

Abstract
Mps1 is one of the several essential kinases whose activation is required for robust mitotic spindle checkpoint signalling. The activity of Mps1 is tightly regulated and increases dramatically during mitosis or in response to spindle damage. To understand the molecular mechanism underlying Mps1 regulation, we determined the crystal structure of the kinase domain of Mps1. The 2.7‐Å‐resolution crystal structure shows that the Mps1 kinase domain adopts a unique inactive conformation. Intramolecular interactions between the key Glu residue in the αC helix of the N‐terminal lobe and the backbone amides in the catalytic loop lock the kinase in the inactive conformation. Autophosphorylation appears to be a priming event for kinase activation. We identified Mps1 autophosphorylation sites in the activation and the P+1 loops. Whereas activation loop autophosphorylation enhances kinase activity, autophosphorylation at the P+1 loop (T686) is associated with the active kinase. Mutation of T686 autophosphorylation site impairs both autophosphorylation and transphosphorylation. Furthermore, we demonstrated that phosphorylation of T676 may be a priming event for phosphorylation at T686. Finally, we identified two critical lysine residues in the loop between helices αEF and αF that are essential for substrate recruitment and maintaining high levels of kinase activity. Our studies reveal critical biochemical mechanisms for Mps1 kinase regulation.
Keywords: Mps1 structure, kinase activation, phosphorylation
Introduction
Aneuploidy is a hallmark of cancer cells in which chromosomes are inappropriately partitioned between daughter cells due to aberrant mitosis. Faithful segregation of chromosomes during each cell division is normally ensured by the mitotic spindle checkpoint, which delays the onset of anaphase until every chromosome has successfully attached to the spindles [1, 2, 3]. The mitotic spindle checkpoint is highly robust as a single unattached kinetochore at the metaphase plate is sufficient to trigger mitotic checkpoint response leading to delay the onset of anaphase. Defects in the mitotic spindle checkpoint pathway are frequently observed in human cancer cells and weakened checkpoint signalling is postulated to be responsible for generating aneuploid cells.
Genetic analysis of the mitotic spindle checkpoint signalling pathway in yeast uncovered many key components of this pathway [1, 4, 5, 6, 7]. Subsequent studies indicated that the mitotic checkpoint is an evolutionarily conserved mechanism that responds to kinetochore attachment or ‘tension’ and possibly spindle alignment. Mammalian homologues of mitotic checkpoint components have been identified, which include Bub1, BubR1, Bub3, Mad1, Mad2 and Mps1 [7, 8]. In addition, the ZW10‐ROD‐zwilch complex, the microtubule motor protein centromere protein E and mitogen‐activated protein kinase also appear to be important for a robust mitotic checkpoint response [8].
The mechanisms by which these signalling components work together to generate the checkpoint signal are incompletely defined. Also unclear is the exact identity of the kinetochore‐produced inhibitory signal(s). Proposed signals include activated Mad2, BubR1 or Bub1 or a complex of Cdc20, Mad2, BubR1 and Bub3 [9, 10, 11]. A prevailing model for mammalian mitotic checkpoint signalling posits that checkpoint activation promotes Mad1‐Mad2 recruitment to the unattached kinetochore where Mad2 is modified and forms a complex with Cdc20 [8, 12]. Release of the Mad2‐Cdc20 complex then inhibits APC/C and thereby ubiquitination of cyclin B. The kinase activities of Bub1, BubR1 and Mps1 are required for efficient recruitment of Mad2 to the unattached kinetochore. The upstream activators for Bub1 and Mps1 have not been identified, although evidence suggests that centromere protein E can bind BubR1 and activate its kinase activity in a manner analogous to cyclin activation of Cdk [13, 14]. Other mechanisms invoke direct phosphorylation and inactivation of Cdc20 by Bub1, BubRI or mitogen‐activated protein kinase may represent a parallel mechanism for inhibition of the APC/C [9, 15].
Mps1/TTK was initially identified as a dual‐specificity protein kinase overexpressed in a number of human tumour cell lines [16, 17, 18]. The biological function of Mps1 was first discovered in yeast, where it is essential for spindle pole body duplication and mitotic spindle checkpoint regulation [6, 19, 20]. The function of Mps1 in mitotic checkpoint regulation is evolutionarily conserved [6, 14, 21, 22, 23, 24]. Depletion of Mps1 in organisms from yeast to human overrides mitotic checkpoint signalling [25]. Mps1 appears to act upstream in the checkpoint signalling pathway as depletion of Mps1 in Xenopus extracts can be overcome by high levels of Mad2 in the checkpoint response [14]. In addition, depletion of Mps1 prevents Mad1 and Mad2 localization to the kinetochore [25].
Mps1 is a mitotic specific kinase because its activity and expression level elevate significantly in mitotic cells. The exact mechanism governing cycle‐dependent regulation of Mps1 is unknown although phosphorylation of Mps1 increases significantly during mitosis and the activation of mitotic checkpoint signalling [22, 23, 26]. Understanding the biochemical mechanism underlying Mps1 kinase activation and regulation is therefore of considerable interest.
Here we report a 2.7‐Å‐resolution crystal structure of the Mps1 kinase domain. We demonstrate that autophosphorylation of Mps1 is a key priming event in activation of Mps1 kinase activity in vitro.
Results
Crystal structure of the Mps1 kinase domain
To understand the molecular mechanism of Mps1 activation, we solved the X‐ray crystal structure of the kinase domain of Mps1. A fragment comprising the kinase domain and the C‐terminal fragment of Mps1 (residues 515–857) was expressed and purified in a baculovirus‐insect cell system (Fig. 1A). The kinase domain contains a mutation, K553R, which disrupts the kinase binding with ATP and therefore ablates its catalytic activity. This mutation is essential for preventing autophosphorylation during protein expression such that homogenous Mps1 for crystallization is obtained. Crystals of Mps1 are monoclinic, with one molecule in the asymmetric unit (Table 1). The structure was solved by single‐anomalous dispersion (SAD). The final model has been refined to a crystallographic R‐value of 22.6% and a free R‐value of 28.5%, using data to a resolution of 2.7 Å (Table 1).
Figure 1.
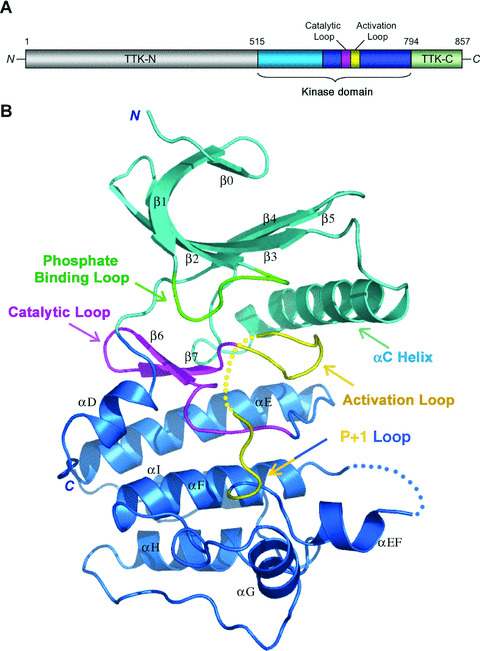
Overview of the Mps1 kinase domain structure. (A) Organization of the Mps1 polypeptide chain. The small and large lobes of the kinase domain are shown in cyan and blue, respectively, and the C‐terminal disordered fragment (residues 795–857) in light green. The catalytic loop and activation loop are highlighted in magenta and yellow, respectively. The N‐terminal region that is not included in the structure determination is coloured in gray. (B) Ribbon diagram of the Mps1 kinase domain. The molecule is coloured as in (A) except for the phosphate‐binding loop in the small lobe is highlighted in green. The dotted lines represent the disordered regions in the activation loop and the loop between αEF and αF.
Table 1.
Crystallographic data and refinement statistics
| Ethyl‐mercury phosphate, MAD | Native | ||
|---|---|---|---|
| Data collection | |||
| Peak | Inflection | ||
| Space group | C2221 | ||
| Cell dimensions | |||
| a, b, c (Å) | 104.253, 104.550, 70.485 | 104.373, 104.653, 70.536 | 105.847, 106.427, 70.682 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 3.2 | 3.2 | 2.7 |
| Completeness (%) | 96.3 (82.4)* | 97.0 (85.2) | 88.9 (60.1) |
| R merge (%) | 0.086 (0.174) | 0.078 (0.201) | 0.039 (0.219) |
| I/σ | 29.5 (7.7) | 28.6 (6.5) | 40.0 (4.2) |
| Redundancy | 7.4 (5.9) | 7.5 (5.7) | 4.5 (2.4) |
| MAD analysis | |||
| Figure of merit acentric/centric | 0.422/0.180 | ||
| Phasing Power | |||
| Isomorphous acentric (centric)/anomalous | 0.354(0.318)/0.762 | ||
| Cullis R | |||
| Isomorphous acentric (centric)/anomalous | 0.907 (0.929)/0.901 | ||
| Refinement | |||
| Resolution (Å) | 50–2.7 | ||
| Reflections (all) | 11300 | ||
| Observed | 10680 | ||
| Working set/free set | 9574/1106 | ||
| Residues | |||
| No. of protein residues | 259 | ||
| No. of water molecules | 19 | ||
| R work/R free | 0.2854/0.2256 | ||
| B factors | |||
| Protein | 78.5 | ||
| Water | 56.2 | ||
| Rmsd | |||
| Bond lengths (Å) | 0.006512 | ||
| Bond angles (°) | 1.20791 |
*Values in parentheses are for highest‐resolution shell. For each structure, one crystal was exposed to X‐ray radiation.
The Mps1 kinase domain adopts the typical protein kinase two‐lobe architecture (Fig. 1B). The N‐terminal small lobe consists of a standard five‐stranded β sheet and a helix (αC) (Fig. 1B). In addition, Mps1 contains an additional β‐strand (β0) at the N terminus of the small lobe, which together with part of β1, covers the twisted β sheet (Fig. 1B). The entire small lobe is substantially more mobile than the C‐terminal large lobe, and residues connecting strands β1 and β2 (the phosphate binding loop) are particularly flexible. The large lobe also possesses a standard structure, composed of a β sheet of two strands (β6 and β7) adjacent to the small lobe, seven α helices, the catalytic loop and the activation loop (Fig. 1B). Electron density is good throughout the kinase domain, except for a portion of the activation loop (residues 672–680) and the loop between helices αEF and αF (residues 700–708). Notably, although included in the protein expression construct, the entire region C‐terminal to the kinase domain (residues 795–857) is invisible in the structure, suggesting that it is either very flexible or largely unstructured.
Comparison of the structure of the Mps1 kinase domain with those of the active protein kinases reveals that the Mps1 kinase domain adopts an inactive conformation (Fig. 2A) [27, 28]. Although the activation loop of Mps1 is partially disordered, its N‐terminus (residues 664–671) and C‐terminal P+1 loop (residues 684–688) assume well‐defined conformation (Fig. 2B). They diverge from the path in the active protein kinase and fold back toward the rear of the catalytic site cleft (Fig. 2A). The folding of the activation loop involves conformational change of the highly flexible residue Gly666 in the conserved motif. The activation loop thus adopts an inhibitory conformation and moves helix C of the small lobe away from its active configuration (Fig. 2A and B). By analogy with all other active state protein kinases, two invariant charged residues, Glu571 from helix C and Lys553 (mutated to Arg in this ‘kinase‐dead’ form) from the small lobe, would form ion‐pairing interactions that are needed to establish the active state of the enzyme (Fig. 2C and D) [27, 28]. Instead, in the inhibitory state of Mps1, the side chain of Glu571 bends towards the large lobe of the kinase and receives three hydrogen bonds from the main chain amides of the inhibitory activation loop (Fig. 2B). This inhibitory configuration is closely reminiscent of the conformation of the activation loop in the inactive state of Ser/Thr protein kinase Pak1 (Fig. S1) [29].
Figure 2.
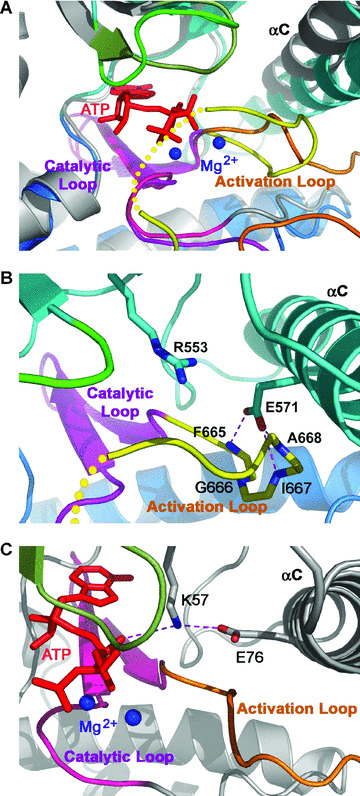
The inhibited conformation of Mps1 kinase domain. Since all the active kinases share similar conformations, here we use the active state structure of Ser/Thr kinase TAO2 as an example of the active state kinases. (A) Superposition of the kinase domains of Mps1 and TAO2 shows that Mps1 adopts an inhibited conformation. Mps1 is coloured as in Fig. 1A. The activation, catalytic and phosphate binding loops of TAO2 are coloured in orange, pink and dark green, respectively, and the rest of the protein coloured in gray with ATP represented in red sticks. The two metal ions found in the TAO2 structure are shown as blue balls. (B) The inhibited ATP‐binding site of Mps1. The activation loop makes a turn, which blocks the contact between Glu571 of Helix C and Lys553Arg and prevents binding of ATP. The hydrogen bonding interactions are shown as magenta dotted lines. (C) The ATP‐binding site of the active TAO1 kinase domain. Glu76 from helix C makes an ion‐pairing interaction with Lys57, which in turn stabilizes the ATP molecule in the active site of TAO1. (D) Structure based sequence alignment of the kinase domains of Mps1 and TAO2. Secondary structure assignments from the Mps1 crystal structure are shown as coloured cylinders and arrows above the aligned sequences. Different regions of the kinase domain (the small and large lobes, the phosphate‐binding loop, the catalytic loop and the activation loop) are coloured as in Figs 1 and 2. Coloured blocks denote the conserved residues between the two kinases. Cyan dots indicate two conserved residues in the small lobe (K553 and E571 in Mps1) that are crucial for the ATP binding in the active kinase conformation (Fig. 2C). In the inactive state, E571 make three hydrogen bonds with the backbone of three residues in the activation loop (F665, I667 and A668; highlighted by yellow dots). Red dots denoted the phosphorylation sites of Mps1 in the activation loop and the red star denotes the novel phosphorylation site (T686) identified in this study.
Autophosphorylation of Mps1 in the activation loop and the P+1 loop
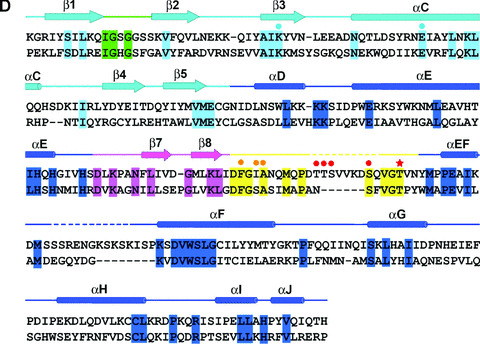
Mps1 kinase activity is elevated 10‐fold upon activation of the mitotic checkpoint and remains hyperphosphorylated during mitosis [22]. The structure of the Mps1 kinase domain suggests that the activation loop of the Mps1 kinase is in an inactive conformation. We therefore hypothesized that phosphorylation and subsequent displacement of the activation loop of Mps1 is likely one of the most critical mechanisms responsible for activation of the Mps1 kinase. The kinase domain of Mps1 purified from insect cells in vitro is autophosphorylated similar to the wild‐type (Fig. 3A). No autophosphorylation of either the full length or kinase domain of Mps1 with the K553R mutation was observed. To determine the residues in the activation loop region that are targeted by autophosphorylation, we performed mass spectrometry analysis of autophosphorylated Mps1 kinase domain purified from insect cells. Four independent phosphopeptides were identified corresponding to autophosphorylation sites at residues T675, T676, S677 and T686 in either the full length or kinase domain of Mps1 (Table 2). T675, T676 and S677 are part of the activation loop and T686 situates on the P+1 loop. Since double or triple phosphorylated peptides containing T675, T676 and S677 were not detected in our analysis, this result implies that Mps1 autophosphorylation in the activation loop is heterogeneous.
Figure 3.
Activation loop autophosphorylation elevates Mps1 kinase activity in vitro. (A) Autophosphorylation of wild‐type Mps1 (GST‐Mps1) or the kinase domain (517–852) of Mps1 (GST‐Mps1CD) with recombinant proteins purified from insect cells in the presence of 32P‐γ‐ATP. The final concentration of enzyme used in the kinase assay is 50 nM. Mutation of K553 of Mps1 to R in either background perturbs the kinase activity. (B) Time courses of Mps1 phosphorylation of Smad2 and Mps1NTD (amino acid 1–275). A total of 50 nM of GST‐Mps1 was incubated with 125 μM of either substrate for indicated times. (C) Activation loop autophosphorylation elevates Mps1 kinase activity in vitro. Untagged Mps1 kinase (50 nM) was pre‐incubated with or without 100 μM ATP at room temperature for 1 hr. After pre‐incubation, the N‐terminal fragment of Mps1 (125 μM) and 32P‐γ‐ATP (1 μCi) were added to both samples. In addition, cold ATP was added to the sample that was pre‐incubated without ATP so that the final concentrations of cold and hot ATP are identical between two samples. The reactions were incubated for the indicated times, followed by kinase activity measurement.
Table 2.
Summary of Mps1 Phosphopeptides near the activation loop
| Peptide | Identified phosphorylation sites |
|---|---|
| 662LIDFGIANQMQPDTTSVVK680 | T675 |
| 662LIDFGIANQMQPDTTSVVK680 | T676 |
| 662LIDFGIANQMQPDTTSVVK680 | T677 |
| 681DSQVGTVNYMPPEAIK696 | T686 |
Autophosphorylation of Mps1 increases kinase activity in vitro
If Mps1 autophosphorylation regulates its kinase activity, we would expect that there is a lag phase to allow Mps1 autophosphorylation in order to reach maximal activity for substrate phosphorylation. Since the authentic substrates for Mps1 kinase have not been identified in mammalian system previously, myelin basic protein is frequently used as a substrate to monitor Mps1 enzymatic activity in vitro. We have recently discovered that Smad2, a downstream signal transducer of TGF‐β signalling pathway, can be phosphorylated by Mps1 at the carboxyl terminal SSXS motif of Smad2 both in vitro and in cultured cells [30]. Since both full length and C‐terminal kinase domain of Mps1 exhibit avid autophosphorylation in vitro (Fig. 3A), we tested whether the N‐terminal fragment of Mps1 is also a substrate of Mps1. To our surprise, the N‐terminal domain of Mps1 (Mps1NTD; residues 1–275) is a good substrate for Mps1 and its phosphorylation kinetics are similar to those of Smad2 (Fig. 3B). Therefore both the N‐terminal and C‐terminal regions of Mps1 are targeted for phosphorylation. Since the N‐terminal domain of Mps1 expresses well and is easy to purify to homogeneity, we developed an assay to monitor Mps1 enzymatic activity using Mps1NTD as the substrate in vitro. Mps1 was pre‐incubated either with or without Mg2+/ATP for 1 hr at room temperature prior to the addition of Mps1NTD, after which a reaction time course of substrate phosphorylation was examined. The activity of Mps1 was found to be higher following pre‐incubation with ATP (Fig. 3C). These findings suggest that autophosphorylation of Mps1 increases its enzymatic activity.
Since the activation loop and the P+1 loop of Mps1 adopts an inactive conformation as seen in the crystal structure, the most obvious candidate phosphorylation sites that regulate Mps1 kinase activity are the ones located in the activation loop and the P+1 loop (Fig. 4A). To determine whether phosphorylation of activation loop serine and threonine residues regulates Mps1 kinase activity in vitro, we constructed Mps1 activation loop (TTS→AAA) and the P+1 loop (T686A) phosphorylation site mutants. These mutants were subsequently expressed in insect cells using a baculoviral expression system (Fig. 4B). Wild‐type and mutant Mps1 kinases were purified and tested for their autophosphorylation activity in vitro. As shown in Fig. 4B, whereas mutation of T675, T676 and T677 reduces kinase activity in vitro, mutation of T686 to alanine resulted in a more marked reduction of autophosphorylation activity. This activity is only slightly above that of the Mps1 D664A mutant, which is considered a kinase‐dead mutant [21]. Time course analysis of wild‐type and activation loop mutant Mps1 auto and transphosphorylation reveals that autophosphorylation at the activation loop appears to have more dramatic effects on transphosphorylation than autophosphorylation (Fig. 4C, D).
Figure 4.
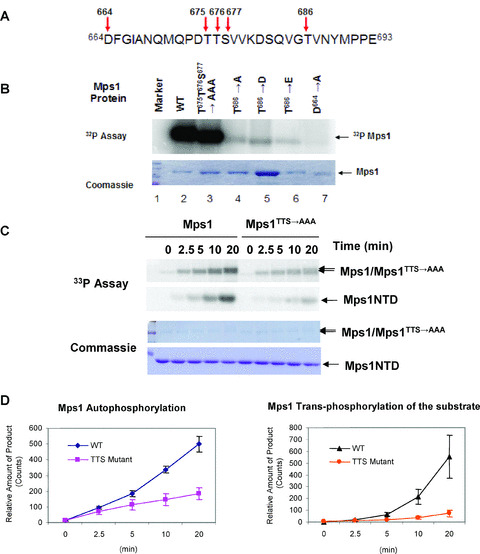
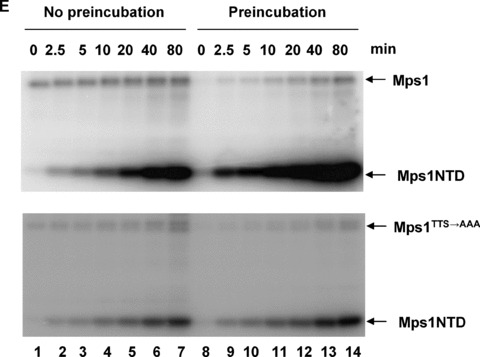
Mutations in the activation loop and the P+1 loop impair kinase activation. (A) The activation loop and the P + 1 loop sequence of human Mps1. Arrows indicate residues that are targeted by phosphorylation and the position of key catalytic residues. (B) Effects of the activation loop and P+1 mutations on Mps1 kinase activity. Recombinant GST‐Mps1 and the indicated mutants were purified from insect cells. A total of 1 μM of each enzyme was incubated with 32P‐γ‐ATP (1 μCi) prior to SDS‐PAGE. The amounts of input proteins were determined by Coomassie blue staining. (C) Direct comparison of auto‐ and transphosphorylation activity of wild‐type and Mps1 activation loop mutant. Wild‐type or mutant Mps1 were incubated with 125 μM of Mps1NTD for indicated times in the presence of 33P‐γ‐ATP (1 μCi) and 100 μM cold ATP without pre‐incubation. Samples were analysed by SDS‐PAGE and stained with Coomassie blue prior to PhosphorImaging. (D) Quantitation of auto‐ and transphosphorylation of wild‐type and mutant Mps1. Three separate experiments were performed and relative amount of 33P‐labeled Mps1 or Mps1NTD was determined by ImageQuant™. Error bars represent standard deviation from three experiments. (E) Wild‐type or the activation loop autophosphorylation‐site‐mutant Mps1 (50 nM) was pre‐incubated either with or without cold ATP (100 μM) for 60 min prior to addition of the substrate. Time course of Mps1 substrate phosphorylation was performed as described in Fig. 3.
Since the Mps1 activation loop (TTS→AAA) mutant retains most but not all of its kinase activity compared to the wild‐type, we tested whether mutation of these autophosphorylation site residues perturbs ATP‐dependent elevation of kinase activity in vitro. Wild‐type and mutant Mps1 were pre‐incubated either with or without ATP prior to addition of the substrate Mps1NTD. As shown in Fig. 4E, mutation of T675, T676 and S678 of Mps1 mitigates the pre‐incubation effects on kinase activity although there is still an observable increase with the mutant kinase. This result suggests that autophosphorylation at the activation loop of Mps1 may contribute to increase in kinase activity.
Autophosphorylation of the Mps1 P+1 loop is associated with the active kinase
The P+1 loop located C‐terminally to the activation loop is highly conserved among protein kinases [28]. For serine/threonine kinases, the P+1 loop usually starts with a conserved serine or threonine residue that links the P+1 loop to the activation loop [28]. In tyrosine kinases, the equivalent residue is a proline. Even though Mps1 is a dual‐specificity kinase capable of phosphorylating both Ser/Thr and Tyr, this key position is occupied by T686. It was rather unexpected to find that T686 is autophosphorylated because phosphorylation at this position has not been previously described for other kinases. Since mutation of T686 to A causes a dramatic reduction in kinase activity, we wondered whether T686D or T686E, which presumably can partially mimic phosphorylation events under certain conditions, could restore the kinase activity.
As shown in Fig. 4B, neither T686D nor T686E can restore the activity, suggesting a stringent requirement for T686 in kinase regulation beyond just negative charge.
Even though we detected T686 phosphorylation by mass spectrometry, it is not clear whether the active kinase is phosphorylated at T686. Furthermore, we cannot discount the possibility that Mps1 without T686 phosphorylation is as active as the phosphorylated form of the kinase. To further address the functional relevance of T686 phosphorylation in kinase regulation, a phospho‐specific antibody against a peptide containing pT686 was raised. This antibody does not react with kinase‐dead Mps1 mutant (D664A) pre‐incubated with or without Mg2+/ATP, as analysed by immunoblot, but it does recognize wild‐type Mps1 under the same conditions (Fig. 5A). The faint band in the lane without ATP may represent a tiny amount of activated Mps1 in the enzyme preparations from insect cells (Fig. 5A). Autophosphorylation of Mps1 at T686 is increased upon incubation with Mg2+/ATP in a time course that parallels its ability to transphosphorylate the substrate (Fig. 5B). Since pre‐incubation of Mps1 enhances kinase activity and mutation of the autophosphorylation sites (T675, T676 and S677) in the activation loop impair this activity, we tested whether autophosphorylation at the activation loop regulates T686 P+1 loop phosphorylation. Wild‐type and Mps1TTS→AAA mutant were incubated with Mg2+/ATP and phosphorylation of T686 was monitored by immunoblotting with the phospho‐T686 antibody. As shown in Fig. 5C, phosphorylation of T686 is increased in wild‐type Mps1 but no significant increase was seen with the activation loop mutant. This result suggests that T686 autophosphorylation may depend on activation loop autophosphorylation, i.e. activation loop phosphorylation could be a priming event for the P+1 loop phosphorylation.
Figure 5.
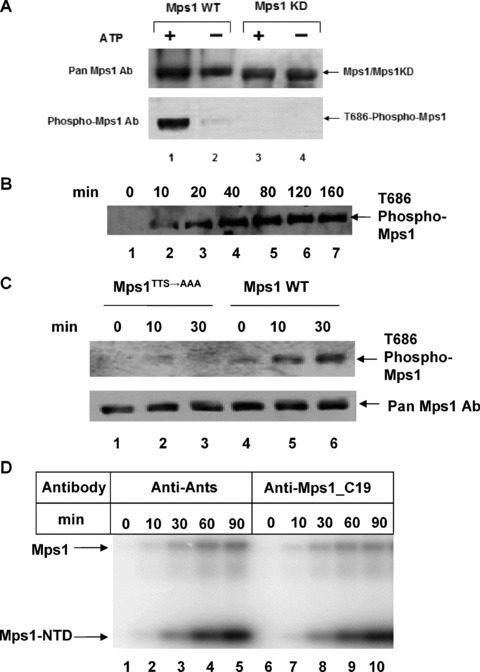
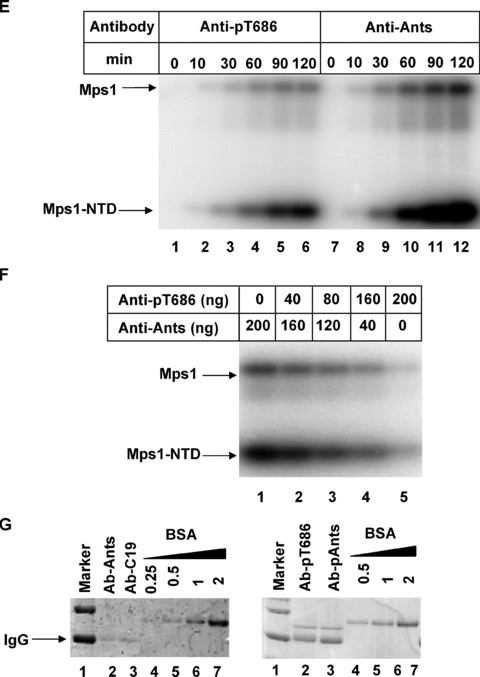
Active Mps1 kinase is phosphorylated in the P+1 loop threonine residue T686. (A) Immunoblotting of wild‐type and kinase dead Mps1 incubated with or without cold ATP for 1 hr at room temperature using a rabbit polyclonal antibody raised against the T686 phosphopeptide (KDSQVG[pThr]VNYMPPE). (B) Time course of Mps1 autophosphorylation at T686 as determined by immunoblotting with the T686 phospho specific antibody. (C) Time course of wild‐type and Mps1activation loop mutant autophosphorylation at T686. (D) Effects of rabbit antibodies against antisocial (Ants) or Mps1 carboxyl terminal domain (C19) on Mps1 auto‐ and transphosphorylation. A total of 200 ng of the indicated antibodies were included in the time course reactions of Mps1. A total of 50 nM of Mps1 and 125 μM of Mps1NTD substrate were used in this experiment (E) Effects of control and phospho‐T686 antibodies on Mps1 auto‐ and transphosphorylation. The amount of antibodies, Mps1 and substrates are identical to (D). (F) An increasing amount of Anti‐pT686 antibody was mixed with a decreasing amount of nonspecific antibody anti‐Ants. Each mixture indicated was incubated with the Mps1 kinase for 1 hr on ice prior to the addition of ATP, Mg2+ and the substrate and incubation at room temperature for 1 hr. A total of 50 nM of Mps1 was used in each reaction. The total amount of input antibodies in each reaction is about 200 ng. (G) The input antibodies used in (D) and (E) were dialysed with 1× kinase reaction buffer. A portion of the dialysed antibodies was analysed by SDS‐PAGE with a known amount of BSA standard and Coomassie blue staining.
If phosphorylation of T686 is associated with the active kinase, we expect that adding the phospho‐specific T686 antibody to the Mps1 in vitro kinase assay would inhibit the kinase if phosphorylation at T686 is important. As a control, we tested whether adding a nonspecific antibody against antisocial (Ants) or a specific antibody raised against the carboxyl terminal region of Mps1 (C19) would have any effect on Mps1 auto or transphosphorylation. Including either antibody in the kinase reaction did not appear to have any adverse effect on Mps1 kinase activity (Fig. 5D).
However, addition of the phosho‐T686 antibody inhibits Mps1 auto‐ and transphosphorylation whereas no effect was observed with the control Ants antibody (Fig. 5E). Next, mixtures of nonspecific (antisocial) and specific (anti‐pT686) antibodies at different ratios were added to the kinase reaction. Mps1 auto‐ and transphosphorylation activities are significantly blocked with increasing concentrations of anti‐pT686 antibody and decreasing concentration of control antibody (Fig. 5F). This result suggests that kinase activity of Mps1 can be blocked by T686 phospho‐specific antibody. Antibodies used in this experiment were dialysed with the kinase reaction buffer and quantified by SDS‐PAGE with the known standard prior to incubations with Mps1 (Fig. 5G). There is a possibility that both T686 unphosphorylated and phosphorylated Mps1 enzymes are active. If so, we would expect that anti‐pT686 only inhibits the phosphorylated Mps1 and unphosphorylated should remain active even in the presence of anti‐pT686. Data shown in Figure 5F indicate that anti‐pT686 largely abrogates Mps1 activity. This observation is consistent with the hypothesis that a majority of active Mps1 kinase is phosphorylated at T686. Hence T686 phosphorylation is associated with the active kinase.
Autophosphorylation increases Mps1 kinase activity in cells
To further address the significance of Mps1 activation loop phosphorylation in a cellular context, we overexpressed wild‐type and activation loop mutants of Mps1 with Smad2 in 293T cells by transient transfection. We have shown that Mps1 phosphorylates Smad2 in vitro and in vivo at the SSMS motif which is normally targeted by the TGF‐β type I receptor kinase [30]. As observed previously, wild‐type but not kinase‐dead Mps1 phosphorylates Smad2 at S465 and S467 determined by immunoblotting with a phospho‐specific Smad2 antibody that specifically reacts with doubly phosphorylated Smad2 (Fig. 6). Among five potential phosphorylation sites on the activation or P+1 loop (T675, T676, S677, S682 and T686), only T686 appears to be absolutely essential for Smad2 phosphorylation. Consistent with the results obtained with purified enzymes in vitro, mutation of T675, T676 and S678 caused a slight reduction in Smad2 phosphorylation by Mps1 while mutation of S682 seems to enhance Smad2 phosphorylation. Autophosphorylation of T686 is completely disrupted by D664A or mutations at T686 to A, D or E. The fact that T686 mutants failed to bind phospho‐T686 antibody further confirmed the specificity of this antibody (Fig. 6). Autophosphorylation of T686 in Mps1 activation loop (TTS→AAA) mutant is reduced while the S682 mutation results in slight increase at this site. This result is consistent with T686 autophosphorylation activity seen with purified enzymes (Fig. 5C). Consequently, autophosphorylation of T686 is correlated with the active form of Mps1 and phosphorylation of Smad2 in cells. Thus, these results suggest that phosphorylation of Mps1 at T686 in cells appears to be required for activation of Mps1.
Figure 6.
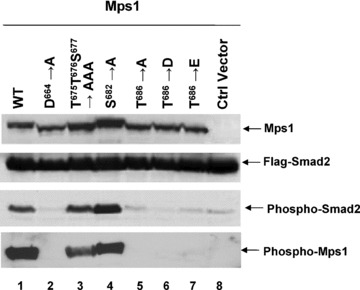
Effects of the activation loop and the P+1 loop mutations on Mps1 transphosphorylation of Smad2 in cells. Mps1 and Mps1 mutants were coexpressed with Flag tagged Smad2 in 293T cells by transient transfection. Forty‐eight hours after transfection, cells were harvested and lysed. Cell lysates were blotted with antibodies against Mps1, Flag or phospho‐Smad2 that recognizes the pSMpS motif of Smad2 or phospho‐Mps1 that recognizes the T686 phosphopeptide.
Importance of the disordered loop between αE and αEF in substrate recruitment and kinase activity
The loop connecting αF and αEF is disordered in the crystal structure. Previous structural studies with a number of protein kinases (Cdk2, Erk2 and PKB) suggest that this loop may be involved in modulating the conformation of activation loop [28]. There is little sequence conservation in this loop between Mps1 from different species except that K710 (human Mps1) is invariant in the Mps1 kinase family. In many kinases that are activated through activation loop phosphorylation, the phosphate group acquired in the activation loop upon kinase activation is involved in stabilizing the active conformation of the kinase through electrostatic interactions with the conserved arginine (R) next to the catalytic aspartate (D) residue (This group of kinases is also known as RD kinases) [28]. Since Mps1 is a non‐RD kinase, Kang et al. proposed that positively charged residues in this region may serve a similar function by interacting with phosphorylated T676 to stabilize the active conformation of Mps1 [26]. Our Mps1 structure data enables us to clearly define the boundary of this loop. There are four positively charged residues within this loop. To determine whether these residues are important for Mps1 auto‐ and transphosphorylation, Mps1 mutants with changes in R702, K706, K708 and K710 were constructed and expressed in 293T cells via transient transfection. These Mps1 mutants were immunoprecipitated from cell lysates and incubated with Mps1NTD in the presence of 32P‐γ‐ATP. As shown in Fig. 7, K708 and K710 but not K706 or R702 showed significant reduction in substrate phosphorylation whereas only slight reduction in autophosphorylation of K708/K710 mutant was observed. Differences in substrate phosphorylation are unlikely due to different expression levels of these mutants in cells as immunoblotting analysis indicates that these mutants were expressed at comparable levels (Fig. 7). To further corroborate the importance of K708 and K710 in substrate phosphorylation and kinase activity, charge reversal mutants were also constructed. Compared to what we observed with alanine substitutions, DD or EE substitutions further compromise the substrate phosphorylation, suggesting K708 and K710 play important roles in substrate recruitment and maintaining high levels of kinase activity.
Figure 7.
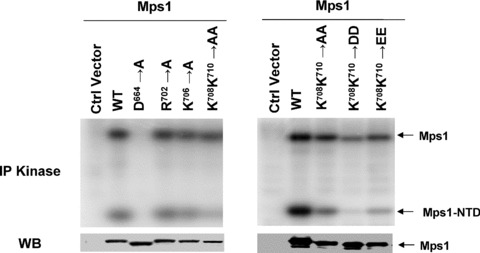
Two lysine residues in the disordered loop connecting αF and αEF of Mps1 are required for optimal kinase activity and substrate recruitment. Flag tagged wild‐type and mutant Mps1 were expressed in 293T cells by transient transfection in 10‐cm culture dishes. Forty‐eight hours after transfection cell lysates were prepared. Expression levels of Mps1 variants were determined by immunoblotting with the anti‐Mps1 antibody (C‐19). Mps1 and Mps1 mutants were immunoprecipitated from cell lysates and incubated with 125 μM of Mps1NTD along with 32P‐γ‐ATP (1μCi) and Mg2+ for 1 hr prior to SDS‐PAGE and phosphorimaging analysis.
Discussion
We report here the crystal structure of Mps1 kinase domain, which shows that the Mps1 kinase domain adopts a unique inactive state. Autophosphorylation of Mps1 appears to be a priming event for kinase activation. Phosphorylation of Ser/Thr residues in the activation loop enhances kinase activity but is not absolutely required for switching to active kinase. In addition, we demonstrated that Mps1 is autophosphorylated at the highly conserved residue T686 in the P+1 loop. This autophosphorylation event is associated with the active kinase. Two positively charged residues in the loop connecting αF and αEF are engaged in substrate recruitment. Based on the crystal structure and biochemical results, we propose that Mps1 autophosphorylation in the activation loop and the P+1 loop are two important events in kinase activation. Autophosphorylation of the activation loop residue is a priming event for Mps1 activation and is likely involved in reorganizing the activation loop, which promotes further Mps1 autophosphorylation at other sites such as T686, which is critical for elevating Mps1 kinase activity.
During mitosis or spindle checkpoint activation, Mps1 kinase activity is strongly activated [22]. The mechanism for Mps1 kinase activation remains elusive although phosphorylation has been postulated to be one of the major priming events to activate Mps1. Conformation change of the activation segments due to phosphorylation is a recurring mechanism that is responsible for activation or inactivation of numerous kinases such as PKA, Cdk2, etc. [28, 31, 32]. Nolen et al. surveyed the conformation diversity of the activation segment which includes the N‐terminal anchor point near DFG, activation loop and the C‐terminal anchor point overlaps with the P+1 loop. By comparing the activation segments of twenty four active and inactive kinase structures, several structural features of the active state of protein kinases have been numerated [28].
Two distinct features revealed from analysing the X‐ray crystal structure of Mps1 kinase domain suggest that unphosphorylated Mps1 adopt an inactive conformation. First, the αC helix rotates from the catalytic site by about 5°, relative to the position it is likely to have in the active conformation. Second, the N‐terminal anchor point of the activation segment forms hydrogen bonds with the conserved glutamate on helix C therefore is predicted to restrict the N‐lobe motion and consequently blocks kinase activation. Furthermore, the conformation of the C‐terminal anchor point (P+1 loop) is also disrupted and distinct from the active conformation of Ser/Thr kinases. Mps1 undergoes extensive yet heterogeneous autophosphorylation in insect cells. To facilitate structural elucidation of Mps1 kinase domain, we made a change at position 553 from Lys to Arg. This residue is equivalent to Lys72 in protein kinase A which is known to be involved in enzyme catalysis [33]. Indeed, mutation of this lysine inactivates Mps1 kinase (Fig. 3A) and made it possible for us to obtain diffractable quality crystals. There is a possibility that Lys553 substitution may alter the equilibrium among several different conformation states and favour the inactive conformation of Mps1 kinase domain under crystal conditions. However, this is unlikely to be the case. First, large‐scale conformational changes have not been observed when this equivalent residue is mutated in other kinase structures [34, 35]. Second, biochemical analysis of enzyme function is consistent with the notion that unphosphorylated Mps1 is likely to be inactive (Fig. 5F). Finally, during the course of our work, Chu et al. reported the inactive human Mps1 kinase domain structures in both apo‐ and inhibitor‐ (SP600125) bound forms [36]. Although crystallized under different solution conditions, in different space groups and having different lattice‐packing environments, our Mps1 structure and two reported Mps1 structures were essentially identical, strongly suggesting that these Mps1 kinase domain structures represent the authentic inactive conformation of the kinase.
Phosphorylation in the activation loop or the anchor points of the activation segment could enable the kinase to adopt active conformation and elevate the kinase activity. That pre‐incubation of Mps1 with ATP leads to an increased in the activity of substrate phosphorylation is consistent with the notion that phosphorylation is a priming event for Mps1 activation. Using mass spectrometry, we have identified that T675, T676 and S677 on the activation loop are autophosphorylated. Autophosphorylation at these sites have also been observed with Mps1 kinase prepared from E. coli[37] and autophosphorylation of T676 has also been detected in Mps1 immunoprecipitated from mitotic HeLa extracts [26]. Our mutational studies suggest that phosphorylation of T676 is involved in elevating kinase activity and the need for phosphorylation around this site accounts for the delay in activating Mps1 kinase in vitro. However, autophosphorylation at this site is not absolutely required for kinase activity. Our results are consistent with the findings by Mattison et al. and Kang et al. in that T676 has a regulatory role in kinase activation and without it the kinase activity decreases by about 4–7 fold [26, 37]. Therefore, autophosphorylation at the activation loop is not the only event crucial for switching kinase to active conformation, suggesting that stabilization of active conformation of Mps1 are achieved by multiple concerted mechanisms. Preliminary kinetic analysis of wild‐type and activation loop mutant of Mps1 reveals that Km of these two enzymes for ATP is comparable but Vmax of the mutant enzyme for auto or transphosphorylation is lower. These results suggest that phosphorylation of T676 is more likely to be involved in catalysis rather than ATP binding. From our analysis of T686 autophosphorylation, it appears that T676 phosphorylation may be a priming event for T686 autophosphorylation. Conceivably phosphorylation at T676 may alter the conformation of the activation loop and further expose T686 for autophosphorylation. In this way, successive phosphorylation at the activation loop and the P+1 loop could be a mechanism for Mps1 kinase activation.
A rather unusual observation in our study is the finding of autophosphorylation at T686 in the P+1 loop of Mps1. Phosphorylation of this site is readily detectable upon incubation with ATP but is not detected in kinase‐dead mutant of Mps1 from insect cells using either mass spectrometry or the T686 phospho‐specific antibody. In this study, we demonstrated that autophosphorylation at T686 tracks the activity of Mps1. Phosphorylation of T686 also occurs in Mps1 expressed in 293T cells and endogenous Mps1 in HeLa cells. Mutation of this residue dramatically reduces both Mps1 autophosphorylation and transphosphorylation activity in vitro and in vivo. The stringent requirement for T686 has also been observed with recombinant Mps1 made in E. coli[36, 37]. This particular residue is not only conserved among all members of Mps1 family of kinases but also invariant in many S/T kinases. In Ser/Thr kinase Cdk2, the hydroxyl group of this residue (T165) forms hydrogen bonds with conserved catalytic residues in the catalytic loop in the active kinase. In tyrosine kinases, the equivalent residue is a highly conserved proline involved in positioning the substrate Tyr residue. If the hydroxyl group of Mps1 T686 functions analogous to T165 of Cdk2, it is expected that phosphorylation of T686 would inhibit Mps1 kinase activity as such modification of hydroxyl group should perturb its interaction with the catalytic loop. The fact that T686 is associated with the active kinase disproves this model. Given that phosphorylation of T686 is an early event in Mps1 activation and is required both for auto and transphosphorylation, we speculate that phosphorylation of this residue in Mps1 could trigger a conformation change that enables Mps1 to correctly position substrate for catalysis. Consequently, modulating the P+1 loop conformation through phosphorylation could be a novel and unique mechanism for Mps1 kinase activation. Furthermore, we speculate that T686 autophosphorylation could alter Mps1 substrate specificity. There is possibility that Mps1 T686 unphosphorylated enzyme has a different substrate preference from T686 phosphorylated Mps1. Future crystallographic studies of the active Mps1 kinase in complex with the substrate may definitively address the role of T686 phosphorylation in Mps1 kinase activation.
The availability of tertiary structure conformation of Mps1 afforded us to identify other potential residues that might be involved in stabilizing the active conformation and recruiting substrate. In kinases that rely on activation loop phosphorylation as a way to regulate their activities, the active conformation is often stabilized through electrostatic interaction between the phospho‐residue in the activation loop and a basic pocket featuring a conserved arginine immediately preceding the conserved catalytic aspartate in the catalytic loop. Mps1 is not a RD kinase and it is apparent that the active site needs to be reorganized to be conducive to substrate binding. The loop connecting αF and αEF is flexible and disordered in the Mps1 structure and thus basic residues in this loop could potentially serve an equivalent function in the RD pocket. Of four basic residues in this loop, only K708 and K710 are required for optimal kinase activity. In our study, mutating these two lysine residues appear to affect Mps1 transphosphorylation more severely than Mps1 autophosphorylation. The effect on autophosphorylation supports the hypothesis of Kang et al. that these two residues might be the binding partners for phospho‐T676 [26]. The more adverse effects on transphosphorylation suggest that they may also involve in substrate binding and positioning during catalysis. Given that Mps1 autophosphorylation appears to occur through an intermolecular mechanism [26], it would be interesting to determine whether different binding interfaces are required for autophosphorylation and transphosphorylation.
Accession number
Atomic coordinates and structure amplitudes of Mps1 kinase domain have been deposited in the Protein Data Bank with accession number 3DBQ.
Materials and methods
Cell culture, transfections and antibodies
All mammalian cell lines were cultured at 37°C in a 5% CO2 atmosphere in Dulbecco’s modified Eagle’s medium supplemented with 10% foetal calf serum (FCS; Gibco‐BRL), penicillin, streptomycin (100 IU/ml and 100 mg/ml, respectively), and L‐glutamine. 293T cells were purchased from ATCC. FuGENE 6 (Roche Applied Sciences) was used for transfection of 293T cells. Antibodies against Mps1 (C19) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA) or Millipore Inc. (Billerica, MA, USA). Anti‐phospho‐Smad2 was a gift of Drs. Ten Djike, Heldin and Moustakas. Phospho‐specific T686 Mps1 antibody was raised against a phospho peptide (KDSQVG[pThr] VNYMPPE) corresponding to the activation loop region of Mps1 by Sigma‐Genosys (The Woodlands, TX, USA). The phospho‐specific antibody was purified using a protein A based antibody purification kit from Biorad followed by depletion of non‐phospho specific immunoglobulins with GST‐Mps1KD (Kinase dead).
Recombinant protein production
Baculoviruses encoding human Mps1 and kinase‐dead version (Mps1_D664A) have been described [30]. Baculoviruses encoding the Mps1 kinase domain (Mps1CD) or inactive Mps1 kinase domain (Mps1CDK553R) were constructed by subcloning the fragment corresponding to Mps1 (517–852) into pFAST‐GST vector described previously [30]. A precision protease (GE Healthcare, Pistcataway, NJ, USA) cleavage site was introduced after GST to facilitate release of untagged Mps1 kinase domain. Point mutations in Mps1 or Mps1 kinase domain were introduced by Quikchange mutagenesis (Stratagene). Recombinant baculoviral stocks were generated and amplified twice in SF9 cells following the manufacturer’s instructions (Invitrogen, San Diego, CA, USA). Hi‐Five cells were infected with the indicated viruses and harvested after 3 days. Extracts of Hi‐Five cells were collected and the corresponding fusion proteins were purified by binding to glutathione‐sepharose beads (GE Healthcare). The N‐terminal domain of Mps1 (M‐NTD) was subcloned into the pET28b‐SUMO vector and expressed in BL21 cells [38]. Untagged M‐NTD was purified by Ni‐NTA affinity purification followed elution with Ulp1 protease as described [38].
Mass spectrometry protein sequencing analysis
GST‐Mps1 or GST‐Mps1CD purified from insect cells were autophosphorylated by incubating 2 μg of each kinase in the 1× kinase buffer in the presence of 100 μM ATP at room temperature for 1 hr. Samples were alkylated with 50 mM iodoacetamide at room temperature for 1 hr prior to adding sequencing grade trypsin (2% w/w) and incubating at 37°C for 12 hrs. Samples were analysed using a 4000 Qtrap (Applied Biosystems, Foster City, CA, USA) interfaced with an Eksigent NanoLC‐2D instrument (Eksigent, Dublin, CA, USA) for nanoflow chromatography, using a 75 μm × 15 cm Zorbax 300SB‐C18 analytical nanocolumn (Agilent Technologies, Palo Alto, CA, USA) for peptide separation. Phosphopeptides were analysed using a polarity switching method to alternate between detection and sequencing of the phosphopeptides in the same run [39]. First, a negative mode precursor ion scan is acquired, monitoring for the marker ion PO3– at –79 m/z, over a mass range of 500–1800 m/z, with Q1 set to low resolution and Q3 set to unit resolution. When the signal intensity of the precursor ion scan is above a threshold of 1000 cps, polarity is switched to positive mode, and a high‐resolution scan is acquired for charge determination and accurate mass measurement of the three most intense ions, followed by positive mode MS/MS sequencing. MS/MS were searched with MASCOT (version 2.0, MatrixScience, London, UK) using a small database of 50 standard proteins including the sequence of the MPS1 fusion protein. Parent mass tolerance was 1.2 Da, MS/MS tolerance was 0.6 Da, with fixed modifications set to carbamidomethyl on cysteine, and variable modifications set for methionine oxidation and phosphorylation on Ser, Thr and Tyr. MS/MS identifications with mascot scores above 20 were the manually validated for quality and phosphorylation site determination.
Crystallization, data collection and structure determination
Mps1 kinase domain was crystallized in solution A (10% of PEG 5K MME, 100 mM Na2HPO4/KH2PO4, pH 6.3, and 150 mM NaCl) at 16°C. Crystals were transferred to solution B (25% PEG 5K MME, 100 mM NaCl, 50 mM Na2HPO4/KH2PO4, pH 6.3) gradually and soaked for 3–4 hrs. Crystals were then transferred to a condition containing solution B plus 25% glycerol and soaked for ∼1 hr. Crystals were then flash frozen in liquid nitrogen. For heavy atom derivatives, crystals were transferred to solution B and soaked in this solution along with the presence of 0.1 mM Ethyl‐Hg‐PO4 for 3–4 hrs. Crystals were then transferred to a condition containing solution B and 25% glycerol without heavy atom and soaked for ∼1 hr before flash frozen in liquid nitrogen. Diffraction data were collected at 100 K to a maximum resolution of 2.5 Å using a CCD detector at beamline 23‐ID‐B of APS. All data were processed using the HKL2000 program [40]. The crystals were in space group C2221, with unit cell dimensions of a = 105.847 Å, b = 106.427 Å, and c = 70.682 Å. Detailed Data collection statistics are presented in Table 1. The cell dimensions were consistent with one molecule per asymmetric unit for a crystal solvent content of 61%. SAD data from the mercury derivative were used to obtain initial phases. Mercury atoms were located and refined, and the SAD phases calculated using SHARP [41], the initial SAD map was significantly improved by solvent flattening. The improved map allowed us to fit a model of the Pak1 kinase domain (PDB ID code 1F3M) as a rigid body. A model of the relevant residues of human Mps1 was built into the density using the program O [42]; the model was then transferred into the native unit cell by rigid‐body refinement and further refined using simulated‐annealing and positional refinement in [43] with manual rebuilding [42].
In vitro kinase assays
The kinase reactions typically included purified recombinant Mps1 or Mps1 immunoprecipitated from cell lysates, 2 μg of GST‐Smad2 or 125 μM of Mps1NTD in 1× kinase reaction buffer containing 50 mM Tris‐HCl (pH 8.0), 10 mM MgCl2 and 1 μCi [32P]‐γ‐ATP. The final concentration of cold ATP varies depending on the experiment and was indicated in the figure legend. Reactions are incubated at room temperature for the indicated times and stopped by adding SDS loading buffer. Reaction mixtures were resolved by SDS‐PAGE and analysed by phosphorimaging. ImageQuant™ or Image J was used to quantify the intensity of bands in the images collected from PhosphorImager or scanned images, respectively.
Supporting information
Figure S1. The inhibited ATP‐binding site of Pak1. The kinase inhibitory (KI) segment of the autoregulatory fragment occupies the cleft between the small and the large lobes of the kinase domain. Lys141 makes hydrogen bonds with Asp407 of the activation loop. Therefore, the activation loop is forced to make a turn, which blocks the contact between Glu355 of Helix C and Lys299Arg and prevents binding of ATP. The hydrogen bonding interactions are shown as magenta‐dotted lines.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgements
We thank Drs. Moustakas, Ten Djike, Heldin and Lima for valuable reagents used in this study. We thank David Clarke for critical readings of the manuscript. This work was supported by grants from the National Institutes of Health (CA107098) to X.L. and American Cancer Society and Sidney Kimmel Foundation to M.L. The General Medicine and Cancer Institutes Collaborative Access Team has been funded in whole or in part with federal funds from the National Cancer Institute (grant Y1‐CO‐1020) and the National Institute of General Medical Science (grant Y1‐GM‐1104). Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. DE‐AC02–06CH11357.
References
- 1. Amon A. The spindle checkpoint. Curr Opin Genet Dev. 1999; 9: 69–75. [DOI] [PubMed] [Google Scholar]
- 2. Draviam VM, Xie S, Sorger PK. Chromosome segregation and genomic stability. Curr Opin Genet Dev. 2004; 14: 120–5. [DOI] [PubMed] [Google Scholar]
- 3. Weaver BA, Cleveland DW. Decoding the links between mitosis, cancer, and chemotherapy: the mitotic checkpoint, adaptation, and cell death. Cancer Cell. 2005; 8: 7–12. [DOI] [PubMed] [Google Scholar]
- 4. Li R, Murray AW. Feedback control of mitosis in budding yeast. Cell. 1991; 66: 519–31. [DOI] [PubMed] [Google Scholar]
- 5. Hoyt MA, Totis L, Roberts BT. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell. 1991; 66: 507–17. [DOI] [PubMed] [Google Scholar]
- 6. Weiss E, Winey M. The Saccharomyces cerevisiae spindle pole body duplication gene MPS1 is part of a mitotic checkpoint. J Cell Biol. 1996; 132: 111–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wassmann K, Benezra R. Mitotic checkpoints: from yeast to cancer. Curr Opin Genet Dev. 2001; 11: 83–90. [DOI] [PubMed] [Google Scholar]
- 8. Kops GJ, Weaver BA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer. 2005; 5: 773–85. [DOI] [PubMed] [Google Scholar]
- 9. Tang Z, Shu H, Oncel D, et al. Phosphorylation of Cdc20 by Bub1 provides a catalytic mechanism for APC/C inhibition by the spindle checkpoint. Mol Cell. 2004; 16: 387–97. [DOI] [PubMed] [Google Scholar]
- 10. Fang G, Yu H, Kirschner MW. The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase‐promoting complex to control anaphase initiation. Genes Dev. 1998; 12: 1871–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sudakin V, Chan GK, Yen TJ. Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J Cell Biol. 2001; 154: 925–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yu H. Regulation of APC‐Cdc20 by the spindle checkpoint. Curr Opin Cell Biol. 2002; 14: 706–14. [DOI] [PubMed] [Google Scholar]
- 13. Mao Y, Abrieu A, Cleveland DW. Activating and silencing the mitotic checkpoint through CENP‐E‐dependent activation/inactivation of BubR1. Cell. 2003; 114: 87–98. [DOI] [PubMed] [Google Scholar]
- 14. Abrieu A, Magnaghi‐Jaulin L, Kahana JA, et al . Mps1 is a kinetochore‐associated kinase essential for the vertebrate mitotic checkpoint. Cell. 2001; 106: 83–93. [DOI] [PubMed] [Google Scholar]
- 15. Chung E, Chen RH. Phosphorylation of Cdc20 is required for its inhibition by the spindle checkpoint. Nat Cell Biol. 2003; 5: 748–53. [DOI] [PubMed] [Google Scholar]
- 16. Mills GB, Schmandt R, McGill M, et al. Expression of TTK, a novel human protein kinase, is associated with cell proliferation. J Biol Chem. 1992; 267: 16000–6. [PubMed] [Google Scholar]
- 17. Lindberg RA, Fischer WH, Hunter T. Characterization of a human protein threonine kinase isolated by screening an expression library with antibodies to phosphotyrosine. Oncogene. 1993; 8: 351–9. [PubMed] [Google Scholar]
- 18. Douville EM, Afar DE, Howell BW, et al. Multiple cDNAs encoding the esk kinase predict transmembrane and intracellular enzyme isoforms. Mol Cell Biol. 1992; 12: 2681–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lauze E, Stoelcker B, Luca FC, et al. Yeast spindle pole body duplication gene MPS1 encodes an essential dual specificity protein kinase. EMBO J. 1995; 14: 1655–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Winey M, Huneycutt BJ. Centrosomes and checkpoints: the MPS1 family of kinases. Oncogene. 2002; 21: 6161–9. [DOI] [PubMed] [Google Scholar]
- 21. Fisk HA, Winey M. The mouse Mps1p‐like kinase regulates centrosome duplication. Cell. 2001; 106: 95–104. [DOI] [PubMed] [Google Scholar]
- 22. Stucke VM, Sillje HH, Arnaud L, Nigg EA. Human Mps1 kinase is required for the spindle assembly checkpoint but not for centrosome duplication. EMBO J. 2002; 21: 1723–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu ST, Chan GK, Hittle JC, et al. Human MPS1 kinase is required for mitotic arrest induced by the loss of CENP‐E from kinetochores. Mol Biol Cell. 2003; 14: 1638–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Poss KD, Nechiporuk A, Stringer KF, et al. Germ cell aneuploidy in zebrafish with mutations in the mitotic checkpoint gene mps1. Genes Dev. 2004; 18: 1527–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fisk HA, Mattison CP, Winey M. A field guide to the Mps1 family of protein kinases. Cell Cycle. 2004; 3: 439–42. [PubMed] [Google Scholar]
- 26. Kang J, Chen Y, Zhao Y, Yu H. Autophosphorylation‐dependent activation of human Mps1 is required for the spindle checkpoint. Proc Natl Acad Sci USA. 2007; 104: 20232–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou T, Raman M, Gao Y, et al. Crystal structure of the TAO2 kinase domain: activation and specificity of a Ste20p MAP3K. Structure. 2004; 12: 1891–900. [DOI] [PubMed] [Google Scholar]
- 28. Nolen B, Taylor S, Ghosh G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell. 2004; 15: 661–75. [DOI] [PubMed] [Google Scholar]
- 29. Lei M, Lu W, Meng W, et al. Structure of PAK1 in an autoinhibited conformation reveals a multistage activation switch. Cell. 2000; 102: 387–97. [DOI] [PubMed] [Google Scholar]
- 30. Zhu S, Wang W, Clarke DC, Liu X. Activation of Mps1 promotes transforming growth factor‐beta‐independent Smad signaling. J Biol Chem. 2007; 282: 18327–38. [DOI] [PubMed] [Google Scholar]
- 31. Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002; 109: 275–82. [DOI] [PubMed] [Google Scholar]
- 32. Harrison SC. Variation on an Src‐like theme. Cell. 2003; 112: 737–40. [DOI] [PubMed] [Google Scholar]
- 33. Knighton DR, Zheng JH, Ten Eyck LF, et al. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate‐dependent protein kinase. Science. 1991; 253: 407–14. [DOI] [PubMed] [Google Scholar]
- 34. Robinson MJ, Harkins PC, Zhang J, et al. Mutation of position 52 in ERK2 creates a nonproductive binding mode for adenosine 5’‐triphosphate. Biochemistry. 1996; 35: 5641–6. [DOI] [PubMed] [Google Scholar]
- 35. Lei M, Robinson MA, Harrison SC. The active conformation of the PAK1 kinase domain. Structure. 2005; 13: 769–78. [DOI] [PubMed] [Google Scholar]
- 36. Chu ML, Chavas LM, Douglas KT, et al. Crystal structure of the catalytic domain of the mitotic checkpoint kinase Mps1 in complex with SP600125. J Biol Chem. 2008; 283: 21495–500. [DOI] [PubMed] [Google Scholar]
- 37. Mattison CP, Old WM, Steiner E, et al. Mps1 activation loop autophosphorylation enhances kinase activity. J Biol Chem. 2007; 282: 30553–61. [DOI] [PubMed] [Google Scholar]
- 38. Mossessova E, Lima CD. Ulp1‐SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol Cell. 2000; 5: 865–76. [DOI] [PubMed] [Google Scholar]
- 39. Williamson BL, Marchese J, Morrice NA. Automated identification and quantification of protein phosphorylation sites by LC/MS on a hybrid triple quadrupole linear ion trap mass spectrometer. Mol Cell Proteomics. 2006; 5: 337–46. [DOI] [PubMed] [Google Scholar]
- 40. Otwinowski Z, Minor W. Processing of x‐ray diffraction data collected in oscillation mode. Academic Press; 1997. pp. 307–26. [DOI] [PubMed]
- 41. La Fortelle E, Bricogne G. Maximum likelihood heavy‐atom parameter refinement for multiple isomorphous replacement and multivavelength anomalous diffraction methods. Orlando : Academic Press; 1997. pp. 472–94. [DOI] [PubMed] [Google Scholar]
- 42. Jones TA, Zou JY, Cowan SW, et al. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta crystallographica. 1991; 47: 110–9. [DOI] [PubMed] [Google Scholar]
- 43. Brunger AT, Adams PD, Clore GM, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998; 54: 905–21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The inhibited ATP‐binding site of Pak1. The kinase inhibitory (KI) segment of the autoregulatory fragment occupies the cleft between the small and the large lobes of the kinase domain. Lys141 makes hydrogen bonds with Asp407 of the activation loop. Therefore, the activation loop is forced to make a turn, which blocks the contact between Glu355 of Helix C and Lys299Arg and prevents binding of ATP. The hydrogen bonding interactions are shown as magenta‐dotted lines.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item