

Abstract
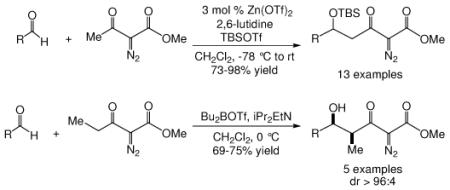
Methyl diazoacetoacetate undergoes zinc triflate catalyzed condensation with a broad selection of aldehydes to produce δ-siloxy-α-diazo-β-ketoalkanoates in good yield, and δ-hydroxy-α-diazo-β-ketoalkanoates are formed with high diastereoselectivity in reactions with α-diazo-β-ketopentanoate promoted by dibutylboron triflate.
Diazoacetoacetate derivatives are among the most commonly used substrates in catalytic diazo decomposition reactions. 1, 2 Their relative stability compared to diazoacetates or vinyldiazoacetates, and their enhanced reaction selectivities compared to diazoacetates, are among their advantages for organic synthesis.1 Traditionally these diazo compounds have been prepared by multi-step synthesis involving diazo transfer as the final step. 3, 4 Alternative methods that involve base-mediated condensation of diazo compounds with various electrophiles5,6 have not been generalizable because they require stoichiometric amounts of base and often occur under relatively harsh reaction conditions.
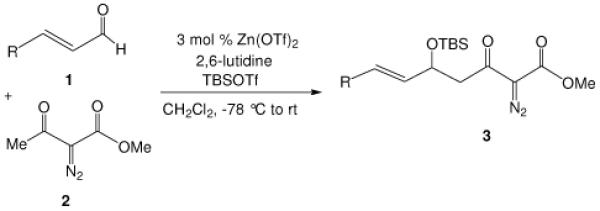
We have reported an efficient Mukaiyama-aldol reaction between methyl 2-diazo-3-(trialkylsilanoxy)-3-butenoate and both aromatic and aliphatic aldehydes catalyzed by scandium(III) triflate7 and, more recently, a process improvement in this reaction using zinc triflate has been described.8 Although methyl 2-diazo-3-(trialkylsilanoxy)-3-butenoate is produced directly from methyl diazoacetoacetate under a standard set of conditions (R3SiOTf, Et3N),9 we set out to examine if the Mukaiyama-aldol reactions between aldehydes and readily accessible diazoacetoacetates, rather than methyl 2-diazo-3-(trialkylsilanoxy)-3-butenoate, using catalytic amounts of Lewis acid under mild conditions could be achieved. Calter and Zhu previously described a stoichiometric version in which ethyl diazoacetoacetate underwent condensation with aldehydes in the presence of stoichiometric amounts of TiCl4 at −78 °C,10 and they also showed that this condensation could be performed in a two step process from ethyl diazoacetoacetate (i: TMSOTf, Et3N in CH2Cl2, −78 °C; ii: ArCHO, BF3•OEt2, −78 °C). We now report a more efficient and general Lewis acid catalyzed one-pot Mukaiyama-aldol reaction between diazoacetoacetates and aldehydes with low catalyst loading of inexpensive zinc triflate. Based on the same strategy but not indicated in prior reports, we also report a highly diastereoselective boron-mediated Mukaiyama-aldol reaction with the use of α-diazo-β-ketopentanoate.
We first screened reaction conditions for the one-pot Mukaiyama-aldol reaction. Reaction between methyl diazoacetoacetate (2) and trans-p-methoxycinnamaldehyde in the presence of a catalytic amount of zinc triflate, triethylamine (3.0 equiv) and TBS triflate (1.2 equiv) at −78 °C yielded both 1,2- and 1,4-addition products in a 5:1 molar ratio (Scheme 1). This result is identical to that obtained from the reaction of methyl 3-tert-butyldimethylsilanyloxy-2-diazobut-3-enoate and trans-4-methoxycinnamaldehyde in the presence of zinc triflate (3 mol %) at room temperature (Scheme 1).
Scheme 1.

Recognizing that the regioselectivity for addition might be dependent on the base employed, a weaker base, 2,6-lutidine,11 was used in place of triethylamine for the reaction with methyl diazoacetoacetate outlined in Scheme 1. Under these conditions the one-pot Mukaiyama-aldol reaction proceeded to produce the 1,2-addition product in 90% isolated yield to the virtual exclusion of the 1,4-addition product. Reactions performed at 0 °C or room temperature, however, produced up to 20% of the 1,4-addition product. Examination of the scope of the reaction with a variety of α,β-unsaturated aldehydes (1), the results for which are presented in Table 1, showed the production of 1,2-addition products in very good yields (Table 1). α,β-Unsaturated ketones do not react with methyl diazoacetoacetate under the same conditions and, with substrates such as 2-cyclohexenone, undergo preferential silyl enolization. That methyl diazoacetoacetate reacted with TBS triflate in the presence of 2,6-lutidine at low temperature to yield the vinyl ether, methyl 3-tert-butyldimethylsilanyloxy-2-diazobut-3-enoate, was confirmed by observing that transformation at low temperature by 1HNMR (see SI for details).
Table 1.
One-pot Mukaiyama-Aldol Reactions Between Methyl Diazoacetoacetate and α,β-Unsaturated Aldehydesa
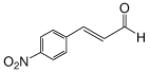
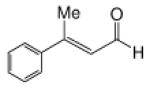
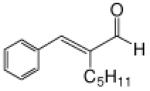
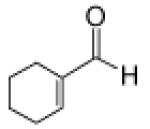
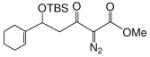
Methyl diazoacetoacetate 2 (1.0 mmol), 2,6-lutidine (3.3 equiv), and aldehyde (1.1 equiv) were added sequentially to zinc triflate (3 mol %) in 5 mL of dry DCM. After cooling to −78 °C, TBSOTf (1.25 equiv) was added dropwise. The mixture was stirred at −78 °C then warmed to room temperature over 16 h.
Yields were obtained by mass following flash column chromatography.
Trace amounts of 1,4-addition product were observed.
This methodology was extended to aromatic and aliphatic aldehydes. As shown in Table 2, reactions between methyl diazoacetoacetate and aromatic aldehydes afforded the addition products in excellent yields and suggest the overall generality of this protocol. Product yields were at least as high as those reported for scandium(III) triflate catalyzed reactions of some of these same aldehydes with methyl 2-diazo-3-(trialkylsilanoxy)-3-butenoate that were previously reported by our group.7 Using 1.0 mol % zinc triflate with entry 1i resulted in the production of 3i in nearly the same yield (91% versus 93%) as when 3 mol % zinc triflate was used. For the aliphatic aldehydes (1l and 1m) the standard conditions (zinc triflate and 2,6-lutidine or zinc triflate and triethylamine) led to less than full conversions over 16 h, so scandium(III) triflate (3.0 mol %) and triethylamine (1.5 equiv) were used and resulted in complete conversion, providing the desired products in good yields (Table 2). Even the enolizable aldehyde (1l) underwent this reaction protocol with excellent results.
Table 2.
One-pot Mukaiyama-Aldol Reactions Between Methyl Diazoacetoacetate and Aromatic/Aliphatic Aldehydesa
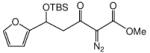
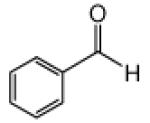
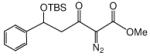
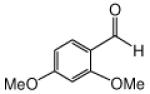
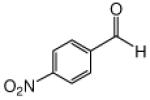
Reactions were performed as described in Table 1.
Reaction catalyzed by 1 mol % Zn(OTf)2 gave the product 3i in 91% yield.
Sc(OTf)3 (3 mol %) and triethylamine (1.5 equiv) were used.
Diastereocontrol in Mukaiyama-aldol reactions is a universal problem whose solutions have been the subject of numerous investigations.12 Using the Mukaiyama-aldol protocol with methyl 2-diazo-3-oxo-pentanoate (4) under the same conditions as those reported in Tables 1 and 2 we discovered that the condensation products were produced as a mixture of two diastereomers (anti/syn ~ 2:1)13 in good yields (Table 3). Similar results were obtained with the use of stoichiometric TiCl4 (80% yield, dr = 77:23). Although a number of approaches have been used to increase diastereoselectivity in aldol reactions,12,14 we selected di-n-butylboryl triflate and diisopropylethylamine because of their demonstrated ability to effect enolization at low temperature15 and because of the success of Evans and coworkers16 in using this combination to generate mainly Z-enol boronates that subsequently react with aldehyde to afford syn aldol products.17
Table 3.
One-pot Mukaiyama-Aldol Reaction between Methyl 2-Diazo-3-oxo-pentanoate and Aromatic/Vinyl Aldehydesa
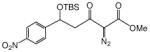
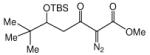
 | |||
|---|---|---|---|
| entry | 5 | anti:syn | yield, % |
| 1c | 72:28 | 95 | |
| 1h | 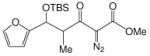 |
69:31 | 82 |
| 1i | 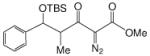 |
69:31 | 97 |
Reactions were performed as described in Table 1.
To enhance the diastereoselectivity, a boron-mediated Mukaiyama-aldol reaction was performed. As shown in Table 4, the reaction between methyl 2-diazo-3-oxopentanoate (4), (n-Bu)2BOTf (1.1 equiv), Hunig's base (2.0 equiv) and aromatic/vinyl/aliphatic aldehydes (1.0 equiv) at 0 °C afforded the syn-aldol product in good yields with very high diastereoselectivity (Table 4). For the investigated aldehydes, the syn/anti is above 96:4. For the reaction with benzaldehyde the same yield and diastereoselectivity was obtained when the reaction was performed at 0 °C as when the reaction was performed at −78 °C. With triethylamine used in place of Hunig's base with entries 1c, 1i, and 1m, 6c, 6i, and 6m were obtained in lower yields (6c 60%, 6i 59%, 6m 51%) but with nearly the same diastereoselectivities (6c, 99:1; 6i, 99:1; 6m, 93:7).
Table 4.
Boron-Mediated Aldol Reaction between Methyl 2-Diazo-3-oxo-pentanoate and Aldehydesa
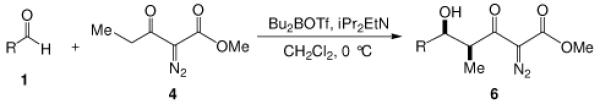
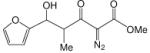
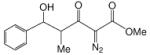
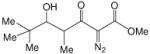
Methyl 2-diazo-3-oxo-pentanoate 4 (2.0 mmol), N,N-diisopropylethylamine (2.0 equiv) and di-n-butylboron triflate (1.1 equiv) were added sequentially to 5 mL of dry DCM. After stirring at 0 °C for 15 min, aldehyde (1.0 equiv) was added. The mixture was stirred at 0 °C for 3 h.
The diastereomer ratio was determined by the integral ratio by 1H NMR of the tertiary C-H bonded to the OH group and/or the integral ratio of 1H NMR signals for the methyl group.
Yields were obtained by mass following flash column chromatography.
Consistent with the prior report of Calter and Zhu,10b rhodium acetate catalyzed diazo decomposition produces the corresponding O-H insertion product in excellent yield. This reaction was explored with Mukaiyama-aldol adduct 6i (Scheme 2). The tetrahydrofuran derivative 7 was formed as ~ 5:3 diastereomeric mixture with the isomer having the methylcarboxylate group trans to the benzene ring predominating.
Scheme 2.

In summary, we have developed an efficient one-pot aldol reaction between diazoacetoacetate with a wide spectrum of aldehydes using catalytic amounts of zinc triflate, and we also report a highly diastereoselective di-n-butylboryl-mediated aldol reaction with the use of substituted diazoacetoacetate. This methodology can be used for the efficient construction of highly substituted diazo compounds. The synthetic utility of the Mukaiyama aldol adduct has been demonstrated with the diazo decomposition/O-H insertion reaction, and we anticipate additional uses of this methodology with the synthesis of more complex α-diazo-β-ketoalkanoates than previously possible. Efforts are continuing to develop the full synthetic potential of this methodology.
Supplementary Material
Acknowledgment
We gratefully acknowledge the financial support provided by the National Institutes of Health (GM46503).
Footnotes
Supporting Information Available: Experimental procedures and spectroscopic data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Doyle MP, McKervey MA, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides. John Wiley & Sons; New York: 1998. [Google Scholar]
- 2.(a) Doyle MP, Duffy R, Ratnikov M, Zhou L. Chem. Rev. doi: 10.1021/cr900239n. ASAP: DOI: 10.1021/cr900239n. [DOI] [PubMed] [Google Scholar]; (b) Merlic CA, Zechman AL. Synthesis. 2003:1137–1156. [Google Scholar]; (c) Davies HML, Beckwith REJ. Chem. Rev. 2003;103:2861–2904. doi: 10.1021/cr0200217. [DOI] [PubMed] [Google Scholar]; (d) Davies HML, Antoulinakis EG. Org. React. 2001;57:1–326. [Google Scholar]; (e) Hodgson DM, Pierard FYTM, Stupple PA. Chem. Soc. Rev. 2001;30:50–61. [Google Scholar]; (f) Doyle MP. In: Catalytic Asymmetric Synthesis. 2nd ed. Ojima I, editor. John Wiley & Sons; New York: 2000. [Google Scholar]; (g) Padwa A, Austin DJ. Angew. Chem. Int. Ed. Engl. 1994;33:1797–1815. [Google Scholar]; (h) Maas G. Top. Curr. Chem. 1987;137:77–253. [Google Scholar]; (i) Burke SD, Grieco PA. Org. React. 1979;26:361–475. [Google Scholar]; Reviews:
- 3.Regitz M, Maas G. Diazo Compounds; Properties and Synthesis. Academic Press; Orlando, FL: 1986. [Google Scholar]; Review:
- 4.(a) Padwa A, Austin DJ, Price AT, Semones MA, Doyle MP, Protopopova MN, Winchester WR, Tran A. J. Am. Chem. Soc. 1993;115:8669–8680. [Google Scholar]; (b) Doyle MP, Westrum LJ, Wolthuis WNE, See MM, Boone WP, Bagheri V, Pearson MM. J. Am. Chem. Soc. 1993;115:958–964. [Google Scholar]; (c) Clemens RJ, Hyatt JA. J. Org. Chem. 1985;50:2431–2435. [Google Scholar]; (d) Clark JS, Middleton MD. Org. Lett. 2002;4:765–768. doi: 10.1021/ol017240j. [DOI] [PubMed] [Google Scholar]
- 5.Zhao Y, Wang J. Synlett. 2005:2886–2892. [Google Scholar]; Review:
- 6.(a) Wenkert E, McPherson CA. J. Am. Chem. Soc. 1972;94:8084–8090. [Google Scholar]; (b) Moody CJ, Morfitt CN. Synthesis. 1998:1039–1042. [Google Scholar]; (c) Kanemasa S, Araki T, Kanai T, Wada E. Tetrahedron Lett. 1999;40:5059–5062. [Google Scholar]; (d) Jiang N, Wang J. Tetrahedron Lett. 2002;43:1285–1287. [Google Scholar]; (e) Sa MM, Silveira GP, Bortoluzzi AJ, Padwa A. Tetrahedron. 2003;59:5441–5447. [Google Scholar]; (f) Erhunmwunse MO, Steel PG. J. Org. Chem. 2008;73:8675–8677. doi: 10.1021/jo8017523. [DOI] [PubMed] [Google Scholar]; (g) Dong C, Mo F, Wang J. J. Org. Chem. 2008;73:1971–1974. doi: 10.1021/jo702275a. [DOI] [PubMed] [Google Scholar]
- 7.Doyle MP, Kundu K, Russell AE. Org. Lett. 2005;7:5171–5174. doi: 10.1021/ol052003s. [DOI] [PubMed] [Google Scholar]
- 8.Liu Y, Zhang Y, Jee N, Doyle MP. Org. Lett. 2008;10:1605–1608. doi: 10.1021/ol800298n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davies HML, Ahmed G, Churchill MR. J. Am. Chem. Soc. 1996;118:10774–10780. [Google Scholar]
- 10.(a) Calter MA, Sugathapala PM, Zhu C. Tetrahedron Lett. 1997;38:3837–3840. [Google Scholar]; (b) Calter MA, Zhu C. J. Org. Chem. 1999;64:1415–1419. [Google Scholar]
- 11.Evans DA, Downey CW, Hubbs JL. J. Am. Chem. Soc. 2003;125:8706–8707. doi: 10.1021/ja035509j. [DOI] [PubMed] [Google Scholar]; For prior use of 2,6-lutidine see:
- 12.(a) Ollevier T, Bouchard J-E, Desyroy V. J. Org. Chem. 2008;73:331–334. doi: 10.1021/jo702085p. [DOI] [PubMed] [Google Scholar]; (b) Jankowska J, Paradowska J, Rakiel B, Mlynarski J. J. Org. Chem. 2007;72:2228–2231. doi: 10.1021/jo0621470. [DOI] [PubMed] [Google Scholar]; (c) Boeckman RK, Jr., Pero JE, Boehmier DJ. J. Am. Chem. Soc. 2006;128:11032–11033. doi: 10.1021/ja063532+. [DOI] [PubMed] [Google Scholar]; (d) Kong K, Romo D. Org. Lett. 2006;8:2909–2912. doi: 10.1021/ol060534q. [DOI] [PubMed] [Google Scholar]; (e) Jankowska J, Mlynarski J. J. Org. Chem. 2006;71:1317–1321. doi: 10.1021/jo0514568. [DOI] [PubMed] [Google Scholar]; (f) Jung ME, van den Heuvel A. Org. Lett. 2003;5:4507–4707. doi: 10.1021/ol0358760. [DOI] [PubMed] [Google Scholar]; (g) Hassfield J, Christmann M, Kalesse M. Org. Lett. 2001;3:3561–3564. doi: 10.1021/ol016677o. [DOI] [PubMed] [Google Scholar]; Representative examples:
- 13.The diastereomeric ratio was determined from 1H NMR integration of the tertiary C-H bonded directly to the OTBS group. The coupling constant J of the tertiary C-H: anti (threo) J = 8.8-9.6 Hz, syn (erythro) J = 6.4-7.6 Hz.
- 14.(a) Ghosh AK, Shevlin M. In: Modern Aldol Reactions. Mahrwald R, editor. Vol. 1. Wiley-VCH; Weinheim: 2004. pp. 63–125. [Google Scholar]; (b) Markert M, Scheffler U, Mahrwald R. J. Am. Chem. Soc. 2009;131:16642–16643. doi: 10.1021/ja907054y. [DOI] [PubMed] [Google Scholar]; (c) Han SB, Hassan A, Krische MJ. Synthesis. 2008;17:2669–2679. doi: 10.1055/s-2008-1067220. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Nishiyama H, Shiomi T. Top. Curr. Chem. 2007;279:105–137. [Google Scholar]; (f) Enders D, Narine AA. J. Org. Chem. 2008;73:7857–7870. doi: 10.1021/jo801374j. [DOI] [PubMed] [Google Scholar]; (e) MacMillan DWC. Nature. 2008;455:304–308. doi: 10.1038/nature07367. [DOI] [PubMed] [Google Scholar]; (g) Bertelsen S, Jorgensen KA. Chem. Soc. Rev. 2009;38:2178–2189. doi: 10.1039/b903816g. [DOI] [PubMed] [Google Scholar]
- 15.Mukaiyama T, Banno K, Narasaka K. J. Am. Chem. Soc. 1974;96:7503–7509. [Google Scholar]
- 16.Evans DA, Nelson JV, Vogel E, Taber TR. J. Am. Chem. Soc. 1981;103:3099–3111. [Google Scholar]
- 17.(a) Brown HC, Dhar RK, Bakshi RK, Pandiarajan PK, Singaram B. J. Am. Chem. Soc. 1989;111:3441–3442. [Google Scholar]; (b) Abiko A. Acc. Chem. Res. 2004;37:387–395. doi: 10.1021/ar030249w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.