

Abstract
Osteoarthritis (OA) has historically been classified as ‘primary’ where no discernible cause was evident and ‘secondary’ where a triggering factor was apparent. Irrespective of the triggering events, late-stage OA is usually characterized by articular cartilage attrition and consequently the anatomical basis for disease has been viewed in terms of cartilage. However, the widespread application of magnetic resonance imaging in early OA has confirmed several different anatomical abnormalities within diseased joints. A key observation has been that several types of primary or idiopathic OA show ligament-related pathology at the time of clinical presentation, so these categories of disease are no longer idiopathic – at least from the anatomical perspective. There is also ample evidence for OA initiation in other structures including menisci and bones in addition to articular cartilage. Therefore, a new classification for OA is proposed, which is based on the anatomical sites of earliest discernible joint structural involvement. The major proposed subgroups within this classification are ligament-, cartilage-, bone-, meniscal- and synovial-related, in addition to disease that is mixed pattern or multifocal in origin. We show how such a structural classification for OA provides a useful reference framework for staging the magnitude of disease. For late-stage or end-stage/whole organ disease, the final common pathway of these different scenarios, joint replacement strategies are likely to remain the only viable option. However, for younger subjects in particular, near the time of clinical disease onset, this scheme has implications for therapy targeted to specific anatomical locations. Thus, in the same way that tumours can be classified and staged according to their tissue of origin and extent of involvement, OA can likewise be anatomically classified and staged. This has implications for therapeutic strategies including regenerative medicine therapy development.
Keywords: bone, cartilage, classification, ligaments, osteoarthritis
Introduction
Well-established or end-stage osteoarthritis (OA) is a whole organ joint disease where the only viable therapeutic option may be joint replacement (Juni et al. 2003). The existing approach to classifying OA is largely based on clinical and radiographic observations made in subjects with this advanced stage of disease (Bellamy et al. 1997; Kellgren & Lawrence, 1957; Schiphof et al. 2008a,b;). A universal feature of end-stage OA is radiographically-evident loss of joint space, usually related to articular cartilage loss that has been corroborated on joint inspection at the time of arthrotomy. Not surprisingly, the anatomical basis for OA and its molecular pathogenesis has centred on the concept that disease initiation and progression are both related to thinning and softening of articular cartilage, with progressive joint deterioration thereafter (Krasnokutsky et al. 2008). Although it is evident that changes in articular cartilage play a crucial role in the pathophysiology of OA, it has been emphasized that considering OA as primarily a disease of articular cartilage is too simplistic (Brandt et al. 2006). It is now generally accepted that complex interactions between various repair and regenerative mechanisms following an initial insult can explain many of the joint changes seen later in the disease (Lories, 2008). Despite this recognition, the emphasis on considering tissues other than articular cartilage as initiators or drivers of OA has focused on subchondral bone, with limited emphasis on the importance of other joint structures (Brandt et al. 2008; Radin et al. 1972).
Given that joint space loss and bone changes represent common radiographic features of OA, the existing disease classification schemes have recognized and focussed on the heterogeneous precipitating factors that can lead to this final common pathway (Table 1). Not surprisingly, the classification of OA has not been evaluated from an anatomical perspective, as historically only bone and joint space loss could be visualized. It remains especially noteworthy that a large group of cases are designated as primary or idiopathic based on the premise that a precipitating cause has not been identified (Table 1). These idiopathic categories are based on the principal site of skeletal involvement including the cervical spine and low back. It is quite clear from an anatomical perspective that some of these structures (e.g. the lumbar intervertebral discs) are totally devoid of articular cartilage, thus confirming that primary OA may not necessarily relate solely to articular cartilage (Table 1). Indeed, this is a contentious area and it has been argued that degenerative disease at such sites should not be categorized as OA (Birrell & Arden, 2008). This is despite the fact that the histological features of joint degeneration of such sites are very reminiscent of joint degeneration elsewhere and such conditions are viewed by some as part of a group of allied disorders (Birrell & Arden, 2008). Indeed, proof of principle for OA initiation in tissues other than articular cartilage comes from observations showing that clinically-evident traumatic injury to the menisci, bone or ligaments may lead to accelerated OA development thereafter (Hunter et al. 2006; Burr, 2004; Louboutin et al. 2009).
Table 1.
Traditional classification of osteoarthritis (OA)
| Primary or idiopathic | |
| Localized | Hands: e.g. nodal OA, erosive OA, first CMC joint OA |
| Feet: e.g. hallux valgus, hallux rigidus, talonavicular OA | |
| Knee: e.g. patello-femoral syndrome, medial/lateral compartment OA | |
| Hip: e.g. diffuse, superior, concentric | |
| Spine: e.g. DISH, spondylosis, intervertebral joints, apophyseal joints | |
| Other: wrist, glenohumoral, acromioclavicular, temporomandibular | |
| Generalized | Includes three or more areas listed above |
| Secondary | |
| Post-traumatic | e.g. fracture, infection, joint surgery |
| Congenital disorders | e.g. congenital hip dislocation, chondral dysplasia |
| Metabolic | Calcium crystal deposition |
| Haemochromatosis | |
| Acromegaly | |
| Paget's disease | |
| Ochronosis | |
| Inflammatory | Any inflammatory arthritis, e.g. rheumatoid arthritis |
| Septic arthritis | |
| Other bone and jointdisorders | Avascular necrosis, neuropathic charcot joints |
CMC, carpometacarpal; DISH, diffuse idiopathic skeletal hyperostosis.
What makes the development of a new classification scheme so timely is the wealth of emerging imaging data that are accruing on OA. In particular, the widespread application of magnetic resonance imaging (MRI) and ongoing improvement in MRI technology have confirmed different anatomical abnormalities, including ligament changes in OA around the time of clinical presentation, in settings where clinical examination and radiography may yield few (if any) positive findings (Conaghan, 2006; Tan et al. 2005, 2006b). Furthermore, recent data from cadaveric studies of age-related joint changes in normal cadaveric tissues also point towards a new anatomical scheme for disease (McGonagle et al. 2008; Benjamin & McGonagle, 2007; Tan et al. 2006b). Combining this emerging knowledge with the extensive data on OA, it is possible to devise a classification scheme that is based on the earliest discernible anatomical abnormality of joints.
The anatomical classification of osteoarthritis
An anatomical classification of OA is set out in Table 2 where the ‘idiopathic’ variety is no longer of unknown origin – at least in terms of the site of the earliest discernable structural abnormalities. The major proposed categories of disease according to this scheme relate to the tissues present in a typical synovial joint (Fig. 1). We thus propose that a distinction is made between cartilage-, ligament-, bone-, meniscal- and synovial-derived OA, together with disease that can be classed as being of multifocal origin. These anatomical subcategories are described further below and the implications of this classification for therapy development are discussed.
Table 2.
Site-specific pathogenic classification of osteoarthritis (OA)
| Type of OA | Recognized causes |
|---|---|
| Chondrogenic OA | Traumatic cartilage injury |
| Hereditary genetic disorders or chondrodysplasias | |
| Ligamentogenic (and enthesogenic) OA | Trauma, e.g. ACL rupture |
| Generalized nodal OA – hand involvement | |
| Cervical spondylosis | |
| Lumbar spondylosis | |
| DISH | |
| Meniscogenic OA | Primary meniscal degeneration |
| Meniscal extrusion | |
| Root horn tears | |
| Synoviogenic OA | Inflammatory, e.g. crystal-, CPPD-, urate- and BCP-related |
| Secondary to inflammatory arthritis | |
| Osteogenic OA | Neuropathic joint disease |
| Avascular necrosis | |
| Bony dysplasia | |
| Joint fracture | |
| Paget's disease | |
| Mixed pattern disease –‘multifocal’ | Proportion of age-related disease with serial insults to different joint structures |
ACL, anterior cruciate ligament; DISH, diffuse idiopathic skeletal hyperostosis; CPPD, calcium pyrophosphate dihydrate; BCP, basic calcium phosphate.
Fig. 1.
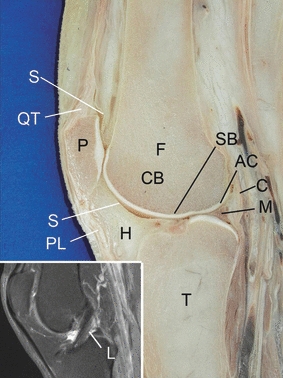
Sagittal section of the knee joint showing the tissues on which the proposed classification of osteoarthritis is based: articular cartilage (AC), subchondral bone (SB), meniscus (M), synovium (S) and (inset) ligament (L). C, capsule; CB, cancellous bone; F, femur; H, Hoffa's fat pad; P, patella; PL, patellar ligament; QT, quadriceps tendon; T, tibia. Anatomical specimen kindly provided by Dr Stefan Milz.
Cartilage-derived osteoarthritis
Functional and anatomical adaptations of cartilage in a normal joint
Most synovial joints are lined with hyaline cartilage, although a few (e.g. the temporomandibular and sternoclavicular joints) are lined with fibrocartilage (Benjamin & McGonagle, 2001). At some locations, the articular cartilage may form part of what has been termed an ‘enthesis organ’ in conjunction with associated capsular specializations (Benjamin et al. 2004a). The superficial part of any hyaline articular cartilage is uncalcified and it normally provides a smooth, wear-resistant articulating surface. It is supported by a thin layer of calcified cartilage that transmits force to the underlying subchondral bone and attaches to it across a highly irregular interface (Oegema et al. 1997). The calcified cartilage also serves to limit diffusion from the underlying bone to the deeper zones of the uncalcified cartilage layer (Oegema et al. 1997). The two zones of cartilage are separated from each other by a calcification front known as the tidemark. Reduplication of this tidemark is a common age-related change and one that is also highly characteristic of OA (Oegema et al. 1997). The formation of additional tidemarks is evidence of mineralization having spread into what was once uncalcified cartilage. The corollary is that the uncalcified cartilage thins and, as a consequence, the force gradient across it is increased (Anderson et al. 1993).
Cartilage of either type (fibrous or hyaline) lines joints because it can withstand compression (and shear) with its compression-tolerance properties stemming mainly from the presence of aggrecan. This large proteoglycan is responsible for attracting incompressible tissue fluid by capillarity into the cartilage (Kiani et al. 2002). The aggrecan molecules are held in position by a network of type II collagen fibres. Because articular cartilage is subject to compression, it is an avascular tissue, as pressure would close any blood vessels. This partly accounts for the lack of a perichondrium that characterizes cartilage in non-skeletal sites such as the respiratory tract. In the trachea, for instance, a fibrous perichondrium not only houses blood vessels but also promotes appositional growth of the cartilage and links it to adjacent structures. By contrast, synovial joint articular cartilage has a superficial surface that is bathed in synovial fluid and is not thus attached to other, more superficially-placed tissues.
The case for cartilage-derived osteoarthritis
Despite occasional objections to this concept, the importance of articular cartilage in the genesis of OA is likely to be crucial in some subgroups of disease (Fig. 2). Traumatic injuries to cartilage in both animal models and human have been associated with the subsequent progression and development of whole organ OA (Guilak et al. 2004). Genetic disorders, such as defects in type 2 collagen and several other chondropathies, where the mutations affect cartilage proteins are associated with early onset OA and rapid progression of the disease (Blom et al. 2009). Human MRI studies have shown that articular cartilage thins with age, which further links age-related cartilage changes to a propensity for OA development (Hudelmaier et al. 2001). In obese subjects, who are prone to large joint OA, cartilage loss is common over the maximal load-bearing regions of the joint (Recnik et al. 2009).
Fig. 2.
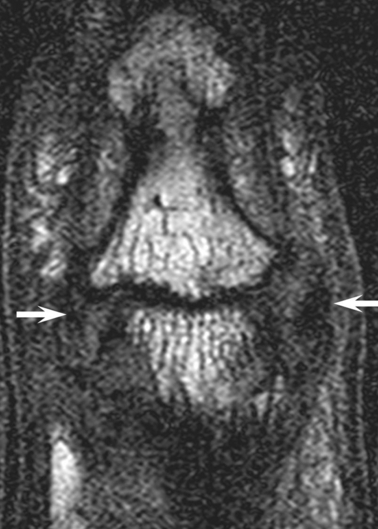
Primary ligamentogenic osteoarthritis (OA) of the distal interphalangeal joint. T1-weighted magnetic resonance imaging of the coronal image of a 51-year-old female with OA of the distal interphalangeal joint. The joint collateral ligaments are abnormal and completely disrupted (arrows). There is also complete loss of articular cartilage. In fact, age-related alternation of the ligaments is common and ligament changes predominate over cartilage changes in early hand OA. Therefore, it is proposed that any intervention solely aimed at cartilage repair in this setting is likely to fail as the primary discernible anatomical derangement is elsewhere within the joint.
For ‘non-traumatic OA’, softening of articular cartilage following breakage of the type II collagen network with increased proteoglycan water absorption has long been considered the initial pathogenic event (Loeser, 2006), with this theory directly stemming from micro-anatomical observations as set out above. The advent of MRI methodologies that give indirect measures of human articular cartilage characteristics in vivo prior to cartilage attrition greatly supports the importance of this molecular mechanism in disease pathogenesis (Gold et al. 2006). Protocols such as T2 and T1ρ mapping highlight the changes in the water content and biochemical changes within cartilage (Blumenkrantz & Majumdar, 2007). The delayed gadolinium diethylenetriamine penta-acetic acid (Gd-DTPA) enhanced MRI of cartilage technique is an MRI technique that allows indirect molecular evidence to be gathered about articular cartilage structure. Both articular cartilage and Gd-DTPA are negatively charged and this electrostatic repulsion prevents the accumulation of Gd-DTPA in healthy cartilage. However, the loss of proteoglycan is associated with Gd-DTPA accumulation. This technique shows high correlation with the biomechanical properties of cartilage (Juras et al. 2009) and has been used to confirm that cartilage proteoglycan loss, but with maintenance of cartilage thickness, is indeed an early harbinger of articular cartilage loss in vivo in human (Ericsson et al. 2009; Tiderius et al. 2003). It has also been used to show that regular exercise appears to preserve the integrity of cartilage and thus supports the concept that articular cartilage conditioning via a programme of exercise may be vital in preventing OA from developing (Roos & Dahlberg, 2005; Seedhom, 2006). Indeed, this cartilage-centric paradigm currently dominates the entire field of OA research, possibly to the detriment of a more holistic understanding of disease, as further discussed below.
Ligament-derived osteoarthritis
The principal functional anatomical adaptations of ligaments in a normal joint
Ligaments are dense fibrous connective tissues of high tensile strength that guide and limit joint movements. They may be in the form of local thickenings of the joint capsule [e.g. the medial collateral ligament (MCL) of the knee joint] or independent structures (e.g. the cruciate ligaments). By virtue of their innervation, they can also play an important role in proprioception. As briefly mentioned above, it is now becoming clear that some ligaments and/or the joint capsules that they reinforce can have complex functional interactions with an adjacent bone surface and with other joint tissues near their entheses and thus form part of an enthesis organ and/or a synovio-entheseal complex (McGonagle et al. 2007b; Benjamin & McGonagle, 2007; Benjamin et al. 2004b). Thus, where a tendon/ligament contacts a bone immediately adjacent to its enthesis, the intermittent compressive loading may be sufficiently high to provoke a chondrogenic metaplasia near the surface of the two contacting tissues. This anatomical configuration may be key to the development of the erosive OA phenotype and joint erosion in general (Grainger et al. 2007; McGonagle et al. 2009). To briefly summarize these observations, it is now emerging that primary changes in ligaments and their insertions can profoundly affect the adjacent bone and synovial tissues.
The case for ligament-derived osteoarthritis
The evidence that ligaments can be primary initiators of damage in the pathogenesis of OA is fairly compelling and has been recently reviewed (McGonagle et al. 2008) (see also Figs 2 and 3). In the experimental setting, several animal models have shown a critical role for ligament abnormalities in disease development (Quasnichka et al. 2005). The earliest structural changes seen in some models of spontaneous OA in the knee joint occur in the cruciate ligaments and changes in other joint tissues, especially articular cartilage, are secondary (Quasnichka et al. 2005). Traumatic injury of ligaments is invariably associated with disease development in animal models and human (Louboutin et al. 2009; Yeow et al. 2008).
Fig. 3.
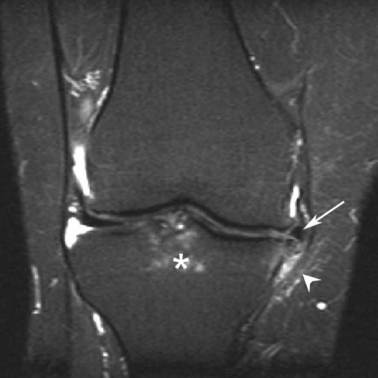
Early ligamentous osteoarthritis (OA)-based disease of the knee joint. Magnetic resonance imaging of a 38-year-old female with knee pain and clinical diagnosis of osteoarthritis, showing bone oedema at the anterior cruciate ligament insertion (asterisk). There is also meniscus extrusion (arrow) and peri-meniscal and high signal within and adjacent to the medial collateral ligament (arrowhead). Note that the articular cartilage is relatively normal. These changes point towards an OA disease process that evolves from the ligament.
In generalized nodal OA, characterized by the formation of Bouchard's and Heberden's nodes, we have noted that the most striking early changes occur in the collateral ligaments (Tan et al. 2005, 2006b). Unlike seronegative spondyloarthritis, where inflammation is centred on the enthesis organ including the immediately adjacent bone, in OA not only are these structures involved but so is the entire collateral ligament (Tan et al. 2006a). The changes are commonplace and range from thickening to frank disruption. Furthermore, the ligaments have a crucial bearing on the phenotypic expression of disease and influence the patterns of node formation, erosion formation and MRI bone oedema (Tan et al. 2005, 2006b; Grainger et al. 2007). In addition, the collateral ligaments appear to be at the epicentre of the inflammatory process and their involvement offers an explanation for the ‘peri-arthritis’ pattern of inflammation evident in hand OA (Tan et al. 2005, 2006b; McGonagle et al. 2008).
The number of distinct clinical entities that are subclassified within the traditional primary or idiopathic OA spectrum (Table 1), where the actual disease process is intimately linked to ligament/capsular pathology rather than articular cartilage or bone, is quite striking and is not only confined to generalized nodal OA (Table 2). For example, lumbar spondylosis or cervical spondylosis affects joint ligaments/capsules at sites that are devoid of synovium or articular cartilage. In addition, the radiographic appearances of diffuse idiopathic skeletal hyperostosis point to a ligamentogenic and capsular disease.
Nodal OA of the hands is often associated with more generalized involvement of the hips and knees and the first metatarsophalangeal joints, in addition to the cervical and lumbar spine as mentioned above. Recently, our studies from the hand have been extended to the knee joint where a systematic assessment of anterior cruciate ligament-related bone pathology was undertaken (Hernandez-Molina et al. 2008). These studies showed a distinct pattern of bone oedema related to the cruciate ligaments that differed from the pattern of bone oedema associated with cartilage loss over the medial tibial plateau (Hernandez-Molina et al. 2008). Historically, the imaging of ligaments has been difficult on 1.5-T field MRI scanners and there has also been a lack of sequences optimized for ligament visualization. Furthermore, MRI scoring systems for knee OA did not include an evaluation of ligaments (Hunter et al. 2008; Peterfy et al. 2004; Kornaat et al. 2005). The recognition of the importance of ligaments combined with improved imaging techniques makes it likely that the role of these structures will become increasingly appreciated in OA, especially in early disease.
It could be argued that some types of ligament-derived OA are better classified more specifically as ‘enthesis-derived’. Perhaps of the greatest translational relevance is the involvement of the entheses of the cruciate ligaments in knee OA, as damage of these is so strongly associated with knee joint degeneration (Hill et al. 2005). From an anatomical perspective, the cruciate ligaments can form closely integrated units (i.e. enthesis organs) with their adjacent tibial spines. Intermittent ligament/bone contact immediately adjacent to the entheses, occurring during the flexion/extension cycle, results in the development of specialized regions of fibrocartilage that dissipate the stress concentration that would otherwise ensue at the attachment site itself (Benjamin & McGonagle, 2001; Benjamin et al. 2004a). The structural changes reported previously in animal models of OA may well affect not only the enthesis itself but also other components of the enthesis organ (Quasnichka et al. 2005). Thus, the cruciate ligament enthesis organ shows a close functional interplay between the under surface of the ligament and the opposing bone surface immediately adjacent to the ligament and synovium. Because of this close functional integration, there is insufficient evidence at this time to split or separate ‘entheses’ and ‘ligaments’ into different groups. However, given the high degree of mechanical stress and the interface tissue nature of the enthesis, it may be a key early site for involvement in ligament-related OA.
Meniscal-derived osteoarthritis
The principal functional anatomical adaptations of menisci in a normal joint
Menisci are a characteristic feature of certain synovial joints including the knee, temporomandibular, sternoclavicular and wrist joints. They are essentially highly modified ligaments that are typically biconcave, poorly vascularized and fibrocartilaginous. They have been suggested to have a number of possible functions including altering the load distribution across the joint, ‘cushioning’ joints and improving their congruity (Messner & Gao, 1998; Aagaard & Verdonk, 1999).
The case for meniscal-derived osteoarthritis
Although not all synovial joints have menisci, we believe that it is important to include this type of OA as the burden of knee joint disease is so great (Fig. 3). It has been known for a long time that the excision of a damaged meniscus, in human and in animal models, substantially increases the risk of progression to joint failure (Yeow et al. 2008; Lanzer & Komenda, 1990; Englund & Lohmander, 2004). What about the role of the meniscus in non-traumatic OA? Meniscal extrusion, rather than articular cartilage loss, has in fact been recognized as a major cause of joint space narrowing for a long time but has been largely overlooked. It has been shown that initial radiographic knee joint space loss was not actually due to thinning of the articular cartilage but was in fact a consequence of extrusion of the medial meniscus from its normal position (Adams et al. 1999). Once the meniscus is displaced, it is easy to conceptualize a series of events including peri-meniscal synovitis, articular cartilage thinning, abnormal ligament stress and subchondral bone oedema, all of which probably contribute to secondary disease progression (Fig. 3). However, meniscal damage without clinical OA is common in asymptomatic older people but the crucial difference may be that the meniscus retains its normal position (Bhattacharyya et al. 2003).
It is important to note that the MCL acts as a restraint to the medial meniscus during flexion and extension of the knee and may play a key role in preventing its extrusion. Abnormalities along the MCL with and without meniscal extrusion have been reported in OA but it remains to be assessed how important these are in the genesis of meniscal extrusion and whether in fact much of medial meniscal-related disease could be related to primary MCL dysfunction (Blankenbaker et al. 2005). Therefore, it must be implicitly acknowledged that the earliest structural abnormalities on MRI, in this case meniscal extrusion, do not exclude the possibility that earlier pre-clinical abnormalities occur in a different anatomical territory, in this particular case the MCL (Table 3; Fig. 3).
Table 3.
Schematic anatomical staging of osteoarthritis for disease of the medial collateral ligament in the knee as an example.
| Stage | Anatomical pathology |
|---|---|
| 1 | Ligament laxity/dysregulation? |
| 2 | Meniscus extrusion + mechanically related peri-meniscal synovitis |
| 3 | Cartilage loss + and ongoing synovitis exacerbating cartilage injury |
| 4 | Bone oedema and joint malalignment with subchondral bone collapse |
| 5 | End-stage disease |
The implications of this staging scenario are that disease emanating in a particular structure, in this case the ligament, may eventually have a secondary synoviogenic or osteogenic phase, or both. Staging of the disease process could lead to the use of therapies to arrest the progression to stage 5 disease, even if it is too late to alter anatomical derangement or displacement of the ligament, which may already be irreversibly established at clinical presentation.
In recent years it has emerged that the meniscus does not have to be displaced to lead to progressive OA as posterior root tears of the lateral meniscus are associated with rapidly progressive OA (Ozkoc et al. 2008; Choi et al. 2009). This probably arises from dynamic knee joint functional perturbations with loss of the normal meniscal ‘hoop stresses’ and abnormal loading (McDermott & Amis, 2006). Indeed, we propose that the small menisco-femoral ligaments attached to the lateral meniscus (the ligaments of Humphrey and Wrisburg) may play a hitherto unappreciated role in ligament-related OA disease pathogenesis and such factors will become fully appreciated with better definition of the functional anatomy of the knee.
Bone-derived osteoarthritis
The principal functional anatomical adaptations of bone in a normal joint
The subchondral bone plate that supports articular cartilage consists of both calcified fibrocartilage and bone. However, the subchondral bone plate associated with the epiphysis of a typical long bone is nowhere near as thick as the cortical bone at midshaft level. The prominence of cancellous bone near synovial joints relates to the shock-absorbing properties of the bone – properties that are essential for protecting the overlying cartilage from damage. This close functional integration probably represents the key reason why the juxta-articular bone may be intimately involved in the OA disease process (Radin et al. 1972).
It is well known from clinical and radiographic studies that subchondral bone sclerosis is frequently linked to degenerative changes in the overlying cartilage. The orientation of trabeculae in the neighbourhood of a joint reflects the predominant direction of the mechanical forces acting on it. An excellent illustration of this stems from the comparative anatomical studies of trabecular bone where clear differences in trabecular organization in the head of the femur relate to different locomotor strategies between athletes involved in jumping activity (‘leapers’) vs. ‘non-leapers’ (Ryan & Ketcham, 2005). These observations underscore the dynamic interplay between bone and cartilage in the normally functioning joint.
The subchondral bone plate can be penetrated by vascular canals that may enter the zone of calcified cartilage and even pass through to the neighbouring uncalcified tissue. However, such vascular invasion is considered to be an indication of ossification rather than improvement of nutrient access to the articular cartilage (Clark, 1990). Whether these vascular configurations at the bone–cartilage interface are of pathophysiological relevance remains unclear but it is conceivable that they could contribute to the undermining of articular cartilage during bone inflammation or damage (Imhof et al. 1997).
The case for bone-derived osteoarthritis
As with injury to other joint tissues, it has long been recognized that trauma to bone resulting in malalignment or juxta-articular fractures predisposes joints to abnormal stress and that this accelerates the development of OA (Fig. 4). Several types of bone dysplasia are associated with altered joint biomechanics and the subsequent development of OA. The accelerated bone remodelling and bone softening that characterize Paget's disease have also been linked to the development of secondary OA (Ralston et al. 2008). Likewise, avascular necrosis of bone is associated with the development of OA (Franchi & Bullough, 1992). These examples and others provide proof of principle that OA may develop in the setting of a primary, bone-based pathology.
Fig. 4.
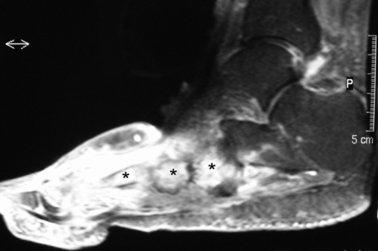
Osteogenic osteoarthritis (OA) of the foot. Fat-suppressed magnetic resonance imaging (MRI) of a 67-year-old patient with type 2 diabetes with neuropathic joint disease resulting in secondary OA affecting the mid foot, showing marked bone oedema affecting the mid foot region (asterisks) as well as adjacent soft tissue oedema. It is of note that there is strong evidence already that this type of proposed osteogenic OA responds well to bisphosphonate therapy (Jude et al. 2001). This raises the possibility that MRI selection of patients with marked bone oedema patterns of disease could help select cases for therapeutic agents that target the bone in osteogenic OA.
But what about the role of bone in OA as an initiator where a secondary bone pathology is not evident? One of the aetiologies for OA that is frequently proposed is that an increase in the stiffness of the subchondral bone plate could initiate cartilage damage, notably fibrillation and fissuring, because the integrity of both tissues is required for normal functioning of the joint (Radin & Rose, 1986; Imhof et al. 1997). It is argued that, if the stiffness of the subchondral plate changes, then so do its shock-absorbing properties. However, one of the difficulties is that subchondral bone stiffness in OA seems to be decreased (Grynpas et al. 1991; Day et al. 2001). In particular, OA bone has a lower mineral content (Grynpas et al. 1991). As subchondral bone is an important shock absorber, forces transmitted to the joint could be instrumental in leading to cartilage damage in OA, particularly if there is excessive force acting on abnormal bone. Intriguingly, the presence of an inverse relationship between bone density in patients with OA and those with osteoporosis suggested that individuals with OA were less likely to have osteoporosis but this theory is still somewhat controversial (Dequeker, 1985).
Although the role of bone as an initiator of OA remains unclear, it is very obvious that progressive joint destruction in OA is associated with bone-based pathology. Baseline MRI bone oedema in idiopathic OA has been associated with the need for future joint replacement in the hip and knee (Scher et al. 2008; Boutry et al. 2002). However, in the knee it is the presence of associated joint malalignment that is most strongly associated with disease progression (Tanamas et al. 2009). Some of the most destructive and rapidly progressive types of OA, namely neuropathic joint diseases, are dominated by extensive bone oedema changes (Greenstein et al. 2002).
More longstanding radiographic data have also assessed the bone in OA. Although radiographic subchondral sclerosis has traditionally been viewed as a secondary consequence of cartilage damage, there are now a number of reports suggesting that it may be a primary change and thus it is actually the bone sclerosis that leads to cartilage damage and not vice versa (Imhof et al. 1997). Furthermore, there are even suggestions that the primary changes occur in the blood supply to the bone and thus that degenerative joint disease could really be regarded as a vascular disease (Imhof et al. 1997). Others, however, have questioned whether there is any relationship at all between subchondral bone changes and cartilage degeneration in certain joints (Raudenbush et al. 2003). These authors found no link between the thickness of the subchondral bone plate (or its porosity) and the degeneration of cartilage on the head of the first metatarsal at the base of the big toe.
To summarize this section, some types of secondary OA are clearly osteogenic in nature and osteogenic changes may be associated with progressive joint deterioration in OA, irrespective of the underlying anatomical site of origin. However, to what extent, if any, ‘non-secondary’ OA commences in the bone anatomical territory remains as controversial today as it did at the time of the seminal hypothesis article of (Radin et al. 1972) on this topic.
Synovial-derived osteoarthritis
The principal functional anatomical adaptations of synovium in a normal joint
The synovial membrane forms the inner layer of the joint capsule, with which it shares the same mesenchymal origin. A distinctive synovium appears relatively late in joint development and this feature, together with its developmental plasticity, probably relates to its propensity for heterotopic cartilage and bone formation (Davies, 1950). Its primary function is well known, i.e. the secretion of synovial fluid that lubricates and nourishes the articular cartilage and synovio-entheseal cartilages (McGonagle et al. 2007b). Synovial fibroblasts secrete collagens (both types I and II), fibronectin and glycosaminoglycans, including hyaluronan and other molecules. The synovial membrane resident cell, the monocyte-derived macrophages, can mount an immune response against foreign antigens and thus form an important line of defence against joint infection. However, there is a downside to this, for it is now clear that an array of self molecules, including necrotic cell material and fragments of extracellular matrix, can also activate innate immune responses thus resulting in synovitis in OA (McGonagle et al. 2007b). Indeed, synovitis has long been linked to both the initiation and progression of OA (Keen et al. 2008; Loeuille et al. 2005; Haywood et al. 2003). The link between the synovium and OA is not merely related to articular cartilage attrition, for many molecules liberated from the synovium may have a direct role in potential cartilage remodelling and these include insulin like growth factor (IGF) and bone morphogenetic protein-2 (BMP-2) (Gelse et al. 2003).
The case for synovial-derived osteoarthritis
Although synovitis may not be a major initiator of the classically described forms of OA, the importance of synovitis and joint inflammation in general as secondary factors involving various pro-inflammatory cytokines that drive progressive joint destruction is well recognized (Vlychou et al. 2009; Weng et al. 2009; Toncheva et al. 2009; Keen et al. 2008; Tan et al. 2005). We propose that synovial-derived OA refers to disease settings where OA is triggered primarily by joint lining inflammation whether primary autoimmune, septic or crystalline arthropathy. Crystals including monosodium urate, pyrophosphate and basic calcium phosphate have been associated with, and could be crucial in, the development of this OA category (Reginato, 1991) and have been included in the original disease classification (Table 1).
We suggest designating the crystal arthropathies within the synovial category of disease since crystals powerfully activate macrophages and neutrophils triggering synovitis (Rose et al. 2006; Weinberger, 1995). However, it is acknowledged that the concept that OA could start in the synovium is somewhat counterintuitive to disease classification as this would imply a primary inflammatory stimulus, most notably rheumatoid arthritis and the spondyloarthritides or sepsis, for example. Indeed, it is evident that some types of synoviogenic disease could actually be designated as a type of systemic inflammatory OA. It is also acknowledged that, in the case of chondrocalcinosis and pyrophosphate arthropathy, the abnormal accumulation of pyrophosphate crystals in the cartilage may be a prerequisite for synovitis and therefore the anatomical classification should place such disease in the chondrogenic OA category.
Multifocal originating osteoarthritis within a given joint
Thus far, we have viewed OA in terms of distinct topographic locations for disease initiation. However, it is also possible that the disease could originate in two or more locations, with major joint trauma being the most obvious example. From the molecular perspective, it is interesting to note that matrilin-3 null mice have a far greater disposition for developing OA than their wild-type counterparts and that the matrilin-3 gene has dual effects on both cartilage and bone (van der Weyden et al. 2006). It is also worth bearing in mind that, in human, in older subjects there is an age-related decline in the quality of all joint structures (Steklov et al. 2009; McDougall et al. 2009), which could contribute to the multifocal pattern of disease.
Therefore, there is a strong case to be made, particularly in older subjects, that disease could be due to successive pathological events in different joint structures, with inadequate or inappropriate joint repair mechanisms. This cumulative damage to cartilage, bone and ligament and other structures could lead to an inexorable progression in disease. The formal confirmation of such a multifocal anatomical subcategory has major implications for putative therapy development strategies, namely that the targeting of a specific anatomical territory or pathway may have limited utility. Indeed, this multifocal originating disease represents the traditional ‘joint decompensation’ model for OA and entails a strong degree of therapeutic nihilism, with the exception of joint replacement strategies.
Implications of an anatomical staging osteoarthritis
As stated, clinical studies and animal models have clearly shown that ligament or meniscal damage, usually post-traumatic, may subsequently be associated with OA disease evolution into end-stage joint disease with the need for joint replacement (Buckwalter & Brown, 2004). Indeed, this has been quietly gaining popularity for some time among orthopaedic surgeons, who define the principal anatomical lesion in some clinical subtypes of early OA, usually post-traumatic, based on clinical and arthroscopic assessments. Such lesions have largely been isolated chondral defects, meniscal or anterior cruciate ligament abnormalities in the knee. Therapy is then targeted to those specific sites with the hope being that such procedures will help to maintain joint function and prevent or at least delay the development of OA. Although the treatment of such lesions (which could be termed stage 1 OA) may indefinitely postpone whole joint organ disease, this only represents a small fraction of the overall disease burden where gross disruption of joint integrity is absent at onset. However, the unparalleled anatomical MRI data that are becoming available in younger subjects with normal radiographic assessment now mean that these early stage lesions may be amenable to therapy development.
For the wider spectrum of established OA there are no clearly defined options but there are tantalizing emerging anatomical targets in disease. For example, the elegant work of Felson's group has already shown that the presence of MRI bone oedema and joint malalignment is associated with progressive knee joint deterioration with the need for future joint replacement (Felson et al. 2003). Irrespective of the anatomical site within a joint of disease initiation, this anatomical combination of bone oedema and malalignment offers the potential for targeting therapies ranging from biophysical to therapeutic agents. Indeed, there are some promising data showing that the correction of joint malalignment may be associated with robust regeneration of articular cartilage, even in aged subjects (Koshino et al. 2003).
Animal models have shown that, when osteitis is blocked, articular cartilage damage can be prevented (Herrak et al. 2004). Interestingly, bisphosphonates (which primarily inhibit osteoclasts) significantly reduce not only bone turnover but also cartilage loss in model settings (Kimura et al. 2008). Disappointingly, however, it has not been possible so far to show efficacy in terms of symptomatic improvement or prevention of radiographic progression in OA (Garnero et al. 2008). However, clinical trials to date have not been designed to use these drugs to target the subgroup with osteogenic OA including those with prominent bone marrow oedema reactions. Selection of patients on the basis of a prominent osteogenic component based on MRI bone oedema will clarify the role of bone-based therapy for OA. Indeed, this strategy appears to be efficacious in the treatment of the rapidly progressive OA associated with diabetic neuropathic joint disease where MRI bone oedema around the ankle joint complex is common (Jude et al. 2001). Furthermore, this class of drug has been associated with the retardation of joint deterioration in avascular necrosis of the hip (Agarwala et al. 2005). This represents another possible example of therapy for osteogenic OA.
The treatment of primary synoviogenic disease (e.g. rheumatoid arthritis) is highly developed with the use of an array of biological drugs to target both the innate and adaptive immunity and are capable of preventing joint destruction (de Vries-Bouwstra et al. 2005). The removal of monosodium urate crystals with allopurinol and other agents attests to the principle that the prevention of synovitis will prevent secondary OA development. Although biologic therapies have shown efficacy in animal models of OA (Goldring, 2001), the role of disease-modifying antirheumatic drugs, including biologic therapies in human for synovitis that is secondary, remains controversial (Magnano et al. 2007; Grunke & Schulze-Koops, 2006).
Staging the osteoarthritis disease process and therapy development
According to this scheme it is evident that OA may target different parts of the joint during disease pathogenesis. For example, intrinsic MCL laxity could be primary and lead to meniscal extrusion, subsequent cartilage loss, bone bruising and joint malalignment that end with progressive joint deterioration (Table 3). The cartilage damage and other factors would be expected to lead to regional synovitis that could further exacerbate tissue destruction. Conceptually, although this is a typecast of meniscal-derived OA, it is clear that the therapeutic strategy might need to change throughout the disease course with initial therapy of ligament or meniscus with attempts to arrest synovitis or MRI-determined bone oedema at a later stage in disease (Table 3). Stated in other terms, the dynamic and evolving nature of disease progression would entail a different therapeutic strategy along the patient's lifetime OA trajectory, and we have already touched on this with respect to the osteogenic component of advanced OA as set out above. In effect, this notion leads to the concept of staging of the OA process as an essential prerequisite for therapy development.
It is currently envisaged that the limited number of joint repair procedures that utilize stem cells or autologous chondrocytes will greatly expand in the coming years with developments in regenerative medicine (McGonagle et al. 2007a). However, from the proposed classification, it is evident that therapy development will have to be broad and challenging. Current therapy development aimed at articular cartilage, meniscus and ligament may be reasonable where traumatic injuries to these structures are evident but it is difficult to currently envisage therapy for a normal meniscus that has been extruded from its weight-bearing location. Likewise, primary ligament abnormalities have been scarcely researched to date. Of potential relevance is the discovery of synovial fluid mesenchymal stem cells, which may be able to contribute to tissue repair (Jones et al. 2004, 2008). The synovial fluid mesenchymal stem cell population is significantly elevated at the earliest stages of OA and, given that they have access to the entire intra-articular compartment including cartilage, ligament and menisci, they could have a hitherto unappreciated role as vehicles for joint repair (Jones et al. 2008). Indeed, the implantation of either ligament or meniscal scaffolds in human within the joint cavity leads to their cellular colonization and appropriate tissue differentiation, which probably reflects the biological effects of synovial fluid mesenchymal stem cells (Yamasaki et al. 2008; Fan et al. 2008).
Conclusion and future direction
An anatomical classification of OA is becoming increasingly critical for helping biomedical investigators who work in affluent societies where patients often present early and where MRI or arthroscopic assessment (or both) may confirm the presence of a single dominant joint structural lesion, usually well in advance of traditional conventional x-ray abnormalities such as joint space loss. Furthermore, as the resolution capabilities of MRI are increasing all the time, information on the importance of ligaments and other joint structures in the pathobiology of OA will become even clearer. An anatomical OA classification permits the development of a logical site-specific approach to both diagnosis and ultimately therapy in early disease without simply giving new molecules or therapies to all new clinical cases. Also, a site-specific anatomical classification will permit workers to evaluate a common therapeutic or orthopaedic strategy for a particular anatomical territory, irrespective of the genesis of disease at that structure.
Of course, a key question to address is whether an anatomical derangement of a particular structure is actually due to disease of that territory. For example, a myriad of factors, including joint malalignment, pro-inflammatory mediators and metalloprotease enzyme pathway dysregulation, all potentially originating elsewhere in the joint, could be key drivers of the cartilage destruction process, even in situations where cartilage structural changes seem primary. There is also the consideration of systemic factors that lead to disease in a specific territory and such factors include the subject's genetic make-up and factors that underline uric acid or basic calcium phosphate crystal production (Molloy et al. 2008).
It must also be pointed out that the key microanatomical functional derangements, namely neuropathy, which may underscore some types of ligament or osteogenic OA may not be discernible on imaging (Konttinen et al. 2006). Likewise, subtle age-related proprioceptive changes within structures such as ligaments or joint capsules as the underlying basis for anatomical derangement will be difficult to prove (Felson et al. 2009).
In conclusion, the crux of the proposed classification scheme is that the designation of some types of primary or idiopathic OA is no longer tenable, at least from an anatomical perspective, as evidence has emerged that shows structural abnormalities at different sites in early disease. In general, researchers and clinicians tend to focus on one specific structure in disease pathogenesis, often neglecting other structures that could sometimes be equally important. Bearing this in mind, our viewpoint is that a more holistic approach using the proposed OA anatomical classification scheme should be adopted. Although OA is a heterogeneous disease process, occasionally multifactorial in origin, disease initiation at specific anatomical locations in different groups offers a new way for disease classification that is of direct translational relevance. This is especially relevant for the targeted development of regenerative medicine strategies for joint failure or for strategies to prevent joint failure.
Author contributions
All authors contributed to the drafting of the manuscript, critical revision of the manuscript and approval of the article.
References
- Aagaard H, Verdonk R. Function of the normal meniscus and consequences of meniscal resection. Scand J Med Sci Sports. 1999;9:134–140. doi: 10.1111/j.1600-0838.1999.tb00443.x. [DOI] [PubMed] [Google Scholar]
- Adams JG, McAlindon T, Dimasi M, et al. Contribution of meniscal extrusion and cartilage loss to joint space narrowing in osteoarthritis. Clin Radiol. 1999;54:502–506. doi: 10.1016/s0009-9260(99)90846-2. [DOI] [PubMed] [Google Scholar]
- Agarwala S, Jain D, Joshi VR, et al. Efficacy of alendronate, a bisphosphonate, in the treatment of AVN of the hip. A prospective open-label study. Rheumatology (Oxford) 2005;44:352–359. doi: 10.1093/rheumatology/keh481. [DOI] [PubMed] [Google Scholar]
- Anderson DD, Brown TD, Radin EL. The influence of basal cartilage calcification on dynamic juxtaarticular stress transmission. Clin Orthop Relat Res. 1993:298–307. [PubMed] [Google Scholar]
- Bellamy N, Kirwan J, Boers M, et al. Recommendations for a core set of outcome measures for future phase III clinical trials in knee, hip, and hand osteoarthritis. Consensus development at OMERACT III. J Rheumatol. 1997;24:799–802. [PubMed] [Google Scholar]
- Benjamin M, McGonagle D. The anatomical basis for disease localisation in seronegative spondyloarthropathy at entheses and related sites. J Anat. 2001;199:503–526. doi: 10.1046/j.1469-7580.2001.19950503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin M, McGonagle D. Histopathologic changes at “synovio-entheseal complexes” suggesting a novel mechanism for synovitis in osteoarthritis and spondylarthritis. Arthritis Rheum. 2007;56:3601–3609. doi: 10.1002/art.23078. [DOI] [PubMed] [Google Scholar]
- Benjamin M, Moriggl B, Brenner E, et al. The “enthesis organ” concept: why enthesopathies may not present as focal insertional disorders. Arthritis Rheum. 2004a;50:3306–3313. doi: 10.1002/art.20566. [DOI] [PubMed] [Google Scholar]
- Benjamin M, Redman S, Milz S, et al. Adipose tissue at entheses: the rheumatological implications of its distribution. A potential site of pain and stress dissipation? Ann Rheum Dis. 2004b;63:1549–1555. doi: 10.1136/ard.2003.019182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya T, Gale D, Dewire P, et al. The clinical importance of meniscal tears demonstrated by magnetic resonance imaging in osteoarthritis of the knee. J Bone Joint Surg Am. 2003;85-A:4–9. doi: 10.2106/00004623-200301000-00002. [DOI] [PubMed] [Google Scholar]
- Birrell F, Arden NK. A view on the pathogenesis of osteoarthritis from the shoulders of giants. Rheumatology (Oxford) 2008;47:1263–1264. doi: 10.1093/rheumatology/ken238. [DOI] [PubMed] [Google Scholar]
- Blankenbaker DG, De Smet AA, Fine JP. Is intra-articular pathology associated with MCL edema on MR imaging of the non-traumatic knee? Skeletal Radiol. 2005;34:462–467. doi: 10.1007/s00256-005-0931-x. [DOI] [PubMed] [Google Scholar]
- Blom AB, Brockbank SM, van Lent PL, et al. Involvement of the Wnt signaling pathway in experimental and human osteoarthritis: prominent role of Wnt-induced signaling protein 1. Arthritis Rheum. 2009;60:501–512. doi: 10.1002/art.24247. [DOI] [PubMed] [Google Scholar]
- Blumenkrantz G, Majumdar S. Quantitative magnetic resonance imaging of articular cartilage in osteoarthritis. Eur Cell Mater. 2007;13:76–86. doi: 10.22203/ecm.v013a08. [DOI] [PubMed] [Google Scholar]
- Boutry N, Paul C, Leroy X, et al. Rapidly destructive osteoarthritis of the hip: MR imaging findings. AJR Am J Roentgenol. 2002;179:657–663. doi: 10.2214/ajr.179.3.1790657. [DOI] [PubMed] [Google Scholar]
- Brandt KD, Radin EL, Dieppe PA, et al. Yet more evidence that osteoarthritis is not a cartilage disease. Ann Rheum Dis. 2006;65:1261–1264. doi: 10.1136/ard.2006.058347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt KD, Dieppe P, Radin EL. Etiopathogenesis of osteoarthritis. Rheum Dis Clin North Am. 2008;34:531–559. doi: 10.1016/j.rdc.2008.05.011. [DOI] [PubMed] [Google Scholar]
- Buckwalter JA, Brown TD. Joint injury, repair, and remodeling: roles in post-traumatic osteoarthritis. Clin Orthop Relat Res. 2004:7–16. [PubMed] [Google Scholar]
- Burr DB. The importance of subchondral bone in the progression of osteoarthritis. J Rheumatol Suppl. 2004;70:77–80. [PubMed] [Google Scholar]
- Choi JW, Chung HW, Ahn JH, et al. Central hole tear of the discoid meniscus of the knee in magnetic resonance imaging: mimicking the bucket-handle tear. J Comput Assist Tomogr. 2009;33:155–159. doi: 10.1097/RCT.0b013e318166d6a7. [DOI] [PubMed] [Google Scholar]
- Clark JM. The structure of vascular channels in the subchondral plate. J Anat. 1990;171:105–115. [PMC free article] [PubMed] [Google Scholar]
- Conaghan P. Is MRI useful in osteoarthritis? Best Pract Res Clin Rheumatol. 2006;20:57–68. doi: 10.1016/j.berh.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Davies DV. The structure and functions of the synovial membrane. Br Med J. 1950;1:92–95. doi: 10.1136/bmj.1.4645.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day JS, Ding M, van der Linden JC, et al. A decreased subchondral trabecular bone tissue elastic modulus is associated with pre-arthritic cartilage damage. J Orthop Res. 2001;19:914–918. doi: 10.1016/S0736-0266(01)00012-2. [DOI] [PubMed] [Google Scholar]
- Dequeker J. The relationship between osteoporosis and osteoarthritis. Clin Rheum Dis. 1985;11:271–296. [PubMed] [Google Scholar]
- Englund M, Lohmander LS. Risk factors for symptomatic knee osteoarthritis fifteen to twenty-two years after meniscectomy. Arthritis Rheum. 2004;50:2811–2819. doi: 10.1002/art.20489. [DOI] [PubMed] [Google Scholar]
- Ericsson YB, Tjornstrand J, Tiderius CJ, et al. Relationship between cartilage glycosaminoglycan content (assessed with dGEMRIC) and OA risk factors in meniscectomized patients. Osteoarth Cartil. 2009;17:565–570. doi: 10.1016/j.joca.2008.10.009. [DOI] [PubMed] [Google Scholar]
- Fan H, Liu H, Wong EJ, et al. In vivo study of anterior cruciate ligament regeneration using mesenchymal stem cells and silk scaffold. Biomaterials. 2008;29:3324–3337. doi: 10.1016/j.biomaterials.2008.04.012. [DOI] [PubMed] [Google Scholar]
- Felson DT, McLaughlin S, Goggins J, et al. Bone marrow edema and its relation to progression of knee osteoarthritis. Ann Intern Med. 2003;139:330–336. doi: 10.7326/0003-4819-139-5_part_1-200309020-00008. [DOI] [PubMed] [Google Scholar]
- Felson DT, Gross KD, Nevitt MC, et al. The effects of impaired joint position sense on the development and progression of pain and structural damage in knee osteoarthritis. Arthritis Rheum. 2009;61:1070–1076. doi: 10.1002/art.24606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchi A, Bullough PG. Secondary avascular necrosis in coxarthrosis: a morphologic study. J Rheumatol. 1992;19:1263–1268. [PubMed] [Google Scholar]
- Garnero P, Aronstein WS, Cohen SB, et al. Relationships between biochemical markers of bone and cartilage degradation with radiological progression in patients with knee osteoarthritis receiving risedronate: the Knee Osteoarthritis Structural Arthritis randomized clinical trial. Osteoarth Cartil. 2008;16:660–666. doi: 10.1016/j.joca.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Gelse K, von der Mark K, Aigner T, et al. Articular cartilage repair by gene therapy using growth factor-producing mesenchymal cells. Arthritis Rheum. 2003;48:430–441. doi: 10.1002/art.10759. [DOI] [PubMed] [Google Scholar]
- Gold GE, Burstein D, Dardzinski B, et al. MRI of articular cartilage in OA: novel pulse sequences and compositional/functional markers. Osteoarth Cartil. 2006;14(Suppl A):A76–A86. doi: 10.1016/j.joca.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Goldring MB. Anticytokine therapy for osteoarthritis. Expert Opin Biol Ther. 2001;1:817–829. doi: 10.1517/14712598.1.5.817. [DOI] [PubMed] [Google Scholar]
- Grainger AJ, Farrant JM, O’Connor PJ, et al. MR imaging of erosions in interphalangeal joint osteoarthritis: is all osteoarthritis erosive? Skeletal Radiol. 2007;36:737–745. doi: 10.1007/s00256-007-0287-5. [DOI] [PubMed] [Google Scholar]
- Greenstein AS, Marzo-Ortega H, Emery P, et al. Magnetic resonance imaging as a predictor of progressive joint destruction in neuropathic joint disease. Arthritis Rheum. 2002;46:2814–2815. doi: 10.1002/art.10532. [DOI] [PubMed] [Google Scholar]
- Grunke M, Schulze-Koops H. Successful treatment of inflammatory knee osteoarthritis with tumour necrosis factor blockade. Ann Rheum Dis. 2006;65:555–556. doi: 10.1136/ard.2006.053272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynpas MD, Alpert B, Katz I, et al. Subchondral bone in osteoarthritis. Calcif Tissue Int. 1991;49:20–26. doi: 10.1007/BF02555898. [DOI] [PubMed] [Google Scholar]
- Guilak F, Fermor B, Keefe FJ, et al. The role of biomechanics and inflammation in cartilage injury and repair. Clin Orthop Relat Res. 2004:17–26. doi: 10.1097/01.blo.0000131233.83640.91. [DOI] [PubMed] [Google Scholar]
- Haywood L, McWilliams DF, Pearson CI, et al. Inflammation and angiogenesis in osteoarthritis. Arthritis Rheum. 2003;48:2173–2177. doi: 10.1002/art.11094. [DOI] [PubMed] [Google Scholar]
- Hernandez-Molina G, Guermazi A, Niu J, et al. Central bone marrow lesions in symptomatic knee osteoarthritis and their relationship to anterior cruciate ligament tears and cartilage loss. Arthritis Rheum. 2008;58:130–136. doi: 10.1002/art.23173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrak P, Gortz B, Hayer S, et al. Zoledronic acid protects against local and systemic bone loss in tumor necrosis factor-mediated arthritis. Arthritis Rheum. 2004;50:2327–2337. doi: 10.1002/art.20384. [DOI] [PubMed] [Google Scholar]
- Hill CL, Seo GS, Gale D, et al. Cruciate ligament integrity in osteoarthritis of the knee. Arthritis Rheum. 2005;52:794–799. doi: 10.1002/art.20943. [DOI] [PubMed] [Google Scholar]
- Hudelmaier M, Glaser C, Hohe J, et al. Age-related changes in the morphology and deformational behavior of knee joint cartilage. Arthritis Rheum. 2001;44:2556–2561. doi: 10.1002/1529-0131(200111)44:11<2556::aid-art436>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Hunter DJ, Zhang YQ, Niu JB, et al. The association of meniscal pathologic changes with cartilage loss in symptomatic knee osteoarthritis. Arthritis Rheum. 2006;54:795–801. doi: 10.1002/art.21724. [DOI] [PubMed] [Google Scholar]
- Hunter DJ, Lo GH, Gale D, et al. The reliability of a new scoring system for knee osteoarthritis MRI and the validity of bone marrow lesion assessment: BLOKS (Boston Leeds Osteoarthritis Knee Score) Ann Rheum Dis. 2008;67:206–211. doi: 10.1136/ard.2006.066183. [DOI] [PubMed] [Google Scholar]
- Imhof H, Breitenseher M, Kainberger F, et al. Degenerative joint disease: cartilage or vascular disease? Skeletal Radiol. 1997;26:398–403. doi: 10.1007/s002560050254. [DOI] [PubMed] [Google Scholar]
- Jones EA, English A, Henshaw K, et al. Enumeration and phenotypic characterization of synovial fluid multipotential mesenchymal progenitor cells in inflammatory and degenerative arthritis. Arthritis Rheum. 2004;50:817–827. doi: 10.1002/art.20203. [DOI] [PubMed] [Google Scholar]
- Jones EA, Crawford A, English A, et al. Synovial fluid mesenchymal stem cells in health and early osteoarthritis: detection and functional evaluation at the single-cell level. Arthritis Rheum. 2008;58:1731–1740. doi: 10.1002/art.23485. [DOI] [PubMed] [Google Scholar]
- Jude EB, Selby PL, Burgess J, et al. Bisphosphonates in the treatment of Charcot neuroarthropathy: a double-blind randomised controlled trial. Diabetologia. 2001;44:2032–2037. doi: 10.1007/s001250100008. [DOI] [PubMed] [Google Scholar]
- Juni P, Dieppe P, Donovan J, et al. Population requirement for primary knee replacement surgery: a cross-sectional study. Rheumatology (Oxford) 2003;42:516–521. doi: 10.1093/rheumatology/keg196. [DOI] [PubMed] [Google Scholar]
- Juras V, Bittsansky M, Majdisova Z, et al. In vitro determination of biomechanical properties of human articular cartilage in osteoarthritis using multi-parametric MRI. J Magn Reson. 2009;197:40–47. doi: 10.1016/j.jmr.2008.11.019. [DOI] [PubMed] [Google Scholar]
- Keen HI, Wakefield RJ, Grainger AJ, et al. An ultrasonographic study of osteoarthritis of the hand: synovitis and its relationship to structural pathology and symptoms. Arthritis Rheum. 2008;59:1756–1763. doi: 10.1002/art.24312. [DOI] [PubMed] [Google Scholar]
- Kellgren JH, Lawrence JS. Radiological assessment of osteo-arthrosis. Ann Rheum Dis. 1957;16:494–502. doi: 10.1136/ard.16.4.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiani C, Chen L, Wu YJ, et al. Structure and function of aggrecan. Cell Res. 2002;12:19–32. doi: 10.1038/sj.cr.7290106. [DOI] [PubMed] [Google Scholar]
- Kimura M, Miyazawa K, Tabuchi M, et al. Bisphosphonate treatment increases the size of the mandibular condyle and normalizes growth of the mandibular ramus in osteoprotegerin-deficient mice. Calcif Tissue Int. 2008;82:137–147. doi: 10.1007/s00223-007-9097-y. [DOI] [PubMed] [Google Scholar]
- Konttinen YT, Tiainen VM, Gomez-Barrena E, et al. Innervation of the joint and role of neuropeptides. Ann N Y Acad Sci. 2006;1069:149–154. doi: 10.1196/annals.1351.013. [DOI] [PubMed] [Google Scholar]
- Kornaat PR, Ceulemans RY, Kroon HM, et al. MRI assessment of knee osteoarthritis: Knee Osteoarthritis Scoring System (KOSS) – inter-observer and intra-observer reproducibility of a compartment-based scoring system. Skeletal Radiol. 2005;34:95–102. doi: 10.1007/s00256-004-0828-0. [DOI] [PubMed] [Google Scholar]
- Koshino T, Wada S, Ara Y, et al. Regeneration of degenerated articular cartilage after high tibial valgus osteotomy for medial compartmental osteoarthritis of the knee. Knee. 2003;10:229–236. doi: 10.1016/s0968-0160(03)00005-x. [DOI] [PubMed] [Google Scholar]
- Krasnokutsky S, Attur M, Palmer G, et al. Current concepts in the pathogenesis of osteoarthritis. Osteoarth Cartil. 2008;16(Suppl 3):S1–S3. doi: 10.1016/j.joca.2008.06.025. [DOI] [PubMed] [Google Scholar]
- Lanzer WL, Komenda G. Changes in articular cartilage after meniscectomy. Clin Orthop Relat Res. 1990:41–48. [PubMed] [Google Scholar]
- Loeser RF. Molecular mechanisms of cartilage destruction: mechanics, inflammatory mediators, and aging collide. Arthritis Rheum. 2006;54:1357–1360. doi: 10.1002/art.21813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeuille D, Chary-Valckenaere I, Champigneulle J, et al. Macroscopic and microscopic features of synovial membrane inflammation in the osteoarthritic knee: correlating magnetic resonance imaging findings with disease severity. Arthritis Rheum. 2005;52:3492–3501. doi: 10.1002/art.21373. [DOI] [PubMed] [Google Scholar]
- Lories RJ. Joint homeostasis, restoration, and remodeling in osteoarthritis. Best Pract Res Clin Rheumatol. 2008;22:209–220. doi: 10.1016/j.berh.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Louboutin H, Debarge R, Richou J, et al. Osteoarthritis in patients with anterior cruciate ligament rupture: a review of risk factors. Knee. 2009;16:239–244. doi: 10.1016/j.knee.2008.11.004. [DOI] [PubMed] [Google Scholar]
- Magnano MD, Chakravarty EF, Broudy C, et al. A pilot study of tumor necrosis factor inhibition in erosive/inflammatory osteoarthritis of the hands. J Rheumatol. 2007;34:1323–1327. [PubMed] [Google Scholar]
- McDermott ID, Amis AA. The consequences of meniscectomy. J Bone Joint Surg Br. 2006;88:1549–1556. doi: 10.1302/0301-620X.88B12.18140. [DOI] [PubMed] [Google Scholar]
- McDougall JJ, Andruski B, Schuelert N, et al. Unravelling the relationship between age, nociception and joint destruction in naturally occurring osteoarthritis of Dunkin Hartley guinea pigs. Pain. 2009;141:222–232. doi: 10.1016/j.pain.2008.10.013. [DOI] [PubMed] [Google Scholar]
- McGonagle D, De Bari C, Arnold P, et al. Lessons from musculoskeletal stem cell research: the key to successful regenerative medicine development. Arthritis Rheum. 2007a;56:714–721. doi: 10.1002/art.22440. [DOI] [PubMed] [Google Scholar]
- McGonagle D, Lories RJ, Tan AL, et al. The concept of a “synovio-entheseal complex” and its implications for understanding joint inflammation and damage in psoriatic arthritis and beyond. Arthritis Rheum. 2007b;56:2482–2491. doi: 10.1002/art.22758. [DOI] [PubMed] [Google Scholar]
- McGonagle D, Tan AL, Grainger AJ, et al. Heberden's nodes and what Heberden could not see: the pivotal role of ligaments in the pathogenesis of early nodal osteoarthritis and beyond. Rheumatology (Oxford) 2008;47:1278–1285. doi: 10.1093/rheumatology/ken093. [DOI] [PubMed] [Google Scholar]
- McGonagle D, Tan AL, Moller Dohn U, et al. Microanatomic studies to define predictive factors for the topography of periarticular erosion formation in inflammatory arthritis. Arthritis Rheum. 2009;60:1042–1051. doi: 10.1002/art.24417. [DOI] [PubMed] [Google Scholar]
- Messner K, Gao J. The menisci of the knee joint. Anatomical and functional characteristics, and a rationale for clinical treatment. J Anat. 1998;193(Pt 2):161–178. doi: 10.1046/j.1469-7580.1998.19320161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy ES, Morgan MP, Doherty GA, et al. Mechanism of basic calcium phosphate crystal-stimulated cyclo-oxygenase-1 up-regulation in osteoarthritic synovial fibroblasts. Rheumatology (Oxford) 2008;47:965–971. doi: 10.1093/rheumatology/ken144. [DOI] [PubMed] [Google Scholar]
- Oegema TR, Jr, Carpenter RJ, Hofmeister F, et al. The interaction of the zone of calcified cartilage and subchondral bone in osteoarthritis. Microsc Res Tech. 1997;37:324–332. doi: 10.1002/(SICI)1097-0029(19970515)37:4<324::AID-JEMT7>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Ozkoc G, Circi E, Gonc U, et al. Radial tears in the root of the posterior horn of the medial meniscus. Knee Surg Sports Traumatol Arthrosc. 2008;16:849–854. doi: 10.1007/s00167-008-0569-z. [DOI] [PubMed] [Google Scholar]
- Peterfy CG, Guermazi A, Zaim S, et al. Whole-Organ Magnetic Resonance Imaging Score (WORMS) of the knee in osteoarthritis. Osteoarth Cartil. 2004;12:177–190. doi: 10.1016/j.joca.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Quasnichka HL, Anderson-MacKenzie JM, Tarlton JF, et al. Cruciate ligament laxity and femoral intercondylar notch narrowing in early-stage knee osteoarthritis. Arthritis Rheum. 2005;52:3100–3109. doi: 10.1002/art.21340. [DOI] [PubMed] [Google Scholar]
- Radin EL, Rose RM. Role of subchondral bone in the initiation and progression of cartilage damage. Clin Orthop Relat Res. 1986:34–40. [PubMed] [Google Scholar]
- Radin EL, Paul IL, Rose RM. Role of mechanical factors in pathogenesis of primary osteoarthritis. Lancet. 1972;1:519–522. doi: 10.1016/s0140-6736(72)90179-1. [DOI] [PubMed] [Google Scholar]
- Ralston SH, Langston AL, Reid IR. Pathogenesis and management of Paget's disease of bone. Lancet. 2008;372:155–163. doi: 10.1016/S0140-6736(08)61035-1. [DOI] [PubMed] [Google Scholar]
- Raudenbush D, Sumner DR, Panchal PM, et al. Subchondral thickness does not vary with cartilage degeneration on the metatarsal. J Am Podiatr Med Assoc. 2003;93:104–110. doi: 10.7547/87507315-93-2-104. [DOI] [PubMed] [Google Scholar]
- Recnik G, Kralj-Iglic V, Iglic A, et al. The role of obesity, biomechanical constitution of the pelvis and contact joint stress in progression of hip osteoarthritis. Osteoarth Cartil. 2009;17:879–882. doi: 10.1016/j.joca.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Reginato AJ. Calcium pyrophosphate dihydrate gout and other crystal deposition diseases. Curr Opin Rheumatol. 1991;3:676–683. doi: 10.1097/00002281-199108000-00020. [DOI] [PubMed] [Google Scholar]
- Roos EM, Dahlberg L. Positive effects of moderate exercise on glycosaminoglycan content in knee cartilage: a four-month, randomized, controlled trial in patients at risk of osteoarthritis. Arthritis Rheum. 2005;52:3507–3514. doi: 10.1002/art.21415. [DOI] [PubMed] [Google Scholar]
- Rose DM, Sydlaske AD, Agha-Babakhani A, et al. Transglutaminase 2 limits murine peritoneal acute gout-like inflammation by regulating macrophage clearance of apoptotic neutrophils. Arthritis Rheum. 2006;54:3363–3371. doi: 10.1002/art.22137. [DOI] [PubMed] [Google Scholar]
- Ryan TM, Ketcham RA. Angular orientation of trabecular bone in the femoral head and its relationship to hip joint loads in leaping primates. J Morphol. 2005;265:249–263. doi: 10.1002/jmor.10315. [DOI] [PubMed] [Google Scholar]
- Scher C, Craig J, Nelson F. Bone marrow edema in the knee in osteoarthrosis and association with total knee arthroplasty within a three-year follow-up. Skeletal Radiol. 2008;37:609–617. doi: 10.1007/s00256-008-0504-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiphof D, Boers M, Bierma-Zeinstra SM. Differences in descriptions of Kellgren and Lawrence grades of knee osteoarthritis. Ann Rheum Dis. 2008a;67:1034–1036. doi: 10.1136/ard.2007.079020. [DOI] [PubMed] [Google Scholar]
- Schiphof D, de Klerk BM, Koes BW, et al. Good reliability, questionable validity of 25 different classification criteria of knee osteoarthritis: a systematic appraisal. J Clin Epidemiol. 2008b;61:1205–1215. doi: 10.1016/j.jclinepi.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Seedhom BB. Conditioning of cartilage during normal activities is an important factor in the development of osteoarthritis. Rheumatology (Oxford) 2006;45:146–149. doi: 10.1093/rheumatology/kei197. [DOI] [PubMed] [Google Scholar]
- Steklov N, Srivastava A, Sung KL, et al. Aging-related differences in chondrocyte viscoelastic properties. Mol Cell Biomech. 2009;6:113–119. [PubMed] [Google Scholar]
- Tan AL, Grainger AJ, Tanner SF, et al. High-resolution magnetic resonance imaging for the assessment of hand osteoarthritis. Arthritis Rheum. 2005;52:2355–2365. doi: 10.1002/art.21210. [DOI] [PubMed] [Google Scholar]
- Tan AL, Grainger AJ, Tanner SF, et al. A high-resolution magnetic resonance imaging study of distal interphalangeal joint arthropathy in psoriatic arthritis and osteoarthritis: are they the same? Arthritis Rheum. 2006a;54:1328–1333. doi: 10.1002/art.21736. [DOI] [PubMed] [Google Scholar]
- Tan AL, Toumi H, Benjamin M, et al. Combined high-resolution magnetic resonance imaging and histological examination to explore the role of ligaments and tendons in the phenotypic expression of early hand osteoarthritis. Ann Rheum Dis. 2006b;65:1267–1272. doi: 10.1136/ard.2005.050112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanamas S, Hanna FS, Cicuttini FM, et al. Does knee malalignment increase the risk of development and progression of knee osteoarthritis? A systematic review. Arthritis Rheum. 2009;61:459–467. doi: 10.1002/art.24336. [DOI] [PubMed] [Google Scholar]
- Tiderius CJ, Olsson LE, Leander P, et al. Delayed gadolinium-enhanced MRI of cartilage (dGEMRIC) in early knee osteoarthritis. Magn Reson Med. 2003;49:488–492. doi: 10.1002/mrm.10389. [DOI] [PubMed] [Google Scholar]
- Toncheva A, Remichkova M, Ikonomova K, et al. Inflammatory response in patients with active and inactive osteoarthritis. Rheumatol Int. 2009;29:1197–1203. doi: 10.1007/s00296-009-0864-0. [DOI] [PubMed] [Google Scholar]
- Vlychou M, Koutroumpas A, Malizos K, et al. Ultrasonographic evidence of inflammation is frequent in hands of patients with erosive osteoarthritis. Osteoarth Cartil. 2009;17:1283–1287. doi: 10.1016/j.joca.2009.04.020. [DOI] [PubMed] [Google Scholar]
- de Vries-Bouwstra JK, Dijkmans BA, Breedveld FC. Biologics in early rheumatoid arthritis. Rheum Dis Clin North Am. 2005;31:745–762. doi: 10.1016/j.rdc.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Weinberger A. Gout, uric acid metabolism, and crystal-induced inflammation. Curr Opin Rheumatol. 1995;7:359–363. doi: 10.1097/00002281-199507000-00017. [DOI] [PubMed] [Google Scholar]
- Weng LH, Wang CJ, Ko JY, et al. Inflammation induction of Dickkopf-1 mediates chondrocyte apoptosis in osteoarthritic joint. Osteoarth Cartil. 2009;17:919–929. doi: 10.1016/j.joca.2008.12.008. [DOI] [PubMed] [Google Scholar]
- van der Weyden L, Wei L, Luo J, et al. Functional knockout of the matrilin-3 gene causes premature chondrocyte maturation to hypertrophy and increases bone mineral density and osteoarthritis. Am J Pathol. 2006;169:515–527. doi: 10.2353/ajpath.2006.050981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki T, Deie M, Shinomiya R, et al. Transplantation of meniscus regenerated by tissue engineering with a scaffold derived from a rat meniscus and mesenchymal stromal cells derived from rat bone marrow. Artif Organs. 2008;32:519–524. doi: 10.1111/j.1525-1594.2008.00580.x. [DOI] [PubMed] [Google Scholar]
- Yeow CH, Cheong CH, Ng KS, et al. Anterior cruciate ligament failure and cartilage damage during knee joint compression: a preliminary study based on the porcine model. Am J Sports Med. 2008;36:934–942. doi: 10.1177/0363546507312645. [DOI] [PubMed] [Google Scholar]