

Abstract
Antibodies against ganglionic acetylcholine receptors (AChR) are implicated as the cause of autoimmune autonomic ganglionopathy (AAG). To characterize ganglionic neurotransmission in an animal model of AAG, evoked and spontaneous excitatory post-synaptic potentials (EPSP) were recorded from neurons in isolated mouse superior cervical ganglia (SCG). In vitro exposure of ganglia to IgG from AAG patients progressively inhibited synaptic transmission. After passive transfer of antibody to mice, evoked EPSP amplitude decreased, and some neurons showed no synaptic responses. EPSP amplitude recovered by day seven despite persistence of ganglionic AChR antibody in the mouse serum. There was a more persistent (at least 14 day) reduction in miniature EPSP amplitude consistent with antibody-mediated reduction in post-synaptic AChR. Although the quantal size was reduced, a progressive increase in the frequency of spontaneous synaptic events occurred, suggesting a compensatory increase in presynaptic efficacy. The quantal size returned to baseline by 21 days while the frequency remained increased for at least four weeks. Ganglionic AChR antibodies cause an impairment of autonomic ganglionic synaptic transmission. Homeostatic plasticity in autonomic neurotransmission could help explain the spontaneous clinical recovery seen in some AAG patients and may also play an important role in regulating normal autonomic reflexes.
Keywords: electrophysiology, EPSP, superior cervical ganglia, mouse, passive transfer, autoimmune
Introduction
Autoimmune autonomic ganglionopathy (AAG) is a form of acquired autonomic failure associated with antibodies specific for the ganglionic nicotinic acetylcholine receptor (AChR). AAG typically presents in a previously healthy individual with symptoms of sympathetic failure (orthostatic hypotension, impaired sweating), parasympathetic failure (dry eyes, dry mouth, fixed pupils, bladder and sexual dysfunction) and gastrointestinal dysmotility (gastroparesis and severe constipation) (Sandroni, et al., 2004, Suarez, et al., 1994, Vernino, et al., 2000). Patients with high levels of ganglionic AChR antibodies usually have a subacute onset of disabling symptoms over a few weeks followed by spontaneous but incomplete recovery. Patients with lower antibody levels may have a chronic insidious presentation or milder, limited forms of autonomic failure. Ganglionic AChR antibodies are detectable in about 50% of patients with subacute AAG. Higher ganglionic AChR antibody levels correlate with greater clinical severity and with greater severity of laboratory measures of autonomic failure (Klein, et al., 2003, Vernino, et al., 2000). In some cases, patients treated with plasma exchange or other immunomodulatory treatments to reduce antibody levels can show dramatic improvement in autonomic function (Gibbons, et al., 2008, Schroeder, et al., 2005). Serum IgG isolated from patients with AAG reduces whole-cell neuronal AChR current in cultured human IMR-32 cells (Wang, et al., 2007).
Animal models of experimental autoimmune autonomic ganglionopathy (EAAG) have been developed. Rabbits immunized against ganglionic AChR produce antibodies and develop autonomic failure that recapitulates most of the clinical features of AAG in man (Lennon, et al., 2003, Vernino, et al., 2003). Although EAAG rabbits often develop chronic disease, they typically show spontaneous partial improvement after initially more severe autonomic deficits (unpublished observation). Passive transfer of ganglionic AChR IgG from rabbits with EAAG to mice can produce transient autonomic deficits (urinary retention, slowed gastrointestinal motility and impaired catecholamine responses to stress). Synaptic transmission in mesenteric ganglia is impaired in rabbits or mice with EAAG although the nature of this synaptic defect has not been characterized in detail (Lennon, et al., 2003, Vernino, et al., 2004). Passive transfer of human IgG from patients with ganglionic AChR antibodies produces only mild and transient autonomic dysfunction in mice (Vernino, et al., 2004). An effect of AAG patient serum on ganglionic synaptic transmission has not been previously demonstrated.
Clinical and experimental observations suggest that AAG is an antibody-mediated disorder caused by reversible disruption of fast synaptic transmission in autonomic ganglia. Microelectrode recordings in isolated mouse superior cervical autonomic ganglia were used to characterize the effect of antibodies from AAG patients on ganglionic neurotransmission. In addition, this passive transfer model of EAAG revealed a novel form of disease-related homeostatic synaptic plasticity.
Materials and Methods
Animal protocols were approved by the UT Southwestern institutional animal care and use committee. Male C57BL/6 mice (6–8 weeks of age, 21–29 gm, from Harlan, Indianapolis, IN) were housed in groups of four in plastic cages with soft bedding and free access to food and water in a 12-h light/dark cycle. Human plasma or serum samples used in these experiments (Table 1) were from six patients with a clinical diagnosis of AAG with subacute onset (five were positive for ganglionic AChR antibodies) and from two healthy control subjects. These same AAG patient samples have been included in previous studies (Gibbons, et al., 2008, Vernino, et al., 2008, Wang, et al., 2007). IgG was isolated from serum or plasma by adsorption to protein A-Sepharose (Vernino, et al., 2004). Eluted IgG samples were dialyzed into physiological saline and sterilized by filtration before use. Passive transfer to mice consisted of two intraperitoneal injections of 0.75 ml of filtered serum or plasma (6 hours apart). Human specimens were collected with informed consent according to a protocol approved by the UT Southwestern Institutional Review Board. A radioimmunoprecipitation assay was used to determine the serum level of ganglionic nAChR binding antibodies in human and mouse samples as described previously (Vernino, et al., 2000).
Table 1.
Characteristics of subjects included in this study.
| Subject | Age/Sex | Diagnosis | Ganglionic AchR Ab (nM) | Source |
|---|---|---|---|---|
| AAG #1 | 35/F | AAG | 33.7 | Plasma |
| AAG #2 | 48/F | AAG | 14.0 | Plasma |
| AAG #3 | 50/F | AAG | 11.9 | Serum |
| AAG #4 | 55/F | AAG | 13.2 | Serum |
| AAG #5 | 45/M | AAG | 2.5 | Plasma |
| SN-AAG | 20/F | AAG | 0.0 | Serum |
| HC1 | 39/M | Healthy Control | 0.0 | Serum |
| HC2 | 24/F | Healthy Control | 0.0 | Serum |
Electrophysiology
The superior cervical ganglia (SCG) are readily identified as paired peripheral autonomic ganglia that lie dorsal to the sheath of the internal carotid artery. The SCG supply sympathetic innervation to target organs located throughout the head (Gibbins, et al., 2000, Gibbins and Morris, 2006). After euthanasia with inhalation of isoflurane and carbon dioxide, blood was collected by cardiac puncture, and the intact mouse superior cervical ganglia and attached preganglionic nerve trunk (3 to 4 cm in length) was carefully isolated. The ganglia preparation was maintained in oxygenated artificial cerebrospinal fluid (aCSF) consisting of (in mM, 130 NaCl, 3.5 KCl, 1.25 NaH2PO4, 24 NaHCO3, 10 dextrose, 1.2 MgCl2 and 1.2 CaCl2, pH=7.3) and exposed to collagenase (1 mg/ml) for 30 minutes. Under a dissecting microscope, the tissue capsule around the ganglion was peeled away with fine forceps. The SCG and attached preganglionic nerve were then placed in the recording chamber and held in place by a fine mesh (Hold-Down, Warner Instruments Hamden, MA) and mounted on the stage of an upright immersion microscope (BX51-WI, Olympus). The ganglia was superfused continuously with oxygenated aCSF at 2 ml/min at room temperature. Neurons within the SCG were directly visualized and impaled with a microelectrode mounted on a hydraulic micromanipulator. Glass microelectrodes were fabricated with a Flaming/Brown micropipette puller (Model P-97, Sutter Instruments) and filled with 3M KCl (input resistance, 50–80 MO). Membrane potential was recorded in zero current clamp mode (MultiClamp 700B, Axon Instruments, Foster City, CA). All data were analyzed with PCLAMP-9 software (Axon Instruments, Sunnyvale, CA).
For data included in the study, the resting membrane potential of SCG neurons was −50 to −70 mV, and the integrity of the neurons was confirmed at the beginning and end of the experiment by current injections of 0.1 to 0.8 nA to evaluate membrane resistance and to estimate the threshold for action potential generation. Neurons with unstable resting membrane potential or failure to show action potential in response to current injection were excluded from analysis. The preganglionic nerve was stimulated using a concentric bipolar needle electrode and an isolated square pulse generator (11–150 mV, 0.25 Hz, S88 Stimulator, Grass Telefactor). For each neuron, the stimulus intensity was gradually increased to obtain the first stable (minimal) excitatory postsynaptic potential (EPSP). All SCG neurons receive one to two “strong” synapses with suprathreshold EPSP amplitude (McLachlan, et al., 1997). Neurons that produced an action potential as the first synaptic response were not included in determination of mean evoked EPSP amplitude.
To analyze synaptic function in more detail, the amplitude and frequency of spontaneous miniature EPSP (mEPSP) were recorded in ganglia neurons. An automated “template search” method (Clements and Bekkers, 1997) was used to automatically detect mEPSPs in 5 to 10 minute recordings. This method identifies miniature synaptic events by rise time and duration rather than simple threshold detection.
Ultrastructural studies
In separate experiments, SCG were collected from euthanized mice three days after injection of serum. Intact SCG were removed, trimmed, and fixed in 2.5% glutaraldehyde in 0.1M cacodylate buffer, post fixed in 1% osmium tetroxide in cacodylate buffer, and incubated in 1% aqueous uranyl acetate overnight at 4°C. Tissues were then rinsed with increasing concentrations of ethanol, rinsed with propylene oxide, and equilibrated in embedding media (Embed-812, Electron Microscopy Sciences, Hatfield, PA). SCG were then placed in fresh embedding material which was polymerized in molds overnight at 70°C. Thick (1μ) sections were cut from the block and examined by light microscopy to identify suitable sections for ultrastructural examination. Thin sections were then cut onto copper grids, and stained with uranyl acetate followed by lead citrate. Images were obtained using a JEOL 1200EX transmission electron microscope. Synaptic areas were generally identified on dendrites between the neuronal cell bodies. Digital images of representative synapses were collected. Tissue preparation and electron microscopy was performed in the Molecular and Cellular Imaging Core facility at UT Southwestern Medical Center.
Statistical analyses
Summary data are presented as mean ± standard error. Under each experimental condition, data from 3 to 5 mice were collected (9 to 24 neurons). Categorical data (i.e. frequency of synaptic failure and frequency of strong synapses) were analyzed using Fisher’s exact test. Continuous variables were analyzed using one-way ANOVA with Dunnett’s post test. Analyses were performed using GraphPad Prism (version 5.02 for Windows, GraphPad Software, San Diego California USA). Experimental data were compared to control data (from untreated ganglia or ganglia exposed to control IgG). P values less than 0.05 were considered significant).
Results
Characteristics of ganglionic synaptic transmission
Stimulation of the preganglionic sympathetic trunk produced a fast synaptic response (EPSP) in every SCG neuron from control mice. Failure of synaptic transmission (i.e. no EPSP produced with the highest stimulus intensity) was never seen in control neurons. Exposure of the ganglia to nicotinic antagonists (200μM hexamethonium or 30μM chlorisondamine) abolished the EPSP while neither atropine (1 μM) nor α-bungarotoxin (100 nM) had any effect on the evoked EPSP. This confirmed that evoked EPSPs recorded in the mouse SCG represent cholinergic synaptic transmission mediated predominantly by heteromeric neuronal ganglionic nicotinic AChR (Brown and Fumagalli, 1977) likely consisting of two α3 neuronal AChR subunits in combination with other subunits (Skok, et al., 1999, Xu, et al., 1999).
With gradual increases in stimulation intensity, the EPSP amplitude increased in a stepwise manner indicating that each neuron receives synaptic input from several distinct preganglionic fibers. In 11 out of 56 control neurons (20%), the initial synaptic response was an action potential, indicating that the minimal stimulus activated either a single strong synapse or multiple EPSPs activated simulataneously. The stimulus intensity was adjusted for each neuron to produce the minimum reproducible EPSP. The minimum EPSP was selected in an effort to isolate the response from a single subthreshold ganglionic synapse in each neuron. With 0.25 Hz stimulation, the amplitude of the EPSP was stable for up to an hour. The mean amplitude of the minimum EPSP in control neurons was 5.2 ± 0.8 mV (n=24).
Effects of IgG on ganglionic synaptic transmission in vitro
After ensuring stability of the minimum evoked EPSP for 10 minutes, purified IgG (1 mg/ml) was added to the aCSF superfusing the SCG. Application of IgG from healthy control subjects or a patient with seronegative AAG had no effect on the EPSP amplitude which was stable for 50 minutes or more (Figure 1A). However, IgG from three patients with ganglionic AChR antibodies all produced a significant reduction in the amplitude of the evoked EPSP (F=71.54, p<0.0001)(Figure 1B). Figure 1C shows the time course of the effect. Exposure to IgG from AAG patient 1 completely eliminated the EPSP within 10 minutes while the others produced a more gradual decline in EPSP amplitude (30 to 55% amplitude reduction in 20 minutes). These findings were consistent with the effect of IgG from these same patients on ganglionic AChR currents in cultured cells (Wang, et al., 2007).
Figure 1. Effect of IgG on ganglionic synaptic transmission.

A) Minimum evoked EPSP (each trace is the average of 5 consecutive responses) from an isolated SCG. The EPSP was stable and did not change after adding IgG (1.0 mg/ml) from a healthy control subject to the perfusing solution. The EPSP at baseline, 20 minutes and 50 minutes after adding IgG are superimposed. B) The average minimum evoked EPSP from an isolated SCG at baseline, 20 minutes and 50 minutes after exposure to IgG from AAG patient #2 (1 mg/ml). There was a progressive decline in the EPSP amplitude over time. C) Time course of the effect of bath-applied IgG on the amplitude of the evoked EPSP. There was no effect of the IgG from the seronegative AAG patient. IgG from AAG patient #1 caused a rapid and complete inhibition of the EPSP within 10 minutes, while IgG from two other AAG patients produced a progressive but incomplete reduction in the EPSP amplitude. Data for AAG#4 was truncated at 20 minutes because there was insufficient data at later time points.
Ganglionic synaptic transmission after passive transfer
Ganglia were collected from mice three days after intraperitoneal injection of serum or plasma from AAG patients and controls. Injection of serum from healthy controls or a seronegative AAG patient had no effect on ganglionic synaptic responses in the mouse (Figure 2A). Three days after injection with serum or plasma containing ganglionic AChR antibody, the mean amplitude of minimum evoked EPSP in ganglionic neurons was reduced by 20 to 85% compared to the mean amplitude in control ganglia (p < 0.0001)(Figure 2A). While synaptic failure was never seen in control ganglia, 26% of ganglionic neurons (13 of 49) from EAAG mice showed no synaptic response even with maximal stimulation of the preganglionic nerve. Additionally, an initial action potential response (“strong synapse”) was seen less often in EAAG ganglia (2 of 49 neurons, 4%) compared to control ganglia (20%, p<0.01, Fisher’s exact test).
Figure 2. Synaptic transmission in mouse SCG after passive transfer.
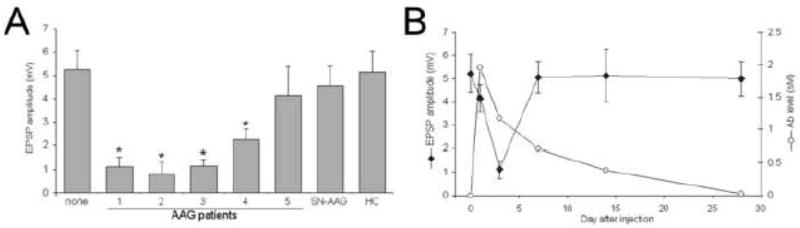
A) Average amplitude of the minimum evoked EPSP from mouse SCG neurons 3 days after injection (mean ± SEM, 8–15 neurons in each group). Ganglia from mice injected with healthy control (HC) or seronegative (SN-AAG) patient sera were not different from EPSP amplitude in untreated mice (labeled “none”). Passive transfer from four of the five AAG patients produced significant reduction in evoked EPSP amplitudes (F=6.183, p<0.0001; * p<0.01 for individual comparisons using Dunnett post-test). B) Time course of serum antibody levels and EPSP amplitude after passive transfer of plasma from AAG patient #1. The evoked EPSP amplitude reaches a nadir at 3 days but recovers by 7 days after injection even though elevated levels of ganglionic AChR antibody are present in the mouse for more than 2 weeks.
The integrity of ganglionic synaptic transmission in passive transfer EAAG mice was qualitatively compared to the level of human ganglionic AChR antibody present in the mouse serum at various time points after injection of plasma from patient 1 (Figure 2B). The amplitude of the minimum evoked EPSP was mildly reduced one day after injection, reached a nadir three days after injection and subsequently recovered by seven days. As expected, the level of serum ganglionic AChR antibody was highest immediately after injection and declined gradually. Ganglionic AChR antibody was still present at high levels in the mouse serum up to 14 days after injection even though the evoked synaptic responses had fully recovered by day 7 (Figure 2B).
Since the amplitude of the evoked EPSP depends on multiple factors, ganglionic synaptic transmission was further assessed by analyzing spontaneous synaptic events. The amplitude of spontaneous miniature EPSPs (mEPSP) is a reflection of the quantal size (i.e. the response of the postsynaptic neuron to release of a single vesicle of acetylcholine) (Vautrin, et al., 1992). In control SCG neurons, mEPSPs occurred at a frequency of 2 to 3 per minute and had mean amplitude of 0.84 mV (Figure 3). Ganglia examined three days after injection with serum from a healthy control subject or seronegative AAG patient showed normal mEPSP amplitude and frequency. Ganglia from EAAG mice, however, showed significantly reduced mEPSP amplitudes (0.44 to 0.68 mV, F=22.95, p<0.0001) indicating a decrease in quantal size, consistent with loss of AChR on the postsynaptic neuron (Figure 3B). In some neurons, spontaneous EPSPs could not be identified, possibly because the mEPSP amplitude was below the threshold of detection (these neurons were not included in the analysis). A significant reduction in mEPSP amplitude was seen in ganglia from mice injected with serum or plasma from all five AAG patients tested. Three days after injection, the frequency of spontaneous mEPSP events in EAAG ganglia was no different than control ganglia.
Figure 3. Analysis of synaptic activity in mouse SCG.

A) Spontaneous miniature EPSP (mEPSP) recorded in an isolated SCG of healthy control mouse (average of 10). Healthy control and seronegative AAG sera had no effect on mEPSP amplitude or frequency. B) Spontaneous mEPSP recorded in an isolated SCG of EAAG mouse (3 days after injection with plasma from AAG patient #1, average of 10). Treatment with serum or plasma from seropositive AAG patients produced a significant reduction in mEPSP amplitude. C,D) Time course of ganglionic synaptic strength in EAAG mice injected with plasma from AAG patient #1. The mEPSP amplitude was reduced by about 50% and remained reduced for at least 14 days after injection, consistent with the presence of ganglionic AChR antibody in the mouse serum (F=7.796. *p<0.01). Neurons with no detectable spontaneous mEPSP were not included in this analysis. D) While the mEPSP amplitude was reduced, the spontaneous mEPSP frequency gradually increased (F=35.26, *p<0.01). The frequency increased more than four fold and then gradually returned to normal after the mEPSP amplitude had recovered. The mEPSP frequency was still increased compared to control four weeks after injection.
If the acetylcholine content of each synaptic vesicle is constant and the synaptic structure is unchanged, a reduction in mEPSP amplitude indicates a reduction in responsiveness of the postganglionic neuron. Qualitative ultrastructural examination of ganglionic synapses from experimental mice did not reveal any differences compared to control ganglia (figure 4). Thus, the reduction in quantal size most likely reflects an antibody-mediated reduction in the number of available synaptic AChR.
Figure 4. Ultrastructural examination of synapses in mouse superior cervical ganglia.

A) Control ganglia collected three days after the mouse was injected with normal human serum were not different from control ganglia from untreated mice (not shown). A typical ganglionic synapse is shown with abundant clear synaptic vesicles and occasional dense core vesicles in the presynaptic terminal (magnification 50,000X). The synapse is identified as a thickening of the apposed membranes.
B) Example of a synapse in ganglia collected three days after the mouse was injected with serum containing ganglionic AChR antibodies (patient 1). The structure of these ganglionic synapses was not different than controls.
Homeostatic plasticity in ganglionic synaptic transmission
To examine the time course of changes in ganglionic synaptic transmission, ganglia were examined from mice at 3, 7, 14, 28, and 49 days after injection with plasma from patient 1. The mEPSP amplitude was reduced to about half of the control amplitude at day 3, and this reduction in quantal size persisted for at least 14 days (F=7.796, p<0.0001)(Figure 3C). The mEPSP amplitude recovered subsequently. The spontaneous mEPSP frequency was not different from controls at day 3 but increased significantly at 7 and 14 days after injection (F=35.26, p<0.0001) (Figure 3D). After recovery of the mEPSP amplitude, the frequency declined but remained significantly higher than control for at least 4 weeks. Seven weeks after injection, both the mEPSP amplitude and frequency had returned to control levels.
Discussion
Autoimmune autonomic ganglionopathy is an antibody-mediated disorder of ganglionic synaptic transmission. Patient serum containing ganglionic AChR antibodies can inhibit synaptic transmission in the mouse autonomic ganglia both in vitro and in vivo. The inhibition of ganglionic transmission is due to a reduction in quantal size consistent with a reduction in the number of functional post-synaptic AChR. A greater inhibitory effect on ganglionic synaptic transmission was associated with higher levels of ganglionic AChR antibody. This antibody-mediated impairment in synaptic function explains the significant impairment in autonomic tone and autonomic reflexes in AAG patients.
We previously showed that high affinity ganglionic AChR antibodies produced in rabbits can cause diffuse autonomic deficits when administered to mice (Vernino, et al., 2004). In that study, a single administration of antibody from human AAG subjects produced only subtle and transient clinical autonomic deficits. Using more sensitive and direct measures of autonomic ganglionic transmission, it is now clear that ganglionic AChR antibodies from AAG patients do have a significant inhibitory effect on ganglionic neurotransmission.
A novel observation in this study was the observation of rapid recovery of synaptic transmission in the passive transfer EAAG mouse. Despite the persistence of circulating ganglionic AChR antibodies and a reduction in quantal size (mEPSP amplitude) for at least two weeks, the evoked EPSP in mouse SCG neurons was reduced for only a few days. The recovery of synaptic function was associated with a dramatic increase in the frequency of spontaneous mEPSP. Although increased mEPSP frequency could be attributed to an increased number of active synapses, it is more likely that this observation reflects plasticity in presynaptic efficacy resulting in an increase in quantal content (i.e. the number of vesicles released during each presynaptic action potential). At experimental days 7 and 14 when the quantal size is reduced by about 50%, the quantal content must double in order for the evoked EPSP amplitude to remain at baseline levels. The ganglionic synaptic plasticity was long-lasting, persisting for several weeks even after recovery of the mEPSP amplitude. Homeostatic plasticity serves to maintain stable ganglionic synaptic strength despite the inhibitory effects of antibody. Similar homeostatic plasticity has been previously described in other experimental settings at neuromuscular junction and neocortical synapses (Davis and Goodman, 1998, Sandrock, et al., 1997, Turrigiano, et al., 1998).
Homeostatic plasticity in ganglionic synaptic transmission may explain some of the clinical features of AAG and the EAAG animal models. Patients with the acute or subacute form of AAG often experience a spontaneous, but incomplete, improvement in autonomic function (Suarez, et al., 1994, Vernino, et al., 2000). Passive transfer of serum antibodies from patients with AAG to mice produces changes in autonomic function that are very transient (Vernino, et al., 2004). A combination of baroreflex-mediated increase in central autonomic output followed by an increase in presynaptic ganglionic synaptic efficacy (summarized in Table 2) serves to prevent any dramatic abnormalities in blood pressure or heart rate in the mouse passive transfer model despite clear disruption of ganglionic synaptic transmission.
Table 2.
Time course of changes in ganglionic synaptic transmission in experimental autoimmune autonomic ganglionopathy
| Time after passive transfer | mEPSP amplitude | mEPSP frequency | Synaptic response (EPSP) | Preganglionic (central) input | Autonomic function |
|---|---|---|---|---|---|
| 0 – 3 days (Acute Ab effect) | ↓ | nl | ↓ | ↑ | Mildly impaired |
| 5 – 14 days (Plasticity) | ↓ | ↑ | nl | nl | Normal |
| 21 – 28 days (Ab clearance) | nl | ↑ | ↑ | ↓ | Mildly enhanced |
Other forms of synaptic plasticity have been described in autonomic ganglia. Use-dependent potentiation of ganglionic synaptic transmission occurs after high-frequency stimulation of the preganglionic nerve (Briggs, et al., 1985) This ganglionic long-term potentiation (gLTP) does not require ganglionic AChR function but appears to depend on serotonergic transmission (Alkadhi, et al., 1996). It is unclear if the homeostatic plasticity observed in EAAG has a similar mechanism to gLTP although it would be reasonable to assume that peripheral autonomic failure would result in an increase in central autonomic drive and increased firing frequency in the preganglionic autonomic fibers. Indeed, in our prior study, mice treated with high titer rabbit ganglionic AChR antibody showed marked inhibition of fast synaptic transmission in superior mesenteric ganglia neurons followed by a significant increase in the frequency of fast synaptic activity during recovery (Vernino, et al., 2004). A compensatory increase in the frequency of firing of the afferent nerves could provide an appropriate stimulus to induce synaptic plasticity.
These studies provide additional strong evidence that patients with circulating ganglionic AchR antibodies have an antibody-mediated disorder of ganglionic synaptic transmission. Despite having a similar clinical phenotype, serum from an AAG patient without ganglionic AchR antibodies had no effect on synaptic transmission. Patients with seronegative AAG, especially those that respond to immunomodulatory treatment, may have an immune response against some component of the autonomic nervous system other than the ganglionic synapse. Further studies are needed to characterize seronegative AAG and to further investigate the mechanisms of ganglionic synaptic plasticity as it relates to normal autonomic function and to disease pathophysiology.
Acknowledgments
The research was supported by NIH P50NS32352. We thank Kim Nickander and Steve Hopkins for excellent technical support and study coordination.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alkadhi KA, Salgado-Commissariat D, Hogan YH, Akpaudo SB. Induction and maintenance of ganglionic long-term potentiation require activation of 5-hydroxytryptamine (5-HT3) receptors. Journal of Physiology. 1996;496:479–489. doi: 10.1113/jphysiol.1996.sp021700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Briggs CA, Brown TH, McAfee DA. Neurophysiology and pharmacology of long-term potentiation in the rat sympathetic ganglion. Journal of Physiology. 1985;359:503–521. doi: 10.1113/jphysiol.1985.sp015599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown DA, Fumagalli L. Dissociation of [alpha]-bungarotoxin binding and receptor block in the rat superior cervical ganglion. Brain Research. 1977;129:165–168. [Google Scholar]
- 4.Clements JD, Bekkers JM. Detection of spontaneous synaptic events with an optimally scaled template. Biophysical Journal. 1997;73:220–229. doi: 10.1016/S0006-3495(97)78062-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davis GW, Goodman CS. Synapse-specific control of synaptic efficacy at the terminals of a single neuron. Nature. 1998;392:82–86. doi: 10.1038/32176. [DOI] [PubMed] [Google Scholar]
- 6.Gibbins IL, Jobling P, Messenger JP, Teo EH, Morris JL. Neuronal morphology and the synaptic organisation of sympathetic ganglia. Journal of the Autonomic Nervous System. 2000;81:104–109. doi: 10.1016/s0165-1838(00)00132-6. [DOI] [PubMed] [Google Scholar]
- 7.Gibbins IL, Morris JL. Structure of peripheral synapses: autonomic ganglia. Cell and Tissue Research. 2006;326:205–220. doi: 10.1007/s00441-006-0233-1. [DOI] [PubMed] [Google Scholar]
- 8.Gibbons CH, Vernino SA, Freeman R. Combined immunomodulatory therapy in autoimmune autonomic ganglionopathy. Archives of Neurology. 2008;65:213–217. doi: 10.1001/archneurol.2007.60. [DOI] [PubMed] [Google Scholar]
- 9.Klein CM, Vernino S, Lennon VA, Sandroni P, Fealey RD, Benrud-Larson L, Sletten D, Low PA. The spectrum of autoimmune autonomic neuropathies. Annals of Neurology. 2003;53:752–758. doi: 10.1002/ana.10556. [DOI] [PubMed] [Google Scholar]
- 10.Lennon VA, Ermilov LG, Szurszewski JH, Vernino S. Immunization with neuronal nicotinic acetylcholine receptor induces neurological autoimmune disease. Journal of Clinical Investigation. 2003;111:907–913. doi: 10.1172/JCI17429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McLachlan EM, Davies PJ, Habler HJ, Jamieson J. On-going and reflex synaptic events in rat superior cervical ganglion cells. Journal of Physiology. 1997;501:165–181. doi: 10.1111/j.1469-7793.1997.165bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sandrock AW, Jr, Dryer SE, Rosen KM, Gozani SN, Kramer R, Theill LE, Fischbach GD. Maintenance of acetylcholine receptor number by neuregulins at the neuromuscular junction in vivo. Science. 1997;276:599–603. doi: 10.1126/science.276.5312.599. [DOI] [PubMed] [Google Scholar]
- 13.Sandroni P, Vernino S, Klein CM, Lennon VA, Benrud-Larson L, Sletten D, Low PA. Idiopathic autonomic neuropathy: comparison of cases seropositive and seronegative for ganglionic acetylcholine receptor antibody. Archives of Neurology. 2004;61:44–48. doi: 10.1001/archneur.61.1.44. [DOI] [PubMed] [Google Scholar]
- 14.Schroeder C, Vernino S, Birkenfeld AL, Tank J, Heusser K, Lipp A, Benter T, Lindschau C, Kettritz R, Luft FC, Jordan J. Plasma exchange for primary autoimmune autonomic failure. New England Journal of Medicine. 2005;353:1585–1590. doi: 10.1056/NEJMoa051719. [DOI] [PubMed] [Google Scholar]
- 15.Skok MV, Voitenko LP, Voitenko SV, Lykhmus EY, Kalashnik EN, Litvin TI, Tzartos SJ, Skok VI. Alpha subunit composition of nicotinic acetylcholine receptors in the rat autonomic ganglia neurons as determined with subunit-specific anti-alpha(181–192) peptide antibodies. Neuroscience. 1999;93:1427–1436. doi: 10.1016/s0306-4522(99)00160-8. [DOI] [PubMed] [Google Scholar]
- 16.Suarez GA, Fealey RD, Camilleri M, Low PA. Idiopathic autonomic neuropathy: Clinical, neurophysiologic, and follow-up studies on 27 patients. Neurology. 1994;44:1675–1682. doi: 10.1212/wnl.44.9.1675. [DOI] [PubMed] [Google Scholar]
- 17.Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- 18.Vautrin J, Kriebel ME, Holsapple J. Further evidence for the dynamic formation of transmitter quanta at the neuromuscular junction. Journal of Neuroscience Research. 1992;32:245–254. doi: 10.1002/jnr.490320214. [DOI] [PubMed] [Google Scholar]
- 19.Vernino S, Ermilov LG, Sha L, Szurszewski JH, Low PA, Lennon VA. Passive transfer of autoimmune autonomic neuropathy to mice. Journal of Neuroscience. 2004;24:7037–7042. doi: 10.1523/JNEUROSCI.1485-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vernino S, Lindstrom J, Hopkins S, Wang Z, Low PA. Characterization of ganglionic acetylcholine receptor autoantibodies. Journal of Neuroimmunology. 2008;197:63–69. doi: 10.1016/j.jneuroim.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vernino S, Low PA, Fealey RD, Stewart JD, Farrugia G, Lennon VA. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. New England Journal of Medicine. 2000;343:847–855. doi: 10.1056/NEJM200009213431204. [DOI] [PubMed] [Google Scholar]
- 22.Vernino S, Low PA, Lennon VA. Experimental autoimmune autonomic neuropathy. Journal of Neurophysiology. 2003;90:2053–2059. doi: 10.1152/jn.00408.2003. [DOI] [PubMed] [Google Scholar]
- 23.Wang Z, Low PA, Jordan J, Freeman R, Gibbons CH, Schroeder C, Sandroni P, Vernino S. Autoimmune autonomic ganglionopathy: IgG effects on ganglionic acetylcholine receptor current. Neurology. 2007;68:1917–1921. doi: 10.1212/01.wnl.0000263185.30294.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu W, Gelber S, Orr-Urtreger A, Armstrong D, Lewis RA, Ou CN, Patrick J, Role L, De Biasi M, Beaudet AL. Megacystis, mydriasis, and ion channel defect in mice lacking the alpha3 neuronal nicotinic acetylcholine receptor. Proc Natl Acad Sci USA. 1999;96:5746–5751. doi: 10.1073/pnas.96.10.5746. [DOI] [PMC free article] [PubMed] [Google Scholar]