

Abstract
Conjugation to glutathione (GSH) by glutathione transferase A4-4 (GSTA4-4) is a major route of elimination for the lipid peroxidation product 4-hydroxynonenal (HNE), a toxic compound that contributes to numerous diseases. Both enantiomers of HNE are presumed to be toxic, and GSTA4-4 has negligible stereoselectivity towards them, despite its high catalytic chemospecificity for alkenals. In contrast to the highly flexible, and substrate promiscuous, GSTA1-1 isoform that has poor catalytic efficiency with HNE, GSTA4-4 has been postulated to be a rigid template that is pre-organized for HNE metabolism. However, the combination of high substrate chemoselectivity and low substrate stereoselectivity is intriguing. The mechanism by which GSTA4-4 achieves this combination is important, because it must metabolize both enantiomers of HNE to efficiently detoxify the biologically formed mixture. The crystal structures of GSTA4-4 and an engineered variant of GSTA1-1 with high catalytic efficiency toward HNE, co-crystallized with a GSH-HNE conjugate analog, demonstrate that GSTA4-4 undergoes no enantiospecific induced fit; instead the active site residue Arg15 is ideally located to interact with the 4-hydroxyl group of either HNE enantiomer. The results reveal an evolutionary strategy for achieving biologically useful stereopromiscuity towards a toxic racemate, concomitant with high catalytic efficiency and substrate specificity towards an endogenously formed toxin.
4-Hydroxynonenal (HNE)1 is a major lipid peroxidation product of oxidative stress, and it plays a causal role in several neurodegenerative diseases, diabetes, atherosclerosis, asthma, cataracts, cancer, and aging (1-4). HNE is an electrophilic toxin, formed as a racemic mixture from arachidonic acid or linoleic acid, which covalently modifies numerous proteins and DNA (5). HNE also acts as a second messenger in signal transduction pathways related to apoptosis (6, 7). Cellular processes that control HNE concentrations likely influence susceptibility to toxic effects of oxidative stress and related diseases (8). Hypothetically, metabolic processes that evolved to clear HNE would, individually or collectively, eliminate both 4R-HNE and 4S-HNE, because although stereoselective interactions occur, both enantiomers are chemically reactive towards cellular macromolecules (9-11). Interestingly, any individual enzyme that evolved to eliminate HNE would need to uncouple substrate chemospecificity from substrate stereoselectivity in order to provide an efficient detoxification pathway. This is a fascinating challenge for an enzyme, to the extent that substrate chemospecificity is presumed to co-evolve with catalytic efficiency; evolutionary steps that increase interactions with the diastereomeric transition state formed from one HNE enantiomer would disfavor interactions with the diastereomeric transition state derived from the other HNE enantiomer. The structural basis for this behavior with enzymes that recognize HNE has not been determined.
A major route of HNE elimination is glutathione transferase (GST)-catalyzed adduction to glutathione (GSH) to form the diastereomeric 3S-glutathionyl 4-hydroxynonanal (3S-GSHNE) conjugates (Figure 1). The GSTA4-4 isoform exhibits remarkable chemoselectivity and high catalytic efficiency towards HNE (12-15) and expression of GSTA4-4 is a critical determinant of an organism's susceptibility to disease and aging. In contrast, the structural homolog GSTA1-1 is an archetypal, substrate promiscuous, detoxification enzyme with 50-fold lower kcat/Km for HNE compared to GSTA4-4 (16). Whereas many crystallographic structures of GSTA1-1 reveal binding interactions with different ligands (17-22), very few structures are available for GSTA4-4, and none include HNE or a GSH-HNE conjugate relevant to the ternary substrate complex (13, 23). Thus, the structural basis for the specificity of GSTA4-4 for HNE is not completely understood. However, rationally engineered mutants of GSTA1-1, including the GSTA1-1 ‘GIMFhelix’ mutant that contains 14 amino acid substitutions (6% of the sequence), have implicated structural elements that contribute; the GIMFhelix mutant is dramatically more active towards HNE than GSTA1-1, but still less efficient than GSTA4-4 (24). The GIMFhelix protein has served as a valuable model for the possible evolution of HNE specificity of GSTA4-4 from the promiscuous GSTA1-1 (24-27). Although low amplitude, fast, dynamics are a clear necessity for effective enzyme catalysis, the available data suggest that GSTA1-1 utilizes induced fit of a conformationally flexible scaffold to achieve promiscuous catalysis, whereas GSTA4-4 is a relatively rigid pre-formed template poised to bind and conjugate the lipid aldehyde HNE.
FIGURE 1.

Reaction of racemic HNE with GSH. 1,4-Addition reaction of HNE with GSH as catalyzed by human GSTA4-4, followed by intramolecular cyclization or reduction by NaBH4.
The stereochemical course of the GSTA4-4 reaction with HNE is paradoxical because it exhibits negligible substrate stereoselectivity, accepting either 4R- or 4S-HNE, but it is completely stereoselective at the level of product formation, allowing attack of GSH from only the si face of the prochiral carbon 3 of HNE ((28), Figure 1). Thus each enantiomeric HNE is efficiently cleared by GSTA4-4, as expected for an enzyme that may have evolved to eliminate a toxic racemic mixture. The difference in the kinetic parameters between nonenal and HNE suggests that the HNE hydroxyl group is exploited within the GSTA4-4 active site (24, 29). However, while the protein flexibility found with GSTA1-1 is easily rationalized as a device to achieve substrate promiscuity including stereochemical aspects, it is unclear how the presumed rigid scaffold of GSTA4-4 achieves high substrate specificity without stereoselectivity, if it does not rearrange to accommodate stereoisomeric substrates. The striking lack of substrate stereoselectivity for HNE could, hypothetically, be due to: 1) a symmetrically placed active site group that interacts with the 4-hydroxyl group in either configuration to yield diastereomeric transition states of equal energy, or 2) two distinct active site groups that each form diastereospecific interactions with the 4-hydroxy group in one of the diastereomeric, energetically degenerate, transition states. In order to understand the structural basis for the high substrate specificity of GSTA4-4 for HNE, coupled with the biologically useful substrate stereopromiscuity, we determined the structures of human GSTA4-4 and the GIMFhelix mutant complexed with the open chain form of the 3S-GSHNE conjugate, 3S-glutathionyl 1,4-dihydroxynonanol (3S-GSDHN). The structures are the first of GSTA4-4 complexed with a biologically relevant ligand that reveals how conformational dynamics of the C-terminus have been completely remodeled to accommodate endogenous alkenal substrates in GSTA4-4 compared to GSTA1-1, and they provide a structural rationalization for the biologically optimal combination of high substrate specificity and catalytic efficiency for racemic HNE without substrate stereoselectivity.
Experimental Procedures
Protein Expression and Purification
Recombinant human GSTA4-4, GSTA1-1, GSTA4-4 Y9F, and GSTA1-1 GIMFhelix were expressed in Escherichia coli and purified via GSH-agarose affinity chromatography as described previously (12, 24) with an additional purification step using a Sephadex® G-75 column washed with 10 mM NaPO4, pH 6.9, 2 mM EDTA, prior to exchange into 10 mM HEPES, pH 7.0, for crystallization screens.
Stereoselectivity Analysis
GSH was pre-incubated with GSTA4-4, GSTA1-1, GSTA4-4 Y9F, GSTA1-1 GIMFhelix, or buffer alone, followed by reaction with racemic HNE. Diastereomer formation was analyzed by LC/MS as described previously (28).
Enzyme Kinetics
The metabolism of racemic HNE (0-160 μM) by the GSTA4-4 Y9F (58 nM) mutant was monitored by a LC/MS product formation activity assay that allows for the concurrent detection of the GSHNE diastereomers derived from S- and R-HNE. The activity assay, preparation and quantification of the GSHNE standards, and LC/MS analyses were performed using previously described methods (28). The apparent catalytic efficiency (kcat/Km) of the GSTA1-1 GIMFhelix mutant (600 pM) was also determined for comparison using the product formation assay with replicates conducted at [HNE] ≪ Km.
Reduction of GSHNE by NaBH4
The diastereomeric 3S-GSHNE conjugates were prepared by incubating GSTA4-4 with GSH and racemic HNE in order to provide the uncyclized and stereochemically relevant 3S-conjugate. 3S-GSHNE was then reduced by NaBH4 and 3S-GSDHN was purified by HPLC and characterized by LC/MS and NMR as described previously (28).
Crystallization and X-ray Diffraction Data Collection
Crystallization screens were conducted using the sitting drop vapor-diffusion method at room temperature with conditions based on published protocols (13, 19, 23) as well as using the Hampton crystallization screen kits, HR2-110 and HR2-112, with protein concentrations of 10 mg/ml in 10 mM HEPES, pH 7.0. Data sets were collected for samples crystallized from: 2 μl GSTA4-4 with a 10-fold molar excess of 3S-GSDHN per subunit mixed with 2 μl reservoir solution containing 24% PEG 4000, 0.1 M sodium acetate trihydrate, pH 4.6, and 0.2 M ammonium sulfate; and 2 μl GSTA1-1 GIMFhelix with a 10-fold molar excess of 3S-GSDHN per subunit and 0.5 μl EtOH mixed with 3 μl reservoir solution containing 16% PEG mme 5000, 0.1 M HEPES, pH 7.5, and 10% isopropyl alcohol. Paraffin oil was used as a cryoprotectant in the case of 3S-GSDHN-GSTA1-1 GIMFhelix and diffraction data were collected at SSRL beamline 11-1 (λ = 0.9795) and processed with HKL2000 (30). Data collection and processing statistics are presented in Table 1.
TABLE 1.
Data collection and processing statistics.
| GST Structure | GSTA4-4-GSDHN | GIMFhelix-GSDHN |
|---|---|---|
| Space Group | P21 | P21 |
| Unit-cell Dimensions (Å,°) | 59.02, 153.04, 60.83; 90.0, 103.1, 90.0 | 95.88, 114.65, 96.23 90.0, 117.7, 90.0 |
| Resolution Limit (Å) | 1.90 | 2.10 |
| Reflections | 79642 | 97225 |
| Completeness (%) | 96.7 | 89.6 |
| Redundancy | 4.7 | 4.5 |
| Rmergea | 0.129 | 0.107 |
| <I> / <σ(I)> | 11.8 | 12.7 |
Rmerge = Σ|(I - <I>)| / ΣI
Molecular Replacement and Refinement
The 3S-GSDHN-GSTA4-4 and 3S-GSDHN-GSTA1-1 GIMFhelix crystal structures were solved using Phaser (31) and the molecular replacement (MR) pipeline, BALBES (32), respectively, with GSTA4-4 and GSTA1-1 structures (PDB codes 1GUL and 1K3Y) selected as the search models. The MR solutions were used to build models that were refined with REFMAC5 (33) in the CCP4 program suite (34) using rigid body refinement followed by restrained refinement. Entire models were checked and adjusted in Xfit (35) after each cycle of refinement and the 3S,4R-GSDHN ligands as well as water molecules were fit to the difference density. The structures were evaluated by Molprobity (36). Coordinates for the GSDHN-GSTA4-4 and GSDHN-GSTA1-1 GIMFhelix models have been deposited in the Protein Data Bank, PDB identifiers 3IK7 and 3IK9, respectively. Refinement and model statistics are presented in Table 2.
TABLE 2.
Model and refinement statistics.
| GST Structure | GSTA4-4-GSDHN | GIMFhelix-GSDHN |
|---|---|---|
| PDB code | 3IK7 | 3IK9 |
| Resolution Limit (Å) | 1.97 | 2.20 |
| R valuea | 0.21 | 0.23 |
| Rfreeb | 0.25 | 0.28 |
| r.m.s.d. bonds (Å) | 0.012 | 0.011 |
| r.m.s.d. angles (°) | 1.33 | 1.30 |
| No. Atoms: | ||
| Protein | 7178 | 14241 |
| Solvent | 208 | 59 |
| Other | 129 | 248 |
| Average B values (Å2): | ||
| Protein | 39.2 | 45.2 |
| Solvent | 36.0 | 28.8 |
| Ligand | 51.1 | 46.2 |
R = Σ‖Fo| - |Fc‖ / Σ|Fo|
Rfree is R over the 5% of the data not included in model refinement.
Results
Stereoselectivity of Product Formation
The stereoselectivity's of GSTA4-4 and GSTA1-1 were compared with the GIMFhelix mutant using LC/MS analyses. While all four diastereomeric products are generated in approximately equal amounts by the nonenzymatic reaction of racemic HNE with GSH, the 3S,4R-GSHNE and 3S,4S-GSHNE diastereomers are stereoselectively derived from the GSTA4-4 catalyzed reaction (28). Shown in Figure 2 are results acquired with the GIMFhelix mutant, which indicate that, although it is more product stereoselective than wild-type GSTA1-1 with regard to the chirality at C3 in the product, it is less product stereoselective than wild-type GSTA4-4. Whereas these experiments indicate that the product stereoselectivity of the GIMFhelix mutant is different from GSTA4-4, enzymatic activity assays indicate that the GIMFhelix mutant is not less substrate stereoselective than GSTA4-4. Based upon the ratio of the apparent catalytic efficiencies (kcat/Km), the GIMFhelix mutant exhibits a preference for S-HNE vs. R-HNE of 1.6, compared to that of 1.5 previously reported for hGSTA4-4 (28).
FIGURE 2.

Comparison of stereoselectivity of product formation. The GSHNE diastereomers were prepared by incubating GSH and (A) buffer alone, (B) GSTA4-4, (C) GSTA1-1 GIMFhelix, or (D) GSTA1-1, with racemic HNE and analyzed by LC/MS (ESI+, m/z 464). The corresponding spontaneous reaction (gray line) is also shown for comparison.
Overall Structure and Model Quality
The 3S-GSDHN-bound human GSTA4-4 and GIMFhelix structures were solved at 1.97 and 2.20 Å, respectively. The final model of the GSTA4-4 complex is composed of two dimers in the asymmetric unit, with a 3S-GSDHN molecule bound to each of the four corresponding active sites. Crystals of the GIMFhelix complex have four dimers in the asymmetric unit, with a 3S-GSDHN molecule bound to each of the eight corresponding actives sites. Each subunit exhibits the canonical GST fold consisting of the βαβαββα motif in the N-terminal domain that yields the highly conserved GSH binding site (G-site) followed by the C-terminal α–helical domain, which contains many of the principal determinants involved in the electrophilic substrate binding site (H-site) (22, 37, 38). Further evaluation of the structures using MolProbity indicates > 99% of the residues lie in the allowed region of the Ramachandran plot, with the majority of outliers identified as Gln67 residues, which recognize the γ-glutamyl portion of GSH and adopt similar dihedral angles in previously characterized GST structures (13, 21, 37). Views of a single subunit and the overall dimer are represented in Figure 3. The subunits in the dimers are related through non-crystallographic pseudo twofold rotation axes that give an average root mean square deviation value of 0.35 and 0.46 Å for the Cα atoms between the pairs of subunits in the GSTA4-4 and GIMF asymmetric units, respectively. Similar to the previously described GSTA4-4 structures, 1GUM and GUL (13), and the first structures of apo and GSH-bound GIMFhelix (PDB's 3I69 and 3I6A) (26), the 3S-GSDHN-bound GSTA4-4 and GIMFhelix structures described here also exhibit C-terminal regions defined in electron density that extend to at least residues 219-220 for all subunits. They also contain the edge-on-face aromatic-aromatic interaction between Phe111 and Tyr217 that reduces local and global dynamics, which aids in pre-organization of the C-terminus for HNE binding as discussed below (14).
FIGURE 3.
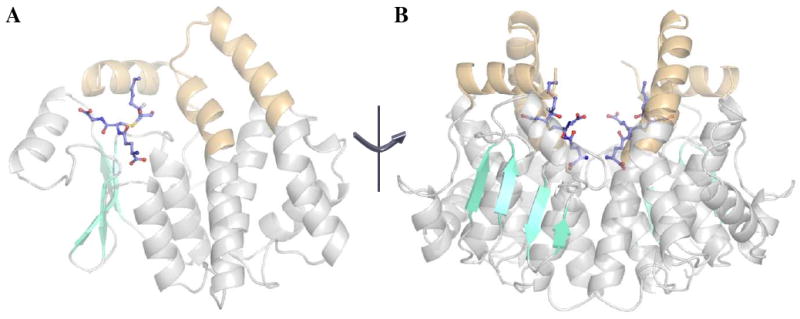
The overall structure of human GSTA4-4 in complex with 3S,4R-GSDHN (3IK7). The views are aligned perpendicular to the two-fold axis of symmetry for the dimer. (A) Structure emphasizing the active site region within one subunit (cyan: β–strands; gray: α–helices except for the C-terminal and α4-α5 tower regions, which are highlighted in orange throughout all figures). (B) Semi-transparent view of the dimer.
Binding of 3S-GSDHN
The GSH portion of 3S-GSDHN maintains all the important contacts with previously described G-site residues (13, 19, 37). The most noticeable difference between the GSTA4-4 and GIMFhelix G-site relates to the glycyl moiety. This portion of the tripeptide extends farther in the GSTA4-4 G-site due to the interaction with Gln45, while the position of the glycyl moiety in GIMFhelix still reflects the presence of the GSTA1-1-derived Arg45 residue. The features of the β1-α1 region, the end of the α4-helix, and the C-terminus have all been proposed to confer specificity of GSTA4-4 toward alkenals and were targeted to afford high alkenal activity to the GIMFhelix mutant (13, 24). The GSTA4-4 crystal structure-based model proposed by Bruns et al., as well as mutagenesis, indicate Tyr212 is essential for activating alkenals (13, 29). As predicted, the hydroxyl group derived from reduction of the aldehyde is positioned at the bottom of the H-site near the hydroxyl group of Tyr212 in the α9-helix of both the GSTA4-4 and GIMFhelix structures. Here the hydroxyl group is actually situated between both the Tyr212 and Tyr9 active site residues. The sulfur atom of the ligand resides near the Tyr9 hydroxyl group and the Arg15 ε-nitrogen while the long alkyl chain that lies in a hydrophobic binding cavity (primarily delineated by Ile107, Met108, and Phe111 in the α4-helix, and Tyr212, Val216, and Tyr217 in the α9-helix) extends into the H-site in a manner that orients the 4-hydroxy substituent toward Arg15 (Figure 4). Although the 3S-GSDHN ligand appears to bind in a similar manner in the GSTA4-4 and GIMFhelix structures (Figure 5A), comparisons of the residues contributing to the active site illustrate an aromatic network of interactions resulting from the presence of a GSTA1-1-derived Phe10 residue into the active site of the GIMFhelix mutant. This residue, which is replaced by a proline in human GSTA4-4, alternates between apo and ligand-bound forms of wild-type GSTA1-1 (18, 19, 21). However, in GSTA4-4 the presence of Tyr212 near the alternate Phe10 position ultimately results in a shift of Phe220 toward the periphery of the active site due to steric hindrance with Phe10 in the region normally occupied by Phe220 in the ligand-bound form of GSTA1-1 (Figure 5B). The locations of Phe10 as well as the 4-hydroxy substituent mentioned above are presumed to have important functional consequences as discussed below.
FIGURE 4.
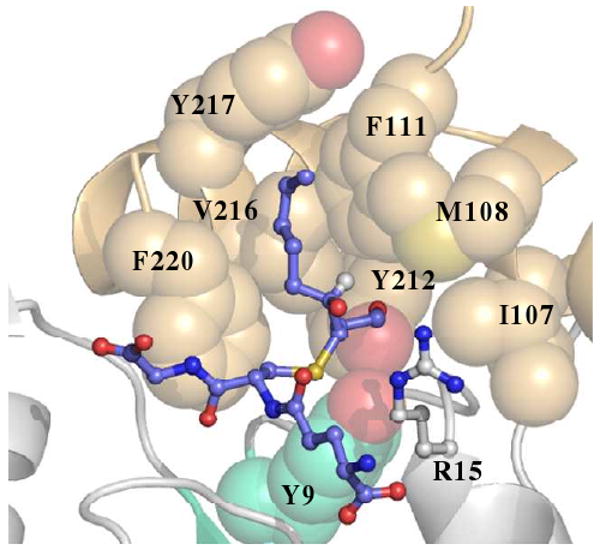
Human GSTA4-4 active site. GSTA4-4 is shown in complex with 3S,4R-GSDHN (3IK7, blue ligand). The conserved G-site is toward the lower half of the figure with Y9 shown in cyan. The 4-hydroxyl group is orientated toward R15 while the aldehyde-derived hydroxyl is in proximity of Y212 at the bottom of the H-site. Other important active site residues are labeled and shown as spheres to illustrate the hydrophobic cavity.
FIGURE 5.
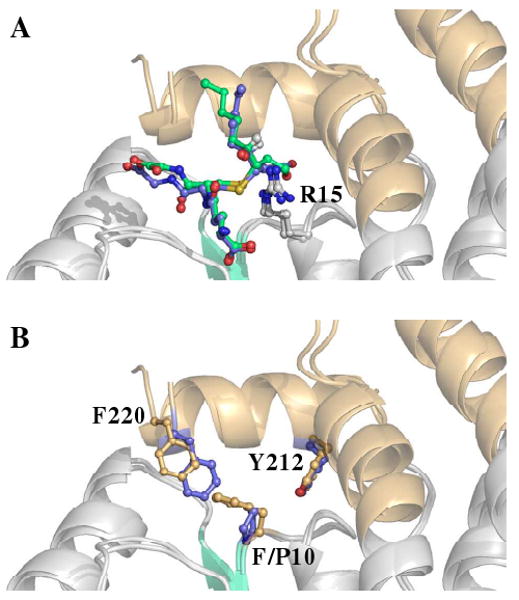
Position of residues 10 and 220 within human A-class GST active sites. (A) Structural superposition of 3S,4R-GSDHN-bound GSTA4-4 (3IK7, blue ligand) and 3S,4R-GSDHN-bound GSTA1-1 GIMFhelix (3IK9, green ligand) (B) Structural superposition contrasting the side chain of F/P10 and F220. The ball and stick representation of residues from GSTA4-4 and GSTA1-1 GIMFhelix are colored blue and orange, respectively. The ligands are not shown for clarity in (B).
Stereoselectivity of the GSTA4-4 Y9F Mutant
Tyr9 is an established active site residue with catalytic importance in GSTs A1-1 and A4-4. Although it is generally categorized among G-site residues, Tyr9 is near the β1-α1 loop that can contribute to the H-site and could in principle also interact with the 4-hydroxyl group of HNE. In order to establish whether the active site Tyr9 residue exerts any affect on the stereoselectivity observed in GSTA4-4, both substrate stereoselectivity and stereoselectivity of product formation were examined using the GSTA4-4 Y9F mutant. LC/MS analyses show that the Y9F mutant conjugates GSH with carbon 3 of HNE in the same stereoselective manner as the wild-type enzyme (Supporting Figure 1). As expected, given the catalytic role of Tyr9 in the G-site, a 71-fold decrease in the kcat was observed compared with the wild-type enzyme (28). However, the apparent kcat /Km for 4S-HNE is 1.5-fold greater than for 4R-HNE with both the GSTA4-4 Y9F mutant and wild-type enzymes (Supporting Figure 2 and (28)) indicating Tyr9 is not a determinant of either substrate or product stereoselectivity, despite its importance in catalysis.
Discussion
Differences in Stereoselectivity of Product Formation
The extra C-terminal α9-helix plays an important role in the different specificities achieved in the structurally related Alpha-class GSTs (13, 14, 24, 27). In general, the C-terminus and the α4-α5 helix-turn-helix “tower” are two of the most dynamic regions in these enzymes. However, the human GSTA4-4 enzyme contains an edge-to-face aromatic-aromatic interaction between Phe111 in the α4-helix and Tyr217 in the α9-helix, which contributes to the closer packing and stability of these regions and increased specificity for HNE (14). Previous results have also implicated this aromatic-aromatic interaction in stereoselectivity, as conformational heterogeneity was found to be inversely correlated with stereoselective product formation in a number of related Alpha-class GSTs (28). Interestingly, while the GSTA4-4 enzyme is completely stereoselective at the level of product formation, the GIMFhelix mutant, which incorporates the Phe111-Tyr217 aromatic-aromatic interaction into the GSTA1-1 scaffold, as well as other key GSTA4-4 active site region interactions, shows intermediate stereoselectivity of product formation that is comparable with the previously described GSTA1-1 V111F/R217Y tower mutant. Given the reported catalytic properties for GIMFhelix and the tower mutant (14, 24, 27), comparisons of the LC/MS results indicate that, while the other H-site mutations present in GIMFhelix are also necessary for increasing activity with alkenals, the single Phe111-Tyr217 interaction is sufficient for achieving the level of product stereoselectivity observed, presumably because it limits local and global dynamics to achieve a higher level of steric control within the active site. There is no corresponding interaction in wild-type GSTA1-1, which shows a great deal of variation in the ordering of the α9-helix (18, 19, 21). The identity and packing of key residues also leads to a differently shaped cavity as illustrated by structural superposition of the 3S-GSDHN ligand within the context of the wild-type GSTA1-1 structure (Supporting Figure 3). However, in light of these results, it is also interesting that simply the removal of this interaction in the previously described GSTA4-4 F111V/Y217R mutant did not result in a drastic decrease in stereoselectivity (28), suggesting that some level of non-additive redundancy important for maintaining stereoselectivity of production formation is distributed throughout the structure of GSTA4-4. Importantly, however, the difference in product stereoselectivity of wild-type GSTA4-4 and GIMFhelix is not correlated with any differences in substrate stereoselectivity: both enzymes exhibit a slight, and equivalent, stereoselectivity for 4S-HNE ((28) and vide infra).
Analysis of the 3S-GSDHN-GIMFhelix crystal structure offers an explanation as to why GSTA4-4 is completely stereoselective in product formation, while the GIMFhelix mutant is not. Initial comparisons of the residues lining a hydrophobic cavity in GSTA4-4 (1GUM and 1GUL) and GIMFhelix (3I69 and 3I6A) suggested that an altered conformation of the Met108 side chain may hinder ligand binding in the putative H-site of GIMFhelix (26). However, superpositions with the 3S-GSDHN-GSTA4-4 structure reported here indicate Met108 is able to occupy alternate locations to accommodate different H-site ligands. Our initial comparisons of GIMFhelix with GSTA4-4 also revealed a unique aromatic network within the mutant's active site that may be responsible for remaining differences in the catalytic efficiencies of GIMFhelix and GSTA4-4, which is confirmed by the ligand-bound structures reported here. Phe220 is a conserved residue in the α9-helix and has been proposed to play a key role in orienting the reacting substrates and guiding them to the transition state (39). However, while Phe220 appears positioned to perform this function without any significant reorganization of active site residues in GSTA4-4, the side chain of Phe10 of GSTA1-1 alternates between apo and ligand-bound forms, and upon ligand binding moves to allow Phe220 to drive C-terminal closure over the active site (19, 21, 39, 40). Position 10 was not among the residues selected for mutation in the construction of GIMFhelix and, analogous to the situation recently described for the first GIMFhelix structures (26), the presence of both Phe10 and Tyr212 within the active site causes Phe10 to maintain its “apo” position, even with both the G- and H-site now occupied by the 3S-GSDHN ligand. In this context Phe10 sterically hinders the optimal pre-organization of the C-terminus of GIMFhelix. This would prevent the maintenance of a consistent HNE orientation with respect to GSH, which is presumably linked with the high substrate specificity of both the H- and G-sites, and the corresponding transition state interactions. Apparently, the pre-organized α9-helix is necessary for complete steric control of the nucleophilic attack that produces only the S-configuration at the site of conjugation in GSTA4-4.
Structural Basis for Coupled Substrate Specificity and Stereopromiscuity
Murine GSTA4-4 was previously co-crystallized with GSHNE (1B48, (23)) but the extrapolation of interactions important for catalysis is complicated not only by key sequence differences between the human and murine proteins, but more importantly by the cyclized structure of the GSHNE ligand. The 3S-GSDHN ligand discussed herein was generated from the NaBH4 reduction of 3S-GSHNE, which was produced using the GSTA4-4-catalyzed conjugation of GSH and racemic HNE (Figure 1). Hence the uncyclized 3S-GSDHN-bound GSTA4-4 crystal structure provides insight into the ternary substrate complex and is the first structural model for the relevant product complex of GSTA4-4 with GSHNE prior to the intramolecular cyclization. Structural superpositions of the apo (1GUM and 3I69) and 3S-GSDHN-bound GSTA4-4 and GIMFhelix structures indicate that substrate and product binding cause negligible structural reorganization. The C-terminus assumes a similar localized structure regardless of ligand occupancy (Figure 6), further emphasizing the pre-organization that specifically accommodates the physiologically relevant ligand within a constricted active site, in contrast to the highly dynamic C-terminus observed in the more promiscuous GSTA1-1 enzyme (18, 21). Such rigid pre-organization of the GSTA4-4 C-terminus suggests that the observed substrate stereopromiscuity does not result from a flexible active site, and must depend on other mechanisms of degenerate substrate recognition.
FIGURE 6.
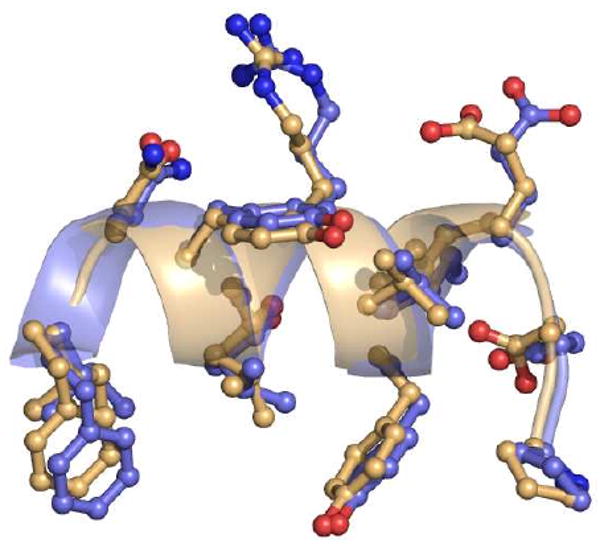
C-terminal regions of apo and ligand-bound human GSTA4-4. Structural superposition of apo (1GUM, orange) and 3S,4R-GSDHN-bound GSTA4-4 (3IK7, blue) illustrating the localized structure of the α9-helix regardless of ligand occupancy. Only residues 208-220 are shown for clarity.
In addition to the extra α9-helix, a distinguishing feature of Alpha-class GSTs is incorporation of a conserved Arg15 residue in a position near the GSH sulfur, which helps to stabilize the thiolate and aids in lowering the pKa value of Tyr9 (22, 41). Beyond its role in the G-site, Arg15 has also been proposed to interact with at least one enantiomer of HNE. Although there is no existing crystal structure with HNE bound to the H-site, a model for the binding of HNE was proposed by Bruns et al. on the basis of GSTA4-4 in complex with the inhibitor S-(2-iodobenzyl)-GSH (13). This model assumed a chirality that would permit hydrogen bonding between the 4-hydroxy substituent and Arg15, though it also anticipates that either enantiomer of HNE should fit into the binding pocket. Likewise, despite a very small preference for 4S-HNE as a substrate, our previously reported kinetic characterization demonstrated GSTA4-4 still exhibits remarkably high apparent efficiencies for both enantiomers (28).
The 3S-GSDHN-bound crystal structures reported here offer the first crystallographic look at a biologically relevant ligand bound to GSTA4-4, and illustrate how the hydroxyl group of either 4R- or 4S-HNE can be positioned equidistant from Arg15 (Figure 4). Because racemic HNE was used to generate the 3S-GSDHN ligand, both configurations at carbon 4 are possible and both 3S,4R-GSDHN and 3S,4S-GSDHN were considered in all the subunits. However, based on the experimental data, a strong preference was not observed for either configuration at carbon 4 so 3S,4R-GSDHN was modeled and refined in all subunits for consistency. Interestingly, even within the constricted and rigid active site, the extension of the GSDHN alkyl chain beyond position 3 could be rotated in a manner that places it toward the “right” side of the hydrophobic binding groove in the H-site cavity (without disrupting the interaction of the aldehyde-derived oxygen with Tyr212 or a 2,3-trans-like arrangement for the ligand). Whereas the binding mode observed in the crystal structure allows the hydroxyl group of both 4R- and 4S-HNE to approach Arg15 without any nearby residues to discriminate between, or disfavor binding of either enantiomer, the additional binding mode reflected in the rotated model would favor an interaction with only 4S-HNE. Occasional binding in this mode could yield the small preference observed for the S-enantiomer in the kinetic assay (28).
Whereas an emerging paradigm assumes that catalytic promiscuity requires protein flexibility, the stereopromiscuity of GSTA4-4 reveals an alternative strategy. Collectively, these results illustrate the structural basis underlying the ability of GSTA4-4 to exploit the hydroxyl group of either 4R- or 4S-HNE, while specifically binding HNE and GSH to maintain high catalytic efficiency linked with stereoselective product formation for both enantiomeric substrates. The symmetrical location of Arg15 between the possible 4-OH orientations of HNE ensures that the biologically formed racemate will be effectively metabolized. The rigid pre-formed structural scaffold including the symmetrically poised Arg15 allows GSTA4-4 to orchestrate this intriguing combination of high substrate specificity and high product stereoselectivity, coupled with low substrate stereoselectivity, that effectively eliminates the toxic racemic mixture produced as a consequence of oxidative stress, while simultaneously achieving complete stereoselectivity of product formation that yields specific GSHNE diastereomers. It is unclear whether the product stereoselectivity has biological utility, or whether it is only a consequence of the rigid environment that optimally aligns reactants for high catalytic efficiency. Interestingly, previous results suggest the GSHNE diastereomers will have different effects and fates in biological tissues (42), which underscores a possible role for the stereoselectivity of GSH conjugation of GSTA4-4. Therefore, in addition to metabolizing both HNE enantiomers, the stereoselectivity exhibited during the generation of the conjugate will conceivably influence other metabolite bioactivity (43) and has further implications regarding stereochemical coordination of the overall detoxification scheme.
Supplementary Material
Acknowledgments
Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program. The authors also gratefully acknowledge Prof. Philip Board (Australian National University) for providing the construct to express hGSTA4-4.
† This work was supported by National Institutes of Health Grants GM62284 and NIHGM32165 and the Swedish Research Council
Footnotes
Abbreviations: HNE, 4-hydroxynonenal; GST, glutathione transferase; GSH, glutathione; GSHNE, glutathionyl 4-hydroxynonanal; GSDHN, glutathionyl 1,4-dihydroxynonanol; MR, molecular replacement; PDB, Protein Data Bank.
Supporting Information Available: Supporting Figures 1-3. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ansari NH, Wang L, Srivastava SK. Role of lipid aldehydes in cataractogenesis: 4-hydroxynonenal-induced cataract. Biochem Mol Med. 1996;58:25–30. doi: 10.1006/bmme.1996.0028. [DOI] [PubMed] [Google Scholar]
- 2.Grimsrud PA, Picklo MJ, Sr, Griffin TJ, Bernlohr DA. Carbonylation of adipose proteins in obesity and insulin resistance: identification of adipocyte fatty acid-binding protein as a cellular target of 4-hydroxynonenal. Mol Cell Proteomics. 2007;6:624–637. doi: 10.1074/mcp.M600120-MCP200. [DOI] [PubMed] [Google Scholar]
- 3.Hu W, Feng Z, Eveleigh J, Iyer G, Pan J, Amin S, Chung FL, Tang MS. The major lipid peroxidation product, trans-4-hydroxy-2-nonenal, preferentially forms DNA adducts at codon 249 of human p53 gene, a unique mutational hotspot in hepatocellular carcinoma. Carcinogenesis. 2002;23:1781–1789. doi: 10.1093/carcin/23.11.1781. [DOI] [PubMed] [Google Scholar]
- 4.Zarkovic K. 4-hydroxynonenal and neurodegenerative diseases. Mol Aspects Med. 2003;24:293–303. doi: 10.1016/s0098-2997(03)00024-4. [DOI] [PubMed] [Google Scholar]
- 5.Carini M, Aldini G, Facino RM. Mass spectrometry for detection of 4-hydroxy-trans-2-nonenal (HNE) adducts with peptides and proteins. Mass Spectrom Rev. 2004;23:281–305. doi: 10.1002/mas.10076. [DOI] [PubMed] [Google Scholar]
- 6.Awasthi YC, Ansari GA, Awasthi S. Regulation of 4-hydroxynonenal mediated signaling by glutathione S-transferases. Methods Enzymol. 2005;401:379–407. doi: 10.1016/S0076-6879(05)01024-4. [DOI] [PubMed] [Google Scholar]
- 7.Yang Y, Sharma R, Sharma A, Awasthi S, Awasthi YC. Lipid peroxidation and cell cycle signaling: 4-hydroxynonenal, a key molecule in stress mediated signaling. Acta Biochim Pol. 2003;50:319–336. [PubMed] [Google Scholar]
- 8.Cheng JZ, Sharma R, Yang Y, Singhal SS, Sharma A, Saini MK, Singh SV, Zimniak P, Awasthi S, Awasthi YC. Accelerated metabolism and exclusion of 4-hydroxynonenal through induction of RLIP76 and hGST5.8 is an early adaptive response of cells to heat and oxidative stress. J Biol Chem. 2001;276:41213–41223. doi: 10.1074/jbc.M106838200. [DOI] [PubMed] [Google Scholar]
- 9.Hashimoto M, Sibata T, Wasada H, Toyokuni S, Uchida K. Structural basis of protein-bound endogenous aldehydes. Chemical and immunochemical characterizations of configurational isomers of a 4-hydroxy-2-nonenal-histidine adduct. J Biol Chem. 2003;278:5044–5051. doi: 10.1074/jbc.M210129200. [DOI] [PubMed] [Google Scholar]
- 10.Hiratsuka A, Hirose K, Saito H, Watabe T. 4-Hydroxy-2(E)-nonenal enantiomers: (S)-selective inactivation of glyceraldehyde-3-phosphate dehydrogenase and detoxification by rat glutathione S-transferase A4-4. Biochem J. 2000;349:729–735. doi: 10.1042/bj3490729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wakita C, Maeshima T, Yamazaki A, Shibata T, Ito S, Akagawa M, Ojika M, Yodoi J, Uchida K. Stereochemical configuration of 4-hydroxy-2-nonenal-cysteine adducts and their stereoselective formation in a redox-regulated protein. J Biol Chem. 2009;284:28810–28822. doi: 10.1074/jbc.M109.019927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Board PG. Identification of cDNAs encoding two human alpha class glutathione transferases (GSTA3 and GSTA4) and the heterologous expression of GSTA4-4. Biochem J. 1998;330:827–831. doi: 10.1042/bj3300827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bruns CM, Hubatsch I, Ridderstrom M, Mannervik B, Tainer JA. Human glutathione transferase A4-4 crystal structures and mutagenesis reveal the basis of high catalytic efficiency with toxic lipid peroxidation products. J Mol Biol. 1999;288:427–439. doi: 10.1006/jmbi.1999.2697. [DOI] [PubMed] [Google Scholar]
- 14.Hou L, Honaker MT, Shireman LM, Balogh LM, Roberts AG, Ng KC, Nath A, Atkins WM. Functional promiscuity correlates with conformational heterogeneity in A-class glutathione S-transferases. J Biol Chem. 2007;282:23264–23274. doi: 10.1074/jbc.M700868200. [DOI] [PubMed] [Google Scholar]
- 15.Hubatsch I, Ridderstrom M, Mannervik B. Human glutathione transferase A4-4: an alpha class enzyme with high catalytic efficiency in the conjugation of 4-hydroxynonenal and other genotoxic products of lipid peroxidation. Biochem J. 1998;330:175–179. doi: 10.1042/bj3300175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao T, Singhal SS, Piper JT, Cheng J, Pandya U, Clark-Wronski J, Awasthi S, Awasthi YC. The role of human glutathione S-transferases hGSTA1-1 and hGSTA2-2 in protection against oxidative stress. Arch Biochem Biophys. 1999;367:216–224. doi: 10.1006/abbi.1999.1277. [DOI] [PubMed] [Google Scholar]
- 17.Adman ET, Le Trong I, Stenkamp RE, Nieslanik BS, Dietze EC, Tai G, Ibarra C, Atkins WM. Localization of the C-terminus of rat glutathione S-transferase A1-1: crystal structure of mutants W21F and W21F/F220Y. Proteins. 2001;42:192–200. doi: 10.1002/1097-0134(20010201)42:2<192::aid-prot60>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 18.Cameron AD, Sinning I, L'Hermite G, Olin B, Board PG, Mannervik B, Jones TA. Structural analysis of human alpha-class glutathione transferase A1-1 in the apo-form and in complexes with ethacrynic acid and its glutathione conjugate. Structure. 1995;3:717–727. doi: 10.1016/s0969-2126(01)00206-4. [DOI] [PubMed] [Google Scholar]
- 19.Grahn E, Novotny M, Jakobsson E, Gustafsson A, Grehn L, Olin B, Madsen D, Wahlberg M, Mannervik B, Kleywegt GJ. New crystal structures of human glutathione transferase A1-1 shed light on glutathione binding and the conformation of the C-terminal helix. Acta Crystallogr D Biol Crystallogr. 2006;62:197–207. doi: 10.1107/S0907444905039296. [DOI] [PubMed] [Google Scholar]
- 20.Kuhnert DC, Sayed Y, Mosebi S, Sayed M, Sewell T, Dirr HW. Tertiary interactions stabilise the C-terminal region of human glutathione transferase A1-1: a crystallographic and calorimetric study. J Mol Biol. 2005;349:825–838. doi: 10.1016/j.jmb.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 21.Le Trong I, Stenkamp RE, Ibarra C, Atkins WM, Adman ET. 1.3-A resolution structure of human glutathione S-transferase with S-hexyl glutathione bound reveals possible extended ligandin binding site. Proteins. 2002;48:618–627. doi: 10.1002/prot.10162. [DOI] [PubMed] [Google Scholar]
- 22.Sinning I, Kleywegt GJ, Cowan SW, Reinemer P, Dirr HW, Huber R, Gilliland GL, Armstrong RN, Ji X, Board PG, et al. Structure determination and refinement of human alpha class glutathione transferase A1-1, and a comparison with the Mu and Pi class enzymes. J Mol Biol. 1993;232:192–212. doi: 10.1006/jmbi.1993.1376. [DOI] [PubMed] [Google Scholar]
- 23.Xiao B, Singh SP, Nanduri B, Awasthi YC, Zimniak P, Ji X. Crystal structure of a murine glutathione S-transferase in complex with a glutathione conjugate of 4-hydroxynon-2-enal in one subunit and glutathione in the other: evidence of signaling across the dimer interface. Biochemistry. 1999;38:11887–11894. doi: 10.1021/bi990468i. [DOI] [PubMed] [Google Scholar]
- 24.Nilsson LO, Gustafsson A, Mannervik B. Redesign of substrate-selectivity determining modules of glutathione transferase A1-1 installs high catalytic efficiency with toxic alkenal products of lipid peroxidation. Proc Natl Acad Sci USA. 2000;97:9408–9412. doi: 10.1073/pnas.150084897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Babbitt PC. Reengineering the glutathione S-transferase scaffold: a rational design strategy pays off. Proc Natl Acad Sci USA. 2000;97:10298–10300. doi: 10.1073/pnas.97.19.10298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balogh LM, Le Trong I, Kripps KA, Tars K, Stenkamp RE, Mannervik B, Atkins WM. Structural analysis of a glutathione transferase A1-1 mutant tailored for high catalytic efficiency with toxic alkenals. Biochemistry. 2009;48:7698–7704. doi: 10.1021/bi900895b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blikstad C, Shokeer A, Kurtovic S, Mannervik B. Emergence of a novel highly specific and catalytically efficient enzyme from a naturally promiscuous glutathione transferase. Biochim Biophys Acta. 2008;1780:1458–1463. doi: 10.1016/j.bbagen.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 28.Balogh LM, Roberts AG, Shireman LM, Greene RJ, Atkins WM. The stereochemical course of 4-hydroxy-2-nonenal metabolism by glutathione S-transferases. J Biol Chem. 2008;283:16702–16710. doi: 10.1074/jbc.M801725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hubatsch I, Mannervik B. A highly acidic tyrosine 9 and a normally titrating tyrosine 212 contribute to the catalytic mechanism of human glutathione transferase A4-4. Biochem Biophys Res Commun. 2001;280:878–882. doi: 10.1006/bbrc.2000.4230. [DOI] [PubMed] [Google Scholar]
- 30.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Colledt in Oscillation Mode. Methods in Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 31.McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr. 2007;63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Long F, Vagin AA, Young P, Murshudov GN. BALBES: a molecular-replacement pipeline. Acta Crystallogr D Biol Crystallogr. 2008;64:125–132. doi: 10.1107/S0907444907050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 34.Collaborative Computational Project, N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 35.McRee DE. A Visual Protein Crystallographic Software System for X11/Xview. J Mol Graphics. 1992;10:44–46. [Google Scholar]
- 36.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Armstrong RN. Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem Res Toxicol. 1997;10:2–18. doi: 10.1021/tx960072x. [DOI] [PubMed] [Google Scholar]
- 38.Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 39.Nilsson LO, Edalat M, Pettersson PL, Mannervik B. Aromatic residues in the C-terminal region of glutathione transferase A1-1 influence rate-determining steps in the catalytic mechanism. Biochim Biophys Acta. 2002;1598:199–205. doi: 10.1016/s0167-4838(02)00362-x. [DOI] [PubMed] [Google Scholar]
- 40.Ibarra C, Nieslanik BS, Atkins WM. Contribution of aromatic-aromatic interactions to the anomalous pK(a) of tyrosine-9 and the C-terminal dynamics of glutathione S-transferase A1-1. Biochemistry. 2001;40:10614–10624. doi: 10.1021/bi010672h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bjornestedt R, Stenberg G, Widersten M, Board PG, Sinning I, Jones TA, Mannervik B. Functional significance of arginine 15 in the active site of human class alpha glutathione transferase A1-1. J Mol Biol. 1995;247:765–773. doi: 10.1016/s0022-2836(05)80154-8. [DOI] [PubMed] [Google Scholar]
- 42.Gueraud F, Crouzet F, Alary J, Rao D, Debrauwer L, Laurent F, Cravedi JP. Enantioselective metabolism of (R)- and (S)-4-hydroxy-2-nonenal in rat. BioFactors. 2005;24:97–104. doi: 10.1002/biof.5520240111. [DOI] [PubMed] [Google Scholar]
- 43.Ramana KV, Bhatnagar A, Srivastava S, Yadav UC, Awasthi S, Awasthi YC, Srivastava SK. Mitogenic responses of vascular smooth muscle cells to lipid peroxidation-derived aldehyde 4-hydroxy-trans-2-nonenal (HNE): role of aldose reductase-catalyzed reduction of the HNE-glutathione conjugates in regulating cell growth. J Biol Chem. 2006;281:17652–17660. doi: 10.1074/jbc.M600270200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.