

Abstract

A number of alkynyl pinacolboronates bearing various functional groups were synthesized according to literature methods. These were then stereoselectively reduced to the cis-alkenyl pinacolboronates via hydroboration with dicyclohexylborane followed by chemoselective protodeboronation using acetic acid. Treatment with potassium hydrogen fluoride smoothly converted these to the corresponding potassium organotrifluoroborates.
The production of stereodefined alkenyl organometallics is a goal of ever-increasing value in synthetic organic chemistry owing to the ability of these organometallics to cross-couple with little if any erosion in stereochemical purity. Although numerous organometallic species take part in cross-coupling reactions, boron-containing reagents are unique for the variety of ways in which they can be synthesized, the relatively mild conditions under which they cross-couple, their atom economy, ease of handling, and low environmental impact.1 Alkenylboron reagents are most commonly synthesized by hydroboration of an appropriate terminal alkyne, and although this is a useful and reliable method, it results in stereoselective production of the trans-alkenylboron derivative. Access to cis-alkenylboron derivatives is well precedented in the literature,2 and yet the vast majority of examples to date possess a conspicuous dearth of functional groups.
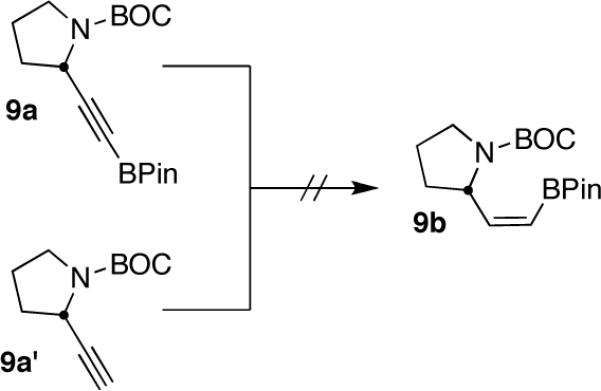
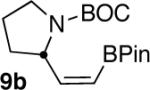
As part of a growing program to create highly functionalized organotrifluoroborates and apply them in the context of natural product synthesis, we sought to synthesize pinacolboronate 9b en route to the corresponding trifluoroborate (Scheme 1).
SCHEME 1.
Lindlar reduction,2a Peterson olefination,2b hydride-mediated inversion,2c and rhodium catalyzed hydroboration of the corresponding alkyne according to the method of Miyaura2d either failed to give the product in synthetically useful yields or gave highly sensitive dialkylboranes as products. We therefore sought a general method for the production of cis-alkenylboron reagents. Noting the large difference in reactivity between pinacolboronates and dialkylboranes, we reasoned that a differentially substituted 1,1-dibora reagent (Figure 1) might undergo selective protodeboronation in the presence of 1 equiv of an appropriate protonating agent.
FIGURE 1.
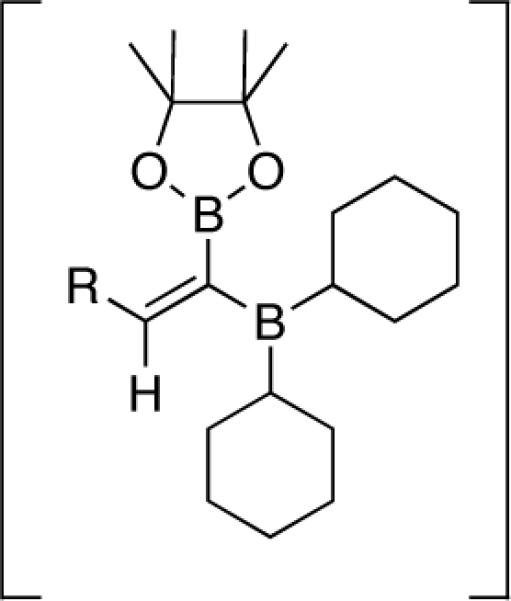
1,1-Dibora Intermediate.
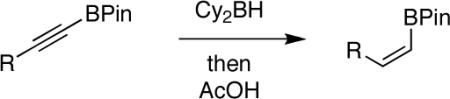
Gratifyingly, hydroboration of 9a with 1 equiv of dicyclohexylborane3 (prepared in advance and stored under an inert atmosphere as the solid) in THF at room temperature led to a homogeneous solution that was then treated with 1 equiv of acetic acid. These unoptimized conditions led to isolation of 9b in 42% yield. Shortly thereafter, a careful search of the literature revealed a similar reduction of 9-BBN-derived alkynylborinates, which also utilized dicyclohexylborane and acetic acid.2e Although the substrate scope in that report was limited to borinates containing simple alkyl groups, certain elements of that procedure proved beneficial in our efforts to expand the chemistry to more elaborate pinacolboronates. Specifically, the use of AcOH at 0 °C, followed by treatment with several equiv of ethanolamine to scavenge unwanted boron side products, greatly simplified the workup while giving the products in both greater purity and yield. The alkenylboronate could be isolated in nearly pure form by simple filtration over Celite. Using this protocol, our initial substrate 9b was isolated in 57% yield with complete stereoselectivity.
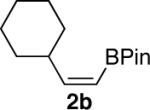
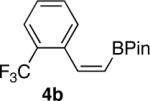
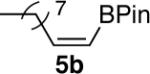
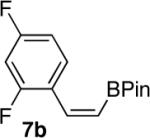
We next explored the scope of this hydroboration/protodeboronation method using a wide range of alkynyl pinacolboronates. These were synthesized in one step from the appropriate alkynes and isopropoxy-1,3,2-dioxabborolane by following the method of Brown.4 The results are summarized in Table 1. Trimethylsilyl, cyclohexyl, and phenyl-bearing substrates underwent reduction to give their respective cis-boronates in high yield (entries 1–3). It is noteworthy that the steric bulk of the trimethylsilyl group had no negative impact on the rate at which the solution became homogeneous, or on the yield of the reaction. Only slightly lower in yield were the reactions of substrates bearing trifluoromethylphenyl, n-alkyl, tert-butyldimethylsilyloxy, and difluorophenyl groups (entries 4–7). As stated above, proline derivative 9b was obtained in a synthetically useful yet somewhat attenuated yield. This is not entirely surprising given the acid-sensitive nature of the tert-butoxylcarbonyl group.
TABLE 1.
Hydroboration/Protodeboronation of Alkynyl Pinacolboronates Bearing Various Functional Groups Pinacolboronates Bearing Various Functional Groupsa
| entry | R | product | % isolated yield |
|---|---|---|---|
| 1 | 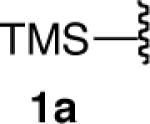 |
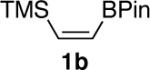 |
86 |
| 2 | 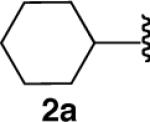 |
 |
84 |
| 3 | 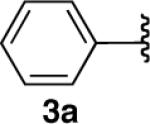 |
 |
83 |
| 4 | 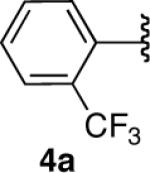 |
 |
78 |
| 5 |  |
 |
77 |
| 6 | 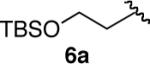 |
77 | |
| 7 | 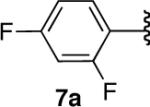 |
 |
71 |
| 8 | 66b | ||
| 9 | 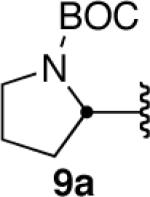 |
 |
57c |
| 10 | 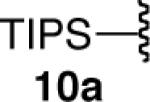 |
 |
93d |
| 11 | 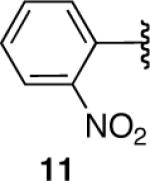 |
-e | |
| 12 | 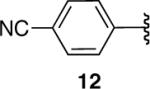 |
-e | |
| 13 | 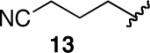 |
-e |
All reactions were carried out on 2 mmol scale in Et2O (4 mL) using 1:1 starting material/ borane. AcOH (2.2 equiv) was added at 0 °C, then ethanolamine at 0 °C and warmed to rt.
The product was isolated as a 90:10 mixture of cisltrans isomers.
The alkynyl pinacolboronate was not stable, necessitating that it be carried through without characterization.
The product was isolated with 10% of protodeboronation byproducts, which were inseparable by chromatrography or distillation.
Decomposition
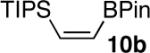
Certain functional substrates proved problematic with the reduction sequence employed. The first of these was ethyne 10a (entry 10). Given the success of the trimethylsilyl substrate 1a in the reaction, we presume that the problem was steric and not electronic. The strain induced by the increased steric bulk of the triisopropylsilyl group likely led to competitive protodeboronation of the otherwise stable pinacolboron moiety cis to it, in the process yielding byproducts that were inseparable by chromatographic methods or bulb-to-bulb distillation.
Nitro-substitution in substrate 11 was also poorly tolerated (entry 11). The Brown procedure4 failed in this case to provide the pinacolboronate without several accompanying impurities. Nevertheless, the semi-pure material was subjected to our optimized hydroboration/protodeboronation sequence. Unfortunately a complex mixture of products was obtained, most likely from competitive reduction of the nitro functional group. Likewise, the Brown method was unable to provide nitrile-containing substrate 12 in a highly pure form. Somewhat surprisingly, the hydroboration of crude 12 in Et2O failed to give a homogeneous solution. Even when 2 equiv of dicyclohexylborane were employed, compound 12 failed to hydroborate and only decomposition products were recovered upon treatment with AcOH followed by the usual workup (entry 12). 11B-NMR confirmed this lack of reactivity in Et2O. Unexpectedly, 11B-NMR experiments in THF indicated complete hydroboration. Addition of AcOH according to the established protocol nevertheless gave a complex mixture of products in THF as well. Identical results were observed with the alkynyl pinacolboronate (13) arising from hex-5-ynenitrile (entry 13).
Given the sensitive nature of alkynyl pinacolboronates to protodeboronation, we sought to advance our initial findings into an operationally simple, one-pot multi-step procedure for the in situ generation and use of the alkynyl pinacol boronates. Early attempts to carry out this procedure by reacting the lithium acetylide with isopropoxy-1,3,2-dioxaborolane according to the method of Brown,4 followed by anhydrous HCl quench and cannulation onto solid dicyclohexylborane failed to give successful hydroboration, even when a sacrificial equivalent of dicyclohexylborane was added to quench the in situ generated isopropanol. The discovery that pinacolborane itself could also serve as the requisite source of boron in the reaction was unexpected, but not unwelcome, and to the best of our knowledge unprecedented (Scheme 2). More surprising still was the discovery that the borohydride (14) resulting from reaction of pinacolborane with the lithium acetylide was stable in Et2O for prolonged periods, even at room temperature, as confirmed by 11B-NMR. HCl quench then produced only hydrogen gas as a byproduct, allowing clean hydroboration using dicyclohexylborane.
SCHEME 2.
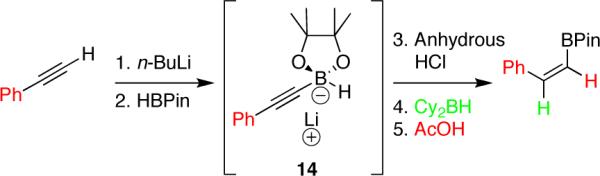
Unfortunately, it was subsequently discovered that the LiCl produced in the third step led to competitive protodeboronation of the pinacolboron moiety upon addition of acetic acid. These problems were quite easily solved via cannula filtration of the heterogeneous solution of alkynyl pinacolboronate before adding it to the hydroborating reagent. A single example, however, was explored as the method gave a moderate yield and proved no easier to implement than our optimized two-step procedure.
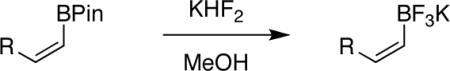
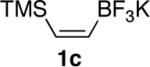
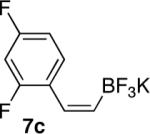
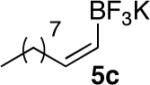
In general, optimized conditions for the hydroboration/protodeboronation sequence led to products of greater than 95% purity, frequently without the need for column chromatography. Even greater levels of purity could be obtained via conversion of the pinacol esters to the corresponding potassium organotrifluoroborates using saturated aqueous potassium hydrogen fluoride according to published methods.1 The tetracoordinate potassium organotrifluoroborates have several advantages over traditional tricoordinate organoboron reagents. In addition to being highly tractable crystalline solids, they are stable toward numerous oxidative and nucleophilic/basic reagents that show little or no tolerance of boronic acids and boronate esters.5 A representative sampling of cis-pinacolboronates were therefore selected for conversion to the corresponding trifluoroborates. The results are summarized in Table 2. All compounds subjected to treatment with potassium hydrogen fluoride gave good to excellent isolated yields. These were conveniently isolated by precipitation from acetone using Et2O or hexane at room temperature. During long-term storage on the bench, the alkenyl pinacolboronates were observed to change color and decompose with a concomitant increase in pinacol-containing impurities by 1H-NMR. The trifluoroborates underwent no such changes. Furthermore, as already stated, conversion to the potassium trifluoroborate was generally accompanied by an increase in purity according to 1H-NMR spectroscopy.
TABLE 2.
Conversion of cis-Alkenyl Pinacolboronates to the Corresponding Trifluoroboratesa
| entry | R | product | % isolated yield |
|---|---|---|---|
| 1 |  |
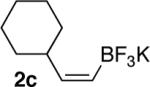 |
83 |
| 2 | 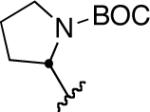 |
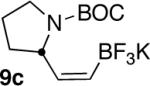 |
83 |
| 3 |  |
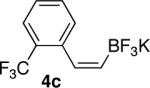 |
82 |
| 4 | 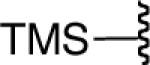 |
 |
78 |
| 5 | 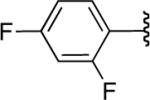 |
 |
78 |
| 6 |  |
 |
72 |
| 7 |  |
 |
62 |
All reactions were carried out using 2 mmol of b in MeOH (0.5 M) and 10 mmol of KHF2 (4.5 M).
In summary, we have developed a mild and general method for the synthesis of cis-alkenyl pinacolboronates and potassium organotrifluoroborates, beginning with the corresponding terminal alkynes. Substrates bearing branched alkyl, n-alkyl, silyl, silyloxy, aryl, and carbamate functional groups were all well tolerated. Nitriles and nitro groups, however, led to complex mixtures of products. Conversion to the corresponding potassium trifluoroborates was uneventful and high yielding in all cases studied. In addition, we have developed a facile, multi-step procedure that produces the cis-pinacolboronate directly from the alkyne without the- need to isolate the intermediate alkynyl pinacolboronate. The reaction proceeds via the intermediacy of a hitherto unreported borohydride. Studies toward the use of cis-alkenyl trifluoroborates for stereospecific coupling in natural product synthesis are ongoing in our laboratories.
Experimental Section
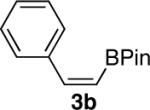
General Procedure for the Hydroboration/Protodeboronation of Alkynyl Pinacolboronates: Preparation of (Z)-4,4,5,5-Tetramethyl-2-styryl-1,3,2-dioxaborolane (3b)
4,4,5,5-Tetramethyl-2-(phenylethynyl)-1,3,2-dioxaborolane (456 mg, 2.0 mmol) and dicyclohexylborane (356 mg, 2.0 mmol) were weighed out together under inert atmosphere. Et2O (4 mL) was then added at rt, and the reaction was stirred for 30 min. The reaction was then cooled to 0 °C, and neat glacial acetic acid (0.126 mL, 2.2 mmol) was added. After stirring for 10 minutes, ethanolamine (0.266 mL, 4.4 mmol) was added and the reaction was brought to rt. Dilution with hexanes was followed by filtration over a silica plug to afford 3b in 83% yield as a clear viscous oil with greater than 95% purity (382 mg, 1.66 mmol). 1H-NMR (500 MHz, CDCl3) δ: 7.54 (d, J = 7.15 Hz, 2H), 7.33–7.18 (m, 4H), 5.59 (d, J = 14.86 Hz, 1H), 1.29 (s, 12H); 13C-NMR (125.8 MHz, CDCl3) δ: 148.1, 138.6, 128.6, 128.0, 127.9, 83.5, 24.8. The spectral data were in agreement with those reported in the literature.2e
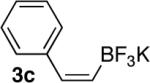
General Procedure for the Conversion of cis-Alkenyl Pinacolboronates to the Corresponding Potassium Trifluoroborates: Preparation of (Z)-2-(Phenyl)ethenyltrifluoroborate (3c)
After dissolving compound 3b (460 mg, 2.0 mmol) in MeOH (4 mL) at rt, a saturated aqueous solution of potassium hydrogen fluoride (~4.5 M, 2.22 mL) was added dropwise and the reaction was allowed to stir at rt for 2 hours. The solvent was then removed in vacuo to afford a mixture of solids that was dried under high vacuum (< 0.5 torr) overnight. Extraction of the solid mixture using acetone (3 × 10 mL), followed by Büchner funnel filtration (3×) afforded a solution of the product in acetone. This solution was then reduced in vacuo to afford a concentrated acetone solution of the product (~2 mL). After addition of Et2O (~30 mL) and Büchner funnel filtration of the precipitate, the product was obtained in 62% yield (260 mg, 1.24 mmol) as a white crystalline solid. mp: > 250 °C. 1H-NMR (500 MHz, Acetone-d6) δ: 7.63 (d, J = 7.3 Hz, 2H), 7.15 (d, J = 7.5 Hz, 2H), 7.03 (t, J = 7.2 Hz, 1H), 6.55 (bs, 1H), 5.64–5.72 (m, 1H); 13C-NMR (125.8 MHz, Acetone-d6) δ: 141.5, 135.5 (q, J = 5.8 Hz), 128.6 (q, J = 2.9 Hz), 127.2, 125.1; 19F-NMR (471 MHz, Acetone-d6) δ: −136.62; 11B-NMR (128.37 MHz, Acetone-d6) δ: 2.01; IR (KBr) = 2930, 1635, 1467 cm−1; HRMS (ESI) calcd. for C8H7BF3 171.0593, found 171.0588.
General Procedure for the one-pot conversion of alkynyl pinacolboronates to cis-alkenyl pinacolboronates: In situ generation of lithium 4,4,5,5-tetramethyl-2-(phenylethynyl)-1,3,2-dioxaborolan-2-ide (14)
Phenylacetylene (0.22 mL, 2 mmol) was dissolved in Et2O (2 mL) and cooled to −78 °C. n-BuLi (2 mmol) was then added dropwise, and the resulting lithium acetylide solution was stirred for 30 min at −78 °C. A solution of 4,4,5,5-tetramethyl-1,3,2-dioxaborolane in Et2O (2 mL) was then added, and after stirring at −78 °C for 10 min the solution of borohydride 14 was allowed to warm to a temperature at which it stirred freely. After 5 min of stirring, anhydrous HCl (4.0 M in dioxane, 2 mmol) was added dropwise, and the heterogeneous solution was allowed to warm to rt. Schlenk filtration over Celite was carried out directly onto solid dicyclohexylborane (356 mg, 2 mmol), and the filter was washed with Et2O (2 mL). The reaction was allowed to stir for an additional 30 min after reaching homogeneity, at which point it was cooled to 0 °C. AcOH and ethanolamine were added according to the standard procedure. The reaction was then filtered over a plug of silica to afford 3b in 52% yield as a clear viscous oil with greater than 95% purity (239 mg, 1.04 mmol). Spectral data were identical to previously isolated material.
Supplementary Material
Acknowledgment
This work was generously supported by the National Institutes of Health (GM035249). We gratefully acknowledge the research group of Professor Patrick Walsh (University of Pennsylvania) for a number of insights and Dr. Rakesh Kohli (University of Pennsylvania) for obtaining high resolution mass spectra of new compounds.
Footnotes
Supporting Information Available: Experimental procedures, spectral characterization, and copies of 1H-, 13C-, 11B-, and 19F-NMR spectra for all compounds prepared by the method described. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Molander GA, Felix LA. J. Org. Chem. 2005;70:3950–3956. doi: 10.1021/jo050286w. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 2.(a) Brown HC, Srebnik M, Bhat N, G. Tetrahedron Lett. 1988;29:2635–2638. [Google Scholar]; (b) Matteson DS, Majumdar D. Organometallics. 1983;2:230–236. [Google Scholar]; (c) Campbell JB, Jr., Molander GA. J. Organomet. Chem. 1978;156:71–79. and references therein. [Google Scholar]; (d) Miyaura N, Ohmura T, Yamamoto Y. J. Am. Chem. Soc. 2000;122:4990–4991. [Google Scholar]; (e) Soderquist JA, Rane AM, Matos K, Ramos J. Tetrahedron Lett. 1995;36:6847–6850. [Google Scholar]; (f) Srebnik M, Deloux L. J. Org. Chem. 1994;59:6871–6873. [Google Scholar]
- 3.Abiko A. Org. Synth. Col. Vol. 2004;10:273. [Google Scholar]
- 4.Brown HC, Bhat NG, Srebnik M. Tetrahedron Lett. 1988;29:2631–2634. [Google Scholar]
- 5.Molander GA, Ellis N. Acc. Chem. Res. 2007;40:275–286. doi: 10.1021/ar050199q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.