

Abstract
Acute lung injury (ALI) is a syndrome with significant morbidity and mortality, but its genetic susceptibility is not clearly understood. In the present study, we characterized functional promoter single nucleotide polymorphisms (SNPs) in the phase II antioxidant gene NQO1 (NAD(P)H:quinone oxidoreductase1) to evaluate its role in susceptibility to ALI. Three previously uncharacterized SNPs in the NQO1 promoter were selected for investigation. Luciferase assays were performed using constructs of each promoter polymorphism to evaluate function. Functional SNPs were genotyped in a prospective cohort of major trauma patients (N= 264) and assessed for association with development of ALI. The A/C SNP at −1221 decreased in vitro transcription of NQO1 at baseline and after exposure to hyperoxia and other oxidant stressors. Patients heterozygous for the −1221 C allele were at significantly lesser risk of ALI after major trauma compared with patients with wild‐type alleles, even after adjustment for APACHE III score, and mechanism of trauma [OR, 0.46 (95% CI 0.23, 0.90); P= 0.024]. This study demonstrated that the AC genotype at position −1221 in the NQO1 gene caused decreased transcription and was associated with a lower incidence of ALI following major trauma. These novel findings may have important implications in diseases with oxidant stress aetiologies.
Keywords: gene, susceptibility, ARDS, acute respiratory distress syndrome, hyperoxia, endotoxin, hydrogen peroxide, oxidant stress, SNP, single nucleotide polymorphism, lung disease
Introduction
Acute lung injury (ALI) is a common and devastating syndrome in the intensive care unit, with mortality rates exceeding 30–50%[1, 2]. ALI occurs secondarily in a number of disease processes, most commonly sepsis, pneumonia, aspiration, and trauma, and host susceptibility likely plays a role in determining the development of ALI following a particular insult [3]. ALI is associated with reactive oxygen species (ROS) formation, where the excessive production of ROS overwhelms the innate antioxidant system, leading to tissue injury [3]. ROS formation also occurs with exposure to hyperoxia, leading to a profound activation of the inflammatory response. The release of inflammatory mediators in response to ROS leads to endothelial cell injury, interstitial and perivascular oedema, epithelial cell hypertrophy and proliferation, and denudation of the alveolar basement membrane [3]. Reactive species can also lead to profound cell damage through direct damage to DNA, lipid peroxidation, protein oxidation and alteration of transcription factors [4, 5]. It has been shown that antioxidants are decreased in the distal airspaces of patients with ALI [6].
NAD(P)H:quinone oxidoreductase 1 (NQO1) is a phase II/antioxidant enzyme involved in the formation of ROS and other free radicals [7]. NQO1 catalyses the two electron reduction of a variety of quinone compounds, which prevents the generation of free radicals and ROS, and protects cells from oxidative damage. Some evidence suggests that NQO1 may also interact directly with ROS, such as hydroxyl radical and hydrogen peroxide, which may influence the oxidant–antioxidant balance [8, 9]. NQO1 expression is induced by oxidant stress, aromatic hydrocarbons, phenols, and certain industrial acrylates and metals [7]. Though NQO1 reduces quinones to hydroquinones, not all of these products are redox‐stable. In some cases, metabolism by NQO1 results in a more active product where redox‐labile hydroquinones can react with molecular oxygen (perhaps in cases of hyperoxia) to form semiquinones. These semiquinones in turn can result in the generation of ROS and cause alkylation of DNA [7].
A number of single nucleotide polymorphisms (SNPs) have been identified in NQO1. Only two of these, Arg139Trp and Pro187Ser [10, 11], have been studied extensively and are implicated in risk of a variety of cancers. NQO1 is highly inducible [12, 13, 14, 15], but little is known about function of promoter SNPs. The role of NQO1 in ALI has not been investigated, but the effects of modulation of NQO1 activity in preliminary studies suggest that alterations in its function could enhance lung injury. Because ALI has been associated with oxidant stress, and a number of investigations have found an association between oxidant stress and NQO1 [13, 16, 17], we tested the hypothesis that NQO1 is an important determinant of ALI. Thus, the overall objective of this investigation was to identify and evaluate functionality of NQO1 promoter SNPs, and determine whether SNPs with in vitro evidence of function are associated with differential risk for ALI in patients who have experienced major trauma.
Methods
Selection of SNPs
NQO1 promoter SNPs were identified through a search of Medline and the SNP database in the National Library of Medicine (http://www.ncbi.nlm.nih.gov), and their positions were localized (Fig. 1; Table 1). SNPs chosen for investigation were within 2 kb of the 5’ end and had a minor allele frequency of >5%. Other NQO1 promoter polymorphisms have been identified (rs2965757, rs689459, rs689458, rs689457, rs12922457) but they were not investigated because they have allele frequencies <5% which limited power to determine their importance in our study population. Primers (Sigma Genosys, The Woodlands, TX, USA) were then designed to amplify the region of interest for the purpose of genotyping genomic DNA from our cohort to identify the presence of polymorphisms (Table 2).
Figure 1.
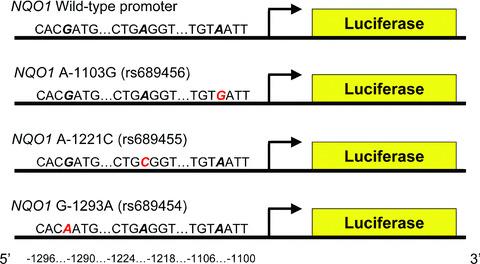
NQO1 promoter polymorphism luciferase constructs. The −1103 G, −1221 C and −1293 A constructs are shown. Red letters indicate the mutant alleles. rs numbers for each mutation are shown in parentheses.
Table 1.
Selected NQO1 promoter polymorphisms, locations and published* heterozygosity
| SNP | Location | Heterozygosity |
|---|---|---|
| rs689454 | G‐1293A | 0.126 |
| rs689455 | A‐1221C | 0.500 |
| rs689456 | A‐1103G | 0.354 |
Table 2.
Primer sequences for sequencing SNPs
| Primer | Sequence (5′→ 3′) |
|---|---|
| NQO1 promoter forward | GGGAAAGTTCTATTGGACAGGG |
| NQO1 promoter reverse | GGTGTGGAGATAGCAGTTATAGGG |
| NQO1 nested promoter forward | GGTCTCACTTTGTTGCTCAGG |
| NQO1 nested promoter reverse | GCAGGAGAATCGCTTGAAC |
Construction of promoter SNPs and transfection into BEAS‐2B cells
Promoter SNP constructs were made utilizing a reporter gene assay. To determine the roles of the −1103, −1221 and −1293 SNPs on NQO1 promoter activity, we cloned separately genomic DNA (−1621 to −781 bp) containing each SNP into pGL3 plasmids containing the luciferase gene lacking a promoter (Promega, Madison, WI, USA) (Fig. 1). The presence of each SNP in the promoter region was confirmed through sequencing. These constructs were then transiently transfected into BEAS‐2B bronchial epithelial cells by using Effectene transfection reagent (Qiagen, Valencia, CA, USA) along with a renilla control plasmid and incubated at 37°C for 24 hrs.
Exposure to oxidant stress
Cells transiently transfected with each promoter construct designed as described above were exposed either to normoxia (air, 21% oxygen) or hyperoxia (90–95% oxygen). The hyperoxia chamber (PRO:OX Model 110, BioSpherix, Ltd., Redfield, NY, USA) was calibrated as per manufacturer’s recommendations and operated with sensors which monitor the constant flow of oxygen. Cells were placed in a tightly sealed chamber and exposed to a mixture of 95% O2/5% CO2 for 8 or 12 hrs. Transfected cells were also separately exposed to hydrogen peroxide (400 μM, Molecular Probes, Carlsbad, CA, USA) for 1.5 hrs, beta‐hydroxy toluene (20 mM, Sigma, St. Louis, MO, USA) for 6 hrs or lipopolysaccharide (100 μg/ml, Sigma) for 6 hrs versus media alone.
Measurement of luciferase activity
After exposure, cells were harvested using passive lysis buffer and samples were processed using the Dual‐Luciferase Reporter Assay System (Promega). Transcriptional activity of the luceriferase reporter gene was then detected by chemiluminescence using a luminometer (Fluoroskan Ascent FL, Thermo‐Fisher Scientific, Inc, Waltham, MA, USA). Luciferase constructs were co‐transfected with a renilla control plasmid to normalize for transfection efficiency using the dual luciferase assay. Each exposure was performed in triplicate and values were averaged and reported as a mean ± 1 standard error of the mean (S.E.M.).
Patient population
A prospective cohort study was performed at the Hospital of the University of Pennsylvania to determine the risk factors for development of ALI following major trauma between 1999 and 2002. This study was approved by the institutional review board at the University of Pennsylvania. There were 278 subjects in this trauma cohort study (142 African American, 136 Caucasian) (Table 3). Subjects met the following inclusion criteria: (1) admission to the surgical intensive care unit (SICU) as a result of acute trauma directly from the hospital’s emergency department; and (2) an Injury Severity Score (ISS) > 16 as calculated on the basis of information available during their first 24 hrs of hospitalization. Exclusion criteria were death or discharge from the SICU in the first 24 hrs, age less than 14 years, current or prior congestive heart failure or recent myocardial infarction, severe chronic respiratory disease, morbid obesity, burns over body surface area of 30% or more, lung transplantation, or bone marrow transplantation, and limitation of major injury to the head and/or neck alone, i.e. an acute injury score of >2 for ‘head/neck’ and <2 for all other body regions.
Table 3.
Baseline characteristics of severe trauma (ISS>15) patients stratified by occurrence of acute lung injury (ALI)
| No ALI (n=185) | ALI (n=93) | P‐value | |
|---|---|---|---|
| Age, years | 35.1 ± 16.9 | 39.4 ± 18.9 | 0.078 |
| Male gender, n (%) | 154 (83) | 76 (83) | 0.895 |
| African American, n (%) | 100 (54) | 42 (46) | 0.188 |
| ISS | 24.5 ± 7.5 | 26.4 ± 8.9 | 0.133 |
| APACHE II | 46.5 ± 16.7 | 57.7 ± 17.9 | <0.001 |
| APACHE II less P(A‐a)O2 | 38.3 ± 13.3 | 43.5 ± 13.7 | 0.002 |
| MAP, mmHga | 66.1 ± 11.3 | 61.7 ± 12.7 | 0.004 |
| Heart rate, bpma | 127 ± 22 | 132 ± 23 | 0.054 |
| Hematocrit,**%a | 28.0 ± 5.6 | 27.8 ± 6.6 | 0.862 |
| Creatinine, g/dla | 1.0 ± 0.6 | 1.1 ± 0.4 | 0.989 |
| Blunt injury, n (%)b | 118 (64) | 63 (68) | 0.474 |
| Long bone fracture, n (%)c | 66 (36) | 35 (39) | 0.627 |
| Pelvic fracture, n (%) | 37 (20) | 19 (21) | 0.864 |
| Pulmonary contusion, n (%) | 43 (24) | 36 (39) | 0.007 |
| Hypertension, n (%)d | 17 (14) | 8 (13) | 0.960 |
| Diabetes mellitus, n (%)d | 7 (5) | 5 (5) | 0.455 |
Abbreviations: ISS, Injury Severity Score; APACHE II, Acute Physiology and Chronic Health Evaluation Score; APACHE II less P(A‐a)O2, Score excluding points contributed by the alveolar‐arterial PO2 difference [P(A‐a)O2]; MAP, mean arterial blood pressure.
aWorst value in first 24 hrs of admission; bAny injury with a blunt component; clong bone fracture defined as fracture to femur, fibula, forearm, humerus or tibia; dfor hypertension and diabetes data, the numbers of patients do not sum to the sample totals because of missing data. Data are expressed as mean ± S.D., unless otherwise specified. P‐value performed with unpaired t‐test, ×2 or Wilcoxon rank sum test.
To qualify as having ALI, subjects had to meet all ALI criteria within the first 5 days following trauma. The standard criteria of the American European Consensus Conference (AECC) definition while tracheally intubated and receiving assisted ventilation are: acute onset, bilateral pulmonary infiltrates on chest radiograph consistent with pulmonary oedema; absence of evidence of left atrial hypertension; and a ratio of arterial oxygen tension to fraction of inspired oxygen (PaO2/FiO2) equal to or less than 300 [18]. Two clinicians reviewed all chest radiographs, and when discordant, a third clinician adjudicated. All three were experienced at interpreting chest radiographs for ALI.
Blood samples from these patients were collected at the time of admission to the emergency department. The samples were centrifuged, and genomic DNA was extracted from thawed buffy coat aliquots using the Qiagen Qiamp 96 DNA Blood Kit (Qiagen). Because no sequence data were obtained for subjects due to either poor amplification during PCR (n= 10) or exhaustion of genomic DNA (n= 11), 257 subjects were included in the analysis.
Sequencing of SNPs in the major trauma cohort
Primers designed to amplify the region of the NQO1 promoter SNPs were used to amplify the region of interest (Table 2). Various sites were amplified from approximately 70 ng of DNA using the Epicentre Failsafe System (Madison, WI, USA), purified with the GenElute PCR Clean‐up Kit (Sigma Chemical Co, St. Louis, MO, USA) and sequencing reactions performed using Big Dye terminator kits (Applied Biosystems, Foster City, CA, USA). Purified products were analysed on an ABI 377 Automated DNA Sequencer (Applied Biosystems). Probes obtained through Applied Biosystems were used to amplify allele‐specific polymorphic regions. Samples were analysed with an Applied Biosystems analyzer using allelic discrimination to determine heterozygosity (Table 4). All samples were genotyped while blinded to the case‐control status of the sample. A random 5% of all genotypes were repeated to validate genotyping procedures and all were concordant. Furthermore, there were not any known unexamined SNPs in the region of the ABI primers, which could have caused genotyping mistakes.
Table 4.
Genotypes, case/control distribution and population‐specific genotype frequencies for three NQO1 promoter SNPs in the major trauma cohort
| SNP | Wild‐type | Heterozygote | Variant | Sequence failed+ | Totals | |||
|---|---|---|---|---|---|---|---|---|
| AFR‐AM* | CAUC | AFR‐AM | CAUC | AFR‐AM | CAUC | |||
| G‐1293A | 126 (39, 87); 94.7 | 108 (39, 69); 87.1 | 4 (0, 4); 3.0 | 8 (2, 6); 6.5 | 3 (1, 2); 2.3 | 8 (3, 5); 6.5 | 10 | 267 |
| A‐1221C | 88 (26, 62); 66.2 | 87 (37, 50); 70.2 | 36 (8, 28); 27.1 | 34 (7, 27); 27.4 | 9 (6, 3); 6.8 | 3 (0, 3); 2.4 | 10 | 267 |
| A‐1103G | 98 (33, 65); 74.8 | 90 (30, 60); 72.6 | 30 (6, 24); 22.9 | 26 (8, 18); 21.0 | 3 (0, 3); 2.3 | 8 (6, 2); 6.5 | 12 | 267 |
*First number is the total patients for each genotype; numbers in parentheses indicate the numbers of cases (first number) and controls for each genotype; numbers after the semicolons are the genotype frequencies for each population.
+ Sequence Failed indicates the number of patients with genotype failure. Abbreviations: AFR‐AM, African American; CAUC, Caucasian.
Statistical analysis
In vitro oxidant exposure study.
Data are expressed as the group mean ± S.E.M. Two‐way analysis of variance (ANOVA) was used to evaluate the effects of exposure (air or unexposed versus LPS, hyperoxia or H2O2) and promoter polymorphism construct (A‐1103G, A‐1221C and G‐1293A) on luciferase activity. Student–Newman–Keuls test was used for a posteriori comparisons of means. All analyses were performed using a commercial statistical analysis package (SigmaStat; Jandel Scientific Software, San Rafael, CA, USA). Statistical significance was accepted at P < 0.05.
Association study.
Allelic distributions at each locus were evaluated for Hardy‐Weinberg equilibrium (HWE) via STATA 9.2 ‘genhw’ command (STATA Data Corp, College Station, TX, USA). The association between individual NQO1 genotype and development of ALI was tested using Chi‐square and logistic regression methods. To assess the potential confounding effects of clinical variables in the relationship of NQO1 genotype with ALI risk, we used multivariable logistic regression models, including potential confounding baseline clinical variables with an association with ALI risk. A two‐sided P‐value of 0.05 was considered significant in all analyses. A study sample size of 267 subjects gave us power to detect a relative risk of 1.9 or higher for genotype prevalences of 10% or greater, assuming an alpha = 0.05, incidence of ALI = 0.30, and beta = 0.80. Chi2 and logistic regression analyses were performed in STATA 9.2 (STATA Data Corp). Haplotype based analyses across the loci were performed using the ‘hapblock’ command in STATA 9.2.
Results
Selection of SNPs
Three promoter polymorphisms in NQO1 were selected for investigation (Table 1) based on potential functional relevance and frequency in the population (see Methods for frequency criteria). Published heterozygosity rates range from 0.126 to 0.500. None of the selected SNPs have been studied previously in the setting of hyperoxia or other oxidant stressors, and their function was unknown.
In vitro luciferase assays
Luciferase constructs containing the wild‐type NQO1 promoter, and promoters containing SNP −1293A, −1221C, or −1103G were transfected into BEAS‐2B bronchial epithelial cells and exposed to hyperoxia, H2O2, BHT or LPS. Luciferase activity in cells transfected with the −1221C construct was significantly lower compared to wild‐type and the other promoter SNPs after exposure to air (P < 0.05; Fig. 2A–D). Relative to air exposed cells, a significant increase in luciferase activity was found in all cells after 8‐ and 12‐hr exposure to hyperoxia (P < 0.05; Fig. 2A). However, luciferase activity in cells with the −1221C genotype promoter construct was significantly less than in wild‐type cells and cells with the other promoter SNPs after 8‐ and 12‐hr exposures to hyperoxia (P < 0.05; Fig. 2A). A significant (P= 0.023) interaction effect was fixed between exposure (air, hyperoxia) and the −1221 SNP on luciferase activity. Hyperoxia‐induced luciferase activity in cells with the −1103 G construct was also significantly reduced at 8 hrs compared to the respective wild‐type control (P < 0.05).
Figure 2.
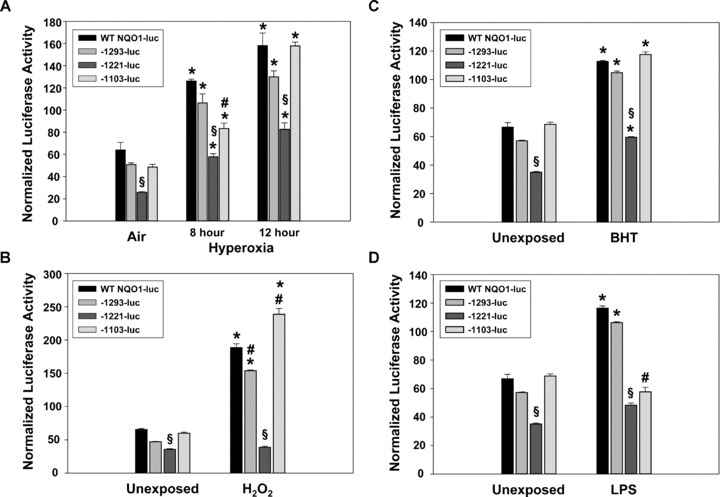
Luciferase activity of promoter polymorphisms A‐1103G, A‐1221C and G‐1293A after exposure to hyperoxia (A), hydrogen peroxide (H2O2; B), beta‐hydroxytoluene (BHT, C) and lipopolysaccharide (LPS, D). Data are presented as means ± S.E.M. n= 3/group. *, significantly different from respective control exposure; §, significantly different from all other constructs within group (P < 0.05); #, significantly different from WT, −1293‐luc and −1221‐luc (P < 0.05).
Cells transfected with each of the promoter luciferase constructs were also exposed to H2O2. As above, luciferase activity was significantly reduced in cells with the −1221C promoter relative to unexposed cells with the wild‐type NQO1 promoter (P < 0.05; Fig. 2B). Luciferase activity increased in cells transfected with wild‐type NQO1 promoter, −1293A promoter and the −1103G promoter after exposure to H2O2 compared to media alone (P < 0.05; Fig. 2B). However, luciferase expression did not increase significantly after exposure to H2O2 in cells transfected with the −1221C promoter. As with hyperoxia, a significant (P < 0.001) interaction effect was found between exposure and the −1221 SNP on luciferase activity. Cells transfected with the −1103G promoter had a significantly heightened response to H2O2 compared to wild‐type cells.
Exposure to beta‐hydroxy toluene (BHT) also caused an increase in luciferase activity in cells transfected with each construct (P < 0.05; Fig. 2C). This increase in luciferase activity induced by exposure to BHT was significantly diminished in cells transfected with SNP −1221C (P < 0.05), but no interaction effect was found.
Transfected cells were also exposed to lipopolysaccharide (LPS) and luciferase activity was significantly increased in the wild‐type construct and SNP −1293A (P < 0.05; Fig. 2D). A minimal increase in luciferase activity was found in cells with the −1221C SNP after exposure to LPS, and a significant (P < 0.001) interaction between exposure and the SNP was found. The SNP −1103G caused significantly decreased expression of luciferase relative to wild‐type cells (P < 0.05).
Association analysis
The trauma cohort was genotyped for each of the NQO1 promoter SNPs and genotype (Table 4) and allelic (Table 5) frequencies were calculated. HWE was calculated for each of the selected SNPs. The A‐1221C SNP conformed to HWE predictions, whereas the G‐1293A and A‐1103G did not (results not shown). Allele frequencies for the trauma cohort did not differ from the published allele frequencies for the G‐1293A and A‐1103G SNPs. Chi‐square analysis of the incidence of ALI among homozygous wild‐type, heterozygote and homozygous variants indicated the A‐1221C SNP was significantly associated with the development of ALI in African Americans (×2= 6.798, P= 0.033), Caucasians (×2= 6.831, P= 0.033) and the combined cohort (×2= 6.541, P= 0.038). These associations were driven by the protective effects of the AC genotype, predominantly in Caucasians. In all patients, 19% of the ALI cases had the AC genotype versus 33% of the controls [OR = 0.42 (95% CI 0.20, 0.82) P= 0.027]; in African‐American subjects, 24% of the ALI cases were AC versus 31% of the controls [OR = 0.68 (0.24, 1.81) P= 0.407]; and in Caucasians, 16% of the ALI cases were AC versus 35% of the controls [OR = 0.35 (0.12, 0.95) P= 0.024]. Therefore, although the direction of effect was the same in the African Americans, there was not statistical significance within that racial category. No significant associations were found for the G‐1293A or A‐1293G SNPs.
Table 5.
Minor allele frequencies of three NQO1 promoter SNPs in dbSNP and the trauma cohort
| SNP | db SNP | Trauma cohort | |
|---|---|---|---|
| African American | Caucasian | ||
| G‐1293A | 0.067 | 0.038 | 0.097 |
| A‐1221C | 0.500 | 0.203 | 0.161 |
| A‐1103G | 0.230 | 0.137 | 0.169 |
Further, logistic regression analysis found the AC genotype at position −1221 was significantly associated with a lower incidence of ALI after major trauma, with an unadjusted odds ratio (OR) of 0.48 relative to the AA genotype (95% CI 0.25, 0.93; P= 0.029) (Table 6). When adjusted for race, APACHE III score, and mechanism of trauma individuals with the NQO1 AC genotype at −1221 remained at lower risk of ALI, OR = 0.46 (95% CI 0.23, 0.90; P= 0.024). Logistic regression analysis of the A‐1221C SNP within racial category indicated significant association in Caucasians (OR 0.362, P= 0.041) when adjusted for APACHE III score and mechanism of trauma. The association of the SNP in African Americans was not significant, although the effect was in the same direction (i.e. protective) as observed in Caucasians (Table 6). There also appeared to be a trend toward a higher incidence of ALI with the CC genotype (6/12, 50%) in all subjects, but no significant difference from wild‐type (AA) genotype was found. Haplotype analyses including the different promoter loci did not add any useful information to association with ALI beyond the single SNP analyses including −1221.
Table 6.
Association of ALI risk relative to the A‐1221C NQO1 SNP in a population of African American and Caucasian patients with major trauma
| Ancestry | Genotype | Odds ratio | Confidence interval | P‐value |
|---|---|---|---|---|
| African American | AA | Referent | — | — |
| AC | 0.582 | 0.226, 1.498 | 0.262 | |
| Caucasian | AA | Referent | — | — |
| AC | 0.362 | 0.137, 0.960 | 0.041 | |
| Combined cohort | AA | Referent | — | — |
| AC | 0.485 | 0.253, 0.928 | 0.029 |
Discussion
ALI leads to significant morbidity and mortality in the intensive care unit, affecting over 190,000 patients in the United States yearly [1, 2]. The risk of developing ALI after a particular insult can be attributed, in part, to genetic predisposition. A number of investigations have implicated genetic polymorphisms in the risk of ALI and ARDS [19, 20, 21, 22]. We previously demonstrated in the same major trauma cohort described herein that individuals with a functional polymorphism in the promoter of the transcription factor nuclear factor‐E2, related factor 2 (NRF2) were at enhanced risk of developing ALI compared to individuals with wild‐type genotype [23]. To further understand the genetic basis of susceptibility to ALI, we asked whether genetic variation in the promoter of NQO1, a highly inducible phase II antioxidant gene, affects risk of ALI. To address this question, we first performed a comprehensive database SNP search and identified potentially functional SNPs in the NQO1 promoter. We then evaluated, using in vitro assays, the impact of the SNPs on transcriptional activity of NQO1 after oxidant challenge. Finally, we determined whether the functional SNPs associated with differences in susceptibility to ALI in major trauma patients. This hypothesis‐directed investigation determined that individuals with a loss of function −1221C allele were at significantly lower risk of ALI compared with individuals who were homozygous wild‐type.
NQO1 is an antioxidant enzyme which has a role in regulating the production of ROS [7]. Two characterized polymorphisms in this enzyme have been shown to predict response to treatment in patients with cancer and myeloid leukaemias [10, 11]. The exact role of NQO1 in response to oxidant stress or ALI is not clearly understood. In this study, we have shown that in vitro, polymorphisms in NQO1 can affect the transcriptional response to oxidant stress. Utilizing promoter luciferase constructs, we investigated the response of three SNPs to oxidant challenge when compared to the wild‐type NQO1 promoter. Interestingly, the SNP A‐1221C caused decreased expression of luciferase basally relative to wild‐type and the A‐1103G and G‐1293A SNPs. Furthermore, after exposure to hyperoxia, hydrogen peroxide, BHT and LPS, luciferase activity with the A‐1221C SNP was significantly reduced relative to the wild‐type NQO1 promoter after each exposure. With the exception of BHT, statistically significant interaction effects were found between all exposures and the SNP on luciferase activity, suggesting that the A‐1221C SNP is unable to induce NQO1 expression in response to various forms of oxidant stress in vitro. Other investigators have also shown that NQO1 alters the redox status of cells after exposure to oxidizing agents [24, 25].
The mechanism by which the A‐1221C SNP affects the promoter activity of NQO1 is not known. Because NRF2 is important in regulation of NQO1 expression [26], a bioinformatic analysis [27] of the 2 kb region of the NQO1 promoter that contains the SNPs was investigated to identify antioxidant response elements (AREs) and evaluate their importance in binding NRF2. These analyses indicated that the A‐1221C SNP is not in a bona fide ARE, so direct effect of the SNP on NRF2 binding should be predicted to be low or nonexistent. Electrophoretic mobility shift analysis (EMSA) found no effect of the A‐1221C SNP on NRF2 binding (data not shown) and confirmed this prediction. Using the most stringent criteria, transcription factor motif analysis (TRANSFAC, [28]) of the transcription factor binding sites that include the −1221 site identified sterol regulatory element‐binding protein 1 (SREBP‐1) as a possible transcription factor. However, further investigation is necessary to confirm a role for SREBP‐1 in the function of the −1221 SNP in NQO1 regulation.
Having demonstrated a functional role for the promoter SNPs, we asked whether the SNPs were associated with risk of developing ALI. Interestingly, we found that the heterozygous genotype for the A‐1221C polymorphism conferred significant protection against developing ALI in patients with major trauma. Our data suggest therefore that decreased production of NQO1, as demonstrated with the variant −1221 polymorphism in the in vitro experiments, results in decreased risk of developing ALI after major trauma. These results may be considered somewhat surprising inasmuch as NQO1 is considered primarily an antioxidant, and is up‐regulated in response to oxidant stressors [16, 26, 29], presumably to reduce oxidant injury. However, NQO1 can also be a pro‐oxidant under certain conditions [7], and may be generating oxidant radicals in the condition of major trauma leading to ALI. It is important to caution that in vitro and in vivo conditions are not always directly translatable, and that the effects of one gene may be dependent on the effects of another (i.e. gene by gene interaction), and that these interactions may also be influenced by the physiological environment. Therefore, understanding of the interaction between NQO1, other antioxidant genes [e.g. glutathione S‐transferase M1 (GSTM1)], and genes for transcription factors [e.g. NRF2 and activator protein 1 (AP‐1)] in conditions that lead to ALI for example, requires addition investigation.
This study has a number of strengths, including a translational study design. We sought initially to validate the function of the NQO1 promoter SNPs before testing for association with ALI phenotype in the trauma cohort. A number of SNPs in the 5′ untranslated region of NQO1 with unknown function have been reported. However, as opposed to the multiple SNP approach that has statistical complications due to inherent obligations to do multiple comparison corrections, we chose an approach that focuses on only the SNPs that have known functional relevance and asked whether they associated with risk of disease. This approach minimizes the possibility that the association of candidate SNP with disease is due to random chance and enables a priori hypothesis generation and testing. This translational approach strengthens considerably the plausibility of an association. Another strength of this investigation is the potential applicability of the functional promoter SNPs to other oxidant‐related diseases. Because the A‐1221C SNP significantly affects the NQO1 luciferase expression basally and in response to LPS, hyperoxia and H2O2, this SNP may also have a role in a variety of other disease processes which involve the dysregulation of ROS, including sepsis, chronic obstructive pulmonary disease and bronchopulmonary dysplasia.
There are also some limitations to this study. First, our models of oxidant stress may have restrictions. For example, hyperoxia has been used in vitro to mimic the changes seen in vivo during ALI, but this model is not without flaws. Trauma leading to ALI may be either due to a systemic inflammatory response and/or ischaemia and reperfusion injury, but may not translate into the same changes in inflammatory mediators and ROS as seen in hyperoxia. Second, our study population was too small to evaluate effects of the homozygous variant at position −1221. Furthermore, although we had adequate power to detect a reasonable relative risk for common genotype prevalences, negative findings in lower prevalence genotypes should be interpreted with caution. This is true of our associations within the African‐American population, which were not statistically significant even though they were consistent in exhibiting a protective effect. Lastly, our study population represents patients who developed ALI after major trauma, and results may not be able to be generalized to patients with ALI due to other causes.
There is also a consideration that the −1221 SNP is in low linkage disequilibrium (LD) with other mutants in this gene or nearby genes that could contribute to the association with ALI. However, using HAPMAP data (http://hapmap.org/) we found weak LD between promoter element SNPs and coding region SNPs in NQO1. Thus the probability that these findings are due to LD alone is unlikely. Furthermore, given the strong functional data for the −1221 SNP, we do not believe that the clinical association is due to LD alone.
This study provides the first evidence of functional significance of the −1221 promoter SNP, which was confirmed in response to a variety of in vitro stimulants. Furthermore, we present evidence that this functional SNP is associated with development of ALI, whereas other SNPs were not. The corroboration of functional and association data supports the validity of the findings. As such, this study provides valuable insights into the function of the NQO1 promoter region, as well as raising interesting questions about the mechanisms of ALI. This information may provide a basis for potential therapies once more is learned about the mechanism of NQO1’s role in ALI.
Acknowledgements
This study was supported by P50 HL60290‐01 (JC, PL), K23 HL04243 (JC), the Doris Duke Charitable Foundation (RA), and the Intramural Program at the National Institute of Environmental Health Sciences, National Institutes of Health, Department of Health and Human Services (AR, SK). We thank Dr. Hye‐Youn Cho, Dr. Xuting Wang, Ms Jacqui Marzec and Mr. Wes Gladwell for assistance with the investigation. We also thank Drs. Donald Cook and Michael Fessler for reviewing the manuscript.
Conflict of Interest statement. The authors declare that they have no competing financial interests.
References
- 1. Goss CH, Brower RG, Hudson LD, et al . Incidence of acute lung injury in the United States. Crit Care Med. 2003; 31: 1607–11. [DOI] [PubMed] [Google Scholar]
- 2. Rubenfeld, GD Caldwell E, Peabody E, et al . Incidence and outcomes of acute lung injury. N Eng J Med. 2005; 353: 1685–93. [DOI] [PubMed] [Google Scholar]
- 3. Piantadosi CA, Schwartz DA. The acute respiratory distress syndrome. Ann Intern Med. 2004; 141: 460–70. [DOI] [PubMed] [Google Scholar]
- 4. Zhang H, Slutsky AS, Vincent JL. Oxygen free radicals in ARDS, septic shock and organ dysfunction. Intensive Care Med. 2000; 26; 474–6. [DOI] [PubMed] [Google Scholar]
- 5. Chow CW, Abreu M, Suzuki T, et al . Perspective: oxidative stress and acute lung injury. Am J Respir Cell Mol Biol. 2003; 29: 427–31. [DOI] [PubMed] [Google Scholar]
- 6. Bowler RP, Velsor L, Duda B, et al . Pulmonary edema fluid antioxidants are depressed in acute lung injury. Crit Care Med. 2003; 31: 2309–15. [DOI] [PubMed] [Google Scholar]
- 7. Ross D, Beall H, Traver RD, et al . Bioactivation of quinines by DT‐diaphorase: molecular, biochemical, and chemical studies. Oncol Res. 1994; 6: 493–500. [PubMed] [Google Scholar]
- 8. Du J, Daniels DH, Asbury C, et al . Mitochondrial production of reactive oxygen species mediate dicumarol‐induced cytotoxicity in cancer cells. J Biol Chem. 2006; 281: 37416–26. [DOI] [PubMed] [Google Scholar]
- 9. Jia Z, Zhu H, Misra HP, et al . Potent induction of total cellular GSH and NQO1 as well as mitocondrial GSH by 3H‐1,2‐dithiole‐3‐thione in SH‐SY5Y neuroblastoma cells and primary human neurons: protection against neurocytotoxicity elicited by dopamine, 6‐hydroxydopamine, 4‐hydroxy‐2‐nonenal, or hydrogen peroxide. Brain Res. 2008; 1197: 159–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Larson RA, Wang Y, Banerjee M, et al . Prevalence of the inactivating 609 C‐T polymorphism in the NAD(P)H:quinine oxidoreductase (NQO1) gene in patients with primary and therapy‐related myeloid leukemia. Blood. 1999; 94: 803–7. [PubMed] [Google Scholar]
- 11. Traver RD, Horikoshi T, Danenberg KD, et al . NAD(P)H:quinone oxidoreductase gene expression in human colon carcinoma cells; characterization of a mutation that modulates DT‐diaphorase activity and mitomycin sensitivity. Cancer Res. 1992; 52: 797–802. [PubMed] [Google Scholar]
- 12. Lim JH, Kim K‐M, Kim SW, et al . Bromocriptine activates NQO1 via Nrf2‐PI3K/Akt signaling: novel cytoprotective mechanism against oxidative damage. Pharmacol Res. 2008; 57: 325–31. [DOI] [PubMed] [Google Scholar]
- 13. Jaiswal AK, Regulation of genes encoding NAD(P)H:quinone oxidoreductases. Free Radic Biol Med. 2000; 29: 254–62. [DOI] [PubMed] [Google Scholar]
- 14. Korashy HM, Brocks DR, El‐Kadi A.O.S. Induction of the NAD(P)H:quinone oxidoreductase 1 by ketoconazole and itraconazole; a mechanism of cancer chemoprotection. Cancer Lett. 2007; 258: 135–43. [DOI] [PubMed] [Google Scholar]
- 15. Zhu H, Jia Z, Mahaney JE, et al . The highly expressed and inducible endogenous NAD(P)H: quinone oxidoreductase 1 in cardiovascular cells acts as a potential superoxide scavenger. Cardiovasc Toxicol. 2007; 7: 202–11. [DOI] [PubMed] [Google Scholar]
- 16. Dinkova‐Kostova AT, Liby KT, Stephenson KK, et al . Extremely potent triterpenoid inducers of the phase 2 response: correlations of protection against oxidant and inflammatory stress. Proc Natl Acad Sci USA. 2005; 102: 4584–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhu H, Zhang L, Xi X, et al . 4‐Hydroxy‐2‐nonenal upregulates antioxidants and phase 2 enzymes in rat H9c2 myocardiac cells: protection against overt oxidative and electrophilic injury. Free Radic Res. 2006; 40: 875–84. [DOI] [PubMed] [Google Scholar]
- 18. Rubenfeld GD, Caldwell E, Granton J, et al . Interobserver variability in applying a radiographic definition for ARDS. Chest. 1999; 116: 1347–53. [DOI] [PubMed] [Google Scholar]
- 19. Gao L, Grant A, Halder I, et al . Novel polymorphisms in the myosin light chain kinase gene confer risk for acute lung injury. Am J Respir Cell Mol Biol. 2006; 34: 487–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gong MN, Zhou W, Williams PL, et al . Polymorphisms in the mannose binding lectin‐2 gene and acute respiratory distress syndrome. Crit Care Med. 2007; 35: 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jerng J‐S, Yu C‐J, Wang H‐C, et al . Polymorphism of the angiotensin‐converting enzyme gene affects the outcome of acute respiratory distress syndrome. Crit Care Med. 2006: 34: 1001–6. [DOI] [PubMed] [Google Scholar]
- 22. Zhai R, Gong MN, Zhou W, et al . Genotypes and haplotypes of the VEGF gene are associated with higher mortality and lower VEGF plasma levels in patients with ARDS. Thorax. 2007; 62: 718–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marzec JM, Christie JD, Reddy SP, et al . Functional polymorphisms in the transcription factor NRF2 in humans increase the risk of acute lung injury. FASEB J. 2007; 21: 2237–46. [DOI] [PubMed] [Google Scholar]
- 24. Siegel D, Gustafson DL, Dehn DL, et al . NAD(P)H:quinone oxidoreductase 1: role as a superoxide scavenger. Mol Pharmacol. 2004; 65: 1238–47. [DOI] [PubMed] [Google Scholar]
- 25. Merker MP, Bongard RD, Krenz GS, et al . Impact of pulmonary arterial endothelial cells on duroquinone redox status. Free Radic Biol Med. 2004; 37: 86–103. [DOI] [PubMed] [Google Scholar]
- 26. Cho HY, Reddy SP, DeBiase A, et al . Gene expression profiling of NRF2‐mediated protection against oxidative injury. Free Radic Biol Med. 2005; 38: 325–43. [DOI] [PubMed] [Google Scholar]
- 27. Wang X, Tomso DJ, Chorley BN, et al . Identification of polymorphic antioxidant response elements in the human genome. Hum Mol Genet. 2007; 16: 1188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Heinemeyer T, Wingender E, Reuter I, et al . Databases on transcriptional regulation: TRANSFAC, TRRD and COMPEL. Nuc Acids Res. 1998; 26: 362–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Katsuoka F, Motohashi H, Ishii T, et al . Genetic evidence that small maf proteins are essential for the activation of antioxidant response element‐dependent genes. Mol Cell Biol. 2005; 25: 8044–51. [DOI] [PMC free article] [PubMed] [Google Scholar]