

Abstract
AIMS
To evaluate the pharmacokinetic interactions between ritonavir and quinine in healthy volunteers.
METHODS
Ten healthy volunteers were each given 600-mg single oral doses of quinine alone, ritonavir alone (200 mg every 12 h for 9 days), and quinine in combination with ritonavir, in a three-period pharmacokinetic nonrandomized sequential design study. Quinine was co-administered with the 15th dose of ritonavir. Blood samples collected at predetermined time intervals were analysed for ritonavir, quinine and its major metabolite, 3-hydroxyquinine, using a validated high-performance liquid chromatography method.
RESULTS
Concurrent ritonavir administration resulted in about fourfold increases in both the Cmax and AUCT[Cmax 2.79 ± 0.22 vs. 10.72 ± 0.32 mg l−1, 95% confidence interval (CI) 7.81, 8.04; AUC 50.06 ± 2.52 vs. 220.47 ± 6.68 mg h−1 l−1, 95% CI 166.3, 175.3], a significant increase (P < 0.01) in the elimination half-life (11.15 ± 0.80 vs. 13.37 ± 0.33 h, 95% CI 1.64, 2.77) and about a 4.5-fold decrease in CL/F (12.01 ± 0.61 vs. 2.71 ± 0.09 l h−1) of quinine. Also, with ritonavir, there was a pronounced reduction of AUC(metabolite)/AUC(unchanged drug) ratio of quinine (1.35 ± 0.10 vs. 0.13 ± 0.02) along with a marked decrease in Cmax (1.80 ± 0.12 vs. 0.96 ± 0.09 mg l−1) and AUC0–48h (62.80 ± 6.30 vs. 25.61 ± 2.44 mg h−1 l−1) of the metabolite. Similarly, quinine caused modest but significant increases (P < 0.01) in the Cmax, AUC and elimination T½ of ritonavir.
CONCLUSIONS
Downward dosage adjustment of quinine appears necessary when concurrently administered with ritonavir.
Keywords: 3-hydroxyquinine, pharmacokinetic interaction, quinine, ritonavir
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Malaria is widespread across some areas of the world, most of which also bear the brunt of the human immunodeficiency virus (HIV) pandemic, resulting in a high incidence of co-infection of both diseases.
Ritonavir, a HIV protease inhibitor, and quinine, an antimalarial agent effective against multidrug-resistant Plasmodium falciparum, are likely to be administered concurrently for treatment of patients with HIV and malaria.
Both drugs are metabolized to a significant extent by CYP3A4 and ritonavir is a potent inhibitor of this enzyme.
WHAT THIS STUDY ADDS
With increasing access to antiretroviral drugs, it is important that potential interactions between therapies for HIV and malaria infections are investigated.
In this study, concurrent administration of ritonavir with quinine was found to be associated with marked elevation in the plasma levels of the antimalarial and a pronounced decrease in plasma concentrations of 3-hydroxyquinine, the major metabolite of quinine.
There was also a modest but significant increase (P < 0.05) in plasma concentrations of ritonavir in the presence of quinine.
Introduction
Malaria is widespread across areas of the world where resources are limited, and most of these areas also bear the brunt of the human immunodeficiency virus (HIV) pandemic. Therefore, there is a wide geographical overlap in occurrence of both diseases, resulting in high incidence of co-infection [1, 2]. With access to antiretroviral drugs increasing, it is important that potential interactions between therapies of these two infections are investigated. Quinine is available in oral and injectable formulations and has tolerable side-effects if used correctly and at the normal therapeutic doses. Development of quinine resistance in Plasmodium falciparum has been relatively slow and incomplete by comparison with those of the other principal antimalarial drugs (e.g. chloroquine, mefloquine, and sulphadoxine-pyrimethamine), and resistance to quinine is rare in Africa [3]. Parenteral quinine is the drug of first choice for severe malaria, and oral quinine is an option for uncomplicated malaria where multidrug resistance is a problem [4]. Although there have been concerns that the efficacy of quinine is declining in some parts of South East Asia, a series of trials to find drugs that are suitable alternatives to quinine has demonstrated that the drug is as effective as artemisinin derivatives, artesunate and artemether [5].
Protease inhibitors such as ritonavir contribute to the improved health of HIV+ individuals, and their inclusion in antiretroviral regimens is commonplace. However, protease inhibitors are often involved in clinically important drug interactions resulting from alteration of cytochrome P450 metabolism [6].
Ritonavir is primarily metabolized by the CYP3A4 isoenzyme and it has a high binding affinity to P-glycoprotein (P-gp) [7]. The drug is also a potent inhibitor of CYP3A4-mediated metabolism and a modest agent for blocking P-gp binding [7, 8]. Studies have shown that ritonavir causes elevation of plasma concentrations of a number of CYP3A4 and P-gp substrates including saquinavir and ketoconazole [7, 9, 10]. Since quinine is mainly metabolized by the same isoenzyme and is also a substrate of P-gp [11], it is plausible for an interaction to occur between these two drugs. Inhibition of quinine metabolism has the potential of increasing the risk of cardiotoxicity of the drug, as was observed in an overlapping treatment with artemether-lumefantrine and quinine, which was reported to result in prolongation of the QTc interval [12]. The metabolism of both lumefantrine and artemether are known to be mediated by CYP3A4 [13]. On the other hand, since ritonavir and quinine are substrates for the same drug-metabolizing enzyme, there is also a possibility for quinine to competitively displace ritonavir from the metabolic site, with resultant increased ritonavir plasma levels and attendant increased risk of toxicity. However, the magnitude and clinical significance of these potential interactions need elucidation since the interaction of ritonavir and CYP enzymes is complex, as it autoinduces its own metabolism on chronic administration [14].
The present study was therefore aimed at determining the bi-directional pharmacokinetic interactions between ritonavir and quinine following co-administration of multiple doses of ritonavir and single doses of quinine. Such a study was deemed important since ritonavir and quinine are likely to be administered concurrently for treatment of patients with HIV and malaria.
Methods
Subjects
Healthy non-smoking men and women who were within 20% of their ideal body weight for height and gender (Metropolitan Life Scale) were regarded eligible to participate in the study. Subjects were excluded if they met one of the following criteria: pregnancy, breast feeding, history of hypersensitivity reactions to protease inhibitors, quinine or similar agents (mefloquine, chloroquine, quinidine, quinolones), serum creatinine >1.5 times the upper limit of normal, liver function test more than three times the upper limit of normal, or evidence by history or physical examination of gastrointestinal, psychiatric, cardiovascular or neurological disorders. Electrocardiograms were also recorded. All subjects gave written informed consent, and the study protocol received the approval of the Obafemi Awolowo University Teaching Hospitals Research Ethics Board and Radiation Safety Committee. Six men and four women who were all certified healthy by a doctor (A.R.O.) in the team completed the study. The mean ± SD age and weight of the subjects were 26.6 ± 2.3 years (range 22–29) and 65.4 ± 4.0 kg (range 60–71), respectively. None of them had received any other drugs for at least 1 month before the study.
Study design, drug administration and blood sampling
This was an open-label, three-period, pharmacokinetic, nonrandomized sequential design study. A scheme depicting the study design is presented in Figure 1. In period 1, after an overnight fast, single oral doses of 600 mg quinine sulphate (two tablets of 300 mg quinine sulphate; Wockhardt, UK Ltd, Wrexham, UK) were given to each of the 10 volunteers. Blood samples (5 ml) were withdrawn by venepuncture from the forearm of each subject prior to and at 1, 2, 4, 6, 8, 12, 24, 36 and 48 h after drug administration into heparinized tubes, for baseline quinine pharmacokinetics evaluation. The blood samples were immediately centrifuged (1500 g for 10 min) to separate plasma, which was stored at −20°C until analysis for quinine and 3-hydroxyquinine. In period 2, which was after a wash-out period of 3 weeks, each of the volunteers received oral doses of 200 mg ritonavir (two capsules of Medavir® 100 mg; Cipla Ltd, Mumbai, India) every 12 h for 13 doses. Blood samples (5 ml) were taken just before the 13th dose (i.e. on day 7) and over 12 h (at 1, 2, 4, 6, 8 and 12 h) within the dosing interval, for ritonavir baseline pharmacokinetics. The blood samples were drawn into heparinized tubes, centrifuged (1500 g) for 10 min and plasma was stored at −20°C until analysis for ritonavir. In period 3, without allowing a wash-out period, subjects continued on the 200 mg ritonavir every 12 h for five additional doses. A single 600-mg quinine sulphate oral dose was given concurrently with the 15th ritonavir dose (day 8) to each of the subjects, and blood samples were collected over 48 h for evaluation of pharmacokinetics of quinine and ritonavir in the presence of each other. No other drugs or alcohol were allowed to be taken throughout the duration of the study.
Figure 1.
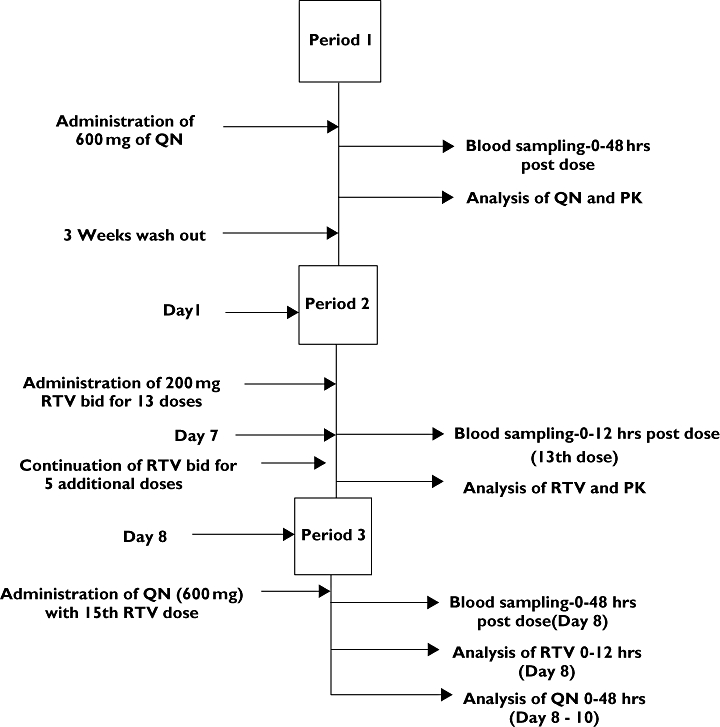
Scheme depicting the study design
Drug analysis
The concentrations of ritonavir, quinine and its major metabolite, 3-hydroxyquinine, in plasma were determined using a new ion-pair reversed-phase high-performance liquid chromatographic method recently developed in our laboratories [15] for simultaneous analysis of the three compounds in human plasma. This involved simple extraction with diethyl-ether under alkaline conditions. Chromatographic separation was achieved on a 5-µm particle size C-18 column (200 × 4.6 mm i.d.) using a mobile phase consisting of methanol:acetonitrile:0.02 M potassium dihydrogen phosphate (15:10:75) containing 75 mmol l−1 perchloric acid (pH 2.8), which was pumped through the column at a flow rate of 1.0 ml min−1. Retention times for ritonavir, 3-hydroxyquinine, quinine and the internal standard (pyrimethamine) were 2.8, 4.0, 7.0 and 12 min, respectively. The limits of detection and validated lower limits of quantification were 10 and 12.5 ng ml−1 for ritonavir while the corresponding values were 5 and 70 ng ml−1 for both quinine and 3-hydroxyquinine, respectively. The coefficients of variation for both the intraday and interday analysis ranged from 1.10 to 8.42% for quinine, 2.75 to 9.20% for 3-hydroxyquinine, and 1.53 to 2.20% for ritonavir. The absolute recovery was >90% for the three compounds. Our laboratory does not participate in an external quality assessment programme for ritonavir or quinine.
Data and statistical analysis
The peak plasma concentrations (Cmax) and time to reach peak concentration (Tmax) were noted directly from the concentration vs. time profiles of quinine, 3-hydroxyquinine and ritonavir. Other pharmacokinetic parameters were calculated from individual plasma concentration vs. time profiles, employing standard noncompartmental methods [16]. For example, the total area under plasma concentration vs. time curve (AUCT) was determined using the linear trapezoidal rule to the last datum and with extrapolation to infinity. The area from the last datum point (Ct) to infinity was obtained as Ct/β. β, the elimination rate constant, was calculated by linear regression analysis of the terminal phase of the log concentration–time profile. The elimination half-life (T1/2β) and oral plasma clearance (CL/F) were evaluated from the ratios of 0.693/β and dose/AUCT, respectively. The pharmacokinetic calculations were done using the pharmacokinetic program package WinNonlin Standard Edition, Version 1.5 (Scientific Consultant Inc., Apex, NC, USA). In the model option for the noncompartmental analysis, the linear trapezoidal rule was used for calculation of the area under the curve (AUC).
The Wilcoxon matched pairs signed ranked test was used to evaluate the difference between any pair of data, i.e. effects of quinine and ritonavir on the pharmacokinetics of each other, or the effect of ritonavir on 3-hydroxyquinine disposition. Results were recorded as mean ± SD. In all, a value of P < 0.05 was considered statistically significant.
Results
Figure 2 shows the mean ± SD plasma concentration vs. time profiles of quinine and 3-hydroxyquinine following oral administration of single doses of 600 mg of quinine sulphate alone, and with concurrent ritonavir, to each of 10 volunteers. Some derived pharmacokinetic parameters of quinine following administration of the drug alone or with ritonavir are presented in Table 1. Co-administration of ritonavir was associated with marked increases (P < 0.001) in the Cmax and AUCT of quinine compared with the values obtained following administration of the antimalarial alone (Table 1). The results show that the Cmax, AUCT and elimination T1/2 of quinine increased by 284% [95% confidence interval (CI) 280, 288], 341% (95% CI 332, 350) and 20% (95% CI 15, 25), respectively, in the presence of ritonavir. Also, the oral plasma clearance (CL/F) of quinine was highly reduced with concurrent ritonavir administration by about 77% (95% CI 74, 81). However, the values of Tmax were comparable, with or without ritonavir (P > 0.1).
Figure 2.
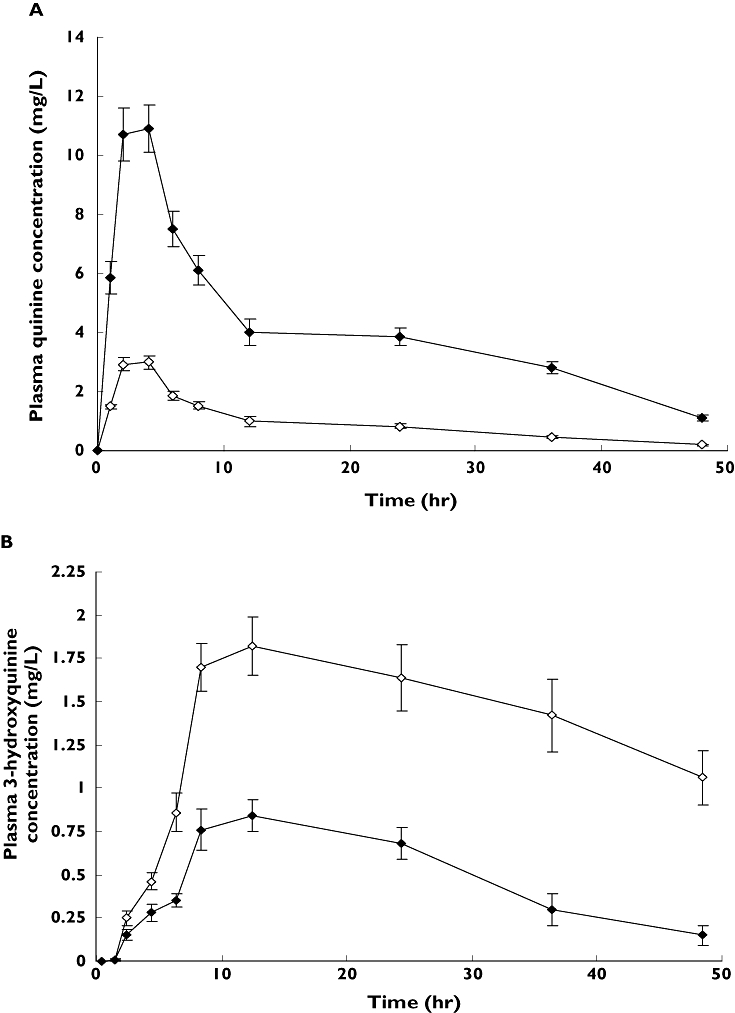
Mean (±) SD plasma concentration vs. time profiles of quinine (a) and 3-hydroxyquinine (b) following administrations of a single 600-mg oral dose of quinine sulphate with or without concurrent multiple oral doses of ritonavir (200 mg 12 hourly for 9 days) to each of 10 healthy volunteers. The quinine dose was co-administered with the 15th dose (day 8) of ritonavir. (A) Quinine alone ( ); Quinine with Ritonavir (
); Quinine with Ritonavir ( ); (B) 3-hydroxyquinine alone (
); (B) 3-hydroxyquinine alone ( ); 3-hydroxyquinine in presence of ritonavir (
); 3-hydroxyquinine in presence of ritonavir ( )
)
Table 1.
Mean pharmacokinetic parameters of quinine and its major metabolite (3-hydroxyquinine) following administration of 600 mg single oral doses of quinine sulphate alone or with multiple doses of ritonavir (200 mg 12 hourly for 9 days) to each of 10 healthy volunteers
| Pharmacokinetic parameter | Quinine alone | Quinine with ritonavir | Mean difference (95% confidence interval of mean difference) |
|---|---|---|---|
| Quinine | 3.20 (2–4) | 3.40 (2–4) | 0.2 (−0.25, 0.65) |
| Tmax (h) | 2.79 ± 0.22 | 10.72 ± 0.32 | 7.92 (7.81, 8.04)* |
| Cmax(mg l−1) | 11.15 ± 0.80 | 13.32 ± 0.33 | 2.21 (1.64, 2.77)* |
| T1/2β (h) | 50.06 ± 4.01 | 220.47 ± 6.68 | 170.78 (166.3, 175.3)* |
| AUCT(mg h−1 l−1) | 12.01 ± 0.61 | 2.71 ± 0.10 | −9.30 (−9.71, −8.89)* |
| CL/F (l h−1) | 8.80 (8–12) | 9.20 (8–12) | 0.40 (−0.51, 1.31) |
| 3-hydroxyquinine | 1.80 ± 0.12 | 0.96 ± 0.09 | −0.84 (−0.94, −0.73)* |
| Tmax (h) | 62.80 ± 6.30 | 25.61 ± 2.44 | −37.49 (−42.12, −32.83)* |
| Cmax (mg l−1) | 1.35 ± 0.10 | 0.13 ± 1.01 | −1.22 (−1.34, −1.11)* |
| AUC0–48h (mg h−1 l−1) | |||
| MR |
Statistically significant (P < 0.01); the pharmacokinetic parameters are as described in the text. Quinine was co-administered with the 15th dose (day 8) of ritonavir. MR, metabolic ratio.
The pharmacokinetic parameters of 3-hydroxyquinine following administration of quinine, with and without ritonavir, are also shown in Table 1. The Cmax and AUC0–48h of the metabolite were significantly diminished in the presence of ritonavir (P < 0.01). The Cmax reduced by 50% (95% CI 41, 52) while AUC0–48h reduced by 60% (95% CI 52, 67). Co-administration of ritonavir also resulted in a pronounced decrease in the ratio of the AUC of quinine metabolite to that of unchanged drug (metabolic ratio) by about 90% (95% CI 82, 99). There was no significant change (P > 0.1) in the Tmax of the metabolite.
The mean ± SD plasma concentration vs. time profiles of ritonavir following oral administration of multiple doses of the drug alone, and with quinine, to each of 10 volunteers are shown in Figure 3, while some of the derived pharmacokinetic parameters of the drug are presented in Table 2. Concurrent quinine administration caused modest but significant increases (P < 0.05) in the Cmax (15% increase; 95% CI 10, 20) and AUC (21% increase; 95% CI 10, 32) of ritonavir. There was also a considerable decrease in plasma clearance of ritonavir evidenced by the elimination half-life, which increased by 32% (95% CI 20, 44). The Cmin, which was the concentration at the end of ritonavir dosing interval of 12 h, showed the highest increase of about 66% (40–72%), while the Tmax values were not significantly altered (P > 0.05).
Figure 3.
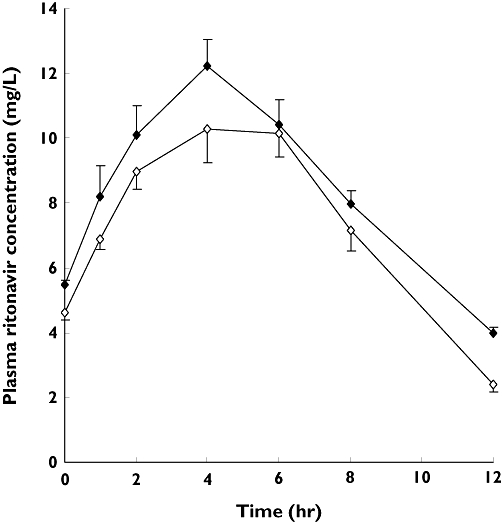
Mean (±) SD plasma concentration vs. time profiles of ritonavir following administration of multiple doses of the drug alone (200 mg 12 hourly for 9 days) or with concurrent single oral doses of quinine (600 mg) to each of 10 healthy volunteers. Quinine was co-administered with the 15th dose (day 8) of ritonavir. Ritonavir alone ( ); Ritonavir in presence of quinine (
); Ritonavir in presence of quinine ( )
)
Table 2.
Some derived mean pharmacokinetic parameters of ritonavir following administration of multiple doses of the drug alone (200 mg 12 hourly for 9 days) or with concurrent single oral doses of quinine (600 mg) to each of 10 healthy volunteers
| Pharmacokinetic parameter | Ritonavir alone | Ritonavir with quinine | Mean difference (95% confidence interval of mean difference) |
|---|---|---|---|
| Tmax (h) | 4.20 (4–6) | 4.0 (2–6) | −0.2 (−0.65, 0.25) |
| Cmax (mg l−1) | 10.35 ± 0.78 | 11.87 ± 0.73 | 1.51 (0.98, 2.05)* |
| Cmin (mg l−1)† | 2.40 ± 0.23 | 3.98 ± 0.44 | 1.58 (0.96, 1.73)* |
| T1/2β (h) | 3.11 ± 0.27 | 4.10 ± 0.64 | 0.996 (0.61, 1.38)* |
| AUC (mg h−1 l−1) | 102.88 ± 5.39 | 124.47 ± 12.44 | 21.59 (30.32, 32.86)* |
Quinine was co-administered with the 15th dose (day 8) of ritonavir.
Statistically significant (P < 0.01).
The concentration at the end of ritonavir dosing interval. The pharmacokinetic parameters are as described in the text.
Discussion
There is a rapid increase in access to antiretroviral therapy in developing countries. However, there is gap in current knowledge that defines interactions with other drugs, especially antimalarial drugs. There is very sparse information on interactions between antimalarial and antiretroviral drugs, including ritonavir. A study has been reported on the pharmacokinetic interaction between mefloquine and ritonavir, where it was observed that the disposition of mefloquine was not significantly altered by multiple dose administration of ritonavir, but the presence of mefloquine reduced the AUC and Cmax of ritonavir by 31 and 36%, respectively [17]. There are no published data on the pharmacokinetic interaction between quinine and ritonavir.
Results from the present study indicate that quinine is rapidly absorbed after oral administration, with an average Tmax of 2–4 h. The pharmacokinetic parameters obtained for quinine when administered alone, such as Tmax, elimination T1/2, CL/F and AUCT, are in general agreement with the findings of other workers [3, 18–21]. This study has demonstrated significant increases in Cmax (a fourfold increase), elimination T1/2 (a 20% increase) and AUCT (a fourfold increase) of quinine following concurrent administration of ritonavir (Figure 1 and Table 1). These indicate a very significant alteration of quinine disposition by ritonavir. Since the major metabolic pathway of quinine is through CYP3A4 subfamily [11], inhibition of the isoenzyme by ritonavir could account for the increased plasma quinine levels. Inhibition of CYP3A4 by ritonavir leading to elevated plasma drug concentrations as well as increased elimination half-life of CYP3A4 substrates have been frequently reported. For example, ritonavir practically completely inhibited the formation of 14-(R)–hydroxyclarithromycin from clarithromycin [22]. The capacity-limited formation of 14-(R)–hydroxyclarithromycin has been shown to be effected by CYP3A4 in human liver microsomes [23]. The AUC for clarithromycin increased by 77% with concomitant ritonavir, and the mean terminal half-life increased form 5 to 14 h [22]. Ritonavir also substantially increased the plasma concentrations of the rifabutin metabolite, 25-O-desacetyl-rifabutin, whose elimination is apparently mediated by CYP3A4 metabolism [24]. Inhibition of CYP3A4 in gastrointestinal (GI) tract enterocytes has the potential also to contribute to increased plasma levels of CYP3A4 substrates. However, studies have shown that grapefruit juice, a potent inhibitor of intestinal CYP3A4, does not affect the disposition of quinine [25]. This suggests that reduction of quinine metabolism is principally through inhibition of CYP3A4 located in the liver and not in the GI tract. In considering other possible mechanisms for quinine–ritonavir interaction, the role of P-gp, a transmembrane protein associated with a phenotype of multidrug resistance, requires attention. There is considerable overlap in substrate specificities with respect to CYP3A4 metabolism and P-gp transport [26, 27]. All protease inhibitors are substrates and inhibitors of P-gp, with ritonavir being the most potent inhibitor of P-gp among the protease inhibitors [7, 8, 28], while quinine had been confirmed as a substrate for P-gp [29]. Therefore, there is a theoretical possibility that P-gp may play a role in quinine–ritonavir interaction at the GI tract wall. However, since quinine is normally well absorbed after oral administration, with a bioavailability of >80% [21, 30], inhibition of intestinal P-gp can not account for the marked increase in the plasma drug levels obtained with concurrent administration of ritonavir. Hence, inhibition of hepatic metabolism must have been the major site of the drug interaction. The marked decreases in the Cmax and AUC0–48h of the major metabolite of quinine (Table 1) lend credence to attributing the elevated plasma quinine levels to inhibition of metabolism of the drug. This assertion is further supported by the occurrence of a pronounced reduction in the ratio of AUC of metabolite to that of unchanged drug, in the presence of ritonavir. This parameter, AUC(metabolite)/AUC(unchanged drug), called metabolic ratio, is a known measure of extent of metabolism of a drug. Since quinine has a narrow therapeutic window [31], the fourfold increases in Cmax and AUCT of the drug by ritonavir would obviously be associated with toxic manifestations of quinine following therapeutic drug administration.
A single 600-mg oral dose of quinine was used in this study so as to minimize adverse reactions that would result from the anticipated increased plasma concentrations of the drug with concurrent administration of ritonavir. For example, a study that investigated amodiaquine pharmacokinetics following co-administration of efavirenz (using multiple doses of amodiaquine) in HIV+ subjects had to be terminated after the first two subjects developed asymptomatic but significant elevations of liver transaminases. Addition of efavirenz increased amodiaquine AUC by 114% in one subject and by 302% in another [32].
This study also investigated the effect of quinine on the pharmacokinetics of ritonavir. The dose of ritonavir selected in the present study was lower than the therapeutic dosage of the drug (600 mg twice daily), due to the apparent lower tolerability of ritonavir in normal volunteers. In addition, ritonavir is now commonly used in combination with other protease inhibitors at a booster dose of 100 or 200 mg [33]. However, the low ritonavir dose still provided strong inhibition of the metabolism of quinine, as it does for other drugs including other protease inhibitors [7, 34]. It has recently been demonstrated that ritonavir produces maximum inhibition of CYP3A4 activity at a dose level of 100 mg [35]. Hence, the dose of 200 mg twice daily as used in the present study would not be expected to result in increased inhibition of quinine metabolism compared with the effect of a 100 mg twice daily regimen. The duration of treatment was adequate to achieve steady state for ritonavir, based on earlier findings that ritonavir auto-induction is minimal at a low dose of 200 mg every 12 h and a steady state is achieved by 1 week with this regimen [14, 36]. A study that investigated the pharmacokinetic interaction between mefloquine and ritonavir in healthy volunteers also dosed ritonavir for 7 days [17].
Studies have shown that none of the known CYP3A4 substrates or inhibitors (e.g. ketoconazole, troleandomycin and 17α-ethinyloestradiol) was able to completely inhibit ritonavir metabolism even at high concentrations [37]. Consistent with the findings in vitro, the present study indicated modest increases (P < 0.05) in the mean values of Cmax, AUC and elimination half-life of ritonavir with concomitant administration of quinine (Table 2). Because of its high affinity to CYP3A4, the metabolism of ritonavir is generally minimally affected by CYP3A4 substrates and inhibitors. For example, following concurrent administration, the AUC of ritonavir was minimally increased by efavirenz (18%) [38], fluconazole (12%) [39] and clarithromycin (12%) [22]. Also, since ritonavir is known to be partially metabolized by CYP2D6 [40] while quinine is an inhibitor of this isozyme [41], the slightly elevated ritonavir concentrations due to quinine co-administration may be partly due to inhibition of CYP2D6 by quinine. Although concurrent administration of a single dose of quinine is associated with a small degree of increase in the plasma levels of ritonavir, the effect of multiple doses of the antimalarial on ritonavir plasma concentrations may be more pronounced. This is because, since competitive displacement from CYP3A4 and inhibition of CYP2D6 mainly mediate the interaction between ritonavir and quinine, a greater effect on changes in the disposition of ritonavir would be expected to result from increased plasma quinine levels. Therefore, there is a possibility of greater inhibition of ritonavir metabolism when multiple doses of quinine are co-administered.
The clinical significance of this study would have been enhanced by investigating the interaction of quinine with low-dose ritonavir (100 mg b.i.d.) in combination with other protease inhibitors as applied in the treatment of HIV-infected patients. The use of only ritonavir is a limitation of the study.
This study was carried out in healthy volunteers, and it is pertinent to note that changes in quinine pharmacokinetics occur in patients with malaria. In malaria, there is an elevated level of α1-acid glycoprotein that is proportional to the severity of the infection. The raised concentrations of this protein account for increased plasma protein binding of quinine with resultant increased plasma quinine levels in malaria [42, 43]. Since the unbound fraction of quinine in plasma is lower in patients with malaria, the degree of interaction between the drug and ritonavir in these patients might be different from that observed in healthy subjects.
In conclusion, this study has demonstrated that concurrent administration of ritonavir, a known inhibitor of CYP3A4, with quinine, a substrate of the same isoenzyme, results in marked increases in plasma levels of the antimalarial, whereas the plasma concentrations of 3-hydroxyquinine, the major metabolite of quinine, are remarkably diminished. The high magnitude of elevation of the plasma concentrations of quinine, with its potential adverse effects, suggests the need for downward adjustment of the dosage of the drug when given concurrently with ritonavir. Similarly, quinine caused modest but statistically significant increases in ritonavir plasma levels that might not warrant dosage adjustment of the protease inhibitor when used at a booster dose.
Competing interests
None to declare.
This work was supported by Obafemi Awolowo University, Ile-Ife, Nigeria, Research Grant No. 11813 AEC.
REFERENCES
- 1.World Health Organization. Malaria and HIV/AIDS interactions and implications: conclusions of a technical consultation convened by WHO. 23–25 June 2004. Available at: http://www.who.int/malaria/malaria_HIV/malaria_hiv_flyer.pdf.
- 2.Grinwade K, French N, Mbatha DD, Zungu DD, Dedicoat M, Gilks CF. HIV infection as a cofactor for severe falciparum malaria in adults living in a region of unstable malaria transmission in South Africa. AIDS. 2004;18:547–9. doi: 10.1097/00002030-200402200-00023. [DOI] [PubMed] [Google Scholar]
- 3.Adam I, Ali DM, Noureldin W, Elbashir MI. Quinine for treatment of chloroquine-resistant falciparum malaria in pregnant and non-pregnant Sudanese women. Ann Trop Med Parasitol. 2005;99:427–9. doi: 10.1179/136485905X36217. [DOI] [PubMed] [Google Scholar]
- 4.Sanjeev K, Nelamangala V, Tim P, Tsiri A, Peter W. Population pharmacokinetics of intramuscular quinine in children with severe malaria. Antimicrob Agents Chemother. 2001;45:1803–9. doi: 10.1128/AAC.45.6.1803-1809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.PrayGod G, Albie F, Eisenhurt M. Artemisinin derivative versus quinine in treating severe malaria in children: a systematic review. Malar J. 2008;7:210. doi: 10.1186/1475-2875-7-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barry M, Gibbons S, Mulcahy F, Back D. Protease inhibitors in patients with HIV disease: clinically important pharmacokinetic considerations. Clin Pharmacokinet. 1997;32:194–209. doi: 10.2165/00003088-199732030-00003. [DOI] [PubMed] [Google Scholar]
- 7.Piscitelli SC, Allicano KDG. Interactions among drugs for HIV and opportunistic infections. N Engl J Med. 2001;344:984–96. doi: 10.1056/NEJM200103293441307. [DOI] [PubMed] [Google Scholar]
- 8.Profit L, Eagling VA, Back DJ. Modulation of P-glycoprotein function in human lymphocytes and Caca-2 cells monolayers by HIV-1 protease inhibitors. AIDS. 1999;13:1623–7. doi: 10.1097/00002030-199909100-00004. [DOI] [PubMed] [Google Scholar]
- 9.Jorga K, Buss NE. Pharmacokinetic drug interaction with saquinavir soft gelatin capsule. Program and abstracts of the 39th Interscience Conference on Antimicrobial Agents and Chemotherapy, San Francisco, September 26–29, 1999. Washington, DC: American Society for Microbiology; 1999: 20. Abstract.
- 10.Khaliq Y, Gallicano K, Venance S, Kravcik S, Cameron DW. Effect of ketoconazole on ritonavir and saquinavir concentrations in plasma and cerebrospinal fluid from patients infected with human immunodeficiency virus. Clin Pharmacol Ther. 2000;68:637–46. doi: 10.1067/mcp.2000.112363. [DOI] [PubMed] [Google Scholar]
- 11.Zhao XJH, Yokoyama K, Chiba S, Wanwimolruk S, Ishizaki T. Identification of human cytochrome P450 isoforms involved in the 3-hydroxylation of quinine by humanlike microsomes and nine recombinant human cytochrome P450. J Pharmacol Exp Ther. 1996;279:1321–34. [PubMed] [Google Scholar]
- 12.Lefevre G, Carpenter P, Souppart C, Schmidli H, Martin JM, Lane A, Ward C, Amakye D. Interaction trial between artemether-lumefantrine (Riamet) and quinine in healthy subjects. J Clin Pharmacol. 2002;42:1147–58. doi: 10.1177/009127002401382632. [DOI] [PubMed] [Google Scholar]
- 13.Lefèvre G, Thomsen MS. Clinical pharmacokinetics of artemether and lumefantrine (Riamet®) Clin Drug Invest. 1999;18:467–80. [Google Scholar]
- 14.Hsu A, Granneman R, Bertz RJ. Ritonavir: clinical pharmacokinetics and interactions with other anti-HIV agents. Clin Pharmacokinet. 1998;35:275–91. doi: 10.2165/00003088-199835040-00002. [DOI] [PubMed] [Google Scholar]
- 15.Soyinka JO, Onyeji CO, Omoruyi SI. Simultaneous liquid chromatographic analysis of ritonavir, quinine and 3-hydroxyquinine in human plasma. J Chromatogr B. 2009;877:441–5. doi: 10.1016/j.jchromb.2008.12.045. [DOI] [PubMed] [Google Scholar]
- 16.Gibaldi M, Perrier D. Pharmacokinetics. 2nd edn. New York: Marcel Dekker; 1982. [Google Scholar]
- 17.Khaliq Y, Gallicano K, Tisdale C, Carignan G, Cooper C, McCarthy A. Pharmacokinetic interaction between mefloquine and ritonavir in healthy volunteers. Br J Clin Pharmacol. 2001;51:591–600. doi: 10.1046/j.1365-2125.2001.01393.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Babalola CP, Bolaji OO, Ogunbona FA. Pharmacokinetics of quinine in African patients with acute falciparum malaria. Pharm World Sci. 1998;20:118–22. doi: 10.1023/a:1008699022244. [DOI] [PubMed] [Google Scholar]
- 19.Mirghani RA, Ericson O, Cook J, Yu P, Gustafsson LL. Simultaneous determination of quinine and four metabolites in plasma and urine by high performance liquid chromatography. J Chromatogr Biomed Sci Appl. 2001;754:57–64. doi: 10.1016/s0378-4347(00)00577-6. [DOI] [PubMed] [Google Scholar]
- 20.Supanaranond W, Davies TME, Prukrittayakemee S, Silamut K. Disposition of oral quinine in acute falciparum malaria. Eur J Clin Pharmacol. 1991;40:49–52. doi: 10.1007/BF00315138. [DOI] [PubMed] [Google Scholar]
- 21.Salako LA, Sowunmi A. Disposition of quinine in plasma, red blood cells and saliva after oral and intravenous administration to healthy adult Africans. Eur J Clin Pharmacol. 1992;42:171–4. doi: 10.1007/BF00278479. [DOI] [PubMed] [Google Scholar]
- 22.Ouellet D, Hsu A, Qian J, Locke CS, Eason CJ, Granneman GR. Pharmacokinetics interaction between ritonavir and clarithromycin. Clin Pharmacol Ther. 1998;64:355–62. doi: 10.1016/S0009-9236(98)90065-0. [DOI] [PubMed] [Google Scholar]
- 23.Rodrigues AD, Roberts EM, Mulford DJ, Yao Y, Ouellet D. Oxidative metabolism of clarithromycin in the presence of human liver microsomes: major role for the cytochrome P450 3A subfamily. Drug Metab Dispos. 1997;25:623–30. [PubMed] [Google Scholar]
- 24.Cato A, Cavanaugh J, Shi H, Hsu A, Leonard J, Granneman R. The effect of multiple doses of ritonavir on the pharmacokinetic of rifabutin. Clin Pharmacol Ther. 1998;63:414–21. doi: 10.1016/S0009-9236(98)90036-4. [DOI] [PubMed] [Google Scholar]
- 25.Ho PC, Chalcroff SC, Coville PF. Grapefruit juice has no effect on quinine pharmacokinetics. Eur J Clin Pharmacol. 1999;55:393–8. doi: 10.1007/s002280050646. [DOI] [PubMed] [Google Scholar]
- 26.Wacher VJ, Silverman JA, Zhang Y, Benet LZ. Role of P-glycoprotein and cytochrome P450 3A in limiting oral absorption of peptides and peptidomimetics. J Pharm Sci. 1998;87:1322–30. doi: 10.1021/js980082d. [DOI] [PubMed] [Google Scholar]
- 27.Kim RB, Wandel C, Leake B. Interrelationship between substrates and inhibitors of human CYP 3A and P-glycoprotein. Pharm Res. 1999;16:408–14. doi: 10.1023/a:1018877803319. [DOI] [PubMed] [Google Scholar]
- 28.Michiharu K, Hitomi N, Hiroto F, Shuichi T. Effect of chronic administration on function of cytochrome P450 3A and P-glycoprotein in rats. Biol Pharm Bull. 2004;28:130–7. doi: 10.1248/bpb.28.130. [DOI] [PubMed] [Google Scholar]
- 29.Eric P, Mourad M, Hubert B. Increased uptake of quinine into the brain by inhibition of P-glycoprotein. Eur J Pharm Sci. 2007;32:123–7. doi: 10.1016/j.ejps.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 30.Paintaud G, Alvan G, Ericsson O. The reproducibility of quinine bioavailability. Br J Clin Pharmacol. 1993;35:305–7. doi: 10.1111/j.1365-2125.1993.tb05698.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Geilling EM, Kelsey FE. Metabolism of quinine. Naunyn-Schmiedeberg's Arch Pharmacol. 2004;205:167–9. doi: 10.1007/BF00244811. [DOI] [PubMed] [Google Scholar]
- 32.German P, Greenhouse B, Coates C, Dorsey G, Rosenthal P, Aweeka F. Drug interaction between Antimalarial Drugs and Efavirenz. Abstract No 577). Clinical Pharmacology in Adults and Children: Issues of Relevance for Developing Countries, California, San Franscisco, US. 2008.
- 33.Kappelhoff BS, Crommentuyn KML, de Maat MMR, Mulder JW, Huitema ADR, Beijnen JH. Practical guidelines to interpret plasma concentrations of antiretroviral drugs. Clin Pharmacokinet. 2004;43:845–53. doi: 10.2165/00003088-200443130-00002. [DOI] [PubMed] [Google Scholar]
- 34.Landmann R, Peytavin G, Leibowitch J. Ritonavir low dosages increases dramatically the saquinavir bioavailability: a PK study in healthy volunteers. abstract no. 42257). Conference Record of the 12th World AIDS Conference, Geneva, Switzerland, 1998, 824.
- 35.Mathias AA, West S, Hui J, Kearney BP. Dose–response of ritonavir on hepatic CYP3A and elvitegravir oral exposure. Clin Pharmacol Ther. 2009;85:64–70. doi: 10.1038/clpt.2008.168. [DOI] [PubMed] [Google Scholar]
- 36.Hsu A, Granneman GR, Witt G. Multiple-dose pharmacokinetics of ritonavir in human immunodeficiency virus-infected subjects. Antimicrobial Agents Chemother. 1997;41:898–905. doi: 10.1128/aac.41.5.898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumar GN, Rodrigues AD, Buko AM, Denissen JF. Cytochrome P450-mediated metabolism of the HIV-1 protease inhibitor ritonavir (ABT-538) in human liver microsomes. J Pharmacol Exp Ther. 1996;277:423–31. [PubMed] [Google Scholar]
- 38.Fiske WD, Benedek IH, Kornhauser DM. Pharmacokinetics of efavirenz (EFV) and ritonavir (RIT) after multiple oral doses in healthy volunteers. 12th World Congress on AIDS; Jun 28-July 3; Geneva, 1998.
- 39.Cato A, III, Cao G, Hsu A. Evaluation of the effect of fluconazole on the pharmacokinetics of ritonavir. Drug Metab Dispos. 1997;25:1104–6. [PubMed] [Google Scholar]
- 40.Guengerich FP, Miller GP, Hanna IH, Martin MV, Léger S, Black C, Chauret N, Silva JM, Trimble LA, Yergey JA, Nicoll-Griffith DA. Diversity in the oxidation of substrates by cytochrome P450 2D6: lack of an obligatory role of aspartate 301−substrate electrostatic bonding. Biochemistry. 2002;41:11025–34. doi: 10.1021/bi020341k. [DOI] [PubMed] [Google Scholar]
- 41.Hutzler JM, Walker GS, Wienkers LC. Inhibition of cytochrome P450 2D6: structure−activity studies using a series of quinidine and quinine analogues. Chem Res Toxicol. 2003;16:450–9. doi: 10.1021/tx025674x. [DOI] [PubMed] [Google Scholar]
- 42.Silamut K, White NJ, Looareesuwan S, Warell DA. Binding of quinine to plasma proteins in falciparum malaria. Am J Trop Med Hyg. 1985;24:681–6. doi: 10.4269/ajtmh.1985.34.681. [DOI] [PubMed] [Google Scholar]
- 43.Silamut K, Molunto P, Ho M, Davis TM, White NJ. Alpha 1-acid glycoprotein and plasma protein binding of quinine in acute falciparum malaria. Br J Clin Pharmacol. 1991;32:311–5. doi: 10.1111/j.1365-2125.1991.tb03904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]