Exp Neurol. 1987b;46:12–27. doi: 10.1097/00005072-198701000-00002. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW, Cipolloni PB, Storey E, Beal MF. Excitotoxin lesions in primates as a model for Huntington’s disease: histopathologic and neurochemical characterization. Exp Neurol. 1993;119:46–71. doi: 10.1006/exnr.1993.1006. [DOI] [PubMed] [Google Scholar]
- Flicker C, Dean RL, Watkins DL, Fisher SK, Bartus RT. Behavioral and neurochemical effects following neurotoxic lesions of a major cholinergic input to the cerebral cortex in the rat. Pharmacol Biochem Behav. 1983;18:973–981. doi: 10.1016/s0091-3057(83)80023-9. [DOI] [PubMed] [Google Scholar]
- Foster AC, Collins JF, Schwarcz R. On the excitotoxic properties of quinolinic acid, 2,3-piperidine dicarboxylic acids and structurally related compounds. Neuropharmacology. 1983;22:1331–1342. doi: 10.1016/0028-3908(83)90221-6. [DOI] [PubMed] [Google Scholar]
- Foster AC, Schwarcz R. Characterization of quinolinic acid phosphoribosyltransferase in human blood and observations in Huntington’s disease. J Neurochem. 1985;45:199–205. doi: 10.1111/j.1471-4159.1985.tb05493.x. [DOI] [PubMed] [Google Scholar]
- Foster AC, Vezzani A, French ED, Schwarcz R. Kynurenic acid blocks neurotoxicity and seizures induced in rats by the related brain metabolite quinolinic acid. Neurosci Lett. 1984;48:273–278. doi: 10.1016/0304-3940(84)90050-8. [DOI] [PubMed] [Google Scholar]
- Foster AC, Whetsell WO, Jr, Bird ED, Schwarcz R. Quinolinic acid phosphoribosyltransferase in human and rat brain: activity in Huntington’s disease and in quinolinate-lesioned rat striatum. Brain Res. 1985b;336:207–214. doi: 10.1016/0006-8993(85)90647-x. [DOI] [PubMed] [Google Scholar]
- Foster AC, White RJ, Schwarcz R. Synthesis of quinolinic acid by 3-hydroxyanthranilic acid oxygenase in rat brain tissue in vitro. J Neurochem. 1986;47:23–30. doi: 10.1111/j.1471-4159.1986.tb02826.x. [DOI] [PubMed] [Google Scholar]
- Foster AC, Zinkand WC, Schwarcz R. Quinolinic acid phosphoribosyltransferase in rat brain. J Neurochem. 1985a;44:446–454. doi: 10.1111/j.1471-4159.1985.tb05435.x. [DOI] [PubMed] [Google Scholar]
- Fukui S, Schwarcz R, Rapoport SI, Takada Y, Smith QR. Blood-brain barrier transport of kynurenines: implications for brain synthesis and metabolism. J Neurochem. 1991;56:2007–2017. doi: 10.1111/j.1471-4159.1991.tb03460.x. [DOI] [PubMed] [Google Scholar]
- Gál EM, Sherman AD. L-Kynurenine: its synthesis and possible regulatory function in brain. Neurochem Res. 1980;5:223–239. doi: 10.1007/BF00964611. [DOI] [PubMed] [Google Scholar]
- Garthwaite G, Hajos F, Garthwaite J. Ionic requirements for neurotoxic effects of excitatory amino acid analogues in rat cerebellar slices. Neuroscience. 1986;18:437–447. doi: 10.1016/0306-4522(86)90164-8. [DOI] [PubMed] [Google Scholar]
- Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: intrinsic immuneffector cell of the brain. Brain Res Bull. 1995;20:269–287. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- Giorgini F. The kynurenine pathway and microglia: Implications for pathology and therapy in Huntington’s disease. In: Outeiro T, editor. Protein Misfolding in Biology and Disease. Transworld Research Network; Kerala, India: 2008. pp. 231–255. [Google Scholar]
- Giorgini F, Guidetti P, Nguyen Q, Bennett SC, Muchowski PJ. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat Gen. 2005;37:526–531. doi: 10.1038/ng1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgini F, Möller T, Kwan W, Zwilling D, Wacker JL, Hong S, Tsai LC, Cheah CS, Schwarcz R, Guidetti P, Muchowski PJ. Histone deacetylase inhibition modulates kynurenine pathway activation in yeast, microglia, and mice expressing a mutant huntingtin fragment. J Biol Chem. 2008;283:7390–7400. doi: 10.1074/jbc.M708192200. [DOI] [PubMed] [Google Scholar]
- Goda K, Hamane Y, Kishimoto R, Ogishi Y. Radical scavenging properties of tryptophan metabolites. Estimation of their radical reactivity. Adv Exp Med Biol. 1999;467:397–402. doi: 10.1007/978-1-4615-4709-9_50. [DOI] [PubMed] [Google Scholar]
- Goldstein LE, Leopold MC, Huang X, Atwood CS, Saunders AJ, Hartshorn M, Lim JT, Faget KY, Muffat JA, Scarpa RC, Chylack LTJ, Bowden EF, Tanzi RE, Bush AI. 3-Hydroxykynurenine and 3-hydroxyanthranilic acid generate hydrogen peroxide and promote α-crystallin cross-linking by metal ion reduction. Biochemistry. 2000;39:7266–7275. doi: 10.1021/bi992997s. [DOI] [PubMed] [Google Scholar]
- Gonzalez A, Varo N, Alegre E, Diaz A, Melero I. Immunosuppression routed via the kynurenine pathway: a biochemical and pathophysiologic approach. Adv Clin Chem. 2008;45:155–197. doi: 10.1016/s0065-2423(07)00007-8. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT, Penney JB, Young AB, D’Amato CJ, Hicks SP, Shoulson I. Alterations in L-glutamate binding in Alzheimer’s and Huntington’s diseases. Science. 1985;227:1496–1499. doi: 10.1126/science.2858129. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT, Young AB, Penney JB. Quantitative autoradiographic distribution of L-[3H]glutamate-binding sites in rat central nervous system. J Neurosci. 1984;4:2133–2144. doi: 10.1523/JNEUROSCI.04-08-02133.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene JG, Greenamyre JT. Bioenergetics and glutamate excitotoxicity. Prog Neurobiol. 1996;48:613–634. doi: 10.1016/0301-0082(96)00006-8. [DOI] [PubMed] [Google Scholar]
- Guidetti P, Bates GP, Graham RK, Hayden MR, Leavitt BR, MacDonald ME, Slow EJ, Wheeler VC, Woodman B, Schwarcz R. Elevated brain 3-hydroxykynurenine and quinolinate levels in Huntington disease mice. Neurobiol Dis. 2006;23:190–197. doi: 10.1016/j.nbd.2006.02.011. [DOI] [PubMed] [Google Scholar]
- Guidetti P, Eastman CL, Schwarcz R. Metabolism of [5–3H]kynurenine in the rat brain in vivo: evidence for the existence of a functional kynurenine pathway. J Neurochem. 1995;65:2621–2632. doi: 10.1046/j.1471-4159.1995.65062621.x. [DOI] [PubMed] [Google Scholar]
- Guidetti P, Luthi-Carter RE, Augood SJ, Schwarcz R. Neostriatal and cortical quinolinate levels are increased in early grade Huntington’s disease. Neurobiol Dis. 2004;17:455–461. doi: 10.1016/j.nbd.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Guidetti P, Reddy PH, Tagle DA, Schwarcz R. Early kynurenergic impairment in Huntington’s disease and in a transgenic animal model. Neurosci Lett. 2000;283:233–235. doi: 10.1016/s0304-3940(00)00956-3. [DOI] [PubMed] [Google Scholar]
- Guidetti P, Schwarcz R. 3-Hydroxykynurenine potentiates quinolinate but not NMDA toxicity in the rat striatum. Eur J Neurosci. 1999;11:3857–3863. doi: 10.1046/j.1460-9568.1999.00806.x. [DOI] [PubMed] [Google Scholar]
- Guillemin GJ, Kerr SJ, Smythe GA, Smith DG, Kapoor V, Armati PJ, Croitoru J, Brew BJ. Kynurenine pathway metabolism in human astrocytes: a paradox for neuronal protection. J Neurochem. 2001;78:842–853. doi: 10.1046/j.1471-4159.2001.00498.x. [DOI] [PubMed] [Google Scholar]
- Hall JG, Hicks TP, McLennan H, Richardson TL, Wheal HV. The excitation of mammalian central neurones by amino acids. J Physiol. 1979;286:29–39. doi: 10.1113/jphysiol.1979.sp012605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann M, Sander SE, Richter A. Effects of the kynurenine 3-hydroxylase inhibitor Ro 61–8048 after intrastriatal injections on the severity of dystonia in the dt sz mutant. Eur J Pharmacol. 2008;586:156–159. doi: 10.1016/j.ejphar.2008.02.052. [DOI] [PubMed] [Google Scholar]
- Harris C, Miranda A, Tanguay J, Boegman R, Beninger R, Jhamandas K. Modulation of striatal quinolinate neurotoxicity by elevation of endogenous brain kynurenic acid. Brit J Pharmacol. 1998;124:391–399. doi: 10.1038/sj.bjp.0701834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassel B, Tessler S, Faull RL, Emson PC. Glutamate uptake is reduced in prefrontal cortex in Huntington’s disease. Neurochem Res. 2008;33:232–237. doi: 10.1007/s11064-007-9463-1. [DOI] [PubMed] [Google Scholar]
- Heyes MP, Achim CL, Wiley CA, Major EO, Saito K, Markey SP. Human microglia convert l-tryptophan into the neurotoxin quinolinic acid. Biochem J. 1996;320:595–597. doi: 10.1042/bj3200595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyes MP, Chen CY, Major EO, Saito K. Different kynurenine pathway enzymes limit quinolinic acid formation by various human cell types. Biochem J. 1997;326:351–356. doi: 10.1042/bj3260351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyes MP, Garnett ES, Brown RR. Normal excretion of quinolinic acid in Huntington’s disease. Life Sci. 1985;37:1811–1816. doi: 10.1016/0024-3205(85)90223-1. [DOI] [PubMed] [Google Scholar]
- Heyes MP, Rubinow D, Lane C, Markey SP. Cerebrospinal fluid quinolinic acid concentrations are increased in acquired immune deficiency syndrome. Ann Neurol. 1989;26:275–277. doi: 10.1002/ana.410260215. [DOI] [PubMed] [Google Scholar]
- Heyes MP, Saito K, Jacobowitz D, Markey SP, Takikawa O, Vickers JH. Poliovirus induces indoleamine-2,3-dioxygenase and quinolinic acid synthesis in macaque brain. FASEB J. 1992;6:2977–2989. doi: 10.1096/fasebj.6.11.1322853. [DOI] [PubMed] [Google Scholar]
- Heyes MP, Saito K, Major EO, Milstien S, Markey SP, Vickers JH. A mechanism of quinolinic acid formation by brain in inflammatory neurological disease. Brain. 1993;116:1425–1450. doi: 10.1093/brain/116.6.1425. [DOI] [PubMed] [Google Scholar]
- Heyes MP, Swartz KJ, Markey SP, Beal MF. Regional brain and cerebrospinal fluid quinolinic acid concentrations in Huntington’s disease. Neurosci Lett. 1991;122:265–269. doi: 10.1016/0304-3940(91)90874-s. [DOI] [PubMed] [Google Scholar]
- Hilmas C, Pereira EF, Alkondon M, Rassoulpour A, Schwarcz R, Albuquerque EX. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: physiopathological implications. J Neurosci. 2001;21:7463–7473. doi: 10.1523/JNEUROSCI.21-19-07463.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honoré T, Davies SN, Drejer J, Fletcher EJ, Jacobsen P, Lodge D, Nielsen FE. Quinoxalinediones: potent competitive non-NMDA glutamate receptor antagonists. Science. 1988;241:701–703. doi: 10.1126/science.2899909. [DOI] [PubMed] [Google Scholar]
- Honoré T, Lauridsen J, Krogsgaard-Larsen P. The binding of [3H]AMPA, a structural analogue of glutamic acid, to rat brain membranes. J Neurochem. 1982;38:173–178. doi: 10.1111/j.1471-4159.1982.tb10868.x. [DOI] [PubMed] [Google Scholar]
- Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Huntington G. On chorea. Medical and Surgical Report. 1872;26:317–321. [Google Scholar]
- Isacson O, Brundin P, Kelly PA, Gage FH, Björklund A. Functional neuronal replacement by grafted striatal neurones in the ibotenic acid-lesioned rat striatum. Nature. 1984;311:458–460. doi: 10.1038/311458a0. [DOI] [PubMed] [Google Scholar]
- Iwata H, Yamagami S, Mizuo H, Baba A. Cysteine sulfinic acid in the central nervous system: uptake and release of cysteine sulfinic acid by a rat brain preparation. J Neurochem. 1982;38:1268–1274. doi: 10.1111/j.1471-4159.1982.tb07900.x. [DOI] [PubMed] [Google Scholar]
- Jackson M, Gentleman S, Lennox G, Ward L, Gray T, Randall K, Morrell K, Lowe J. The cortical neuritic pathology of Huntington’s disease. Neuropathol Appl Neurobiol. 1995;21:18–26. doi: 10.1111/j.1365-2990.1995.tb01024.x. [DOI] [PubMed] [Google Scholar]
- Johnston MV. Excitotoxicity in perinatal brain injury. Brain Pathol. 2005;15:234–240. doi: 10.1111/j.1750-3639.2005.tb00526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas P, Spruston N. Mechanisms shaping glutamate-mediated excitatory postsynaptic currents in the CNS. Curr Opin Neurobiol. 1994;4:366–372. doi: 10.1016/0959-4388(94)90098-1. [DOI] [PubMed] [Google Scholar]
- Kalia LV, Kalia SK, Salter MW. NMDA receptors in clinical neurology: excitatory times ahead. Lancet Neurol. 2008;7:742–755. doi: 10.1016/S1474-4422(08)70165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler M, Terramani T, Lynch G, Baudry M. A glycine site associated with N-methyl-D-aspartic acid receptors: characterization and identification of a new class of antagonists. J Neurochem. 1989;52:1319–1328. doi: 10.1111/j.1471-4159.1989.tb01881.x. [DOI] [PubMed] [Google Scholar]
- Kido R, Noguchi T. Kynurenine pyruvate transaminase and its specific inhibitor in rat intestine. Acta Vitam Enzyol. 1975;29:310–312. [PubMed] [Google Scholar]
- Kim JS, Kornhuber HH, Holzmüller B, Schmid-Burgk W, Mergner T, Krzepinski G. Reduction of cerebrospinal fluid glutamic acid in Huntington’s chorea and in schizophrenic patients. Arch Psych Nervenkr. 1980;228:7–10. doi: 10.1007/BF00365738. [DOI] [PubMed] [Google Scholar]
- Kiss C, Ceresoli-Borroni G, Guidetti P, Zielke CL, Zielke HR, Schwarcz R. Kynurenate production by cultured human astrocytes. J Neural Transm. 2003;110:1–14. doi: 10.1007/s00702-002-0770-z. [DOI] [PubMed] [Google Scholar]
- Kita T, Morrison PF, Heyes MP, Markey SP. Effects of systemic and central nervous system localized inflammation on the contributions of metabolic precursors to the L-kynurenine and quinolinic acid pools in brain. J Neurochem. 2002;82:258–268. doi: 10.1046/j.1471-4159.2002.00955.x. [DOI] [PubMed] [Google Scholar]
- Klancnik JM, Cuenod M, Gahwiler BH, Jiang ZP, Do KQ. Release of endogenous amino acids, including homocysteic acid and cysteine sulphinic acid, from rat hippocampal slices evoked by electrical stimulation of schaffer collateral-commissural fibres. Neuroscience. 1992;49:557–570. doi: 10.1016/0306-4522(92)90226-r. [DOI] [PubMed] [Google Scholar]
- Köhler C, Schwarcz R, Fuxe K. Intrahippocampal injections of ibotenic acid provide histological evidence for a neurotoxic mechanism different from kainic acid. Neurosci Lett. 1979;15:223–228. doi: 10.1016/0304-3940(79)96117-2. [DOI] [PubMed] [Google Scholar]
- Lapin IP. Stimulant and convulsive effects of kynurenines injected into brain ventricles in mice. J Neural Transm. 1978;42:37–43. doi: 10.1007/BF01262727. [DOI] [PubMed] [Google Scholar]
- Lehrmann E, Molinari A, Speciale C, Schwarcz R. Immunohistochemical visualization of newly formed quinolinate in the normal and excitotoxically lesioned rat striatum. Exp Brain Res. 2001;141:389–397. doi: 10.1007/s002210100887. [DOI] [PubMed] [Google Scholar]
- Leipnitz G, Schumacher C, Dalcin KB, Scussiato K, Solano A, Funchal C, Dutra-Filho CS, Wyse AT, Wannmacher CM, Latini A, Wajner M. In vitro evidence for an antioxidant role of 3-hydroxykynurenine and 3-hydroxyanthranilic acid in the brain. Neurochem Int. 2007;50:83–94. doi: 10.1016/j.neuint.2006.04.017. [DOI] [PubMed] [Google Scholar]
- Leist M, Fava E, Montecucco C, Nicotera P. Peroxynitrite and nitric oxide donors induce neuronal apoptosis by eliciting autocrine excitotoxicity. Eur J Neurosci. 1997;9:1488–1498. doi: 10.1111/j.1460-9568.1997.tb01503.x. [DOI] [PubMed] [Google Scholar]
- Lima S, Kumar S, Gawandi V, Momany C, Phillips RS. Crystal structure of the Homo sapiens kynureninase-3-hydroxyhippuric acid inhibitor complex: insights into the molecular basis of kynureninase substrate specificity. J Med Chem. 2009;52:389–396. doi: 10.1021/jm8010806. [DOI] [PubMed] [Google Scholar]
- Lopez AS, Alegre E, LeMaoult J, Carosella E, Gonzalez A. Regulatory role of tryptophan degradation pathway in HLA-G expression by human monocyte-derived dendritic cells. Mol Immunol. 2006;43:2151–2160. doi: 10.1016/j.molimm.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Ma L, Morton AJ, Nicholson LF. Microglia density decreases with age in a mouse model of Huntington’s disease. Glia. 2003;43:274–280. doi: 10.1002/glia.10261. [DOI] [PubMed] [Google Scholar]
- Mangano RM, Schwarcz R. Platelet glutamate and aspartate uptake in Huntington’s disease. J Neurochem. 1981;37:1072–1074. doi: 10.1111/j.1471-4159.1981.tb04502.x. [DOI] [PubMed] [Google Scholar]
- Mangano RM, Schwarcz R. Huntington’s disease. Glutamate and aspartate metabolism in blood platelets. J Neurol Sci. 1982;53:489–500. doi: 10.1016/0022-510x(82)90245-3. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Mason ST, Sanberg PR, Fibiger HC. Amphetamine-induced locomotor activity and stereotypy after kainic acid lesions of the striatum. Life Sci. 1978;22:451–460. doi: 10.1016/0024-3205(78)90424-1. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Calcium and neurodegeneration. Aging Cell. 2007;6:337–350. doi: 10.1111/j.1474-9726.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- May PC, Gray PN. L-Homocysteic acid as an alternative cytotoxin for studying glutamate-induced cellular degeneration of Huntington’s disease and normal skin fibroblasts. Life Sci. 1985;37:1483–1489. doi: 10.1016/0024-3205(85)90179-1. [DOI] [PubMed] [Google Scholar]
- McBean GJ, Roberts PJ. Neurotoxicity of L-glutamate and DL-threo-3-hydroxyaspartate in the rat striatum. J Neurochem. 1985;44:247–254. doi: 10.1111/j.1471-4159.1985.tb07137.x. [DOI] [PubMed] [Google Scholar]
- McDonald JW, Silverstein FS, Johnston M., V Neurotoxicity of N-methyl-D-aspartate is markedly enhanced in developing rat central nervous system. Brain Res. 1988;459:200–203. doi: 10.1016/0006-8993(88)90306-x. [DOI] [PubMed] [Google Scholar]
- McGeer EG, McGeer PL. Duplication of biochemical changes of Huntington’s chorea by intrastriatal injections of glutamic and kainic acids. Nature. 1976;263:517–519. doi: 10.1038/263517a0. [DOI] [PubMed] [Google Scholar]
- McGeer EG, McGeer PL, Singh K. Kainate-induced degeneration of neostriatal neurons: dependency upon corticostriatal tract. Brain Res. 1978;139:381–383. doi: 10.1016/0006-8993(78)90941-1. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG, Fibiger HC. Choline acetylase and glutamic acid decarboxylase in Huntington’s chorea. A preliminary study. Neurology. 1973;23:912–917. doi: 10.1212/wnl.23.9.912. [DOI] [PubMed] [Google Scholar]
- Minghetti L, Greco A, Potenza RL, Pezzola A, Blum D, Bantubungi K, Popoli P. Effects of the adenosine A2A receptor antagonist SCH 58621 on cyclooxygenase-2 expression, glial activation, and brain-derived neurotrophic factor availability in a rat model of striatal neurodegeneration. J Neuropathol Exp Neurol. 2007;66:363–371. doi: 10.1097/nen.0b013e3180517477. [DOI] [PubMed] [Google Scholar]
- Miranda AF, Boegman RJ, Beninger RJ, Jhamandas K. Protection against quinolinic acid-mediated excitotoxicity in nigrostriatal dopaminergic neurons by endogenous kynurenic acid. Neuroscience. 1997;78:967–975. doi: 10.1016/s0306-4522(96)00655-0. [DOI] [PubMed] [Google Scholar]
- Monaghan DT, Beaton JA. Quinolinate differentiates between forebrain and cerebellar NMDA receptors. Eur J Pharmacol. 1991;194:123–125. doi: 10.1016/0014-2999(91)90134-c. [DOI] [PubMed] [Google Scholar]
- Monaghan DT, Holets VR, Toy DW, Cotman CW. Anatomical distributions of four pharmacologically distinct 3H-L-glutamate binding sites. Nature. 1983;306:176–179. doi: 10.1038/306176a0. [DOI] [PubMed] [Google Scholar]
- Moroni F. Tryptophan metabolism and brain function: focus on kynurenine and other indole metabolites. Eur J Pharmacol. 1999;375:87–100. doi: 10.1016/s0014-2999(99)00196-x. [DOI] [PubMed] [Google Scholar]
- Moroni F, Russi P, Gallo-Mezo MA, Moneti G, Pellicciari R. Modulation of quinolinic and kynurenic acid content in the rat brain: effects of endotoxins and nicotinylalanine. J Neurochem. 1991;57:1630–1635. doi: 10.1111/j.1471-4159.1991.tb06361.x. [DOI] [PubMed] [Google Scholar]
- Müller AC, Dairam A, Limson JL, Daya S. Mechanisms by which acyclovir reduces the oxidative neurotoxicity and biosynthesis of quinolinic acid. Life Sci. 2007;80:918–925. doi: 10.1016/j.lfs.2006.11.031. [DOI] [PubMed] [Google Scholar]
- Müller N, Schwarz MJ. The immune-mediated alteration of serotonin and glutamate: towards an integrated view of depression. Mol Psych. 2007;12:988–1000. doi: 10.1038/sj.mp.4002006. [DOI] [PubMed] [Google Scholar]
- Myers RH, Vonsattel JP, Paskevich PA, Kiely DK, Stevens TJ, Cupples LA, Richardson EP, Jr, Bird ED. Decreased neuronal and increased oligodendroglial densities in Huntington’s disease caudate nucleus. J Neuropathol Exp Neurol. 1991;50:729–742. doi: 10.1097/00005072-199111000-00005. [DOI] [PubMed] [Google Scholar]
- Nadler JV, Evenson DA, Cuthbertson GJ. Comparative toxicity of kainic acid and other acidic amino acids toward rat hippocampal neurons. Neuroscience. 1981;6:2505–2517. doi: 10.1016/0306-4522(81)90096-8. [DOI] [PubMed] [Google Scholar]
- Nakagami Y, Saito H, Katsuki H. 3-Hydroxykynurenine toxicity on the rat striatum in vivo. Jpn J Pharmacol. 1996;71:183–186. doi: 10.1254/jjp.71.183. [DOI] [PubMed] [Google Scholar]
- Nakao N, Grasbon-Frodl EM, Widner H, Brundin P. Antioxidant treatment protects striatal neurons against excitotoxic insults. Neuroscience. 1996;73:185–200. doi: 10.1016/0306-4522(96)00034-6. [DOI] [PubMed] [Google Scholar]
- Németh H, Toldi J, Vecsei L. Role of kynurenines in the central and peripheral nervous systems. Curr Neurovasc Res. 2005;2:249–260. doi: 10.2174/1567202054368326. [DOI] [PubMed] [Google Scholar]
- Németh H, Toldi J, Vecsei L. Kynurenines, Parkinson’s disease and other neurodegenerative disorders: preclinical and clinical studies. J Neural Transm. 2006;70:285–304. doi: 10.1007/978-3-211-45295-0_45. [DOI] [PubMed] [Google Scholar]
- Nicotera P, Lipton SA. Excitotoxins in neuronal apoptosis and necrosis. J Cereb Blood Flow Metab. 1999;19:583–591. doi: 10.1097/00004647-199906000-00001. [DOI] [PubMed] [Google Scholar]
- O’Brien RJ, Lau LF, Huganir RL. Molecular mechanisms of glutamate receptor clustering at excitatory synapses. Curr Opin Neurobiol. 1998;8:364–369. doi: 10.1016/s0959-4388(98)80062-7. [DOI] [PubMed] [Google Scholar]
- O’Connor JC, Lawson MA, Andre C, Moreau M, Lestage J, Castanon N, Kelley KW, Dantzer R. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol Psych. 2008 doi: 10.1038/sj.mp.4002148. epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto H, Hayaishi O. Solubilization and partial purification of kynurenine hydroxylase from mitochondrial outer membrane and Its electron donors. Arch Biochem Biophys. 1969;131:603–608. doi: 10.1016/0003-9861(69)90435-4. [DOI] [PubMed] [Google Scholar]
- Okuda S, Nishiyama N, Saito H, Katsuki H. Hydrogen peroxide-mediated neuronal cell death induced by an endogenous neurotoxin, 3-hydroxykynurenine. Proc Natl Acad Sci (USA) 1996;93:12553–12558. doi: 10.1073/pnas.93.22.12553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda S, Nishiyama N, Saito H, Katsuki H. 3-Hydroxykynurenine, an endogenous oxidative stress generator, causes neuronal cell death with apoptotic features and region selectivity. J Neurochem. 1998;70:299–307. doi: 10.1046/j.1471-4159.1998.70010299.x. [DOI] [PubMed] [Google Scholar]
- Olney JW. Toxic effects of glutamate and related amino acids on the developing central nervous system. In: Nyhan WH, editor. Heritable Disorders of Amino Acid Metabolism. John Wiley; New York: 1974. pp. 501–512. [Google Scholar]
- Olney JW, de Gubareff T. Extreme sensitivity of olfactory cortical neurons to kainic acid toxicity. In: McGeer EG, WOlney J, McGeer PL, editors. Kainic Acid as a Tool in Neurobiology. Raven Press; New York: 1978a. pp. 201–217. [Google Scholar]
- Olney JW, de Gubareff T. Glutamate neurotoxicity and Huntington’s chorea. Nature. 1978b;271:557–559. doi: 10.1038/271557a0. [DOI] [PubMed] [Google Scholar]
- Olney JW, de Gubareff T, Labruyere J. alpha-Aminoadipate blocks the neurotoxic action of N-methyl aspartate. Life Sci. 1979b;25:537–540. doi: 10.1016/0024-3205(79)90567-8. [DOI] [PubMed] [Google Scholar]
- Olney JW, Fuller T, de Gubareff T. Acute dendrotoxic changes in the hippocampus of kainate treated rats. Brain Res. 1979a;176:91–100. doi: 10.1016/0006-8993(79)90872-2. [DOI] [PubMed] [Google Scholar]
- Olney JW, Ho OL, Rhee V. Cytotoxic effects of acidic and sulphur containing amino acids on the infant mouse central nervous system. Exp Brain Res. 1971;14:61–76. doi: 10.1007/BF00234911. [DOI] [PubMed] [Google Scholar]
- Olney JW, Labruyere J, Collins JF, Curry K. D-Aminaphosphonovalerate is 100-fold more powerful than D-alpha-aminoadipate in blocking N-methylaspartate neurotoxicity. Brain Res. 1981;221:207–210. doi: 10.1016/0006-8993(81)91076-3. [DOI] [PubMed] [Google Scholar]
- Olney JW, Misra CH, de Gubareff T. Cysteine-S-sulfate: brain damaging metabolite in sulfite oxidase deficiency. J Neuropathol Exp Neurol. 1975;34:167–177. doi: 10.1097/00005072-197503000-00005. [DOI] [PubMed] [Google Scholar]
- Olney JW, Price MT, Salles KS, Labruyere J, Ryerson R, Mahan K, Frierdich G, Samson L. L-homocysteic acid: an endogenous excitotoxic ligand of the NMDA receptor. Brain Res Bull. 1987;19:597–602. doi: 10.1016/0361-9230(87)90077-3. [DOI] [PubMed] [Google Scholar]
- Olney JW, Rhee V, Ho OL. Kainic acid: a powerful neurotoxic analogue of glutamate. Brain Res. 1974;77:507–512. doi: 10.1016/0006-8993(74)90640-4. [DOI] [PubMed] [Google Scholar]
- Olney JW, Sharpe LG. Brain lesions in an infant rhesus monkey treated with monsodium glutamate. Science. 1969;166:386–388. doi: 10.1126/science.166.3903.386. [DOI] [PubMed] [Google Scholar]
- Oxenkrug GF. Genetic and hormonal regulation of tryptophan kynurenine metabolism: implications for vascular cognitive impairment, major depressive disorder, and aging. Ann N Y Acad Sci. 2007;1122:35–49. doi: 10.1196/annals.1403.003. [DOI] [PubMed] [Google Scholar]
- Pearson SJ, Meldrum A, Reynolds GP. An investigation of the activities of 3-hydroxykynureninase and kynurenine aminotransferase in the brain of Huntington’s disease. J Neural Transm. 1995;102:67–73. doi: 10.1007/BF01276566. [DOI] [PubMed] [Google Scholar]
- Pearson SJ, Reynolds GP. Determination of 3-hydroxykynurenine in human brain and plasma by high-performance liquid chromatography with electrochemical detection. J Chromatogr. 1991;565:436–440. doi: 10.1016/0378-4347(91)80406-3. [DOI] [PubMed] [Google Scholar]
- Pearson SJ, Reynolds GP. Increased brain concentrations of a neurotoxin, 3-hydroxykynurenine, in Huntington’s disease. Neurosci Lett. 1992;144:199–201. doi: 10.1016/0304-3940(92)90749-w. [DOI] [PubMed] [Google Scholar]
- Pellegrini-Giampietro DE, Pulsinelli WA, Zukin RS. NMDA and non-NMDA receptor gene expression following global brain ischemia in rats: effect of NMDA and non-NMDA receptor antagonists. J Neurochem. 1994;62:1067–1073. doi: 10.1046/j.1471-4159.1994.62031067.x. [DOI] [PubMed] [Google Scholar]
- Pellicciari R, Natalini B, Costantino G, Mahmoud MR, Mattoli L, Sadeghpour BM, Moroni F, Chiarugi A, Carpenedo R. Modulation of the kynurenine pathway in search for new neuroprotective agents. Synthesis and preliminary evaluation of (m-nitrobenzoyl)alanine, a potent inhibitor of kynurenine-3-hydroxylase. J Med Chem. 1994;37:647–655. doi: 10.1021/jm00031a015. [DOI] [PubMed] [Google Scholar]
- Pérez-De La Cruz V, González-Cortés C, Galván-Arzate S, Medina-Campos ON, Pérez-Severiano F, Ali SF, Pedraza-Chaverrí J, Santamaría A. Excitotoxic brain damage involves early peroxynitrite formation in a model of Huntington’s disease in rats: protective role of iron porphyrinate 5,10,15,20-tetrakis (4-sulfonatophenyl)porphyrinate iron (III) Neuroscience. 2005;135:463–474. doi: 10.1016/j.neuroscience.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Pérez-De La Cruz V, Santamaria A. Integrative hypothesis for Huntington’s disease: a brief review of experimental evidence. Physiol Res. 2007;56:513–526. doi: 10.33549/physiolres.931049. [DOI] [PubMed] [Google Scholar]
- Perry TL, Hansen S. What excitotoxin kills striatal neurons in Huntington’s disease? Clues from neurochemical studies. Neurology. 1990;40:20–24. doi: 10.1212/wnl.40.1.20. [DOI] [PubMed] [Google Scholar]
- Plaitakis A, Berl S, Yahr MD. Abnormal glutamate metabolism in an adult-onset degenerative neurological disorder. Science. 1982;216:193–196. doi: 10.1126/science.6121377. [DOI] [PubMed] [Google Scholar]
- Pláteník J, Stopka P, Vejrazka M, Stípek S. Quiniolinic acid - iron (II) complexes: slow autoxidation, but enhanced hydroxyl radical production in the fenton reaction. Free Rad Res. 2001;34:445–459. doi: 10.1080/10715760100300391. [DOI] [PubMed] [Google Scholar]
- Rassoulpour A, Wu HQ, Ferré S, Schwarcz R. Nanomolar concentrations of kynurenic acid reduce extracellular dopamine levels in the striatum. J Neurochem. 2005;93:762–765. doi: 10.1111/j.1471-4159.2005.03134.x. [DOI] [PubMed] [Google Scholar]
- Reiner A, Del Mar N, Deng YP, Meade CA, Sun Z, Goldowitz D. R6/2 neurons with intranuclear inclusions survive for prolonged periods in the brains of chimeric mice. J Comp Neurol. 2007;505:603–629. doi: 10.1002/cne.21515. [DOI] [PubMed] [Google Scholar]
- Reinhard JF., Jr Pharmacological manipulation of brain kynurenine metabolism. Ann N Y Acad Sci. 2004;1035:335–349. doi: 10.1196/annals.1332.020. [DOI] [PubMed] [Google Scholar]
- Reinhard JFJ, Erickson JB. Quinolinic acid in neurological disease: opportunities for novel drug discovery. Adv Pharmacol. 1994;30:85–127. doi: 10.1016/s1054-3589(08)60173-8. [DOI] [PubMed] [Google Scholar]
- Reynolds GP, Pearson SJ, Halket J, Sandler M. Brain quinolinic acid in Huntington’s disease. J Neurochem. 1988;50:1959–1960. doi: 10.1111/j.1471-4159.1988.tb02503.x. [DOI] [PubMed] [Google Scholar]
- Rios C, Santamaria A. Quinolinic acid is a potent lipid peroxidant in rat brain homogenates. Neurochem Res. 1991;16:1139–1143. doi: 10.1007/BF00966592. [DOI] [PubMed] [Google Scholar]
- Roberts RC, Du F, McCarthy KE, Okuno E, Schwarcz R. Immunocytochemical localization of kynurenine aminotransferase in the rat striatum: a light and electron microscopic study. J Comp Neurol. 1992;326:82–90. doi: 10.1002/cne.903260107. [DOI] [PubMed] [Google Scholar]
- Rosas HD, Salat DH, Lee SY, Zaleta AK, Hevelone N, Hersch SM. Complexity and heterogeneity: what drives the ever-changing brain in Huntington’s disease? Ann N Y Acad Sci. 2008;1147:196–205. doi: 10.1196/annals.1427.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röver S, Cesura AM, Huguenin P, Kettler R, Szente A. Synthesis and biochemical evaluation of N-(4-phenylthiazol-2-yl)benzenesulfonamides as high-affinity inhibitors of kynurenine 3-hydroxylase. J Med Chem. 1997;40:4378–4385. doi: 10.1021/jm970467t. [DOI] [PubMed] [Google Scholar]
- Ruddick JP, Evans AK, Nutt DJ, Lightman SL, Rook GA, Lowry CA. Tryptophan metabolism in the central nervous system: medical implications. Expert Rev Mol Med. 2006;8:1–27. doi: 10.1017/S1462399406000068. [DOI] [PubMed] [Google Scholar]
- Sacco RL, DeRosa JT, Haley EC, Levin B, Ordronneau P, Phillips SJ, Rundek T, Snipes RG, Thompson JLP. Glycine antagonist in neuroprotection for patients with acute stroke. JAMA. 2001;285:1719–1728. doi: 10.1001/jama.285.13.1719. [DOI] [PubMed] [Google Scholar]
- Saito K, Lackner A, Markey SP, Heyes MP. Cerebral cortex and lung indoleamine-2,3-dioxygenase activity is increased in type-D retrovirus infected macaques. Brain Res. 1991;540:353–356. doi: 10.1016/0006-8993(91)90536-5. [DOI] [PubMed] [Google Scholar]
- Saito K, Nowak TS, Jr, Markey SP, Heyes MP. Mechanism of delayed increases in kynurenine pathway metabolism in damaged brain regions following transient cerebral ischemia. J Neurochem. 1993;60:180–192. doi: 10.1111/j.1471-4159.1993.tb05836.x. [DOI] [PubMed] [Google Scholar]
- Salt TE, Eaton SA. Functions of ionotropic and metabotropic glutamate receptors in sensory transmission in the mammalian thalamus. Prog Neurobiol. 1996;48:55–72. doi: 10.1016/0301-0082(95)00047-x. [DOI] [PubMed] [Google Scholar]
- Samuel D, Errami M, Nieoullon A. Localization of N-methyl-D-aspartate receptors in the rat striatum: effects of specific lesions on the [3H]3-(2-carboxypiperazin-4-yl)propyl-1-phosphonic acid binding. J Neurochem. 1990;54:1926–1933. doi: 10.1111/j.1471-4159.1990.tb04893.x. [DOI] [PubMed] [Google Scholar]
- Sanberg PR, Pisa M, Fibiger HC. Avoidance, operant and locomotor behavior in rats with neostriatal injections of kainic acid. Pharmacol Biochem Behav. 1979;10:137–144. doi: 10.1016/0091-3057(79)90179-5. [DOI] [PubMed] [Google Scholar]
- Santamaría A, Jiménez-Capdeville ME, Camacho A, Rodríguez-Martínez E, Flores A, Galván-Arzate S. In vivo hydroxyl radical formation after quinolinic acid infusion into rat corpus striatum. Neuroreport. 2001;12:2693–2696. doi: 10.1097/00001756-200108280-00020. [DOI] [PubMed] [Google Scholar]
- Santamaría A, Ríos C. MK-801, an N-methyl-D-aspartate receptor antagonist, blocks quinolinic acid-induced lipid peroxidation in rat corpus striatum. Neurosci Lett. 1993;159:51–54. doi: 10.1016/0304-3940(93)90796-n. [DOI] [PubMed] [Google Scholar]
- Santiago-López D, Vázquez-Román B, Pérez-de La Cruz V, Barrera D, Rembao D, Salinas-Lara C, Pedraza-Chaverrí J, Galván-Arzate S, Ali SF, Santamaría A. Peroxynitrite decomposition catalyst, iron metalloporphyrin, reduces quinolinate-induced neurotoxicity in rats. Synapse. 2004;54:233–238. doi: 10.1002/syn.20084. [DOI] [PubMed] [Google Scholar]
- Sapko MT, Guidetti P, Yu P, Tagle DA, Pellicciari R, Schwarcz R. Endogenous kynurenate controls the vulnerability of striatal neurons to quinolinate: Implications for Huntington’s disease. Exp Neurol. 2006;197:31–40. doi: 10.1016/j.expneurol.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Sapp E, Kegel KB, Aronin N, Hashikawa T, Uchiyama Y, Tohyama K, Bhide PG, Vonsattel JP, DiFiglia M. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J Neuropathol Exp Neurol. 2001;60:161–172. doi: 10.1093/jnen/60.2.161. [DOI] [PubMed] [Google Scholar]
- Savvateeva E, Popov A, Kamyshev N, Bragina J, Heisenberg M, Senitz D, Kornhuber J, Riederer P. Age-dependent memory loss, synaptic pathology and altered brain plasticity in the drosophila mutant cardinal accumulating 3-hydroxykynurenine. J Neural Transm. 2000;107:581–601. doi: 10.1007/s007020070080. [DOI] [PubMed] [Google Scholar]
- Schlossberger HG, Kochen W, Linzen B, Steinhart H. Progress in Tryptophan and Serotonin Research. Walter de Gruyter; Berlin: 1984. [Google Scholar]
- Schröcksnadel K, Wirleitner B, Winkler C, Fuchs D. Monitoring tryptophan metabolism in chronic immune activation. Clin Chim Acta. 2006;364:82–90. doi: 10.1016/j.cca.2005.06.013. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Collins JF, Parks DA. alpha-Amino-omega-phosphono carboxylates block ibotenate but not kainate neurotoxicity in rat hippocampus. Neurosci Lett. 1982;33:85–90. doi: 10.1016/0304-3940(82)90134-3. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Coyle JT. Striatal lesions with kainic acid: neurochemical characteristics. Brain Res. 1977;127:235–249. doi: 10.1016/0006-8993(77)90538-8. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Creese I, Coyle JT, Snyder SH. Dopamine receptors localised on cerebral cortical afferents to rat corpus striatum. Nature. 1978;271:766–768. doi: 10.1038/271766a0. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Foster AC, French ED, Whetsell WO, Jr, Köhler C. Excitotoxic models for neurodegenerative disorders. Life Sci. 1984;35:19–32. doi: 10.1016/0024-3205(84)90148-6. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Fuxe K. 3H-kainic acid binding: relevance for evaluating the neurotoxicity of kainic acid. Life Sci. 1979;24:1471–1480. doi: 10.1016/0024-3205(79)90030-4. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Fuxe K, Agnati LF, Hökfelt T, Coyle JT. Rotational behaviour in rats with unilateral striatal kainic acid lesions: a behavioural model for studies on intact dopamine receptors. Brain Res. 1979a;170:485–495. doi: 10.1016/0006-8993(79)90966-1. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Fuxe K, Hökfelt T, Andersson K, Coyle JT. Dopamine and Huntington’s disease: assessment using the kainic acid model. In: Fuxe K, Calne DB, editors. Dopaminergic Ergot Derivatives and Motor Function. Pergamon Press; New York: 1979c. pp. 115–125. [Google Scholar]
- Schwarcz R, Hökfelt T, Fuxe K, Jonsson G, Goldstein M, Terenius L. Ibotenic acid-induced neuronal degeneration: a morphological and neurochemical study. Exp Brain Res. 1979b;37:199–216. doi: 10.1007/BF00237708. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Köhler C. Differential vulnerability of central neurons of the rat to quinolinic acid. Neurosci Lett. 1983;38:85–90. doi: 10.1016/0304-3940(83)90115-5. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Meldrum B. Excitatory aminoacid antagonists provide a therapeutic approach to neurological disorders. Lancet. 1985;2:140–143. doi: 10.1016/s0140-6736(85)90238-7. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Okuno E, White RJ. Basal ganglia lesions in the rat: effects on quinolinic acid metabolism. Brain Res. 1989;490:103–109. doi: 10.1016/0006-8993(89)90435-6. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Okuno E, White RJ, Bird ED, Whetsell WO., Jr 3-Hydroxyanthranilate oxygenase activity is increased in the brains of Huntington disease victims. Proc Natl Acad Sci (USA) 1988b;85:4079–4081. doi: 10.1073/pnas.85.11.4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarcz R, Pellicciari R. Manipulation of brain kynurenines: glial targets, neuronal effects, and clinical opportunities. J Pharmacol Exp Ther. 2002;303:1–10. doi: 10.1124/jpet.102.034439. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Tamminga CA, Kurlan R, Shoulson I. Cerebrospinal fluid levels of quinolinic acid in Huntington’s disease and schizophrenia. Ann Neurol. 1988a;24:580–582. doi: 10.1002/ana.410240417. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Whetsell WO, Jr, Mangano RM. Quinolinic acid: an endogenous metabolite that produces axon-sparing lesions in rat brain. Science. 1983;219:316–318. doi: 10.1126/science.6849138. [DOI] [PubMed] [Google Scholar]
- Seeburg PH. The TINS/TIPS lecture - The molecular biology of mammalian glutamate receptor channels. Trends Neurosci. 1993;16:359–365. doi: 10.1016/0166-2236(93)90093-2. [DOI] [PubMed] [Google Scholar]
- Sharpe NA, Tepper JM. Postnatal development of excitatory synaptic input to the rat neostriatum: an electron microscopic study. Neuroscience. 1998;84:1163–1175. doi: 10.1016/s0306-4522(97)00583-6. [DOI] [PubMed] [Google Scholar]
- Sheardown MJ, Nielsen EO, Hansen AJ, Jacobsen P, Honoré T. 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline: a neuroprotectant for cerebral ischemia. Science. 1990;247:571–574. doi: 10.1126/science.2154034. [DOI] [PubMed] [Google Scholar]
- Simmons DA, Casale M, Alcon B, Pham N, Narayan N, Lynch G. Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington’s disease. Glia. 2007;55:1074–1084. doi: 10.1002/glia.20526. [DOI] [PubMed] [Google Scholar]
- Simon RP, Swan JH, Griffiths T, Meldrum BS. Blockade of N-methyl-D-aspartate receptors may protect against ischemic damage in the brain. Science. 1984;226:850–852. doi: 10.1126/science.6093256. [DOI] [PubMed] [Google Scholar]
- Singhrao SK, Neal JW, Morgan BP, Gasque P. Increased complement biosynthesis by microglia and complement activation on neurons in Huntington’s disease. Exp Neurol. 1999;159:362–376. doi: 10.1006/exnr.1999.7170. [DOI] [PubMed] [Google Scholar]
- Singhrao SK, Thomas P, Wood JD, MacMillan JC, Neal JW, Harper PS, Jones AL. Huntingtin protein colocalizes with lesions of neurodegenerative diseases: an investigation in Huntington’s, Alzheimer’s, and Pick’s diseases. Exp Neurol. 1998;150:213–222. doi: 10.1006/exnr.1998.6778. [DOI] [PubMed] [Google Scholar]
- Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- Speciale C, Schwarcz R. Uptake of kynurenine into rat brain slices. J Neurochem. 1990;54:156–163. doi: 10.1111/j.1471-4159.1990.tb13296.x. [DOI] [PubMed] [Google Scholar]
- Speciale C, Wu HQ, Cini M, Marconi M, Varasi M, Schwarcz R. (R,S)-3,4-dichlorobenzoylalanine (FCE 28833A) causes a large and persistent increase in brain kynurenic acid levels in rats. Eur J Pharmacol. 1996;315:263–267. doi: 10.1016/s0014-2999(96)00613-9. [DOI] [PubMed] [Google Scholar]
- Stachowski E, Sathyasaikumar KV, Guidetti P, Muchowski PJ, Schwarcz R. Dysfunction of kynurenine pathway metabolism in the R6/2 mouse model of Huntington’s disease. Soc Neurosci Abstr. 2008;33:642.20. [Google Scholar]
- Stahl WL, Ward CB, Casper JB, Bird TD. Effects of L-glutamate on viabilities of cultured diploid skin fibroblasts and lymphocytes increased toxicity not observed in Huntington’s disease. J Neurol Sci. 1984;66:183–191. doi: 10.1016/0022-510x(84)90006-6. [DOI] [PubMed] [Google Scholar]
- Steiner HX, McBean GJ, Köhler C, Roberts PJ, Schwarcz R. Ibotenate-induced neuronal degeneration in immature rat brain. Brain Res. 1984;307:117–124. doi: 10.1016/0006-8993(84)90467-0. [DOI] [PubMed] [Google Scholar]
- Stípek S, Stastný F, Pláteník J, Crkovská J, Zima T. The effect of quinolinate on rat brain lipid peroxidation is dependent on iron. Neurochem Int. 1997;30:233–237. [PubMed] [Google Scholar]
- Stoll L, Hall J, Van Buren N, Hall A, Knight L, Morgan A, Zuger S, Van Deusen H, Gentile L. Differential regulation of ionotropic glutamate receptors. Biophys J. 2007;92:1343–1349. doi: 10.1529/biophysj.106.089896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone TW, Darlington LG. Endogenous kynurenines as targets for drug discovery and development. Nat Rev Drug Discov. 2002;1:609–620. doi: 10.1038/nrd870. [DOI] [PubMed] [Google Scholar]
- Stone TW, Perkins MN. Quinolinic acid: a potent endogenous excitant at amino acid receptors in CNS. Eur J Pharmacol. 1981;72:411–412. doi: 10.1016/0014-2999(81)90587-2. [DOI] [PubMed] [Google Scholar]
- Stoy N, Mackay GM, Forrest CM, Christofides J, Egerton M, Stone TW, Darlington LG. Tryptophan metabolism and oxidative stress in patients with Huntington’s disease. J Neurochem. 2005;93:611–623. doi: 10.1111/j.1471-4159.2005.03070.x. [DOI] [PubMed] [Google Scholar]
- Swartz KJ, During MJ, Freese A, Beal MF. Cerebral synthesis and release of kynurenic acid: an endogenous antagonist of excitatory amino acid receptors. J Neurosci. 1990;10:2965–2973. doi: 10.1523/JNEUROSCI.10-09-02965.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai YF, Pavese N, Gerhard A, Tabrizi SJ, Barker RA, Brooks DJ, Piccini P. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain. 2007;130:1759–1766. doi: 10.1093/brain/awm044. [DOI] [PubMed] [Google Scholar]
- Tsai G, Gastfriend DR, Coyle JT. The glutamatergic basis of human alcoholism. Am J Psychiat. 1995;152:332–340. doi: 10.1176/ajp.152.3.332. [DOI] [PubMed] [Google Scholar]
- Uemura T, Hirai K. L-kynurenine 3-monooxygenase from mitochondrial outer membrane of pig liver: purification, some properties, and monoclonal antibodies directed to the enzyme. J Biochem. 1998;123:253–262. doi: 10.1093/oxfordjournals.jbchem.a021930. [DOI] [PubMed] [Google Scholar]
- Ungerstedt U. Striatal dopamine release after amphetamine or nerve degeneration revealed by rotational behaviour. Acta Physiol Scand Suppl. 1971;367:49–68. doi: 10.1111/j.1365-201x.1971.tb10999.x. [DOI] [PubMed] [Google Scholar]
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Vazquez S, Garner B, Sheil MM, Truscott RJ. Characterisation of the major autoxidation products of 3-hydroxykynurenine under physiological conditions. Free Rad Res. 2000;32:11–23. doi: 10.1080/10715760000300021. [DOI] [PubMed] [Google Scholar]
- Villmann C, Becker CM. On the hypes and falls in neuroprotection: targeting the NMDA receptor. Neuroscientist. 2007;13:594–615. doi: 10.1177/1073858406296259. [DOI] [PubMed] [Google Scholar]
- Vincent SR, McGeer EG. Kainic acid binding to membranes of striatal neurons. Life Sci. 1979;24:265–270. doi: 10.1016/0024-3205(79)90228-5. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EPJ. Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- Walsh JL, Wu HQ, Ungerstedt U, Schwarcz R. 4-Chloro-3-hydroxyanthranilate inhibits quinolinate production in the rat hippocampus in vivo. Brain Res Bull. 1994;33:513–516. doi: 10.1016/0361-9230(94)90076-0. [DOI] [PubMed] [Google Scholar]
- Wenk GL. A primate model of Alzheimer’s disease. Behav Brain Res. 1993;57:117–122. doi: 10.1016/0166-4328(93)90127-c. [DOI] [PubMed] [Google Scholar]
- Wenk GL, Cribbs B, McCall L. Nucleus basalis magnocellularis: optimal coordinates for selective reduction of choline acetyltransferase in frontal neocortex by ibotenic acid injections. Exp Brain Res. 1984;56:335–340. doi: 10.1007/BF00236289. [DOI] [PubMed] [Google Scholar]
- Wheeler VC, White JK, Gutekunst CA, Vrbanac V, Weaver M, Li XJ, Li SH, Yi H, Vonsattel JP, Gusella JF, Hersch S, Auerbach W, Joyner AL, MacDonald ME. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum Mol Genet. 2000;9:503–513. doi: 10.1093/hmg/9.4.503. [DOI] [PubMed] [Google Scholar]
- Whetsell WO, Jr, Schwarcz R. Prolonged exposure to submicromolar concentrations of quinolinic acid causes excitotoxic damage in organotypic cultures of rat corticostriatal system. Neurosci Lett. 1989;97:271–275. doi: 10.1016/0304-3940(89)90609-5. [DOI] [PubMed] [Google Scholar]
- Wieloch T. Hypoglycemia-induced neuronal damage prevented by an N-methyl-D-aspartate antagonist. Science. 1985;230:681–683. doi: 10.1126/science.2996146. [DOI] [PubMed] [Google Scholar]
- Wolfensberger M, Amsler U, Cuénod M, Foster AC, Whetsell WO, Jr, Schwarcz R. Identification of quinolinic acid in rat and human brain tissue. Neurosci Lett. 1983;41:247–252. doi: 10.1016/0304-3940(83)90458-5. [DOI] [PubMed] [Google Scholar]
- Wu HQ, Guidetti P, Goodman JH, Varasi M, Ceresoli-Borroni G, Speciale C, Scharfman HE, Schwarcz R. Kynurenergic manipulations influence excitatory synaptic function and excitotoxic vulnerability in the rat hippocampus in vivo. Neuroscience. 2000;97:243–251. doi: 10.1016/s0306-4522(00)00030-0. [DOI] [PubMed] [Google Scholar]
- Wyttenbach A, Sauvageot O, Carmichael J, Diaz-Latoud C, Arrigo AP, Rubinsztein DC. Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum Mol Genet. 2002;11:1137–1151. doi: 10.1093/hmg/11.9.1137. [DOI] [PubMed] [Google Scholar]
- Xu H, Zhang GX, Ciric B, Rostami A. IDO: a double-edged sword for T(H)1/T(H)2 regulation. Immunol Lett. 2008;121:1–6. doi: 10.1016/j.imlet.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]