

Abstract
Novel approaches to prevent and reverse ultraviolet (UV) damage are needed to combat rising sunlight-induced skin cancer rates. Mouse studies have shown that oral or topical caffeine promotes elimination of UV-damaged keratinocytes via apoptosis and markedly inhibits subsequent skin cancer development. This potentially important therapeutic effect has not been studied in human skin cells. Here we use primary human keratinocytes to examine which of several caffeine effects mediates this process. In these cells, caffeine more than doubled apoptosis after 75 mJ/cm2 of UVB. Selectively targeting two of caffeine’s known effects did not alter UVB-induced apoptosis: inhibition of ATM (ataxia-telangiectasia mutated) and augmentation of cyclic AMP levels. In contrast, siRNA against ATR (ataxia-telangiectasia and Rad3-related) doubled apoptosis after UV through a p53-independent mechanism. Caffeine did not further augment apoptosis after UVB in cells in which ATR had been specifically depleted, suggesting a key target of caffeine in this effect is ATR. Inhibition of a central ATR target, Chk1, via siRNA or a novel highly specific inhibitor (PF610666) also augmented UVB-induced apoptosis. These data suggest that a relevant target of caffeine is the ATR-Chk1 pathway and that inhibiting ATR or Chk1 may have promise in preventing or reversing UV damage.
Keywords: ATR, Chk1, UV, apoptosis, keratinocytes
INTRODUCTION
Non-melanoma skin cancer is the most common form of cancer in humans with an annual incidence of over one million new cases in the United States. Epidemiologic and molecular studies suggest exposure to ultraviolet (UV) light is the most important etiologic factor (Brash et al., 2008). With an increase in recreational sun exposure, the use of UV tanning beds, and deterioration of the ozone layer, the incidence of non-melanoma skin cancer is expected to increase. There is thus a need to identify and develop novel approaches to target UV-induced skin cancers.
Much of the mutagenic and carcinogenic effects of UV radiation are a consequence of DNA damage in the form of cyclobutane pyrimidine dimers and 6–4 pyrimidine-pyrimidone photoproducts (Brash et al., 2008). UV exposure activates diverse cellular responses in human cells, including cell cycle arrest, DNA repair, and apoptosis. To ameliorate the effects of UV radiation, cells rely on an intricate network of signal transduction pathways known as cell cycle checkpoints. Cell cycle checkpoints are biochemical surveillance pathways that arrest cell cycle progression pending the completion of essential events and/or the repair of damaged DNA (Sancar et al., 2004). By coordinating cell cycle progression with DNA repair, cell cycle checkpoints help maintain genetic stability and prevent carcinogenesis (Abraham, 2001). However, recent studies have suggested that cell cycle checkpoints that are operational in cancer cells may reduce the effectiveness of many chemotherapeutic agents that function by inducing DNA damage (Zhou and Bartek, 2004). Indeed, chemical or genetic inhibition of cell cycle checkpoint function sensitizes cancer cells to a wide variety of DNA damaging agents, including UV (Nghiem et al., 2001), ionizing radiation (Syljuasen et al., 2004), alkylating agents (Bunch and Eastman, 1996), and topoisomerase inhibitors (Shao et al., 1997).
In humans, consumption of coffee or tea is associated with lower incidences of non-melanoma skin cancers in several epidemiologic studies (Abel et al., 2007; Hakim et al., 2000; Jacobsen et al., 1986; Rees et al., 2007). In a recent study of 93,676 Caucasian women, each additional cup of caffeinated coffee ingested was associated with a 5% decreased risk of developing non-melanoma skin cancer while drinking de-caffeinated coffee showed no such benefit (Abel et al., 2007). These studies imply that caffeine, one of the shared constituents of coffee and tea, might inhibit skin carcinogenesis in humans. To examine this possibility, the inhibitory effect of these beverages on skin carcinogenesis has been investigated in mice. Indeed, oral administration of regular tea but not decaffeinated tea was found to protect mice from UV carcinogenesis which led to subsequent direct tests of caffeine’s effects on this process (Huang et al., 1997; Lou et al., 1999). Hairless mice treated with UV twice weekly for twenty weeks had normal-appearing skin, but were at a higher risk for developing subsequent papillomas, keratoacanthomas and squamous cell carcinomas (Lou et al., 1999; Lu et al., 2002b). This animal model resembles humans that are chronically exposed to UV early in life and have an increased risk of developing skin cancer later in life. Using this model, Conney and colleagues have shown that caffeine, when administered orally or topically, inhibits tumor formation in these high risk mice (Lou et al., 1999; Lu et al., 2002b).
One potential mechanism for the inhibitory effect of caffeine is the induction of apoptosis in UV-damaged keratinocytes. Importantly, even long after completion of UV exposure, topical application of caffeine selectively induced apoptosis in nonmalignant tumors and squamous cell carcinomas without affecting neighboring normal “non-tumor” mouse skin (Lu et al., 2002b). Moreover, the effect of caffeine does not require p53 as p53 mutant patches of keratinocytes were selectively killed upon topical application of caffeine (Lu et al., 2005b). Although caffeine is a known inhibitor of cyclic adenosine monophosphate (cAMP) phosphodiesterase (Butcher and Sutherland, 1962), the inhibition of ATM (ataxia-telangiectasia mutated) kinase and in particular ATR (ataxia-telangiectasia and Rad3-related) kinase (Blasina et al., 1999; Sarkaria et al., 1999) might account for the observed effects of caffeine in the hairless mouse model (Lu et al., 2008).
The response to UV in human keratinocytes (HKC) is significantly different from that of other cell types that are more easily grown and more widely used in UV studies (D’Errico et al., 2005; D’Errico et al., 2003) as detailed in the Discussion. Therefore, it is important to investigate the response to UV in HKC, the relevant cellular target for UV-induced skin cancers. In addition, it is important to use the physiologically relevant wavelengths of UV (ultraviolet-B, UVB, 290–320 nm), which penetrate the atmosphere and are the major carcinogenic wavelengths of the light spectrum. The goals of this study were to test the effect of caffeine on UVB-irradiated HKC and to determine the target of caffeine in this response.
We found that caffeine does indeed augment UVB-induced apoptosis in primary cultures of HKC. We show the relevant mechanism of caffeine is not likely to be its inhibitory effect on cAMP phosphodiesterase because a cell-permeable analog of cAMP had no effect on UVB-induced apoptosis. Instead, we found that the ATR-Chk1 pathway is the probable target of caffeine in this response because inhibition of ATR or Chk1 (checkpoint kinase 1) each phenocopied the effect of caffeine and augmented UVB-induced apoptosis in HKC. Consistent with the observed effects of caffeine in the mouse, we found that replication checkpoint inhibition in primary human keratinocytes augmented cell death in a p53-independent manner. To our knowledge, these are the first data to demonstrate that primary human keratinocytes are sensitized to apoptosis by ATR pathway inhibition and also the most detailed dissection of the mechanism by which caffeine eliminates UV-damaged cells.
RESULTS
Caffeine augments UVB-induced apoptosis in human keratinocytes
Treatment of primary human keratinocytes (HKC) with 75 mJ/cm2 of UVB induced an apoptotic phenotype within 8 h including cleavage of caspase 3 (Figures 1a and S1a). Additionally, UVB induced an increase in poly (adenosine diphosphate-ribose) polymerase (PARP) cleavage, a response that is dependent on caspase activation and the induction of apoptosis. Consistent with the activation of caspase 3, propidium iodide (Figures 1b and S1b) and Annexin V (Figure S1c) staining of UVB-treated HKC revealed an increase in the percentage of cells undergoing apoptosis. Taken together, activation of the caspase cascade and an increase in the percentage of cells with fragmented DNA suggests that UVB induces apoptosis in HKC.
Figure 1. Caffeine augments UVB-induced apoptosis in human keratinocytes.
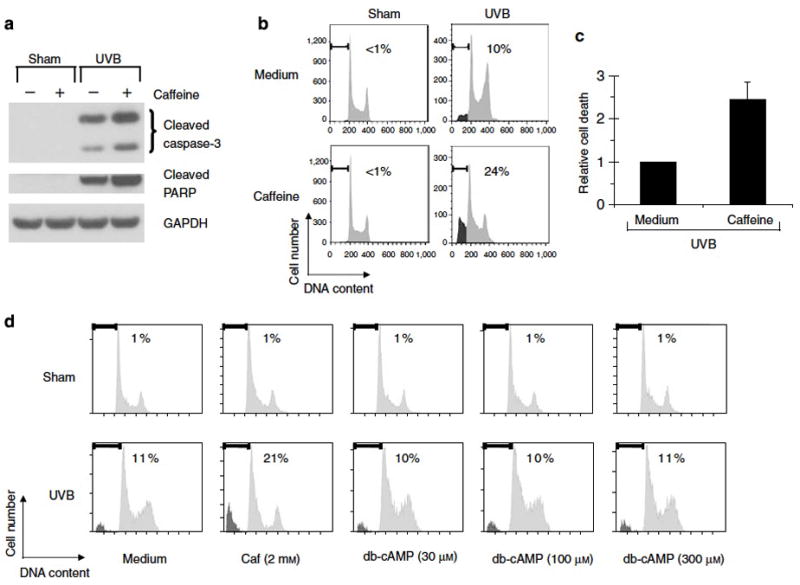
(a–c) Human keratinocytes were treated with vehicle (medium) or 2 mM of caffeine 30 min prior to 75 mJ/cm2 of UVB irradiation. (a) Western blots using the indicated antibodies. Cells were harvested 8 h after UVB irradiation. (b) Percentage of sub-2N DNA content measured by flow cytometry. Cells were harvested 24 h after UVB irradiation and stained with propidium iodide. (c) Relative cell death was calculated by comparing percentage of sub-2N DNA content in caffeine/UVB-treated cells with that in medium/UVB-treated cells in each experiment. Average of relative cell death is shown (n=4). Error bar, standard error of the mean. (d) Percentage of sub-2N DNA content measured by flow cytometry. Human keratinocytes were treated with caffeine (Caf) or dibutyryl cyclic AMP (db-cAMP) at the indicated doses 30 min prior to 75 mJ/cm2 of UVB irradiation. Cells were harvested 24 h after UVB irradiation and stained with propidium iodide.
To investigate the effect of caffeine on the response of HKC to UVB, cells were treated with 2 mM caffeine 30 min prior to irradiation with UVB. Treatment with caffeine augmented UVB-induced cleavage of caspase 3 and PARP (Figure 1a). Using flow cytometry to quantitate apoptotic cells, the proportion of cells undergoing apoptosis increased from 10% in the UVB treated keratinocytes to 24% in cells treated with caffeine (Figure 1b). While the absolute number of cells undergoing UVB-induced apoptosis varied depending on the growth characteristics of the HKC culture (in particular, the passage number), we observed a reproducible two- to three-fold increase in apoptosis when cells were pretreated with caffeine (Figure 1c).
To better understand the mechanism of the observed caffeine effect, we investigated the contribution of several known mechanisms of action for caffeine. Caffeine is an inhibitor of cAMP phosphodiesterase (IC50 = 3 mM) (Butcher and Sutherland, 1962) and it thus effectively increases levels of cAMP in cells. To determine if elevated levels of intracellular cAMP could phenocopy the observed effects of caffeine, HKC were pretreated with varying doses of dibutyryl cAMP, a cell-permeable analog of cAMP. While caffeine augmented UVB-induced apoptosis in HKC, doses of dibutyryl cAMP up to 300 μM had no effect (Figure 1d). It is highly likely that caffeine’s effect on apoptosis is not dependent on cAMP because processes that are cAMP-dependent can typically be mimicked by significantly lower concentrations of dibutyryl cAMP (typically < 100 μM) (Wang et al., 2006b). These data suggest that caffeine augments UVB-induced apoptosis in HKC through a separate mechanism than cAMP augmentation via phosphodiesterase inhibition.
ATR siRNA mimics caffeine: inhibition of Chk1 phosphorylation and augmentation of apoptosis after UV
To investigate whether caffeine inhibits ATR function in HKC following UVB treatment, we examined the phosphorylation of the ATR-dependent serine 345 residue of Chk1 in the presence and absence of caffeine. Indeed, treatment with 2 mM caffeine inhibited UVB-induced Chk1 phosphorylation at serine 345 (Figure 2a) suggesting that caffeine inhibits ATR under these conditions. To investigate a possible role for ATR in the response to UVB in HKC, we used siRNA to inhibit ATR expression. Keratinocytes were electroporated with siRNA against ATR or a non-targeting/scrambled control and treated with UVB 48 h later. siRNA against ATR reduced the abundance of ATR protein by >90% and interfered with ATR signaling as judged by marked attenuation of UVB-induced Chk1 phosphorylation (Figure 2b). Knockdown of ATR phenocopied the effects of caffeine in HKC and augmented the UVB-induced increase in caspase 3 and PARP cleavage (Figure 2b). To determine if the observed caffeine effect was through ATR inhibition, we treated ATR siRNA-electroporated HKC with medium or 2 mM caffeine as shown in Fig 2c. UVB treatment of HKC in which ATR had been inhibited by siRNA induced 50% apoptosis. However, there was no additive effect of caffeine on ATR-inhibited HKC (50% without caffeine, 48% with caffeine). The modest observed difference from 42% with caffeine alone to 48% with caffeine plus ATR-siRNA can be attributed to the more effective inhibition of ATR signaling via siRNA as compared with chemical inhibition by 2 mM caffeine (compare p-Chk1-Ser345 in panels 2a and 2b at 1 h timepoint). The lack of an additive effect of caffeine treatment on ATR siRNA-treated HKC suggests that the majority of the caffeine effect under these conditions is through ATR inhibition. Taken together, these data suggest that ATR is involved in the response to UVB in HKC and is a relevant target of caffeine.
Figure 2. ATR siRNA mimics caffeine: inhibition of Chk1 phosphorylation and augmentation of apoptosis after UV.
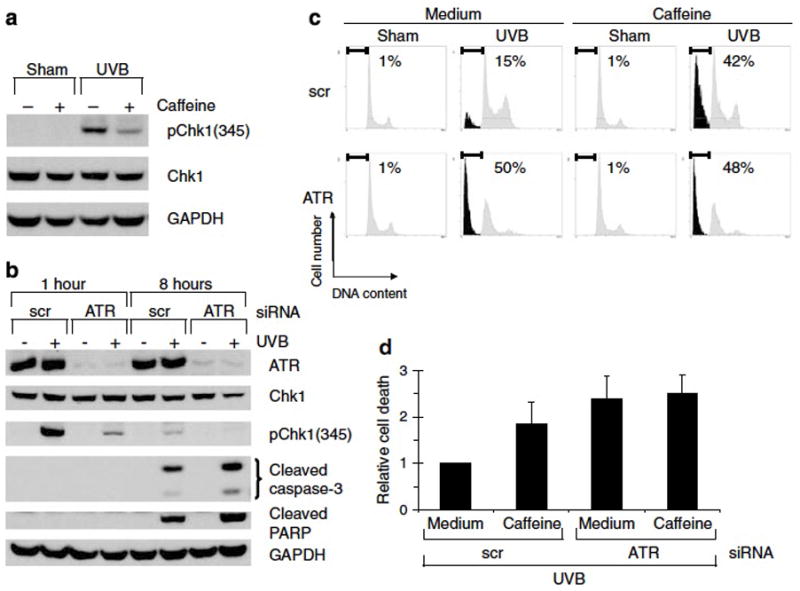
(a) Western blots using the indicated antibodies. Human keratinocytes were treated with vehicle (medium) or 2 mM of caffeine 30 min prior to 75 mJ/cm2 of UVB irradiation. Cells were harvested 2 h after UVB irradiation. (b–d) Human keratinocytes were transfected by nucleofection with siRNA against ATR or a non-targeting/scrambled control (scr). Forty-eight hours later, cells were exposed to 75 mJ/cm2 of UVB. (b) Western blots using the indicated antibodies. Cells were harvested 1 or 8 h after UVB irradiation as indicated. (c) Percentage of sub-2N DNA content measured by flow cytometry. Cells were treated with vehicle (medium) or 2 mM of caffeine 30 min prior to UVB irradiation. Cells were harvested 24 h after UVB irradiation and stained with propidium iodide. (d) Relative cell death was calculated by comparing percentage of sub-2N DNA content in scr + caffeine/UVB-, ATR + medium/UVB-, or ATR + caffeine/UVB-treated cells with that in scr + medium/UVB-treated cells in each experiment. Average of relative cell death is shown (n=3). Error bar, standard error of the mean.
ATM inhibition does not augment UVB-induced apoptosis
Caffeine inhibits not only ATR, but also ATM even more potently (IC50 for ATM = 0.2 mM, IC50 for ATR = 1.1 mM) (Sarkaria et al., 1999). The fact that caffeine addition did not further increase UVB-induced apoptosis in HKC depleted of ATR by siRNA suggests that ATM is not involved in this UV-apoptosis pathway (Figure 2c and d). In addition, we depleted cells of ATM and/or ATR using siRNA to further investigate a possible role for ATM in this pathway. Knockdown of each protein kinase was monitored by Western blot analysis (Figure 3a). Consistent with the findings in Figure 2, ATR knockdown once again potentiated the apoptotic effect of UVB (Figure 3). By targeting ATM with siRNA, protein expression was suppressed by roughly 70%, and the induction by UVB of active ATM (phospho-Serine 1981 ATM) was inhibited by roughly 50% (Figure S2). siRNA against ATM did not affect damage-induced phosphorylation of Thr-68 on Chk2 (a known target of ATM), but ATR is known to compensate for ATM in this process (Helt et al., 2005; Wang et al., 2006a). Indeed, double knockdown of ATM & ATR showed a marked suppression of Chk2 phosphorylation suggesting that ATM function was significantly inhibited by siRNA in these cells (Figure S2). In no case, however, did the addition of siRNA against ATM affect UVB-induced apoptosis (Figure 3b and c). Taken together, these data suggest that ATM is not the relevant target of caffeine for this process in HKC.
Figure 3. ATM inhibition does not augment UVB-induced apoptosis.

Human keratinocytes were transfected by nucleofection with siRNA against ATR and/or ATM, or a non-targeting/scrambled control (scr). Forty-eight hours later, cells were exposed to 75 mJ/cm2 of UVB. (a) Western blots using the indicated antibodies. Cells were harvested 8 h after UVB irradiation. (b) Percentage of sub-2N DNA content measured by flow cytometry. Cells were harvested 24 h after UVB irradiation and stained with propidium iodide. (c) Relative cell death was calculated by comparing percentage of sub-2N DNA content in ATR/UVB-, ATM/UVB-, or ATR+ATM/UVB-treated cells with that in scr/UVB-treated cells in each experiment. Average of relative cell death is shown (n=3). Error bar, standard error of the mean.
Genetic and chemical inhibition of Chk1 augments UVB-induced apoptosis
In response to DNA damage, ATR mediates much of its checkpoint signaling through its downstream effector kinase Chk1. To investigate whether or not Chk1 inhibition would potentiate UVB-induced apoptosis in HKC, we abrogated Chk1 function both genetically and chemically. Electroporation of HKC with siRNA directed against Chk1 reduced protein expression by nearly 90% (Figure 4a). HKC with reduced expression of Chk1 were significantly sensitized to apoptosis induced by UVB (Figure 4a), however, Chk1 inhibition by siRNA did not augment UVB-induced apoptosis to the same degree as caffeine or as ATR siRNA. We observed a 60% increase in cell death when comparing siRNA against Chk1 to scrambled siRNA controls.
Figure 4. Genetic and chemical inhibition of Chk1 augments UVB-induced apoptosis.

(a) Human keratinocytes were transfected by nucleofection with siRNA against Chk1 or a non-targeting/scrambled control (scr). Cells were exposed to 75 mJ/cm2 of UVB 48 h after transfection. Cells were harvested 24 h after UVB irradiation and stained with propidium iodide. Percentage of sub-2N DNA content was measured by flow cytometry. Relative cell death was calculated by comparing percentage of sub-2N DNA content in Chk1/UVB-treated cells with that in scr/UVB-treated cells in each experiment. Average of relative cell death is shown (n=2). Error bar, standard error of the mean. Western blots using the indicated antibodies are shown in inset. Cells were harvested 48 h after transfection. (b) Front view of the docked pose of PF610666 into Chk1. See text for description of how the compound binds in the kinase catalytic domain. Chemical structure of PF610666 is shown in inset. (c) Western blots using the indicated antibodies. Human keratinocytes were treated with PF610666 at the indicated doses 30 min prior to 75 mJ/cm2 of UVB irradiation. Cells were harvested 8 h after UVB irradiation. (d) Relative cell death was calculated by comparing the percentage of cells with sub-2N DNA content in each experiment among cells that received UVB plus no PF610666 to those that received UVB plus 500 or 1000 nM PF610666. Cells were harvested 24 h after UVB irradiation and stained with propidium iodide. Percentage of sub-2N DNA content was measured by flow cytometry. Average of relative cell death is shown (n=3). Error bar, standard error of the mean.
Chemical inhibition of Chk1 has typically been carried out with UCN-01, originally identified as an inhibitor of protein kinase C (Busby et al., 2000; Wang et al., 1996). UCN-01 has been shown to have inhibitory effects toward several protein kinases, including Chk1, PKC, CDKs. Thus a major effort in the field is to generate more selective Chk1 inhibitors. Here we describe the characteristics and first use of a potent, highly selective Chk1 inhibitor, PF610666 (Figure 4b). The docking pose of PF610666 with the Chk1 enzyme (Figure 4b) was obtained by modifying the Chk1 co-crystal structure (Chen et al., 2000) with PF279192, which only differs by a halogen atom that does not contact the active site (a chloride in PF610666 and a bromide in PF279192). The compound binds to the ATP binding pocket through hydrogen bonds with Cys87 and Glu85 in the hinge region (Figure 4b, white asterisk on left). The phenylbutanamide tail (Figure 4b, white asterisk on right) is immediately adjacent to the key catalytic residue Asp148. In this manner, PF610666 prevents Chk1 from phosphorylating its protein substrates. The inhibitory effect of PF610666 was tested against a panel of human kinases and shown to be highly selective against Chk1 (IC50 = 7 nM; Table 1). Effective concentration 50 (EC50) of PF610666 for cellular activity measured as the ability to override the camptothecin-induced checkpoint was 70 nM.
Table 1.
IC50 values of PF610666 and UCN-01 for selected kinases
| IC50 [nM] | ||
|---|---|---|
| Kinase Analyzed | PF610666 | UCN-01 |
| CHK1 | 7 | 11 |
| CHK2 | > 1000 | 1040 |
| CDK1 | > 1000 | 31 |
| CDK2 | > 1000 | 30 |
| PKC | > 1000 | 7 |
| RAF | > 1000 | 620 |
| JNK | > 1000 | ND |
| MEK1 | > 1000 | ND |
| PLK3 | > 1000 | ND |
| mTOR | > 1000 | ND |
The concentration of the indicated compound at which the maximum in vitro activity of the indicated kinase has been inhibited to 50% (IC50) is shown in nM. IC50 values for PF610666 were determined as described in Methods. IC50 values for UCN-01 were obtained from previous studies (Busby et al., 2000; Kawakami et al., 1996). “IC50 > 1000 nM” means that the kinase inhibition was less than 50% at 1000 nM. ND, not determined.
The fraction of maximum in vitro kinase activity that was inhibited for each of the following kinases in the presence of 1 μM PF610666 is listed below (“100%” means kinase activity was entirely blocked by 1 μM of the compound): 100% for Chk1, 38% for Chk2, 14% for Raf, and less than 10% for each of these kinases: CDK1, CDK2, PKC, JNK, MEK1, PLK3 and mTOR.
Abbreviations used in Table 1 are: CHK, checkpoint kinase; CDK, cyclin-dependent kinase; PKC, protein kinase C; JNK, c-Jun N-terminal kinase; MEK1, Mitogen activated protein kinase/Extracellular signal-regulated kinase Kinase-1; PLK3, polo-like kinase 3; mTOR, mammalian target of rapamycin.
To complement our genetic studies, we treated HKC with varying doses of PF610666. Treatment of HKC with PF610666 augmented UVB-induced PARP cleavage at 8 h (Figure 4c) and increased the percentage of cells with sub-2N DNA content at 24 h (Figure 4d). Taken together, genetic and chemical inhibition of Chk1 could each phenocopy the effect of caffeine and of ATR inhibition providing further support for the importance of this pathway in the UV response and in caffeine’s effect on this response.
ATR-induced apoptosis is largely p53-independent in human keratinocytes
p53 is frequently mutated in non-melanoma skin cancers and therapies that induce apoptosis independent of p53 are of great interest. To investigate the contribution of p53 in the response of HKC to UVB, we inhibited p53 expression using siRNA. HKC were electroporated with siRNA against p53 and/or ATR, and then UV irradiated 48 h later (Figure 5a). Treatment of HKC with 75 mJ/cm2 UVB strongly induced p53 protein levels at 8 h. Knockdown of p53 not only blocked this induction, but also suppressed p53 protein levels below the pre-UVB treatment baseline (Figure 5a). As in the previous experiments, knockdown of ATR markedly augmented the number of HKC that underwent UVB-induced apoptosis (Figure 5b and c). Blocking p53 protein induction, however, had no effect on UVB-induced apoptosis in HKC and only modestly reduced the ability of ATR inhibition to augment apoptosis.
Figure 5. ATR-induced apoptosis is largely p53-independent in human keratinocytes.

Human keratinocytes were transfected by nucleofection with siRNA against ATR and/or p53, or a non-targeting/scrambled control (scr). Forty-eight hours later, cells were exposed to 75 mJ/cm2 of UVB. (a) Western blots using the indicated antibodies. Cells were harvested 8 h after UVB irradiation. (b) Percentage of sub-2N DNA content measured by flow cytometry. Cells were harvested 24 h after UVB irradiation and stained with propidium iodide. (c) Relative cell death was calculated by comparing percentage of sub-2N DNA content in ATR/UVB-, p53/UVB-, or ATR+p53/UVB-treated cells with that in scr/UVB-treated cells in each experiment. Average of relative cell death is shown (n=3). Error bar, standard error of the mean.
DISCUSSION
In the study presented here we have used the relevant primary human cell type to examine the mechanism by which caffeine affects the response to UV. We found that genetic or chemical inhibition of the ATR-Chk1 pathway augmented UVB-induced apoptosis in a largely p53-independent manner in human keratinocytes. In contrast, other known and plausible targets of caffeine were not involved in the UV response including ATM and the regulation of cAMP levels. Our finding that cAMP levels were not involved in UV signaling in human keratinocytes agrees well with a prior study that concluded that augmenting cAMP does not mimic the effects of caffeine on lowering UV-induced carcinogenesis in mouse skin (Zajdela and Latarjet, 1978).
The effects of caffeine on skin cancer are of interest due to human epidemiologic studies linking coffee and tea consumption with lower rates of non-melanoma skin cancer (Abel et al., 2007; Hakim et al., 2000; Jacobsen et al., 1986; Rees et al., 2007) as well as animal studies showing that caffeine inhibits tumor formation in mice chronically treated with UV (Huang et al., 1997; Lu et al., 2002b; Zajdela and Latarjet, 1978). We have previously shown that topical caffeine application to mouse skin after a single dose of UVB augmented the number of apoptotic keratinocytes as evaluated by sunburn cell formation and other markers of programmed cell death (Koo et al., 2007; Lu et al., 2002a). These findings suggest that topical application of caffeine to mouse skin after UV irradiation augments the deletion of DNA-damaged keratinocytes, and may provide protection from UV-induced skin cancer development.
Non-melanoma skin cancers arise via the transformation of keratinocytes induced by UV. Thus, it is important to investigate the cellular responses to UVB in the relevant cell type that gives rise to UV-induced skin cancers. A recent study comparing primary human keratinocytes and dermal fibroblasts revealed that keratinocytes are far more susceptible to UVB-induced apoptosis than fibroblasts; yet they are more UVB-resistant than fibroblasts in terms of their proliferative capacity as measured by colony survival assays (D’Errico et al., 2003). Moreover, keratinocytes have a greater capacity (via robust global genome repair) for UV-DNA repair than fibroblasts, as might be expected from cells that are ‘professional’ UV-responsive cells (D’Errico et al., 2005). Indeed, we found that caffeine markedly augmented UVB-induced apoptosis in human keratinocytes (Figure 1). This finding suggests that caffeine’s promotion of UVB-apoptosis described in mouse keratinocytes also occurs in human primary keratinocytes.
Caffeine concentration is an important issue when comparing studies in mice and humans. Oral administration of caffeine as drinking fluid (0.4 mg/ml) inhibits UV-induced skin carcinogenesis in mice (Huang et al., 1997). The plasma and epidermal concentrations of caffeine in mice receiving the cancer-preventive dose of caffeine in drinking water (0.4 mg/ml, 2 mM) is 16 μM and 16 nmol/g tissue, respectively, on average in a 24-hour-period (Conney et al., 2007). The observed plasma concentration of caffeine in mice is consistent with the plasma concentration in humans (3.2 mg/l, 16 μM) drinking 2–5 cups of coffee per day (de Leon et al., 2003). Recently, Conney and colleagues showed that caffeine administered orally (0.4 mg/ml, 2 mM) with drinking water markedly inhibited the UVB-induced phosphorylation of Chk1 on Ser345 in the epidermis of mice (Lu et al., 2008). Because Ser345-phosphorylation of Chk1 has been shown to be highly dependent on ATR function, this means that in vivo in the mouse, plasma concentrations of 16 μM appear to be sufficient to inhibit ATR function in keratinocytes. Our in vitro studies using keratinocytes are similar to other studies using cells in culture in which effective ATR inhibition requires caffeine doses closer to those required in cell-free in vitro kinase assays where the IC50 against ATR is 1.1 mM for caffeine (Sarkaria et al., 1999). These differences in the ability of caffeine to inhibit ATR function in vitro versus in vivo have yet to be explained but may be related to the non-physiologic conditions that are required to initiate ATR activity in vitro (Sarkaria et al., 1999).
We also investigated the involvement of Chk1, which is phosphorylated by ATR in response to UV. These experiments were carried out because if ATR is the relevant target of caffeine, its effects would likely be mediated by Chk1 and also because novel specific Chk1 inhibitors are on the horizon for human therapy. UV irradiation induced phosphorylation of Chk1 maximally at 1 h, and the phosphorylation signal was nearly lost by 8 h (Figure 2b and S1a). Similar Chk1 phosphorylation kinetics have been observed in a recent study and attributed to PPM1D, a serine/threonine phosphatase whose expression is transcriptionally induced shortly after UV irradiation, and which dephosphorylates phospho-Chk1 at later time points (8 h or later post-UV) (Lu et al., 2005a).
Augmentation of UVB-induced apoptosis by siRNA against Chk1 (Figure 4a) was not as robust as that by siRNA against ATR (Figure 2d). A possible explanation is that incomplete Chk1 knockdown by siRNA allowed sufficient Chk1 function to partially activate the checkpoint. Another possibility is that ATR targets other than Chk1 also play a role in UV-induced apoptosis and ATR knockdown suppresses their function as well as Chk1’s function. Similarly, Chk1 knockdown was not as strong as 1 μM PF610666 (Figure 4d) in augmenting UVB-induced apoptosis and this may also be due to more effective inhibition of Chk1 by PF610666 or by this compound inhibiting kinases in addition to Chk1 (Chk2 and other kinases are partially inhibited in vitro at this concentration of PF610666 (Table 1)).
Pharmacologic inhibition of Chk1 has been extensively used to study the contribution of Chk1 in the cellular responses to DNA damage. UCN-01, originally identified as an inhibitor of protein kinase C, has been shown to be a potent Chk1 inhibitor (Busby et al., 2000; Wang et al., 1996). Studies have shown that UCN-01 abrogates Chk1-dependent cellular responses and sensitizes cancer cells to several DNA damaging agents, including mitomycin C (Akinaga et al., 1993), cisplatin (Bunch and Eastman, 1996), camptothecin (Shao et al., 1997), and 5-fluorouracil (Hsueh et al., 1998). Due to the known off-target inhibitory effects (ie: PKC and CDKs) of UCN-01 (Busby et al., 2000; Kawakami et al., 1996) and an interest in therapeutic applications for Chk1 inhibition, novel inhibitors of this kinase have been developed. Here we show that a newly developed compound that is far more selective than UCN-01 for Chk1 inhibition (Table 1), potently augmented UVB-induced apoptosis (Figure 4c and d). The ability of this compound to potentiate the effects of DNA damage has led to the development of its sister compound as a cancer therapeutic (Blasina et al., 2008).
Prior studies in human cancer cell lines (Nghiem et al., 2001) and mouse skin (Lu et al., 2005b; Lu et al., 2004) have shown that p53 disruption sensitizes cells to inhibition of ATR. In contrast, we found that p53 knockdown did not augment UVB-induced apoptosis by ATR inhibition in HKC (Figure 5b and c). This discrepancy might be explained by one or more of the following possibilities: (i) apoptosis signaling mechanisms may differ between these cell types; (ii) differences in methodology: p53 expression itself was suppressed in this study, while p53 function was disrupted via over-expression of MDM2 or E6 in the human cancer cell line study. What is clear and in agreement between these studies is that the majority of the apoptosis augmentation by ATR inhibition is p53-independent.
Normal human skin carries clonal patches of p53-mutated keratinocytes in as much as 4% of the epidermis representing thousands of such clones per person (Jonason et al., 1996). Sun exposure increases the frequency and size of the p53-mutant clones in skin. These findings suggest that p53-mutant clones might be resistant to UV-induced apoptosis. There is thus a need for a p53-independent therapy against UV-damaged keratinocytes. Topical applications of caffeine to mice with multiple patches of p53-mutant skin cells enhanced the elimination of these cells in the absence of further UVB irradiation (Lu et al., 2005b). Here we have shown that inhibition of the ATR-Chk1 pathway by caffeine or a selective Chk1 inhibitor promotes culling of DNA-damaged human primary keratinocytes regardless of p53 status. These are the most detailed studies yet to determine the mechanism by which caffeine augments UV-apoptosis. These data suggest that topical application of caffeine or another ATR-Chk1 pathway inhibitor, perhaps in a sunscreen or after-sun preparation, could be investigated as an approach to minimize or reverse the effects of UV damage in human skin.
MATERIALS AND METHODS
Cell Lines and Culture Conditions
Normal human keratinocytes (HKC) were isolated from adult abdominal epidermis harvested from two female patients (age 37 and 66) as previously described (Boswell et al., 2007; Kitano and Okada, 1983; Normand and Karasek, 1995) in a protocol approved by the Massachusetts General Hospital Institutional Review Board. Written, informed patient consent was obtained in adherence to the Helsinki Guidelines. Briefly, fresh specimens were washed in phosphate buffered saline (PBS) and disinfected with 70% ethanol. After removing adipose and dermal tissue, the remaining skin was digested in dispase solution [Hanks’ balanced salt solution (Invitrogen, Carlsbad, CA) containing 10 mM HEPES, 0.075% sodium bicarbonate, and 50 μg/ml gentamicin mixed 1:1 with Dispase II solution (Roche, Indianapolis, IN)] at 4°C for 18 h. The epidermis was then peeled off and trypsinized to isolate the keratinocytes, which were plated in Defined Keratinocyte-SFM (serum-free medium) (Invitrogen) on collagen I (Inamed, Fremont, CA)-coated plates and maintained at 37 °C in a humidified atmosphere of 5% CO2. The stock culture was aliquoted, frozen, and thawed as needed. Experiments were conducted on logarithmically growing HKC between passages 2–5.
Chemicals
Caffeine (Sigma, St. Louis, MO) was dissolved in PBS to a final concentration of 100 mM. Thirty minutes prior to UVB treatment, the appropriate volume of caffeine was added to culture medium to a final concentration of 2 mM. N6,2′-O-Dibutyryladenosine 3′,5′-cyclic monophosphate sodium salt (dibutyryl cAMP) (Sigma) was dissolved in purified water to a final concentration of 100 mM. Thirty minutes prior to UVB treatment, the appropriate volume of dibutyryl cAMP was added to culture medium. PF610666 was synthesized by Pfizer (San Diego, CA). PF610666 was dissolved in DMSO and stored at room temperature. Thirty minutes prior to UVB treatment, the appropriate volume of PF610666 or vehicle (DMSO) was added to culture medium.
PF610666 Inhibitor Potency and Kinase Selectivity
The inhibitory potency of PF610666 against the human, recombinant Chk1 kinase domain was determined in a biochemical activity-based assay. Chk1 activity was measured by a pyruvate kinase-lactate dehydrogenase coupled, continuous spectrophotometric assay where the phosphorylation of a Chk1 peptide substrate (Syntide-2, PLARTLSVAGLPGKK) was coupled to the oxidation of NADH and the corresponding change in absorbance intensity was measured at 340 nm using a SpectraMax plate reader (Molecular Devices Corp, Sunnyvale, CA). The assay was performed in a 96-well plate for 20 minutes at 30oC in 0.1 mL of assay buffer containing 50 mM TRIS pH 7.5, 0.4 M NaCl, 4 mM PEP, 0.15 mM NADH, 28 units of lactate dehydrogenase/mL, 16 units of pyruvate kinase/mL, 3 mM DTT, 0.125 mM Syntide-2, 0.15 mM ATP and 25 mM magnesium chloride. Assays were initiated with 1 nM of Chk1 kinase domain. The inhibition of Chk1 activity was determined by measuring initial velocities in the presence of varying concentrations of PF610666 (Blasina et al., 2008). The data was analyzed using Enzyme Kinetic and Excel software and fit to a kinetic model for competitive inhibition to obtain a Ki value. The kinase selectivity of PF610666 was evaluated by screening the compound at 1 or 10 μM against a panel of about 100 protein kinases. These kinase selectivity screens were performed in-house at Pfizer or by the kinase screening laboratories at Upstate/Millipore and the Division of Signal Transduction Therapy, University of Dundee. Effective concentration 50 (EC50) was defined as the concentration of a Chk1 inhibitor that overcame a camptothecin-induced S-phase checkpoint and caused mitosis (as measured by histone H3 phosphorylation at Ser10 in dot blot assay) in 50% of the cell population as compared to a nocodazole-only treated positive control.
RNA Interference
SMARTpool siRNAs were synthesized by Dharmacon (Lafayette, CO) against human ATM, p53, and Chk1. An siRNA duplex against human p53 was also synthesized using the following target sequence: AAGACTCCAGTGGTAATCTAC. ATR knockdown was accomplished using an siRNA duplex against the following target sequence: AACCTCCGTGATGTTGCTTGA. An siCONTROL non-targeting siRNA (Dharmacon) was used as a non-specific control. Cells were transfected by nucleofection (Amaxa, Gaithersburg, MD) using the human dermal fibroblast nucleofector kit and program T-24. Cells were harvested by trypsinization, resuspended in nucleofector solution at approximately 1–4 × 106 cells per 100 μl of solution, transfected with 140 picomoles of siRNA, and split to the appropriate number of collagen I-coated plates. Cells were allowed to recover for 48 h prior to treatment. Knockdown of targeted gene products was monitored by Western immunoblot analysis.
UV Irradiation
Prior to treatment with UVB, culture medium (containing caffeine, dibutyryl cAMP or PF610666 as indicated) was removed and reserved. Cultures were washed once with warm PBS, and then placed uncovered under a panel of four UVB bulbs (RPR-3000, Southern New England Ultraviolet, Branford, CT), emitting radiation centered around 311 nm. A Kodacel filter (K6808, Eastman Kodak, Rochester, NY) was used to eliminate any UVC light (<290 nm). Cells were exposed to 75 mJ/cm2 of UVB. UV dose was monitored with a Photolight IL1400A radiometer equipped with a SEL240/UVB detector (International Light Technologies, Peabody, MA). Following irradiation, the reserved medium was replaced, and the cultures were incubated for the indicated periods of time. Sham-treated cultures were handled exactly the same way, except that they were not exposed to UVB.
Western Immunoblot Analyses
Cells were harvested by trypsinization, washed once in PBS, and resuspended in RIPA (10 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, 0.25% Na-deoxycholate) supplemented with Complete Protease Inhibitor Cocktail (Roche). After clarifying the extract by centrifugation, protein concentration was determined using the Bradford Assay Reagent (Bio-Rad, Hercules, CA). Samples containing equal amounts of protein were mixed with 4x NuPAGE LDS Sample Buffer (Invitrogen) containing 5% β-mercaptoethanol, boiled, and separated by SDS-PAGE. Proteins were transferred to PVDF membrane and probed with antibodies against Chk1 (G-4), Actin (I-19) (both from Santa Cruz Biotechnology, Santa Cruz, CA); Chk2 (Ab-1) (NeoMarkers, Fremont, CA); p53 (1C12), phospho-Chk1 (Ser345) (133D3), phospho-Chk2 (Thr68), phospho-ATM (Ser1981) (10H11.E12), cleaved PARP (Asp214) (19F4), cleaved Caspase-3 (Asp175) (5A1) (all from Cell Signaling Technology, Danvers, MA); GAPDH (ab9485) (Abcam, Cambridge, MA); ATM (2C1) (GeneTex, San Antonio, TX), and an ATR rabbit polyclonal antibody generated using a peptide spanning amino acids 1–20 of human ATR (Nghiem et al., 2001).
Flow Cytometry
For measuring sub-2N DNA content, cells were trypsinized and harvested 24 h post UV irradiation and fixed with 70% ethanol for 1 h or overnight at 4°C. Cells were washed once with PBS, treated with 1 mg/ml ribonuclease A (Sigma) for 30 min at 37°C, and stained with 50 μg/ml propidium iodide (Sigma). For measuring Annexin V-positive cells, cells were stained with Annexin V-FITC (Assay Designs, Ann Arbor, MI) following the manufacturer’s protocol. Cells were analyzed on a FACSCalibur (BD Biosciences, San Jose, CA) or Cytomics FC 500 (Beckman Coulter, Fullerton, CA) flow cytometer. The acquired data were analyzed using the FlowJo 6.3.4 software package (Tree Star, Inc., Ashland, OR) or CXP Analysis 2.2 (Beckman Coulter).
Supplementary Material
Human keratinocytes were exposed to 75 mJ/cm2 of UVB and harvested at the indicated time points after UVB irradiation. (a) Western blots using the indicated antibodies. (b) Percentage of sub-2N DNA content measured by flow cytometry. Cells were treated with vehicle (medium) or 2 mM of caffeine 30 min prior to UVB irradiation. Cells were stained with propidium iodide. (c) Percentage of Annexin V-positive cells measured by flow cytometry. Cells were treated with vehicle (medium) or 2 mM of caffeine 30 min prior to UVB irradiation. Cells were stained with Annexin V-FITC (Assay Designs, Ann Arbor, MI) following the manufacturer’s protocol.
{kind=link}
Human keratinocytes were transfected by nucleofection with siRNA against ATR and/or ATM, or a non-targeting/scrambled control (scr). Forty-eight hours later, cells were exposed to 75 mJ/cm2 of UVB. Cells were harvested 1 h post UVB treatment and proteins were analyzed by Western blotting using the indicated antibodies.
Acknowledgments
We thank Drs. Anna Mandinova and Sarah Boswell for advice and technical assistance with primary keratinocyte isolation and culture. This work was supported by NIH grants R01-AR049832 and K02-AR50993 (PN), Harvard Skin Cancer SPORE Career Development Award (PN), NIH grant R01-CA114442 (AC), and Shiseido Corporation support to the Cutaneous Biology Research Center at Massachusetts General Hospital.
Abbreviations
- ATM
ataxia-telangiectasia mutated
- ATR
ataxia-telangiectasia and Rad3-related
- Chk1
checkpoint kinase 1
- UV
ultraviolet
- UVB
ultraviolet-B
- HKC
human keratinocytes
- cAMP
cyclic adenosine monophosphate
- PARP
poly (adenosine diphosphate-ribose) polymerase
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest.
References
- Abel EL, Hendrix SO, McNeeley SG, Johnson KC, Rosenberg CA, Mossavar-Rahmani Y, et al. Daily coffee consumption and prevalence of nonmelanoma skin cancer in Caucasian women. Eur J Cancer Prev. 2007;16:446–452. doi: 10.1097/01.cej.0000243850.59362.73. [DOI] [PubMed] [Google Scholar]
- Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- Akinaga S, Nomura K, Gomi K, Okabe M. Enhancement of antitumor activity of mitomycin C in vitro and in vivo by UCN-01, a selective inhibitor of protein kinase C. Cancer Chemother Pharmacol. 1993;32:183–189. doi: 10.1007/BF00685833. [DOI] [PubMed] [Google Scholar]
- Blasina A, Hallin J, Chen E, Arango ME, Kraynov E, Register J, et al. Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol Cancer Ther. 2008;7:2394–2404. doi: 10.1158/1535-7163.MCT-07-2391. [DOI] [PubMed] [Google Scholar]
- Blasina A, Price BD, Turenne GA, McGowan CH. Caffeine inhibits the checkpoint kinase ATM. Curr Biol. 1999;9:1135–1138. doi: 10.1016/s0960-9822(99)80486-2. [DOI] [PubMed] [Google Scholar]
- Boswell SA, Ongusaha PP, Nghiem P, Lee SW. The protective role of a small GTPase RhoE against UVB-induced DNA damage in keratinocytes. J Biol Chem. 2007;282:4850–4858. doi: 10.1074/jbc.M610532200. [DOI] [PubMed] [Google Scholar]
- Brash DE, Heffernan T, Nghiem P. Carcinogenesis: Ultraviolet Radiation. In: Wolff K, Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, editors. Fitzpatrick’s Dermatology in General Medicine. 7. Vol. 1. United States of America: McGraw-Hill; 2008. pp. 999–1006. [Google Scholar]
- Bunch RT, Eastman A. Enhancement of cisplatin-induced cytotoxicity by 7-hydroxystaurosporine (UCN-01), a new G2-checkpoint inhibitor. Clin Cancer Res. 1996;2:791–797. [PubMed] [Google Scholar]
- Busby EC, Leistritz DF, Abraham RT, Karnitz LM, Sarkaria JN. The radiosensitizing agent 7-hydroxystaurosporine (UCN-01) inhibits the DNA damage checkpoint kinase hChk1. Cancer Res. 2000;60:2108–2112. [PubMed] [Google Scholar]
- Butcher RW, Sutherland EW. Adenosine 3′,5′-phosphate in biological materials. I. Purification and properties of cyclic 3′,5′-nucleotide phosphodiesterase and use of this enzyme to characterize adenosine 3′,5′-phosphate in human urine. J Biol Chem. 1962;237:1244–1250. [PubMed] [Google Scholar]
- Chen P, Luo C, Deng Y, Ryan K, Register J, Margosiak S, et al. The 1.7 A crystal structure of human cell cycle checkpoint kinase Chk1: implications for Chk1 regulation. Cell. 2000;100:681–692. doi: 10.1016/s0092-8674(00)80704-7. [DOI] [PubMed] [Google Scholar]
- Conney AH, Zhou S, Lee MJ, Xie JG, Yang CS, Lou YR, et al. Stimulatory effect of oral administration of tea, coffee or caffeine on UVB-induced apoptosis in the epidermis of SKH-1 mice. Toxicol Appl Pharmacol. 2007;224:209–213. doi: 10.1016/j.taap.2006.11.001. [DOI] [PubMed] [Google Scholar]
- D’Errico M, Teson M, Calcagnile A, Nardo T, De Luca N, Lazzari C, et al. Differential role of transcription-coupled repair in UVB-induced response of human fibroblasts and keratinocytes. Cancer Res. 2005;65:432–438. [PubMed] [Google Scholar]
- D’Errico M, Teson M, Calcagnile A, Proietti De Santis L, Nikaido O, Botta E, et al. Apoptosis and efficient repair of DNA damage protect human keratinocytes against UVB. Cell Death Differ. 2003;10:754–756. doi: 10.1038/sj.cdd.4401224. [DOI] [PubMed] [Google Scholar]
- de Leon J, Diaz FJ, Rogers T, Browne D, Dinsmore L, Ghosheh OH, et al. A pilot study of plasma caffeine concentrations in a US sample of smoker and nonsmoker volunteers. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:165–171. doi: 10.1016/s0278-5846(02)00348-2. [DOI] [PubMed] [Google Scholar]
- Hakim IA, Harris RB, Weisgerber UM. Tea intake and squamous cell carcinoma of the skin: influence of type of tea beverages. Cancer Epidemiol Biomarkers Prev. 2000;9:727–731. [PubMed] [Google Scholar]
- Helt CE, Cliby WA, Keng PC, Bambara RA, O’Reilly MA. Ataxia telangiectasia mutated (ATM) and ATM and Rad3-related protein exhibit selective target specificities in response to different forms of DNA damage. J Biol Chem. 2005;280:1186–1192. doi: 10.1074/jbc.M410873200. [DOI] [PubMed] [Google Scholar]
- Hsueh CT, Kelsen D, Schwartz GK. UCN-01 suppresses thymidylate synthase gene expression and enhances 5-fluorouracil-induced apoptosis in a sequence-dependent manner. Clin Cancer Res. 1998;4:2201–2206. [PubMed] [Google Scholar]
- Huang MT, Xie JG, Wang ZY, Ho CT, Lou YR, Wang CX, et al. Effects of tea, decaffeinated tea, and caffeine on UVB light-induced complete carcinogenesis in SKH-1 mice: demonstration of caffeine as a biologically important constituent of tea. Cancer Res. 1997;57:2623–2629. [PubMed] [Google Scholar]
- Jacobsen BK, Bjelke E, Kvale G, Heuch I. Coffee drinking, mortality, and cancer incidence: results from a Norwegian prospective study. J Natl Cancer Inst. 1986;76:823–831. [PubMed] [Google Scholar]
- Jonason AS, Kunala S, Price GJ, Restifo RJ, Spinelli HM, Persing JA, et al. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc Natl Acad Sci U S A. 1996;93:14025–14029. doi: 10.1073/pnas.93.24.14025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K, Futami H, Takahara J, Yamaguchi K. UCN-01, 7-hydroxyl-staurosporine, inhibits kinase activity of cyclin-dependent kinases and reduces the phosphorylation of the retinoblastoma susceptibility gene product in A549 human lung cancer cell line. Biochem Biophys Res Commun. 1996;219:778–783. doi: 10.1006/bbrc.1996.0310. [DOI] [PubMed] [Google Scholar]
- Kitano Y, Okada N. Separation of the epidermal sheet by dispase. Br J Dermatol. 1983;108:555–560. doi: 10.1111/j.1365-2133.1983.tb01056.x. [DOI] [PubMed] [Google Scholar]
- Koo SW, Hirakawa S, Fujii S, Kawasumi M, Nghiem P. Protection from photodamage by topical application of caffeine after ultraviolet irradiation. Br J Dermatol. 2007;156:957–964. doi: 10.1111/j.1365-2133.2007.07812.x. [DOI] [PubMed] [Google Scholar]
- Lou YR, Lu YP, Xie JG, Huang MT, Conney AH. Effects of oral administration of tea, decaffeinated tea, and caffeine on the formation and growth of tumors in high-risk SKH-1 mice previously treated with ultraviolet B light. Nutr Cancer. 1999;33:146–153. doi: 10.1207/S15327914NC330205. [DOI] [PubMed] [Google Scholar]
- Lu X, Nannenga B, Donehower LA. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev. 2005a;19:1162–1174. doi: 10.1101/gad.1291305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YP, Lou YR, Li XH, Xie JG, Lin Y, Shih WJ, et al. Stimulatory effect of topical application of caffeine on UVB-induced apoptosis in mouse skin. Oncol Res. 2002a;13:61–70. [PubMed] [Google Scholar]
- Lu YP, Lou YR, Liao J, Xie JG, Peng QY, Yang CS, et al. Administration of green tea or caffeine enhances the disappearance of UVB-induced patches of mutant p53 positive epidermal cells in SKH-1 mice. Carcinogenesis. 2005b;26:1465–1472. doi: 10.1093/carcin/bgi086. [DOI] [PubMed] [Google Scholar]
- Lu YP, Lou YR, Peng QY, Xie JG, Conney AH. Stimulatory effect of topical application of caffeine on UVB-induced apoptosis in the epidermis of p53 and Bax knockout mice. Cancer Res. 2004;64:5020–5027. doi: 10.1158/0008-5472.CAN-04-0760. [DOI] [PubMed] [Google Scholar]
- Lu YP, Lou YR, Peng QY, Xie JG, Nghiem P, Conney AH. Effect of caffeine on the ATR/Chk1 pathway in the epidermis of UVB-irradiated mice. Cancer Res. 2008;68:2523–2529. doi: 10.1158/0008-5472.CAN-07-5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YP, Lou YR, Xie JG, Peng QY, Liao J, Yang CS, et al. Topical applications of caffeine or (−)-epigallocatechin gallate (EGCG) inhibit carcinogenesis and selectively increase apoptosis in UVB-induced skin tumors in mice. Proc Natl Acad Sci U S A. 2002b;99:12455–12460. doi: 10.1073/pnas.182429899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nghiem P, Park PK, Kim Y, Vaziri C, Schreiber SL. ATR inhibition selectively sensitizes G1 checkpoint-deficient cells to lethal premature chromatin condensation. Proc Natl Acad Sci U S A. 2001;98:9092–9097. doi: 10.1073/pnas.161281798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normand J, Karasek MA. A method for the isolation and serial propagation of keratinocytes, endothelial cells, and fibroblasts from a single punch biopsy of human skin. In Vitro Cell Dev Biol Anim. 1995;31:447–455. doi: 10.1007/BF02634257. [DOI] [PubMed] [Google Scholar]
- Rees JR, Stukel TA, Perry AE, Zens MS, Spencer SK, Karagas MR. Tea consumption and basal cell and squamous cell skin cancer: results of a case-control study. J Am Acad Dermatol. 2007;56:781–785. doi: 10.1016/j.jaad.2006.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, et al. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999;59:4375–4382. [PubMed] [Google Scholar]
- Shao RG, Cao CX, Shimizu T, O’Connor PM, Kohn KW, Pommier Y. Abrogation of an S-phase checkpoint and potentiation of camptothecin cytotoxicity by 7-hydroxystaurosporine (UCN-01) in human cancer cell lines, possibly influenced by p53 function. Cancer Res. 1997;57:4029–4035. [PubMed] [Google Scholar]
- Syljuasen RG, Sorensen CS, Nylandsted J, Lukas C, Lukas J, Bartek J. Inhibition of Chk1 by CEP-3891 accelerates mitotic nuclear fragmentation in response to ionizing Radiation. Cancer Res. 2004;64:9035–9040. doi: 10.1158/0008-5472.CAN-04-2434. [DOI] [PubMed] [Google Scholar]
- Wang Q, Fan S, Eastman A, Worland PJ, Sausville EA, O’Connor PM. UCN-01: a potent abrogator of G2 checkpoint function in cancer cells with disrupted p53. J Natl Cancer Inst. 1996;88:956–965. doi: 10.1093/jnci/88.14.956. [DOI] [PubMed] [Google Scholar]
- Wang XQ, Redpath JL, Fan ST, Stanbridge EJ. ATR dependent activation of Chk2. J Cell Physiol. 2006a;208:613–619. doi: 10.1002/jcp.20700. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kim PK, Peng X, Loughran P, Vodovotz Y, Zhang B, et al. Cyclic AMP and cyclic GMP suppress TNFalpha-induced hepatocyte apoptosis by inhibiting FADD up-regulation via a protein kinase A-dependent pathway. Apoptosis. 2006b;11:441–451. doi: 10.1007/s10495-005-4293-6. [DOI] [PubMed] [Google Scholar]
- Zajdela F, Latarjet R. Ultraviolet light induction of skin carcinoma in the mouse; influence of cAMP modifying agents. Bull Cancer. 1978;65:305–313. [PubMed] [Google Scholar]
- Zhou BB, Bartek J. Targeting the checkpoint kinases: chemosensitization versus chemoprotection. Nat Rev Cancer. 2004;4:216–225. doi: 10.1038/nrc1296. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Human keratinocytes were exposed to 75 mJ/cm2 of UVB and harvested at the indicated time points after UVB irradiation. (a) Western blots using the indicated antibodies. (b) Percentage of sub-2N DNA content measured by flow cytometry. Cells were treated with vehicle (medium) or 2 mM of caffeine 30 min prior to UVB irradiation. Cells were stained with propidium iodide. (c) Percentage of Annexin V-positive cells measured by flow cytometry. Cells were treated with vehicle (medium) or 2 mM of caffeine 30 min prior to UVB irradiation. Cells were stained with Annexin V-FITC (Assay Designs, Ann Arbor, MI) following the manufacturer’s protocol.
Human keratinocytes were transfected by nucleofection with siRNA against ATR and/or ATM, or a non-targeting/scrambled control (scr). Forty-eight hours later, cells were exposed to 75 mJ/cm2 of UVB. Cells were harvested 1 h post UVB treatment and proteins were analyzed by Western blotting using the indicated antibodies.