

Abstract
One general principle of gene regulation is that DNA-binding transcription factors modulate transcription by recruiting cofactors that modify histones and chromatin structure. A second implicit principle is that a particular cofactor is necessary at all the target genes where the cofactor is recruited. Increasingly, these principles do not appear to be absolute, as experimentally defined relationships between transcription, cofactors, and chromatin modification grow in complexity. The KAT3 histone acetyltransferases CREB binding protein (CBP) and p300 have at least 400 interacting protein partners, thereby acting as hubs in gene regulatory networks. Studies using mutant primary cells indicate that the occurrence of CBP and p300 at any given target gene sometimes correlates with, rather than dictates transcription. This suggests that there are unexpected levels of redundancy between CBP/p300 and other unrelated coactivators, or that CBP/p300 recruitment may sometimes be coincidental. A transcription factor may therefore recruit the same group of coactivators as part of its “toolbox”, but it is the characteristics of the individual target gene that determine which coactivation “tools” are required for its transcription.
Keywords: CBP, p300, histones, chromatin, transcription, coactivators, HAT
Traditional models of transcriptional activator and coactivator function
The transcriptional activation and repression of thousands of genes dictates and defines the differentiated state of the hundreds of cell types in mammals. Along with about 2,000 DNA-binding transcription factors, there are estimated to be more than 100 transcriptional coactivators and corepressors that regulate the expression of about 20,000 protein coding genes in the human genome.1 At present, it is not entirely clear why there is so much potential for combinatorial complexity in mammalian transcriptional regulation. Probably a large variety of available coactivators provides redundancy, combinatorial regulatory potential, and tissue- and signal-specific regulation.2–4
Specific DNA sequences and the transcription factors that bind to them are the major regulators of gene expression. Bound at the gene promoter or enhancer, the activation (or repression) domains of the transcription factor recruit transcriptional cofactors such as coactivators or corepressors by protein-protein interactions. It is generally thought that the correctly positioned enzymatic and protein-binding activities of these coactivators stimulate transcription.5 Patterns of gene expression then arise largely from synergistic or antagonistic interactions that occur when various combinations of DNA-bound transcription factors and cofactors assemble at each gene. In this scenario, transcriptional coactivators are largely utilitarian, providing enzymatic or adaptor functions to the regulatory-specificity defined by DNA-bound transcription factors. Implicit in these traditional models of transcription factor function is the idea that coactivators contribute to the transcription of all the target genes to which they are recruited. Hence the recent interest in mapping the occurrence of transcription factors, cofactors, and histone modifications in the genome using methods such as chromatin immunoprecipitation coupled with deep DNA sequencing (ChIP-seq).6–8
Less-traditional models of coactivator function
In contrast to traditional models of transactivation above, increasing evidence supports a model where coactivators may be recruited to promoters but are unequally utilized for the expression of genes in mammals. Recently for example, Toll-like receptor-responsive promoters that are rich in CpG dinucleotides were found to assemble unstable nucleosomes, which reduces their dependence on SWI/SNF nucleosome remodeling complexes.9 In the case of CBP and p300, some hypoxia-responsive genes recruit but do not appear to require these histone acetyltransferases (HATs) for expression, and some NF-κB targets recruit CBP/p300 when not transcribed.3, 10, 11 A specific interaction surface on CBP/p300 for the cAMP-responsive factor CREB is also differentially required for individual cAMP-responsive genes.12 That coactivators like CBP/p300 provide gene-specific functions suggests another reason for cofactor diversity in mammals. Moreover, such non-customary models for coactivation are unexpected given how cofactors like CBP and p300 are believed to function biochemically. In the next several sections we will review aspects of HATs and histone acetylation to help place them in the context of these unconventional models of coactivation function.
CBP and p300 constitute the KAT3 family of HATs
There are four main multi-gene families of mammalian HATs based on sequence similarity (Ensembl database): GCN5 and PCAF (Gcn5l2 and Pcaf in mice), the MYST family (Htatip, Myst1, Myst2, Myst3, and Myst4), the nuclear (or steroid) receptor coactivator family (Ncoa1, Ncoa2, Ncoa3, and sometimes including Clock) and the CBP and p300 family (Crebbp and Ep300) (Fig. 1).13 In 2007, HATs were reclassified as KATs (lysine or K-acetyltransferases) to reflect their varied protein substrates and grouped into families similar to that in Ensembl (Fig. 1).13 While HAT family members tend to share a high degree of sequence similarity, HAT domain sequences (and that of most other domains) are very dissimilar between families.14 Such divergence between HAT families suggests that they evolved for functions distinct from just the acetylation of histones.
Figure 1.
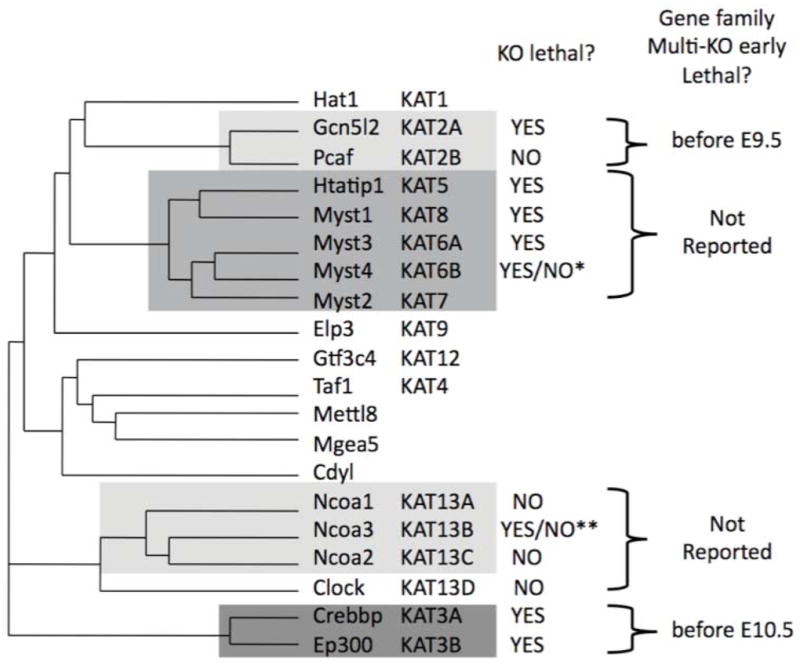
Phylogram of 20 mammalian HAT proteins by clustalW sequence alignment and their KAT nomenclature. Shaded boxes indicate the gene families as defined by Ensembl. Reported lethality phenotypes are indicated for individual gene knockout mouse models and where possible for multi-gene family knockouts. * 2/3 of mice die by 1 month of age. **Increased mortality throughout lifespan.
CBP (Crebbp) and p300 (Ep300) encode highly related protein acetyltransferases that possess several conserved protein-binding domains [i.e., RID, CH1 (TAZ1), KIX, Bromodomain, PHD, HAT, ZZ, CH3 (TAZ2), and IBiD (NCBD)] that bind a variety of transcriptional regulators and other proteins (Figure 2).15, 16 Indeed, distinct activators require different regions of p300 in vitro.17 Both CBP and p300 occur in mammals, whereas Drosophila and C. elegans have only CBP, and yeast has neither.
Figure 2.

CBP and p300 are closely related HATs that possess unique protein binding domains. Principal protein-binding domains of CBP and p300: nuclear receptor interaction domain (RID), the Cys/His-rich region 1 (CH1), the CREB-binding domain (KIX), bromodomain (Br), plant homeodomain (PHD), histone acetyltransferase domain (HAT), zinc-binding domain near the dystrophin WW domain (ZZ), the Cys/His-rich region 3 (CH3), and the interferon response factor-binding domain (IBiD).
The CBP-p300 interactome includes 400 interacting protein partners
CBP and p300 have at least 400 described interacting protein partners, making them among the most heavily connected nodes in the known mammalian protein-protein interactome (Table 1, internet search “CBP-p300 interactome” for an updated list with references). Analysis of global transcription networks in model organisms indicates that proteins that act as nodes or “hubs” are more likely to be encoded by essential genes.18 Indeed, consistent with a role as hubs, both CBP and p300 are required for normal development and have been implicated in human disease.
Table 1.
The CBP and p300 interactome. 400 mammalian and viral proteins reported to interact physically or functionally with the KAT3 family members of CBP and p300. An updated and referenced list can be downloaded at www.stjude.org/brindle.
| 53BP1 | CtBP1 | H4 | MN1 | PGC-1a | SREBP1a |
| A-Myb | Ctbp2 | HBZ | MR | PHOX2B | SREBP2 |
| A238L | Cyclin D1 | HCV core protein | Mre11 | PIAS1 | SRF |
| Actin | Cyclin E-Cdk2 | HDAC1 | MSG1 | PIAS3 | STAT1a |
| AhR | Dach1 | HDAC6 | Msh2 | PIMT | STAT2 |
| AIRE | Daxx | HEB | Msh6 | Pit-1 | STAT3 |
| Akt | DBP | Hes6 | Msx1 | PLAG1 | STAT4 |
| AMF-1 | DDX24 | hHR23A | Msx3 | PLAGL2 | STAT5a |
| AML1 | dHAND | Hic-5 | MTF-1 | PLZF | STAT5b |
| AP-2 | E1A | HIF-1a | MyoD | PML | STAT6 |
| Ap1b1 | E2 | HIPK2 | mZac1b | Pol beta | Strap |
| APC5 | E2F-1 | HLF | N-Cadherin | POLR2A | Sug1 |
| APC7 | E2F-5 | HNF-6 | N-CoR | Polyoma large T | SV40 large T |
| APE1 | E47 | HNF1a | NAP-1 | pp90 RSK | SYT |
| AR | E6 | HNF1beta | NAP-2 | PPARa | T-bet |
| Arnt | EBF | HNF4a | NBS1 | PPARg | TAF1 |
| ATF-1 | EBNA2 | HOXA10 | NEDD1 | PR | TAFII68 |
| ATF-2 | EBNA3C | HOXA11 | NEMO | PRIC320 | TAL1 |
| ATF-4 | Egr-1 | HOXA9 | Neuro D | PRMT5 | Tat |
| ATG7 | EID-1 | HOXB1 | NF-E2 | ProTa | Tax |
| ATR | EID-2 | HOXB2 | NF-kB p50 | Prox-1 | TAZ |
| atrophin-1 | EID3 | HOXB3 | NF-kB p65 | PRS1 | TBP |
| b-catenin | Eif2b1 | HOXB4 | NF-YA | PTEN | TCF-4N |
| B-Myb | EKLF | HOXB6 | NFAT1 | Ptf1a | TCL1 |
| B56gamma3 | Elk-1 | HOXB7 | NFATc1 | PU.1 | TDG |
| BAF47 | Emb | HOXB9 | NFATc4 | pVHL | TFIIB |
| Bat3 | ER81 | HOXD10 | NFI | Rad50 | TFIIEb |
| BCL6 | ERa | HOXD12 | NFY-B | RAR | TFIIF |
| beta-TrCp1/Fbw1a | ERK2 | HOXD13 | Ngn1 | Rb | TFIIIC |
| bICP0 | ESE-1 | HOXD4 | Nished | RbAp48 | TIF2 |
| BRCA1 | Ets-1 | HPV E7 | Notch-1 | RBCK1 | TIP-1 |
| BRG1 | Ets-2 | HSF1 | Nrf2 | Rch1 | TIP-2 |
| Bridge-1 | EVI1 | htt | NS1 | RECQL4 | TIP-3 |
| BRLF1 | EWS | IKK-alpha | Nup93 | RIP140 | TIP-4 |
| Brn-2 | Eya1 | importin-a7 | Nup98 | RNA helicase A | TIP-5 |
| c-Fos | Eya3 | ING2 | Nur77 | ROCK2 | TIP-6 |
| c-Jun | Fbx3 | IRF-1 | OGG1 | RORalpha | TIP-7 |
| c-Maf | Fen1 | IRF-2 | OLIG2 | RSK2 | TIP-8 |
| c-Myb | FGFR1 | IRF-3 | P-Lim | Runx2 | TLS |
| c-Myc | FHL2 | IRF-7 | P/CAF | Runx3 | TLX1 |
| C/EBPa | FKLF2 | JDP2 | p/CIP | RXR | TP2 |
| C/EBPb | FOG-2 | JMY | p100 | Sam68 | TR beta |
| C/EBPd | FoxA2 | JunB | p220(NPAT) | Sap-1a | TRBP |
| C53 | Foxl1 | K8/K-bZIP | p28ING5 | SATB1 | TReP-132 |
| CARM1 | FoxM1B | KLF2 | p29ING4 | SF-1 | Trip10 |
| Cart1 | FOXO1a | KLF4 | p30(II) | SGK1 | TSG101 |
| CAS | FOXO3a | KLF5 | p33ING1b | Sh3gl1 | TTF-1 |
| CD44 | FOXO4 | KLF6 | p34SEI-1 | SIRT1 | Twist |
| Cdc25B | FXR | Ku70 | p35srj | SIRT2 | UBF |
| CDK8 | GABPa | MafG | p38 MAPK | SKIP | VDR |
| CDP | Gak | MAM | p53 | Skn-1a | vIRF-1 |
| cdx-2 | GAPDH | MAML2 | P532 | Skp2 | VP16 |
| CFIm25 | GATA-1 | mArnt3 | p63 | Smad1 | Vpr |
| CHC | GATA-2 | MASH1 | p68 RNA helicase | Smad2 | VRK1 |
| CIITA | GATA-3 | MDC1 | p73 | Smad3 | VRK2 |
| CITED4 | GATA-4 | MDM2 | p8 | Smad4 | WRN |
| CK2 | GATA-5 | MED25 | PAP | Smad7 | WT1 |
| Cnot3 | GATA-6 | MEF-2D | PARP-1 | SMIF | XAF1 |
| CoAA | GCMa | MEF2C | pax-6 | SNIP1 | YAP |
| CoCoA | GCN5 | MEKK1 | Pax3 | Sox-2 | YB-1 |
| COUP-TF II | Gli3 | mGPBP | Pax5 | SOX4 | YY1 |
| CPAP | GMEB-1 | Mi | Pax8 | Sox9 | ZBP-89 |
| CREB | GMEB-2 | Mi-2beta | PC4 | Sp1 | ZBTB2 |
| CREM | GR | MIER1 | PCNA | Sp3 | ZEB-1 |
| CREST | H2A | Miz-1 | PDX-1 | Spi-B | ZNF639 |
| CRTC2 | H2B | MKP-1 | PEA3 | SRC1 | |
| CRX | H3 | MLL | PELP1 | SRCAP |
CBP and p300 mutations in Rubinstein-Taybi Syndrome (RTS)
RTS is a congenital developmental disorder, characterized by mental retardation, broad toes and thumbs, short stature, and facial anomalies.19 In 1995, Petrij et al. identified RTS patients with heterozygous mutations in CBP, indicating that RTS is caused by a partial deficiency of CBP protein (i.e. haploinsufficiency).20 A screen of 92 RTS patients revealed that 36 had mutations in CBP (including five missense mutations in the conserved HAT region) and three had mutations in p300.21 The insufficient or altered functions of mutant CBP (and p300) that lead to RTS are unclear, although loss of HAT activity may be important.22
Consistent with critical roles in human development, CBP or p300 nullizygous mice die during embryogenesis (day E8–E11), as do compound heterozygotes.23, 24 The latter observation indicates that the combined amount of the two proteins is limiting. p300+/− mice tend to be small and less thrifty but are otherwise grossly normal (unpublished data).24 Conversely, CBP+/− mice exhibit traits in common with RTS, including growth retardation and craniofacial anomalies, showing that many CBP developmental functions are conserved in mice and man.25–30 Collectively, these studies support CBP and p300 as having gene network hub functions and reveal that they can have distinct roles in development.
Cells that lack CBP and p300
A cell type that can be stably sustained in the absence of both CBP and p300 has not yet been reported. The prevailing assumption is that some CBP or p300 protein is required for cell viability or proliferation, as shown for mouse lymphocytes in vivo.31, 32 Consistent with this view, RNA interference (RNAi) mediated knockdown of CBP and p300 in immortal HeLa cells results in cell death due to “mitotic catastrophe” and “chromosome shredding.”33 Similarly, RNAi knockdown of dCBP in Drosophila kc cells also leads to cell death.34 These RNAi studies indicate that this approach is not generally applicable for knocking down CBP/p300 in cell lines for transcription experiments. Two recent studies have transiently reduced CBP and p300 levels by RNAi knock-down to investigate histone acetylation.35, 36 Interestingly, these two papers show that the acetylation of histone H3 lysines K18 and K56 is highly dependent on CBP/p300, although effects on transcription were not reported.
Conditional knockout of CBP and p300
Since homozygous null mutations of either coactivator cause early embryonic lethality, the role of CBP and p300 in adult cell lineages remains largely unknown. Studies using conditional knockout CBPflox and p300flox alleles indicate that both proteins play essential but distinct roles in hematopoiesis. Both genes contribute to antigen receptor signaling-responsive gene expression in T and B cells.31, 32, 37 CBP and p300 are highly essential collectively but not individually for peripheral B cell homeostasis.31 However, deletion of p300 before the pro-B-cell stage, using a Mx-Cre transgene, remarkably reduced B-cell numbers. In contrast, loss of either CBP or p300 during early T cell development results in a decrease in CD4 CD8 double-positive thymocytes.32 Moreover, CBP mutant mice exhibit an increase in CD8 single positive thymocytes not seen in p300 mutants.32 In fact, CBP appears to be vital to demarcate conventional and innate CD8+ T-cell development.37 Conditional deletion of CBP has also provided insight into how it may function as a tumor suppressor. The T cell lymphomagenesis that results from loss of CBP in the T-cells of MMTV-Cre;CBPflox/flox mice occurs in synergy with p27 Kip1 insufficiency.38 In addition to immune cell function, CBP and/or p300 also play essential roles in renin cells and primordial germ cells.39, 40
Hypomorphic mutations in CBP and p300 show that their genome-unique domains are necessary for many but not all target genes
As CBP and p300 are essential for early mouse development, knock-in mutations in mice have been useful to further define their functions. Mutations that cause the loss of CBP or p300 histone acetyltransferase activity are dominant lethals that are detrimental to mouse development and transcription.41, 42 Mice have also been created with point mutations in the KIX domains of CBP and p300 that inhibit their ability to bind the hematopoietic determining factor c-Myb and the cyclic-AMP- and calcium-responsive factor CREB.43 The KIX domain of p300 is especially important for hematopoiesis, preventing the overproduction of platelets and megakaryocytes.43 An independent study revealed that the increased platelets and megakaryocytes exhibited by ENU-induced Plt6 mutant mice can be attributed to a Tyr to Asn substitution within the p300 KIX domain.44 Targeted point mutations in the CBP KIX domain43 highlight its importance in learning and memory, which are CREB-mediated processes.45–47 Investigation of cAMP-inducible genes in primary mouse embryonic fibroblasts (MEFs) entirely deficient for normal KIX domains, reveals an unexpected spectrum of transcriptional responses.12 While some cAMP-inducible genes are highly sensitive to the KIX mutations, others show only partial loss of activity or are unaffected. However, ChIP assays showed that cAMP-inducible recruitment of CBP or p300 to CREB target promoters was only partially blocked by the KIX mutation.12 Therefore, KIX domain-independent recruitment of CBP/p300 to CREB may provide sufficient coactivation function to genes that are less transcriptionally affected by the KIX mutation.
Mutation of Ser436 in the CBP CH1 domain has been shown to increase CREB activity and liver gluconeogenesis in mice, suggesting this residue may negatively control the interaction between CREB and CBP.48, 49 Indeed, phosphorylation of Ser436 is thought to be critical for lowering blood glucose in response to insulin or the anti-diabetic drug metformin by inhibiting the CREB-mediated transcription of gluconeogenic enzyme genes in the liver.50 Interestingly however, MEFs homozygous for a deletion mutation in the CBP CH1 domain, but that retain Ser436 showed no obvious effect on CREB activity in transient assays.11 In contrast, MEFs carrying the same knock-in deletions in both the CBP and p300 CH1 domain suggest that this region contributes to a sizable fraction of hypoxia-responsive gene expression.11 Moreover, the CH1 domain is critical for the efficient recruitment of CBP/p300 to HIF-target genes in response to hypoxia. Together, studies of CBP and p300 mutants have established their crucial roles in development and transcription, and have revealed that endogenous target genes do not have a uniform requirement for certain functions of the coactivators.
Mechanisms of transactivation by CBP and p300
It is thought that CBP and p300 can modulate transcription by five main mechanisms: acetylation of lysines in the N-terminal tails of histones, polyubiquitination51 and acetylation of specific lysines on other transcriptional regulators, recruiting components of the Pol II machinery, and acting as adaptors to recruit other cofactors (e.g., coactivators). Currently, histone acetylation is considered to be the most important, or at least the most universal, of these mechanisms. However, which of these (or other) mechanisms are critical for the transactivation of endogenous target genes remains uncertain.
Histone acetylation correlates strongly with active transcription
More than 100 different modifications have been detected for the histones H2A, H2B, H3, and H4 that form the octamer of the typical nucleosome.52 These include the eight main types of histone post-translational modifications, which are thought to alter chromatin structure or affect the recruitment of non-histone proteins: acetylation, methylation, phosphorylation, ubiquitinylation, SUMOylation, ADP ribosylation, deimination, and proline isomerization.53 The many different histone acetylation marks positively correlate with transcription52 and in human T cells, the global recruitment of CBP, p300, and other HATs correlates strongly with both histone acetylation and the level of gene expression.8
One popular model of transcription states that a series of histone modifications occurs during transcription initiation and elongation, and that each modification is necessary for full gene expression.54 Of all known modifications, acetylation would seem the most likely to “loosen” chromatin and facilitate transcription because it neutralizes the positive charge of lysines, thereby reducing the binding of histones to negatively charged DNA.53 In this charge-neutralization model, the total amount of histone acetylation rather than the modification of particular residues would be critical. More recently a cofactor recruitment model was described where the acetylation of lysines 5, 8, and 12 of histone H4 recruits bromodomain containing protein-4 (Brd4), which is thought to be important for the induced expression of primary signal-responsive genes.55
Histone modifications in mammals – essential for transcription or just correlative?
There is broad consensus that certain histone modifications correlate strongly with gene activity but recent models incorporate increasing complexity to explain how histone marks regulate transcription.54, 56 The multitude of histone modifications observed may indicate that they have one or more of these characteristics: 1) gene or context-restricted functions; 2) functional redundancy; or 3) status as non-functional “bystander” marks caused by enzymes that modify nonhistone proteins or are performing other functions.52 Multiple alleles of each histone gene in mammals makes testing these models in vivo by mutagenesis extremely difficult, if not currently impossible.
Effects of histone mutations on gene expression in yeast
The extent to which any of the characteristics proposed above applies to histone acetylation in mammals is unclear. However, clues to the importance of histone modification for gene expression can be found from studies in baker’s yeast. The viability of yeast strains with certain histone N-terminal tail deletion mutations, argues against a universal requirement for histone acetylation in gene regulation.57–64 Moreover, expression defects are typically more gene-specific than genome-wide in these yeast mutants. Mutation of individual residues in histones H2A, H2B, H3, and H4 can also result in surprisingly moderate or specific defects in transcription.61, 62, 65, 66 As for histone lysine acetylation, the individual mutation of K5, K8, or K12 of histone H4 in yeast has minor effects on transcription, whereas K16 has more unique functions.65, 67 Combining these H4 mutations leads to cumulative changes in the expression of a group of genes, suggesting that acetylation can also act by affecting overall histone charge. Extrapolating these findings to mammals suggests that histone acetylation and (by inference) HATs are also not universally required for transcription.
Histone acetylating enzymes and activated transcription
What about the roles of histone-acetylating enzymes if it is uncertain why histone acetylation correlates strongly with active transcription? There are 13 known HAT genes in yeast, but only ELP3 and TAF1 are essential for cell viability, indicating that most, or perhaps all, HATs have redundant or specialized transcriptional functions (source: Saccharomyces Genome Database). It is unknown whether this is also true for mice. To date, mutant phenotypes for only 12 of the 20 known HAT genes have been reported (source: Jackson Laboratories, Mouse Genome Informatics (MGI)). Since mammalian HATs are often individually essential for embryo development (which may be for a specific requirement in an essential cell type or for widespread requirements in many cells and processes), investigating the transcriptional roles of entire HAT families using conventional gene knockouts has been problematic or impossible. Certainly, the MGI database only reports phenotypes for mice with mutations in HAT genes that belong to the four major families defined in Figure 1. Moreover, the single instance in which both members of a HAT family were knocked out (GCN5 and PCAF) resulted in early embryonic lethality, preventing transcriptional analysis (Fig. 1).68 Individually, CBP and p300 knockouts (and compound heterozygotes) lead to embryonic lethality by E10.5 and anecdotal reports indicate the double-null phenotype is more severe, also preventing transcriptional analysis of this entire family.24
Conclusions
Histone N-terminal tail acetylation is a class of chromatin modification that strongly correlates with active transcription. Yet, the crucial functions that are fulfilled by HATs to stimulate transcription remain uncertain in mammals. There are at least 20 known HATs in mice and 13 of these belong to four multi-gene families. These HAT families share surprisingly little sequence identity within the acetyltransferase domain, and they have major differences in other functional domains. This diversity amongst HATs suggests that the bulk acetylation of histones is not their key function in gene activation. Moreover the recruitment of HATs such as CBP and p300 at a gene may not equate with a functional requirement for transcription. This suggests there is an unexpected level of redundancy between seemingly dissimilar transactivating cofactors and mechanisms.
Acknowledgments
We apologize to those whose work could not be cited because of limited space. Supported by NIH grant DE018183 (P.K.B), Cancer Center (CORE) support grant P30 CA021765, and the American Lebanese Syrian Associated Charities of St. Jude Children’s Research Hospital. The authors do not have any conflicts of interest to declare.
References
- 1.Messina DN, Glasscock J, Gish W, Lovett M. An ORFeome-based analysis of human transcription factor genes and the construction of a microarray to interrogate their expression. Genome Res. 2004;14:2041–7. doi: 10.1101/gr.2584104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spiegelman BM, Heinrich R. Biological control through regulated transcriptional coactivators. Cell. 2004;119:157–67. doi: 10.1016/j.cell.2004.09.037. [DOI] [PubMed] [Google Scholar]
- 3.Kasper LH, Brindle PK. Mammalian gene expression program resiliency: the roles of multiple coactivator mechanisms in hypoxia-responsive transcription. Cell Cycle. 2006;5:142–6. doi: 10.4161/cc.5.2.2353. [DOI] [PubMed] [Google Scholar]
- 4.Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006;20:1405–28. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- 5.Ptashne M. Words. Curr Biol. 2007;17:R533–5. doi: 10.1016/j.cub.2007.05.071. [DOI] [PubMed] [Google Scholar]
- 6.Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–8. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–17. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 8.Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, et al. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–31. doi: 10.1016/j.cell.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramirez-Carrozzi VR, Braas D, Bhatt DM, Cheng CS, Hong C, Doty KR, et al. A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell. 2009;138:114–28. doi: 10.1016/j.cell.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leung TH, Hoffmann A, Baltimore D. One nucleotide in a kappaB site can determine cofactor specificity for NF-kappaB dimers. Cell. 2004;118:453–64. doi: 10.1016/j.cell.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 11.Kasper LH, Boussouar F, Boyd K, Xu W, Biesen M, Rehg J, et al. Two transactivation mechanisms cooperate for the bulk of HIF-1-responsive gene expression. Embo J. 2005;24:3846–58. doi: 10.1038/sj.emboj.7600846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu W, Kasper LH, Lerach S, Jeevan T, Brindle PK. Individual CREB-target genes dictate usage of distinct cAMP-responsive coactivation mechanisms. Embo J. 2007;26:2890–903. doi: 10.1038/sj.emboj.7601734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allis CD, Berger SL, Cote J, Dent S, Jenuwien T, Kouzarides T, et al. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131:633–6. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 14.Marmorstein R. Structure of histone acetyltransferases. J Mol Biol. 2001;311:433–44. doi: 10.1006/jmbi.2001.4859. [DOI] [PubMed] [Google Scholar]
- 15.Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–77. [PubMed] [Google Scholar]
- 16.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 17.Kraus WL, Manning ET, Kadonaga JT. Biochemical analysis of distinct activation functions in p300 that enhance transcription initiation with chromatin templates. Mol Cell Biol. 1999;19:8123–35. doi: 10.1128/mcb.19.12.8123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeong H, Mason SP, Barabasi AL, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001;411:41–2. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- 19.Cantani A, Gagliesi D. Rubinstein-Taybi syndrome. Review of 732 cases and analysis of the typical traits. Eur Rev Med Pharmacol Sci. 1998;2:81–7. [PubMed] [Google Scholar]
- 20.Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RCM, Masuno M, et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–51. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- 21.Roelfsema JH, White SJ, Ariyurek Y, Bartholdi D, Niedrist D, Papadia F, et al. Genetic heterogeneity in Rubinstein-Taybi syndrome: mutations in both the CBP and EP300 genes cause disease. Am J Hum Genet. 2005;76:572–80. doi: 10.1086/429130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kalkhoven E, Roelfsema JH, Teunissen H, den Boer A, Ariyurek Y, Zantema A, et al. Loss of CBP acetyltransferase activity by PHD finger mutations in Rubinstein-Taybi syndrome. Hum Mol Genet. 2003;12:441–50. doi: 10.1093/hmg/ddg039. [DOI] [PubMed] [Google Scholar]
- 23.Tanaka Y, Naruse I, Hongo T, Xu M, Nakahata T, Maekawa T, et al. Extensive brain hemorrhage and embryonic lethality in a mouse null mutant of CREB-binding protein. Mech Dev. 2000;95:133–45. doi: 10.1016/s0925-4773(00)00360-9. [DOI] [PubMed] [Google Scholar]
- 24.Yao TP, Oh SP, Fuchs M, Zhou ND, Ch’ng LE, Newsome D, et al. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell. 1998;93:361–72. doi: 10.1016/s0092-8674(00)81165-4. [DOI] [PubMed] [Google Scholar]
- 25.Tanaka Y, Naruse I, Maekawa T, Masuya H, Shiroishi T, Ishii S. Abnormal skeletal patterning in embryos lacking a single Cbp allele: a partial similarity with Rubinstein-Taybi syndrome. Proc Natl Acad Sci U S A. 1997;94:10215–20. doi: 10.1073/pnas.94.19.10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korzus E, Rosenfeld MG, Mayford M. CBP Histone Acetyltransferase Activity Is a Critical Component of Memory Consolidation. Neuron. 2004;42:961–72. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alarcon JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, et al. Chromatin Acetylation, Memory, and LTP Are Impaired in CBP(+/−) Mice; A Model for the Cognitive Deficit in Rubinstein-Taybi Syndrome and Its Amelioration. Neuron. 2004;42:947–59. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 28.Bourtchouladze R, Lidge R, Catapano R, Stanley J, Gossweiler S, Romashko D, et al. A mouse model of Rubinstein-Taybi syndrome: defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4. Proc Natl Acad Sci U S A. 2003;100:10518–22. doi: 10.1073/pnas.1834280100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oike Y, Hata A, Mamiya T, Kaname T, Noda Y, Suzuki M, et al. Truncated CBP protein leads to classical Rubinstein-Taybi syndrome phenotypes in mice: implications for a dominant-negative mechanism. HumMolGenet. 1999;8:387–96. doi: 10.1093/hmg/8.3.387. [DOI] [PubMed] [Google Scholar]
- 30.Kung AL, Rebel VI, Bronson RT, Ch’ng LE, Sieff CA, Livingston DM, et al. Gene dose-dependent control of hematopoiesis and hematologic tumor suppression by CBP. Genes Dev. 2000;14:272–7. [PMC free article] [PubMed] [Google Scholar]
- 31.Xu W, Fukuyama T, Ney PA, Wang D, Rehg J, Boyd K, et al. Global transcriptional coactivators CREB-binding protein and p300 are highly essential collectively but not individually in peripheral B cells. Blood. 2006;107:4407–16. doi: 10.1182/blood-2005-08-3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kasper LH, Fukuyama T, Biesen MA, Boussouar F, Tong C, de Pauw A, et al. Conditional knockout mice reveal distinct functions for the global transcriptional coactivators CBP and p300 in T-cell development. Mol Cell Biol. 2006;26:789–809. doi: 10.1128/MCB.26.3.789-809.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stauffer D, Chang B, Huang J, Dunn A, Thayer M. p300/CREB-binding protein interacts with ATR and is required for the DNA replication checkpoint. J Biol Chem. 2007;282:9678–87. doi: 10.1074/jbc.M609261200. [DOI] [PubMed] [Google Scholar]
- 34.Smolik S, Jones K. Drosophila dCBP Is Involved in Establishing the DNA Replication Checkpoint. Mol Cell Biol. 2007;27:135–46. doi: 10.1128/MCB.01283-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horwitz GA, Zhang K, McBrian MA, Grunstein M, Kurdistani SK, Berk AJ. Adenovirus small e1a alters global patterns of histone modification. Science. 2008;321:1084–5. doi: 10.1126/science.1155544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Das C, Lucia MS, Hansen KC, Tyler JK. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature. 2009;459:113–7. doi: 10.1038/nature07861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fukuyama T, Kasper LH, Boussouar F, Jeevan T, van Deursen J, Brindle PK. Histone acetyltransferase CBP is vital to demarcate conventional and innate CD8+ T-cell development. Mol Cell Biol. 2009;29:3894–904. doi: 10.1128/MCB.01598-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kang-Decker N, Tong C, Boussouar F, Baker DJ, Xu W, Leontovich AA, et al. Loss of CBP causes T cell lymphomagenesis in synergy with p27(Kip1) insufficiency. Cancer Cell. 2004;5:177–89. doi: 10.1016/s1535-6108(04)00022-4. [DOI] [PubMed] [Google Scholar]
- 39.Gomez RA, Pentz ES, Jin X, Cordaillat M, Sequeira Lopez ML. CBP and p300 are essential for renin cell identity and morphological integrity of the kidney. Am J Physiol Heart Circ Physiol. 2009;296:H1255–62. doi: 10.1152/ajpheart.01266.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Elliott AM, de Miguel MP, Rebel VI, Donovan PJ. Identifying genes differentially expressed between PGCs and ES cells reveals a role for CREB-binding protein in germ cell survival. Dev Biol. 2007;311:347–58. doi: 10.1016/j.ydbio.2007.08.029. [DOI] [PubMed] [Google Scholar]
- 41.Roth JF, Shikama N, Henzen C, Desbaillets I, Lutz W, Marino S, et al. Differential role of p300 and CBP acetyltransferase during myogenesis: p300 acts upstream of MyoD and Myf5. Embo J. 2003;22:5186–96. doi: 10.1093/emboj/cdg473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shikama N, Lutz W, Kretzschmar R, Sauter N, Roth JF, Marino S, et al. Essential function of p300 acetyltransferase activity in heart, lung and small intestine formation. Embo J. 2003;22:5175–85. doi: 10.1093/emboj/cdg502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kasper LH, Boussouar F, Ney PA, Jackson CW, Rehg J, van Deursen JM, et al. A transcription-factor-binding surface of coactivator p300 is required for haematopoiesis. Nature. 2002;419:738–43. doi: 10.1038/nature01062. [DOI] [PubMed] [Google Scholar]
- 44.Kauppi M, Murphy JM, de Graaf CA, Hyland CD, Greig KT, Metcalf D, et al. Point mutation in the gene encoding p300 suppresses thrombocytopenia in Mpl−/− mice. Blood. 2008;112:3148–53. doi: 10.1182/blood-2007-10-119677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oliveira AM, Abel T, Brindle PK, Wood MA. Differential Role for CBP and p300 CREB-Binding Domain in Motor Skill Learning. Behav Neurosci. 2006;120:724–9. doi: 10.1037/0735-7044.120.3.724. [DOI] [PubMed] [Google Scholar]
- 46.Wood MA, Attner MA, Oliveira AM, Brindle PK, Abel T. A transcription factor-binding domain of the coactivator CBP is essential for long-term memory and the expression of specific target genes. Learn Mem. 2006;13:609–17. doi: 10.1101/lm.213906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, et al. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB: CBP-dependent transcriptional activation. J Neurosci. 2007;27:6128–40. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou XY, Shibusawa N, Naik K, Porras D, Temple K, Ou H, et al. Insulin regulation of hepatic gluconeogenesis through phosphorylation of CREB-binding protein. Nat Med. 2004;10:633–7. doi: 10.1038/nm1050. [DOI] [PubMed] [Google Scholar]
- 49.Hussain MA, Porras DL, Rowe MH, West JR, Song WJ, Schreiber WE, et al. Increased pancreatic beta-cell proliferation mediated by CREB binding protein gene activation. Mol Cell Biol. 2006;26:7747–59. doi: 10.1128/MCB.02353-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.He L, Sabet A, Djedjos S, Miller R, Sun X, Hussain MA, et al. Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell. 2009;137:635–46. doi: 10.1016/j.cell.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi D, Pop MS, Kulikov R, Love IM, Kung AL, Grossman SR. CBP and p300 are cytoplasmic E4 polyubiquitin ligases for p53. Proc Natl Acad Sci U S A. 2009;106:16275–80. doi: 10.1073/pnas.0904305106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rando OJ, Chang HY. Genome-wide views of chromatin structure. Annual review of biochemistry. 2009;78:245–71. doi: 10.1146/annurev.biochem.78.071107.134639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 54.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–12. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 55.Hargreaves DC, Horng T, Medzhitov R. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell. 2009;138:129–45. doi: 10.1016/j.cell.2009.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rando OJ. Global patterns of histone modifications. Current opinion in genetics & development. 2007;17:94–9. doi: 10.1016/j.gde.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 57.Durrin LK, Mann RK, Kayne PS, Grunstein M. Yeast histone H4 N-terminal sequence is required for promoter activation in vivo. Cell. 1991;65:1023–31. doi: 10.1016/0092-8674(91)90554-c. [DOI] [PubMed] [Google Scholar]
- 58.Thompson JS, Ling X, Grunstein M. Histone H3 amino terminus is required for telomeric and silent mating locus repression in yeast. Nature. 1994;369:245–7. doi: 10.1038/369245a0. [DOI] [PubMed] [Google Scholar]
- 59.Wallis JW, Rykowski M, Grunstein M. Yeast histone H2B containing large amino terminus deletions can function in vivo. Cell. 1983;35:711–9. doi: 10.1016/0092-8674(83)90104-6. [DOI] [PubMed] [Google Scholar]
- 60.Kayne PS, Kim UJ, Han M, Mullen JR, Yoshizaki F, Grunstein M. Extremely conserved histone H4 N terminus is dispensable for growth but essential for repressing the silent mating loci in yeast. Cell. 1988;55:27–39. doi: 10.1016/0092-8674(88)90006-2. [DOI] [PubMed] [Google Scholar]
- 61.Parra MA, Wyrick JJ. Regulation of gene transcription by the histone H2A N-terminal domain. Mol Cell Biol. 2007;27:7641–8. doi: 10.1128/MCB.00742-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parra MA, Kerr D, Fahy D, Pouchnik DJ, Wyrick JJ. Deciphering the roles of the histone H2B N-terminal domain in genome-wide transcription. Mol Cell Biol. 2006;26:3842–52. doi: 10.1128/MCB.26.10.3842-3852.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Blackwell JS, Jr, Wilkinson ST, Mosammaparast N, Pemberton LF. Mutational analysis of H3 and H4 N termini reveals distinct roles in nuclear import. J Biol Chem. 2007;282:20142–50. doi: 10.1074/jbc.M701989200. [DOI] [PubMed] [Google Scholar]
- 64.Formosa T, Ruone S, Adams MD, Olsen AE, Eriksson P, Yu Y, et al. Defects in SPT16 or POB3 (yFACT) in Saccharomyces cerevisiae cause dependence on the Hir/Hpc pathway: polymerase passage may degrade chromatin structure. Genetics. 2002;162:1557–71. doi: 10.1093/genetics/162.4.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dion MF, Altschuler SJ, Wu LF, Rando OJ. Genomic characterization reveals a simple histone H4 acetylation code. Proc Natl Acad Sci U S A. 2005;102:5501–6. doi: 10.1073/pnas.0500136102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martin AM, Pouchnik DJ, Walker JL, Wyrick JJ. Redundant roles for histone H3 N-terminal lysine residues in subtelomeric gene repression in Saccharomyces cerevisiae. Genetics. 2004;167:1123–32. doi: 10.1534/genetics.104.026674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annual review of biochemistry. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- 68.Yamauchi T, Yamauchi J, Kuwata T, Tamura T, Yamashita T, Bae N, et al. Distinct but overlapping roles of histone acetylase PCAF and of the closely related PCAF-B/GCN5 in mouse embryogenesis. Proc Natl Acad Sci U S A. 2000;97:11303–6. doi: 10.1073/pnas.97.21.11303. [DOI] [PMC free article] [PubMed] [Google Scholar]