

Abstract
A detailed account on Simmons-Smith cyclopropanations of allenamides en route to amido-spiro[2.2]pentanes is described here. While the diastereoselectivity was low when using unsubstituted allenamides, the reaction is overall efficient and general, representing the most direct synthesis of both chemically and biologically interesting amido-spiro[2.2]pentane systems. With α-substituted allenamides, while the diastereoselectivity could be improved significantly based on a series of conformational analysis, both mono- and bis-cyclopropanation products were observed. Consequently, several structurally intriguing amido-methylene cyclopropanes could also be prepared.
Introduction
Spiro[2.2]pentanes,1 the smallest member of the triangulane or oligo-spirocyclopropane family, represent a unique structural topology with both rigidity and orthogonality that have found applications in a number of biological contexts.2 In particular, simple α-spiropentyl acetic acid [Figure 1] has been shown to mimic α-(methylenecyclopropyl) acetic acid, a well-known inhibitor against acyl-CoA dehydrogenase that is critical in the fatty acid oxidation pathway. In addition, α-(methylenecyclopropyl) acetic acid itself has also been identified as a toxic metabolite of the natural amino acid hypoglycine A found in the fruits of Jamaican ackee trees.3–7 Consequently, it is a key cause of vomiting sickness when ingesting the Jamaican ackee fruit due to the resulting deficiency it causes in the acyl-CoA dehydrogenase activity.8,9 Moreover, amino-spiro[2.2]pentanes have received much attention recently for an array of other purposes ranging from constructing deoxyribonucleotide analogs10 to exploring the chemistry and biology of spiro[2.2]pentane amino acid derivatives.11,12
Figure 1.

Spiro[2.2]Pentanes.
Despite these biological interests, and despite a number of elegant approaches toward spiro[2.2]pentanes in literature, the overall synthetic effort toward amino-spiro[2.2]pentanes has been limited.2,13 Few involve direct bis-cyclopropanations of allenes14 with many adopting mono-cyclopropanations of methylene cyclopropanes prepared through other means.15 To the best of our knowledge, preparation of amino-spiro[2.2]pentanes directly through bis-cyclopropanations of 1-amino-allenes is not known.2,10–13,16 Our recent interest17,18 in cyclopropanations of enamides13,19,20 en route to optically enriched amino-cyclopropanes21,22 coupled with our decade long efforts in developing the chemistry of allenamides23–26 allowed us to envision the possibility of developing a direct construction of amido-spiro[2.2]pentanes via Simmons-Smith cyclopropanation of allenamides 1 [Scheme 1]. Based on our previous work on a number of different stereoselective cycloaddition manifolds employing allenamides,27 we anticipated that this cyclopropanation could proceed stereoselectively in which the zinc carbenoid can approach the bottom π-face of the more favored conformer 1a [Scheme 1]. This would lead to methylene cyclopropane 2, and while 2 is useful in its own right,28 an ensuing second cyclopropanation would provide 3a as the major amino-spiro[2.2]pentane isomer with 3b being derived from cyclopropanation of the minor conformer 1b. We report here details of these investigations.
Scheme 1.

Simmons-Smith Cyclopropanations of Allenamides.
Results and Discussions
1. Cyclopropanations of α-Unsubstituted Allenamides
The feasibility for Simmons-Smith cyclopropanations of allenamides was quickly established as shown in Scheme 2. By using 5.0 equiv Et2Zn and 10.0 equiv ICH2-X, cyclopropanation of achiral allenamide 4 proceeded in excellent yields to give amido-spiro[2.2]pentane 629 with no difference between using ICH2-I and ICH2-Cl respectively in CH2Cl2 and ClCH2CH2Cl [DCE]. We did not observe any mono-cyclopropanation product 5 after 12 h, suggesting that the second cyclopropanation via 5 took place rapidly in these reactions with Zn(CH2X)2 serving as the highly reactive cyclopropanating species.30
Scheme 2.

Cyclopropanation of Achiral Allenamide 4.
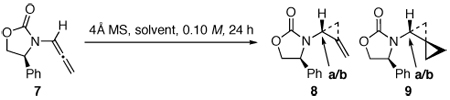
We turned our attention to chiral allenamides using specifically 7 [Table 1], and while the cyclopropanation was equally effective in providing de novo amido-spiro[2.2]pentane 9, the diastereomeric ratio was not desirable. We explored a range of conditions13a including different temperatures [entries 1–4] and solvents [entries 5–7], while featuring Zn(CH2Cl)2 as the cyclopropanating species. In addition, we examined the nature of cyclopropanating species such as Zn(CH2I)2 either without [entry 8] or with the addition of a chelating solvent such as DME [rendering the zinc cyclopropanating reagent more nucleophilic] [entry 9]. Finally, we explored Furukawa type reagents [entries 10–12]31,32 as well as Yamamoto’s AlMe3 activation of ICH2-I [entry 13].33
Table 1.
Cyclopropanations of Chiral Allenamide 7.
 | |||||
|---|---|---|---|---|---|
| entry | cyclopropanat. agent | temp [°C] | solventa | yield [%] [ratios]b | |
| 1 | Zn(CH2Cl)2c | − 20 | DCE | - | -d |
| 2 | Zn(CH2Cl)2 | 25 | DCE | - | 67 [1.4:1] |
| 3 | Zn(CH2Cl)2 | 25 | DCEe | 22 [2.8:1] | 45 [1.2:1] |
| 4 | Zn(CH2Cl)2 | 85 | DCE | - | 50 [1:1] |
| 5 | Zn(CH2Cl)2 | 25 | DME | - | -d |
| 6 | Zn(CH2Cl)2 | 25 | THF | - | -d |
| 7 | Zn(CH2Cl)2 | 25 | Tol | - | 50 [1:1] |
| 8 | Zn(CH2I)2 f | 25 | CH2Cl2 | - | 40 [1.5:1] |
| 9 | Zn(CH2I)2-DMEf | 25 | DCE | 10 [1.5:1] | -d |
| 10 | CF3CO2ZnCH2Ig | 25 | CH2Cl2 | - | - |
| 11 | EtZnCH2Ih | 25 | CH2Cl2 | - | -d |
| 12 | EtZnCH2I-DMEh | 25 | CH2Cl2 | - | -d |
| 13 | R2AlCH2Ii | 25 | CH2Cl2 | - | -d |
DCE: 1,2-Dichloroethane. DME: 1,2-Dimethoxyethane.
NMR yields for 8 and Isolated yields for 9. The ratio in brackets denotes a:b with a being the major isomer as shown in the scheme, and was assigned via NMR.
Employing 5.0 equiv Et2Zn and 10.0 equiv ICH2-Cl.
Recovering 20–65% of the starting allenamide 7.
conc. = 0.05 M.
Employing 5.0 equiv Et2Zn and 10.0 equiv ICH2-I. For entry 9, 5.0 equiv of DME was added.
Employing 5.0 equiv each of Et2Zn, ICH2-I, and CF3CO2H.
Employing 5.0 equiv Et2Zn and 5.0 equiv ICH2-I. For entry 12, 5.0 equiv of DME was added.
Employing 5.0 equiv Me3Al and 5.0 equiv ICH2-I.
We did not continue to pursue other cyclopropanating species such as Molander’s Sm-Hg activation of ICH2-Cl,34 as we recognized that we were not going to improve the diastereomeric ratio of amido-spiro[2.2]pentane 9 via bis-cyclopropanation of 7 by only using different cyclopropanating species. It is noteworthy that this effort allowed us to observe by 1H NMR the mono-cyclopropanation product 8, although not isolated [entries 3 and 9]. This implies that the 2nd cyclopropanation is slower for chiral allenamides. Stereochemically, both the major and minor diastereomers of 9 were unambiguously assigned using single-crystal X-ray structures as shown in Figure 2. The ability to access both diastereomers of these structurally very interesting and novel amido-spiro[2.2]pentanes renders this none-stereoselective aspect of this reaction an opportunity and less of a limitation.
Figure 2.

X-Ray Structures of 9a [left] and 9b [Right].
Subsequently, a number of chiral auxiliaries on the allenamide were probed in an attempt to improve the diastereoselectivity. As shown in Scheme 3, amido-spiro[2.2]pentanes such as 10, 11, 14, and 15 could be attained in good yields through bis-cyclopropanations of their respective allenamides. However, the diastereomeric ratio remained low, and when employing the more bulky Sibi’s auxiliary35 or Seebach’s auxiliary,36 the reaction appeared to be shut down, as only trace amounts of amido-spiro[2.2]pentanes 12 and 16 could be found. Close’s auxiliary37 gave the best ratio in 17 but with a lower yield. There is essentially no difference in the level of stereoselectivity between using auxiliaries containing just the mono-substitution alpha to the amido-nitrogen atom [see 10–12] and those with vicinal substitutions on the oxazolidinone ring [see 14–17].
Scheme 3.

Effect of Chiral Auxiliaries on Stereoselectivity.
2. A Comparison with Chiral Enamides
While the lack of diastereoselectivity was frustrating, it intrigued us mechanistically. We had previously examined Simmons-Smith cyclopropanations of chiral enamides and achieved a much greater success in stereochemical control.17 As shown in Scheme 4, chiral E-enamides such as 18 and E-19 gave amido-cyclopropanes 20 and trans-21 with diastereomeric ratios of 95:5 and 83:17, respectively, while chiral Z-enamides such as 22 and Z-19 led to even higher diastereomeric ratios of ≥95:5 in each case. These results are in direct contrast to our current cyclopropanation work
Scheme 4.

Cyclopropanations of E- and Z-Enamides.
To rationalize the above stereochemical outcome, we examined conformations of these enamides through both X-ray structures [see structures of E-19 and Z-19 in Figure 3] and PM3 calculations via Spartan Model.™ Both the X-ray structure [see E-19] and computation model revealed that E-enamides [R = alkyl or aryl] assume the more favorable conformation E1 [Scheme 5], which was what we had speculated earlier in some epoxidation work.38,39 The other locally minimized conformation is E2 but it is less favored than E1 by 1.17 kcal mol−1. In both conformers, the olefin is approaching co-planarity with the oxazolidinone ring, allowing delocalization of the nitrogen lone pair into the olefin. Being devoid of actual transition state calculations, we will make an assumption here that these cyclopropnations proceed through the major enamide [or allenamide] conformer with the awareness that Curtin-Hamette could very well be in play here, and that we are only attempting to identif a model with some consistent rationale and predictative power at this juncture.
Figure 3.

X-Ray Structures of Enamides E-19 [Left] and Z-19 [Right].
Scheme 5.

A Model for the Enamide Cyclopropanation.
Based on this assumption, if the cyclopropanation proceeds through the more favored conformation E1, the necessary π-facial differentiation in E1 would provide the excellent stereochemical outcome with E2 being a possible source for the minor diastereomer. On the other hand, in both the X-ray structure [see Z-19 in Figure 3] and computation model of Z-enamides, there is a distinct shift from a coplanar motif in conformation Z1 to the more favored Z2 [ΔE = −1.03 to −1.28 kcal mol−1]. This is likely due to the oxazolidinone ring rotating along the C-N bond toward the direction so that the Ph substituent could be shifted away from the R group to alleviate the allylic strain. Despite such conformational change relative to E-enamides, the bottom π-face in the more favored conformation Z2 remains sterically more accessible, thereby providing the same sense of facial selectivity in the cyclopropanation as for the E-enamide.
Although we have not examined this in detail, the greater diastereoselectivity attained for Z-enamides relative to those of E-enamides could be a result of a greater shielding of the top face by the phenyl ring, and/or a possible chelation of the oxazolidinone carbonyl oxygen with the zinc reagent in a directed cyclopropanation manner.
In contrast, while chiral allenamides assume a similar set of conformations40 as shown in Scheme 6, calculations [PM3 calculations via Spartan Model™] suggest that the energetic difference between conformers 24a and 24b [see ΔE = −0.21 kcal mol−1 for R = H] appears to be relatively much smaller than those from enamides. In addition, we also find that the first cyclopropanation is very facile relative to the cyclopropanation of enamides. In general, the starting allenamides are consumed within 1–2 h at 0 °C [or rt] based on monitoring by NMR, leading to 25-Ma and 25-Mb, whereas cyclopropanation of enamides in most cases required 24–72 h.17 A mixture of mono- and bis-cyclopropanation products with a ratio of 1:1.5 was usually seen after 3 h at 0 °C, and the long reaction time is associated with the second cyclopropanation, leading to 25-Ba and 25-Bb.
Scheme 6.

A Comparison with the Enamide Cyclopropanation.
Consequently, again, based on the assumption that these cyclopropnations also proceed through the major allenamide conformer, the lack of diastereoselectivity observed in Simmons-Smith cyclopropanations of allenamides could be due to a facile cyclopropanation through an almost equal distribution of unsubstituted allenamide conformers 24a and 24b [for R = H]. While this proposed model is based on ground state energetic difference, if valid, α-substituted allenamides [for R ≠ H] would then lead to an improved selectivity because 24a is now favored by 1.05 kcal mol−1 [for R = Me] over 24b due to its the enhanced allylic strain.
3. Cyclopropanations of α-Substituted Allenamides
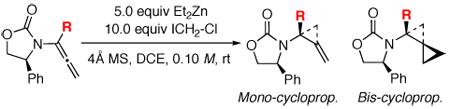
Based on the above conformational model, we prepared α-substituted achiral allenamides 26–28 [Scheme 7] through α-alkylation of the respective unsubstituted allenamides.41 Cyclopropanations of allenamides 26–28 were not only feasible, but also led to the observation and isolation of a substantial amount of mono-cyclopropanation products 29-M through 31-M. In the case of allenamide 28, we isolated 60% of mono-cyclopropane 31-M. These results suggest that α-substituted allenamides further impede the second cyclopropanation compared to unsubstituted chiral allenamides. The unique structural motif of the amido-methylene cyclopropane 31-M is displayed in Figure 4 through its single-crystal X-ray structure.
Scheme 7.

Mono- Versus Bis-Cyclopropanation of Allenamides.
Figure 4.

X-Ray Structures of 31-M [Left] and 37-B [Right].
We proceeded to examine α-substituted chiral allenamides 32–35 as shown in Table 2. In all cases, we isolated both mono-[36-M through 39-M] and bis-cyclopropanation products [36-B through 39-B]. A longer reaction time usually resulted in more of the respective bis-cyclopropanation product. In accord with our conformational analysis, the diastereomeric ratio was indeed improved with a dependence on the size of the R groups. The stereochemistry of 37-B was unambiguously assigned using X-ray structural analysis [Figure 4].
Table 2.
Cyclopropanations of Chiral α-Substituted Allenamides.
 | ||||
|---|---|---|---|---|
| entry | allenamide | R = | time [h] | yield [%] [ratios]a |
| 1 | 32 | Me | 3 | 36-M: 38 [3.5:1] 36-B: 26 [3.5:1] |
| overall dr = 3.5:1b | ||||
| 2 | 32 | Me | 16 | 36-M: 19c [10.0:1] 36-B: 36 [3.0:1] |
| overall dr = 4.1:1b | ||||
| 3 | 33 | n-Bu | 3 | 37-M: 37d [5.0:1] 37-B: 18 [≥20:1] |
| overall dr = 7.9:1b | ||||
| 4 | 33 | n-Bu | 16 | 37-M: 30d [6.0:1] 37-B: 22 [≥20:1] |
| overall dr = 11.1:1b | ||||
| 5 | 34 | Bn | 16 | 38-M: 32 [7.0:1] 38-B: 20 [≥20:1] |
| overall dr = 12.0:1b | ||||
| 6 | 35 | CH2C6H4[o-Ph] | 3 | 39-M: 36 [5.0:1] 39-B: 23 [≥20:1] |
| overall dr = 8.8:1b | ||||
Isolated yields. Dr ratios are in the bracket with the respective major diastereomer being shown in the scheme and all ratios were assigned using crude 1H NMR.
Overall dr ratios represent the combined dr for the first cyclopropanation.
NMR yield.
See reference 42.
To ensure that major isomers of mono- and bis-scyclopropanation product 37-M and 37-B in fact possess the same stereochemistry at the carbon bearing the amido group, amido-methylene cyclopropane 37-M was subjected to the same cyclopropanation conditions [Scheme 8]. While the reaction was slow and incomplete, we found a 43% yield of 37-B [as a single isomer], thereby confirming that the major isomer of bis-cyclopropanation products indeed comes from a second cyclopropanation of the major isomer of the respective mono-cyclopropanation products. This assessment would then translate the individual ratios of mono- and bis-cyclopropanation into an excellent overall or combined diastereoselectivity for the first cyclopropanation [see numbers in red] that correlates well overall with increasing in the size of the R group, and provide a solid support for the conformational model proposed above.
Scheme 8.

Assignment of Mono-Cyclopropane 37-M.
Lastly, the rate of the second cyclopropanation appears to be directly correlated with the degree of steric crowding of either π-face of the methylene cyclopropane intermediate. As shown in Scheme 9, in the case of unsubstituted allenamides, both p-faces of the olefin in achiral amido-methylene cyclopropane 5 are open for the second cyclopropanation, whereas chiral amido-methylene cyclopropane ent-8 is blocked on the bottom p-face with the top still available. Thus, we did not observe amido-methylene cyclopropane 5 but saw ent-8 in 12 h under the same reaction conditions. For α-substituted allenamides, both π-faces of amido-methylene cyclopropanes such as 29–31 and 36-M through 39-M are now sterically more encumbered. Consequently, the second cyclopropanation of 29–31 and 36-M through 39-M should be slower relative to those of 5 and ent-8, leading to the observation and/or isolation of methylene cyclopropanes for α-substituted allenamides.
Scheme 9.

Rate Comparisons for the Second Cyclopropanation.
Conclusion
We have described here Simmons-Smith cyclopropanations of allenamides in the synthesis of amido-spiro[2.2]pentanes. While the diastereoselectivity was low when using unsubstituted allenamides, the reaction is overall efficient and general, leading to an array of amido-spiro[2.2]pentanes. With α-substituted allenamides, while the diastereoselectivity could be improved significantly based on a conformational analysis, both mono- and bis-cyclopropanations were observed in these cases. Consequently, several structurally intriguing amido-methylene cyclopropanes could also be prepared. With allenamides being readily accessible, these efforts have yielded the most straightforward protocol in constructing chemically and biologically intriguing amido-spiro[2.2]pentane systems.
Supplementary Material
Acknowledgment
Authors thank NIH-NIGMS [GM066055] for funding. We thank Mr. Benjiman E. Kucera and Dr. Vic Young of University of Minnesota for providing X-ray structural analysis.
Footnotes
Experimental Electronic supplementary information (ESI) available: experimental procedures, 1H NMR spectra, characterizations for all new compounds, and X-ray structural data.
References
- 1.For the earliest example, see: Gustavson G. J. Prakt. Chem. 1896;54:97. For early accounts, see: Rogowski F. Chem. Ber. 1939;72B:2021. Murray MJ, Stevenson EH. J. Am. Chem. Soc. 1944;66:812. Donohue J, Humphrey GL, Schomaker V. J. Am. Chem. Soc. 1945;67:332. doi: 10.1021/ja01199a041.
- 2.For a leading review see: de Meijere A, Kozhushkov SI. Chem. Rev. 2000;100:93. doi: 10.1021/cr960153y.
- 3.Tserng KY, Jin S-J, Hoppel CL. Biochemistry. 1991;30:10755. doi: 10.1021/bi00108a021. [DOI] [PubMed] [Google Scholar]
- 4.Li D, Zhou H-Q, Dakoji S, Shin I, Oh E, Liu H-W. J. Am. Chem. Soc. 1998;120:2008. [Google Scholar]
- 5.(a) Osmundsen H, Sherratt HSA. FEBS Lett. 1975;55:81. doi: 10.1016/0014-5793(75)80951-3. [DOI] [PubMed] [Google Scholar]; (b) Kean EA. Biochem. Biophys. Acta. 1976;422:81. doi: 10.1016/0005-2744(76)90003-6. [DOI] [PubMed] [Google Scholar]; (c) Abeles RH. In: Enzyme Activated Irreversible Inhibitors. Seiler N, Jung MJ, Koch-Weser J, editors. Amsterdam: Elsevier-North-Holland Press; 1978. p. 1. [Google Scholar]; (d) Ghisla S, Wenz A, Thorpe C. In: Enzyme Inhibitors. Brodbeck U, editor. Weinheim, Germany: Verlag Chim Press; 1980. p. 43. [Google Scholar]; (e) Wenz A, Thorpe C, Ghisla S. J. Biol. Chem. 1981;256:9809. [PubMed] [Google Scholar]; (f) Ghisla S, Melde K, Zeller HD, Boschert W. In: Fatty Acid Oxidation; Clinical, Biochemical, and Molecular Aspects. Tanaka K, Coates PM, Alan R, editors. New York: Liss, Inc. Press; 1990. p. 185. [Google Scholar]
- 6.(a) Lai M-T, Li D, Oh E, Liu H-W. J. Am. Chem. Soc. 1993;115:1619. [Google Scholar]; (b) Lai M-T, Liu H-W. J. Am. Chem. Soc. 1992;114:3160. [Google Scholar]; (c) Lai M-T, Liu H-W. J. Am. Chem. Soc. 1991;113:7388. [Google Scholar]; (d) Lai M-T, Liu H-W. J. Am. Chem. Soc. 1990;112:4034. [Google Scholar]; (e) Lenn ND, Shih Y, Stankovich MT, Liu H-W. J. Am. Chem. Soc. 1989;111:3065. [Google Scholar]
- 7.(a) Baldwin JE, Adlington RM, Bebbinton D, Riussell AT. Tetrahedron. 1994;50:12015. [Google Scholar]; (b) Baldwin JE, Widdison WC. J. Am. Chem. Soc. 1992;114:2245. [Google Scholar]; (c) Baldwin JE, Ostrander RL, Simon CD, Widdison WC. J. Am. Chem. Soc. 1990;112:2021. [Google Scholar]; (d) Baldwin JE, Parker DW. J. Org. Chem. 1987;52:1475. [Google Scholar]
- 8.(a) Tanaka K, Isselbacher KJ, Shih V. Science. 1972;175:69. doi: 10.1126/science.175.4017.69. [DOI] [PubMed] [Google Scholar]; (b) Holt C, von Holt M, Bohm H. Biochim. Biophys. Acta. 1966;125:11. doi: 10.1016/0005-2760(66)90139-1. [DOI] [PubMed] [Google Scholar]; (c) Black DK, Landor SR. J. Chem. Soc. C. 1968:288. [Google Scholar]
- 9.(a) Tanaka K. In: Handbook of Clinical Neurology. Vinken PJ, Bruyn GW, editors. Vol. 37. Amsterdam: Elsevier-North Holland Press; 1979. pp. 511–513. [Google Scholar]; (b) Stuart KL. In: Hypoglycin. Kean EA, editor. New York: Academic Press; 1975. p. 39. [Google Scholar]
- 10.Guan H-P, Ksebati MB, Cheng Y-C, Drach JC, Kern ER, Zemlicka J. J. Org Chem. 2000;65:1280. doi: 10.1021/jo991030r. [DOI] [PubMed] [Google Scholar]
- 11.de Meijere A, Ernst K, Zuck B, Brandl M, Kozhushkov SI, Tamm M, Yufit DS, Howard JAK, Labahn T. Eur. J. Org. Chem. 1999:3105. [Google Scholar]
- 12.Pellicciari R, Marinozzi M, Camaioni E, del Carmen Nnez M, Costantino G, Gasparini F, Giorgi G, Macchiarulo A, Subramanian N. J. Org. Chem. 2002;67:5497. doi: 10.1021/jo020138v. [DOI] [PubMed] [Google Scholar]
- 13.For a leading review on cyclopropanations, see: Lebel H, Marcoux J-F, Molinaro C, Charette AB. Chem. Rev. 2003;103:977. doi: 10.1021/cr010007e. For other reviews on cyclopropanations, see: Doyle MP. Chem. Rev. 1986;86:919. Padwa A, Krumpe KE. Tetrahedron. 1992;48:5385. Calter MA. Curr. Org. Chem. 1997;1:37. Doyle MP, Forbe DC. Chem. Rev. 1998;98:911. doi: 10.1021/cr940066a. Davies HML, Autoulinakis E. Org. React. 2003;57:1. Maas G. Chem. Soc. Rev. 2004;33:183. doi: 10.1039/b309046a. Doyle MP. J. Org. Chem. 2006;71:9253. doi: 10.1021/jo061411m. Brackmann F, de Meijere A. Chem. Rev. 2007;107:4493. doi: 10.1021/cr078376j. Pellissier H. Tetrahedron. 2008;64:7041.
- 14.For some examples, see: Charette AB, Jolicoeur E, Bydlinski GAS. Org. Lett. 2001;3:3293. doi: 10.1021/ol0165140. Lautens M, Delanghe PHM. J. Am. Chem. Soc. 1994;116:8526. Lautens M, Delanghe PHM. Org. Chem. 1993;58:5037. Russo JA, Price WA. J. Org. Chem. 1993;58:3589. Zefirov NS, Kozhushkov SI, Kuznetsova TS, Lukin KA, Kazimirchik IV. Zh. Org. Khim. 1988;24:673. For earlier work, see: Ullman E, Fanshave WJ. J. Am. Chem. Soc. 1961;83:2379. Rahman W, Kuivila HG. J. Org. Chem. 1966;31:772. Noyori R, Takaya H, Nakanisi Y, Nozaki H. Can. J. Chem. 1969;47:1242. (i) Also see reference 4.
- 15.For some examples, see: Gajewski JJ, Burka LT. J. Org. Chem. 1970;35:2190. Trost BM, Bogdanowicz MJ. J. Am. Chem. Soc. 1973;95:5307. Zefirov NS, Kozhushkov SI, Ugrak BI, Lukin KA, Kokoreva OV, Yufit DS, Struchkov YT, Zoellner S, Boese R, Demeijere A. J. Org. Chem. 1992;57:701. Weber M, de Meijere A. Chem. Ber. 1986;118:2450. Akhachinskauya IV, Donskaya NA, Kalykina IV, Oprunenko YF, Shabarov YS. Zh. Org. Khim. 1989;25:1645. Cheng C, Shimo T, Somekawa K, Kawaminami M. Tetrahedron Lett. 1997;52:9005. Muehling O, Wessig P. Photochemical & Photobiological Sciences. 2006;5:1000. doi: 10.1039/b610181j. (h) Also see reference 4.
- 16.For some other examples of amino-spiro[2.2]pentane synthesis, see: Yashin NV, Averina EB, Grishin YK, Kuznetsova TS, Zefirov NS. Synthesis. 2006:279. Applequist DE, Johnston MR, Fisher F. J. Am. Chem. Soc. 1970;92:4614. Konzelma LM, Conley RT. J. Org. Chem. 1968;33:3828.
- 17.Song Z, Lu T, Hsung RP, Al-Rashid ZF, Ko C, Tang Y. Angew. Chem. Int. Ed. 2007;46:4069. doi: 10.1002/anie.200700681. [DOI] [PubMed] [Google Scholar]
- 18.Lu T, Song Z, Hsung RP. Org. Lett. 2008;10:541. doi: 10.1021/ol702824s. [DOI] [PubMed] [Google Scholar]
- 19.For some examples of cyclopropanations of enamides, see: Melby T, Hughes E, Hansen T. Synlett. 2007:2277. Aggarwal VK, Vicente J, Bonnert RV. Org. Lett. 2001;3:2785. doi: 10.1021/ol0164177. Voigt J, Noltemeyer M, Reiser O. Synlett. 1997;202 Jiménez J, Rifé J, Ortuño RM. Tetrahedron Asym. 1996;7:537. Arenare L, De Carprariis P, Marinozzi M, Natalini B, Pellicciari R. Tetrahedron Lett. 1994;35:1425. Akiba T, Tamura O, Hashimoto M, Kobayashi Y, Katoh T, Nakatani K, Kamada M, Hayakawa I, Terashima S. Tetrahedron. 1994;50:3905. Paulini K, Reissig H-U. Liebigs Ann. Chem. 1991:455. Davies HML, Saikali E, Young WB. J. Org. Chem. 1991;56:5696. Seebach D, Stucky G, Pfammatter E. Chem. Ber. 1989;122:2377. Seebach D, Stucky G. Angew. Chem. Int. Ed. 1988;27:1351. Wenkert E, Hudlicky T. J. Org. Chem. 1988;53:1953.
- 20.For reviews of chemistry of enamides, see: Rappoport Z. The Chemistry of Enamines in The Chemistry of Functional Groups. New York: John Wiley and Sons Press; 1994. Whitesell JK, Whitesell MA. Synthesis. 1983;517 Hickmott PW. Tetrahedron. 1982;38:1975–3363. (d) For a review on the synthesis of enamides, see: Tracey MR, Hsung RP, Antoline JE, Kurtz KCM, Shen L, Slafer BW, Zhang Y. In: Science of Synthesis, Houben-Weyl Methods of Molecular Transformations. Steve M, editor. Weinreb, Georg Thieme Verlag KG Press; 2005. Chapter 21.4.
- 21.For leading reviews on amino-cyclopropanes, see: Gnad F, Reiser O. Chem. Rev. 2003;103:1603. doi: 10.1021/cr010015v. Seebach D, Matthews JL. Chem. Commun. 1997:2015. Gellman SH. Acc. Chem. Res. 1998;31:173. Burgess K, Ho KK, Moyesherman D. Synlett. 1994:575. Alami A, Calmes M, Daunis J, Jacquier R. Bull. Soc. Chim. Fr. 1993;130:5.
- 22.For recent examples, see: Faler CA, Joullié MM. Org. Lett. 2007;9:1987. doi: 10.1021/ol0705907. Zeng XZ, Wei XD, Farina V, Napolitano E, Xu YB, Zhang L, Haddad N, Yee NK, Grinberg N, Shen S, Senanayake CH. J. Org. Chem. 2006;71:8864. doi: 10.1021/jo061587o. Begis G, Sheppard TD, Cladingboel DE, Motherwell WB, Tocher DA. Synthesis. 2005:3186.
- 23.For a leading review on allenamides, see: Hsung RP, Wei L-L, Xiong H. Acc. Chem. Res. 2003;36:773. doi: 10.1021/ar030029i.
- 24.For a compendium on the chemistry of allenes, see: Krause N, Hashmi ASK. Modern Allene Chemistry. vol. 1 and 2. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA Press; 2004. For general reviews on the chemistry of allenes, see: Brummond KM, Kent JL. Tetrahedron. 2000;56:3263. Ma S. Chem. Rev. 2005;105:2829. doi: 10.1021/cr020024j.
- 25.For recent reports on allenamide chemistry, see: Skucas E, Zbieg JR, Krische MJ. J. Am. Chem. Soc. 2009;131:5054. doi: 10.1021/ja900827p. Armstrong A, Emmerson DPG. Org. Lett. 2009;11:1547. doi: 10.1021/ol900146s. Beccalli EM, Broggini G, Clerici F, Galli S, Kammerer C, Rigamonti M, Sottocornola S. Org. Lett. 2009;11:1563. doi: 10.1021/ol900171g. Broggini G, Galli S, Rigamonti M, Sottocornola S, Zecchi G. Tetrahedron Lett. 2009;50:1447. Brummond KM, Yan B. Synlett. 2008:2303. Fuwa H, Tako T, Ebine M, Sasaki M. Chem. Lett. 2008;37:904. González-Gómez Á, Añorbe L, Poblador A, Domínguez G, Pérez-Castells J. Eur. J. Org. Chem. 2008:1370. Navarro-Vázquez A, Rodríguez D, Martínez-Esperón MF, García A, Saá C, Domínguez D. Tetrahedron Lett. 2007;48:2741. Fuwa H, Sasaki M. Org. Biomol. Chem. 2007;5:2214. doi: 10.1039/b707338k. Watanabe T, Oishi S, Fuji N, Ohno H. Org. Lett. 2007;9:4821. doi: 10.1021/ol702179n. Hyland CJT, Hegedus LS. J. Org. Chem. 2006;71:8658. doi: 10.1021/jo061340r. Parthasarathy K, Jeganmohan M, Cheng C-H. Org. Lett. 2006;8:621. doi: 10.1021/ol0527936. Fenández I, Monterde MI, Plumet J. Tetrahedron Lett. 2005;46:6029. Alouane N, Bernaud F, Marrot J, Vrancken E, Mangeney P. Org. Lett. 2005;7:5797. doi: 10.1021/ol0522743. de los Rios C, Hegedus LS. J. Org. Chem. 2005;70:6541. doi: 10.1021/jo050813b. Hyland CJT, Hegedus LS. J. Org. Chem. 2005;70:8628. doi: 10.1021/jo051385c. Trost BM, Dylan ST. Org. Lett. 2005;7:2117. doi: 10.1021/ol050395x.
- 26.For our recent efforts, see: Song Z, Hsung RP, Lu T, Lohse AG. J. Org. Chem. 2007;72:9722. doi: 10.1021/jo7017922. Song Z, Hsung RP. Org. Lett. 2007;9:2199. doi: 10.1021/ol070791a. Huang J, Ianni JC, Antoline JE, Hsung RP, Kozlowski MC. Org. Lett. 2006;8:1565. doi: 10.1021/ol0600640. Shen L, Hsung RP. Org. Lett. 2005;7:775. doi: 10.1021/ol047572z. Berry CR, Hsung RP, Antoline JE, Petersen ME, Rameshkumar C, Nielson JA. J. Org. Chem. 2005;70:4038. doi: 10.1021/jo0503411. Shen L, Hsung RP, Zhang Y, Antoline JE, Zhang X. Org. Lett. 2005;7:3081. doi: 10.1021/ol051094q. Huang J, Hsung RP. J. Am. Chem. Soc. 2005;127:50. doi: 10.1021/ja044760b.
- 27.For [4 + 3] cycloadditions, see: Antoline JE, Hsung RP. Synlett. 2008:739. Antoline JE, Hsung RP, Huang J, Song Z, Li G. Org. Lett. 2007;9:1275. doi: 10.1021/ol070103n. Xiong H, Hsung RP, Berry CR, Rameshkumar C. J. Am. Chem. Soc. 2001;123:7174. doi: 10.1021/ja0108638. Huang J, Hsung RP. J. Am. Chem. Soc. 2005;127:50. doi: 10.1021/ja044760b. Rameshkumar C, Hsung RP. Angew. Chem. Int. Ed. 2004;43:615. doi: 10.1002/anie.200352632. Xiong H, Huang J, Ghosh S, Hsung RP. J. Am. Chem. Soc. 2003;125:12694. doi: 10.1021/ja030416n. For hetero-[4 + 2] cycloadditions, see: Berry CR, Hsung RP. Tetrahedron. 2004;60:7629. Wei L-L, Hsung RP, Xiong H, Mulder JA, Nkansah NT. Org. Lett. 1999;1:2145. For epoxidations, see: Rameshkumar C, Xiong H, Tracey M, Berry CR, Yao LJ, Hsung RP. J. Org. Chem. 2002;67:1339. doi: 10.1021/jo011048d.
- 28.Without citing specific examples, for leading reviews, see: Rubin M, Rubina M, Gevorgyan V. Chem. Rev. 2007;107:3117. doi: 10.1021/cr050988l. Brandi A, Cicchi S, Cordero FM, Goti A. Chem. Rev. 2003;103:1213. doi: 10.1021/cr010005u.
- 29.See Supporting Information.Crystallographic data for 9a: C14H15NO2, M = 229.27, triclinic, P1, a = 6.2295(4), b = 11.8164(7), c = 12.8744(8) Å, α = 84.5470(10), β = 86.5820(10), γ = 76.4120(10)°, V = 916.30(10) Å3, T = 173(2) K, Z = 3, µ = 0.083 mm−1, 3730(Rint=0.0242), final R indices[ I > 2σ(I)], R1 = 0.0320, wR2 = 0.0732, R indeces (all data) R1 = 0.0424, wR2 = 0.0793.Crystallographic data for 9b: C14H15NO2, M = 229.27, orthorhombic, P212121, a = 6.4331(18), b = 11.657(3), c = 15.860(4) Å, α = 90, β = 90, γ = 90 °, V = 1189.4(6) Å3, T = 173(2) K, Z = 4, µ = 0.086 mm−1, 1265(Rint=0.0431), final R indices[ I > 2σ (I)], R1 = 0.0329, wR2 = 0.0756, R indeces (all data) R1 = 0.0416, wR2 = 0.0815.Crystallographic data for E-19: C17H15NO2, M = 265.30, orthorhombic, P212121, a = 6.0167(5), b = 10.2379(9), c = 22.355(2) Å, α = 90, β = 90, γ = 90 °, V = 1377.0(2) Å3, T = 173(2) K, Z = 4, µ = 0.084 mm−1, 1790(Rint=0.0439), final R indices[ I > 2σ(I)], R1 = 0.0329, wR2 = 0.0761, R indeces (all data) R1 = 0.0460, wR2 = 0.0803.Crystallographic data for Z-19: C17H15NO2, M = 265.30, monoclinic, C2, a = 30.294(8), b = 6.4077(17), c = 7.0525(19) Å, α = 90, β = 100.238(4), γ = 90 °, V = 1347.2(6) Å3, T = 173(2) K, Z = 4, µ = 0.086 mm−1, 1492(Rint=0.0503), final R indices[ I > 2σ(I)], R1 = 0.0394, wR2 = 0.0868, R indeces (all data) R1 = 0.0553, wR2 = 0.0922.Crystallographic data for 31M: C20H19NO2, M = 305.36, monoclinic, P21/n, a = 10.3033(13), b = 10.2448(13), c = 15.332(19) Å, α = 90, β = 95.088(2), γ = 90 °, V = 1612.0(4) Å3, T = 173(2) K, Z = 4, µ = 0.081 mm−1, 3678(Rint=0.0269), final R indices[ I > 2σ(I)], R1 = 0.0415, wR2 = 0.1043, R indeces (all data) R1 = 0.0584, wR2 = 0.1178.Crystallographic data for 37B: C18H23NO2, M = 285.37, monoclinic, P21/n, a = 6.2307(11), b = 16.094(3), c = 8.0544(15) Å, α = 90, β = 96.906(2), γ = 90 °, V = 801.8(3) Å3, T = 173(2) K, Z = 2, µ = 0.076 mm−1, 1704(Rint=0.0328), final R indices[ I > 2σ(I)], R1 = 0.0342, wR2 = 0.0811, R indeces (all data) R1 = 0.0425, wR2 = 0.0861
- 30.Denmark SE, Edwards JP. J. Org. Chem. 1991;56:6974. [Google Scholar]
- 31.(a) Furukawa J, Kawabata N, Nishimura J. Tetrahedron Lett. 1966;7:3353. [Google Scholar]; (b) Furukawa J, Kawabata N, Nishimura J. Tetrahedron. 1968;24:53. [Google Scholar]
- 32.Yang Z, Lorenz JC, Shi Y. Tetrahedron Lett. 1998;39:8621. [Google Scholar]
- 33.Maruoka K, Fukutani Y, Yamamoto H. J. Org. Chem. 1985;50:4412. [Google Scholar]
- 34. Molander GA, Harring LS. J. Org. Chem. 1989;54:3525. Molander GA, Etter JB. J. Org. Chem. 1987;52:3942. Also see: Lautens M, Ren Y. J. Org. Chem. 1996;61:2210.
- 35.(a) Sibi MP, Deshpande PK, Ji JG. Tetrahedron Lett. 1995;36:8965. [Google Scholar]; (b) Sibi MP, Ji JG. Angew. Chem., Int. Ed. Engl. 1996;35:190. [Google Scholar]; (c) Sibi MP, Porter NA. Acc. Chem. Res. 1999;32:163. [Google Scholar]
- 36.For a leading reference, see: Hintermann T, Seebach D. Helv. Chim. Acta. 1998;81:2093.
- 37.Close WJ. J. Org. Chem. 1950;15:1131. [Google Scholar]
- 38.Xiong H, Hsung RP, Shen L, Hahn JM. Tetrahedron Lett. 2002;43:4449. [Google Scholar]
- 39.Adam independently reported their work thereafter, see: Adam W, Bosio SG, Wolff BT. Org. Lett. 2003;5:819. doi: 10.1021/ol0273194. For subsequent work with Turro, see: Sivaguru J, Saito H, Poon T, Omonuwa T, Franz R, Jockusch S, Hooper C, Inoue Y, Adam W, Turro NJ. Org. Lett. 2005;7:2089. doi: 10.1021/ol0502230. and references cited therein.
- 40.Tracey MR, Grebe TP, Brennessel WW, Hsung RP. Acta Cryst. 2004;C60:o830. doi: 10.1107/S0108270104023121. [DOI] [PubMed] [Google Scholar]
- 41.Xiong H, Hsung RP, Wei L-L, Berry CR, Mulder JA, Stockwell B. Org. Lett. 2000;2:2869. doi: 10.1021/ol000181+. [DOI] [PubMed] [Google Scholar]
-
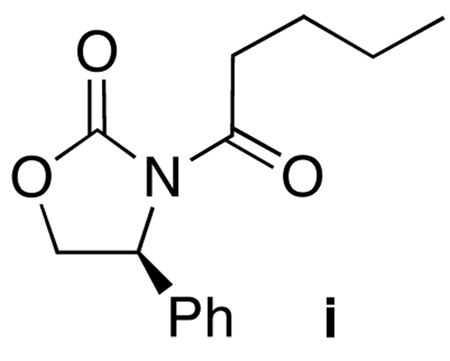
42.An unexpected oxidative product i [see below] was found in the reaction mixture. Compound i was also seen during the purification of chiral allenamide 33, but we are not certain of its origin at this point.

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.